Label: CONTRAVE EXTENDED-RELEASE- naltrexone hydrochloride and bupropion hydrochloride tablet, extended release

- NDC Code(s): 51267-890-07, 51267-890-99
- Packager: Nalpropion Pharmaceuticals LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated November 10, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CONTRAVE® safely and effectively. See full prescribing information for CONTRAVE.
CONTRAVE (naltrexone hydrochloride and bupropion hydrochloride) extended-release tablets, for oral use
Initial U.S. Approval: 2014WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
See full prescribing information for complete boxed warning
- Increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants for major depressive disorder and other psychiatric disorders. (5.1)
- Monitor for worsening and emergence of suicidal thoughts and behaviors. (5.1)
- CONTRAVE is not approved for use in pediatric patients. (5.1)
INDICATIONS AND USAGE
CONTRAVE is a combination of naltrexone, an opioid antagonist, and bupropion, an aminoketone antidepressant, indicated in combination with a reduced-calorie diet and increased physical activity to reduce excess body weight and maintain weight reduction long term in adults with obesity or overweight in the presence of at least one weight-related comorbid condition. (1)
Limitations of Use:
DOSAGE AND ADMINISTRATION
CONTRAVE dose escalation schedule (2.1):
Morning Dose Evening Dose Week 1 1 tablet None Week 2 1 tablet 1 tablet Week 3 2 tablets 1 tablet Week 4 – Onward 2 tablets 2 tablets DOSAGE FORMS AND STRENGTHS
Extended-Release Tablets: 8 mg naltrexone hydrochloride (HCl) /90 mg bupropion HCl (3)
CONTRAINDICATIONS
- Uncontrolled hypertension (4)
- Seizure disorders, anorexia nervosa or bulimia, or undergoing abrupt discontinuation of alcohol, benzodiazepines, barbiturates, and antiepileptic drugs (4)
- Use of other bupropion-containing products (4)
- Chronic opioid use (4)
- During or within 14 days of taking monoamine oxidase inhibitors (MAOI) (4)
- Known allergy to any of the ingredients in CONTRAVE (4)
WARNINGS AND PRECAUTIONS
- Suicidal Behavior and Ideation: Monitor for depression or suicidal thoughts. Discontinue CONTRAVE if symptoms develop. (5.1)
- Neuropsychiatric Adverse Events During Smoking Cessation: Postmarketing reports of serious or clinically significant neuropsychiatric adverse events have included changes in mood (including depression and mania), psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, agitation, anxiety, and panic, as well as suicidal ideation, suicide attempt, and completed suicide. Observe patients taking CONTRAVE for the occurrence of such symptoms and instruct them to discontinue CONTRAVE and contact a healthcare provider if they experience such adverse events. (5.2)
- Risk of seizure may be minimized by adhering to the recommended dosing schedule and avoiding coadministration with high-fat meal. (5.3)
- Increase in Blood Pressure and Heart Rate: Monitor blood pressure and heart rate in all patients, especially those with cardiac or cerebrovascular disease. (5.5)
- Hepatotoxicity: Cases of hepatitis and clinically significant liver dysfunction observed with naltrexone exposure. (5.7)
- Angle-closure glaucoma: Angle-closure glaucoma has occurred in patients with untreated anatomically narrow angles treated with antidepressants. (5.9)
- Use of Antidiabetic Medications: Weight loss may cause hypoglycemia. Monitor blood glucose. (5.10)
ADVERSE REACTIONS
- Most common adverse reactions (greater than or equal to 5%): nausea, constipation, headache, vomiting, dizziness, insomnia, dry mouth and diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Currax Pharmaceuticals LLC at 1-800-793-2145 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- MAOIs: Increased risk of hypertensive reactions can occur when used concomitantly. (7.1)
- Drugs Metabolized by CYP2D6: Bupropion inhibits CYP2D6 and can increase concentrations of antidepressants, (e.g., selective serotonin reuptake inhibitors and many tricyclics), antipsychotics (e.g., haloperidol, risperidone and thioridazine), beta-blockers (e.g., metoprolol) and Type 1C antiarrhythmics (e.g., propafenone and flecainide). Consider dose reduction when using with CONTRAVE. (7.3)
- Digoxin: May decrease plasma digoxin levels. Monitor digoxin levels. (7.3)
- Concomitant Treatment with CYP2B6 Inhibitors (e.g., ticlopidine or clopidogrel) can increase bupropion exposure. Do not exceed one tablet twice daily when taken with CYP2B6 inhibitors. (2.5, 7.4)
- CYP2B6 Inducers (e.g., ritonavir, lopinavir, efavirenz, carbamazepine, phenobarbital, and phenytoin) may reduce efficacy by reducing bupropion exposure, avoid concomitant use. (7.4)
- Drugs that Lower Seizure Threshold: Dose CONTRAVE with caution. (5.3, 7.5)
- Dopaminergic Drugs (levodopa and amantadine): CNS toxicity can occur when used concomitantly with CONTRAVE. (7.6)
- Drug-Laboratory Test Interactions: CONTRAVE can cause false- positive urine test results for amphetamines. (7.8)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dose Adjustment in Patients with Renal Impairment
2.3 Dose Adjustment in Patients with Hepatic Impairment
2.4 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Antidepressant
2.5 Concomitant Use with CYP2B6 Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Behavior and Ideation
5.2 Neuropsychiatric Adverse Events and Suicide Risk in Smoking Cessation Treatment
5.3 Seizures
5.4 Patients Receiving Opioid Analgesics
5.5 Increase in Blood Pressure and Heart Rate
5.6 Allergic Reactions
5.7 Hepatotoxicity
5.8 Activation of Mania
5.9 Angle-Closure Glaucoma
5.10 Potential Risk of Hypoglycemia in Patients with Type 2 Diabetes Mellitus on Antidiabetic Therapy
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Monoamine Oxidase Inhibitors (MAOI)
7.2 Opioid Analgesics
7.3 Potential for CONTRAVE to Affect Other Drugs
7.4 Potential for Other Drugs to Affect CONTRAVE
7.5 Drugs That Lower Seizure Threshold
7.6 Dopaminergic Drugs (Levodopa and Amantadine)
7.7 Use with Alcohol
7.8 Drug-Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
SUICIDALITY AND ANTIDEPRESSANT DRUGS
CONTRAVE® is not approved for use in the treatment of major depressive disorder or other psychiatric disorders. CONTRAVE contains bupropion, the same active ingredient as some antidepressant medications (including, but not limited to, WELLBUTRIN, WELLBUTRIN SR, WELLBUTRIN XL, and APLENZIN). Antidepressants increased the risk of suicidal thoughts and behavior in children, adolescents, and young adults in short-term trials. These trials did not show an increase in the risk of suicidal thoughts and behavior with antidepressant use in subjects over age 24; there was a reduction in risk with antidepressant use in subjects aged 65 and older. In patients of all ages who are started on CONTRAVE, monitor closely for worsening, and for the emergence of suicidal thoughts and behaviors. Advise families and caregivers of the need for close observation and communication with the prescriber. CONTRAVE is not approved for use in pediatric patients [see Warnings and Precautions (5.1), Use in Specific Populations (8.4)].
-
1 INDICATIONS AND USAGE
CONTRAVE is indicated in combination with a reduced-calorie diet and increased physical activity to reduce excess body weight and maintain weight reduction long term in adults with obesity or overweight in the presence of at least one weight-related comorbid condition.
Limitations of Use:
- The effect of CONTRAVE on cardiovascular morbidity and mortality has not been established.
- CONTRAVE contains naltrexone and bupropion. Coadministration with other naltrexone-containing products is not recommended. Coadministration with other bupropion-containing products is contraindicated.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Initiate and escalate the dosage of CONTRAVE according to the schedule in Table 1:
Table 1. Dosage Initiation and Escalation Schedule for CONTRAVE Morning Dose Evening Dose Week 1 1 tablet None Week 2 1 tablet 1 tablet Week 3 2 tablets 1 tablet Week 4 – Onward 2 tablets 2 tablets A total daily dosage of 32 mg of naltrexone hydrochloride (HCl) and 360 mg bupropion HCl (two CONTRAVE 8 mg/90 mg tablets twice daily) is reached at the start of Week 4.
CONTRAVE should be taken by mouth in the morning and in the evening. The tablets should not be cut, chewed, or crushed. Total daily doses greater than 32 mg/360 mg per day (two tablets twice daily) are not recommended. In clinical trials, CONTRAVE was administered with meals. However, CONTRAVE should not be taken with a high-fat meal because of a resulting significant increase in bupropion and naltrexone systemic exposure [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
Patients may develop elevated blood pressure or heart rate during CONTRAVE treatment; the risk may be greater during the initial three months of therapy [see Warnings and Precautions (5.6)]. Because patients with hypertension may be at increased risk for developing blood pressure elevations, such patients should be monitored for this potential effect when initiating treatment with CONTRAVE.
Response to therapy should be evaluated after 12 weeks at the maintenance dosage. If a patient has not lost at least 5% of baseline body weight, discontinue CONTRAVE, as it is unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment.
2.2 Dose Adjustment in Patients with Renal Impairment
In patients with moderate or severe renal impairment, the maximum recommended daily dose for CONTRAVE is two tablets (one tablet each morning and evening). CONTRAVE is not recommended for use in patients with end-stage renal disease [see Use in Specific Population (8.6) and Clinical Pharmacology (12.3)].
2.3 Dose Adjustment in Patients with Hepatic Impairment
In patients with moderate hepatic impairment, the maximum recommended daily maintenance dose of CONTRAVE is two tablets (one tablet each morning and evening). CONTRAVE is not recommended for use in patients with severe hepatic impairment [see Use in Specific Population (8.7) and Clinical Pharmacology (12.3)].
2.4 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Antidepressant
At least 14 days should elapse between discontinuation of an MAOI intended to treat depression and initiation of therapy with CONTRAVE. Conversely, at least 14 days should be allowed after stopping CONTRAVE before starting an MAOI antidepressant [see Contraindications (4) and Drug Interactions (7.1)].
2.5 Concomitant Use with CYP2B6 Inhibitors
During concomitant use with CYP2B6 inhibitors (e.g., ticlopidine or clopidogrel), the maximum recommended daily dose of CONTRAVE is two tablets (one tablet each morning and evening) [see Drug Interactions (7.4) and Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
CONTRAVE is contraindicated in
- Uncontrolled hypertension [see Warnings and Precautions (5.5)]
- Seizure disorder or a history of seizures [see Warnings and Precautions (5.3)]
- Use of other bupropion-containing products (including, but not limited to, WELLBUTRIN, WELLBUTRIN SR, WELLBUTRIN XL, APLENZIN and ZYBAN)
- Bulimia or anorexia nervosa, which increase the risk for seizure [see Warnings and Precautions (5.3)]
- Chronic opioid or opiate agonist (e.g., methadone) or partial agonists (e.g., buprenorphine) use, or acute opiate withdrawal [see Warnings and Precautions (5.4) and Drug Interactions (7.2)]
- Patients undergoing an abrupt discontinuation of alcohol, benzodiazepines, barbiturates, and antiepileptic drugs [see Warnings and Precautions (5.3) and Drug Interactions (7.7)]
- Concomitant administration of monoamine oxidase inhibitors (MAOI). At least 14 days should elapse between discontinuation of MAOI and initiation of treatment with CONTRAVE. There is an increased risk of hypertensive reactions when CONTRAVE is used concomitantly with MAOIs. Starting CONTRAVE in a patient treated with reversible MAOIs such as linezolid or intravenous methylene blue is also contraindicated [see Dosage and Administration (2.4), Drug Interactions (7.1)]
- Known allergy to bupropion, naltrexone or any other component of CONTRAVE. Anaphylactoid/anaphylactic reactions and Stevens-Johnson syndrome have been reported with bupropion [see Warnings and Precautions (5.6)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Behavior and Ideation
CONTRAVE contains bupropion, a dopamine and norepinephrine re-uptake inhibitor that is similar to some drugs used for the treatment of depression; therefore, the following precautions pertaining to these products should be considered when treating patients with CONTRAVE.
Patients with major depressive disorder, both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment.
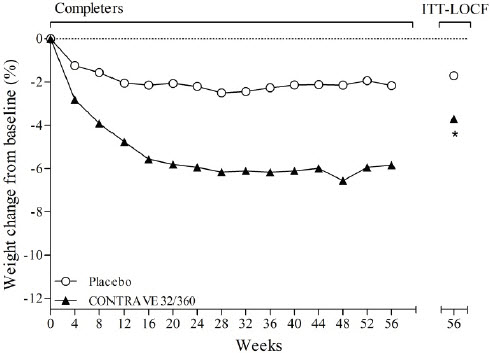
In placebo-controlled clinical trials with CONTRAVE for the treatment of obesity in adult patients, no suicides or suicide attempts were reported in studies up to 56 weeks duration with CONTRAVE (equivalent to bupropion doses of 360 mg/day). In these same studies, suicidal ideation was reported by 3 (0.20%) of 1,515 patients treated with placebo compared with 1 (0.03%) of 3,239 treated with CONTRAVE.
Pooled analyses of short-term placebo-controlled trials of antidepressant drugs (selective serotonin re-uptake inhibitors [SSRIs] and others) show that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18 to 24) with major depressive disorder (MDD) and other psychiatric disorders. Short-term clinical trials did not show an increase in the risk of suicidality with antidepressants compared with placebo in adults beyond age 24; there was a reduction with antidepressants compared with placebo in adults aged 65 and older. CONTRAVE is not approved for use in pediatric patients.
The pooled analyses of placebo-controlled trials of antidepressant drugs in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term trials of nine antidepressant drugs in over 4,400 patients. The pooled analyses of placebo-controlled trials in adults with MDD or other psychiatric disorders included a total of 295 short-term trials (median duration of two months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk differences (drug vs placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1,000 patients treated) are provided in Table 2.
Table 2. Risk Differences in the Number of Suicidality Cases by Age Group in the Pooled Placebo-Controlled Trials of Antidepressants in Pediatric and Adult Subjects Age Range Drug-Placebo Difference in Number of Cases of Suicidality per 1,000 Patients Treated Increases Compared to Placebo <18 14 additional cases 18 to 24 5 additional cases Decreases Compared to Placebo 25 to 64 1 fewer case ≥65 6 fewer cases No suicides occurred in any of the antidepressant pediatric trials. There were suicides in the adult antidepressant trials, but the number was not sufficient to reach any conclusion about drug effect on suicide.
It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled trials in adults with depression that the use of antidepressants can delay the recurrence of depression.
All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases. This warning applies to CONTRAVE because one of its components, bupropion, is a member of an antidepressant class.
The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient's presenting symptoms.
Families and caregivers of patients being treated with antidepressants for major depressive disorder or other indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of anxiety, agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to healthcare providers. Such monitoring should include daily observation by families and caregivers. Prescriptions for CONTRAVE should be written for the smallest quantity of tablets consistent with good patient management, in order to reduce the risk of overdose.
5.2 Neuropsychiatric Adverse Events and Suicide Risk in Smoking Cessation Treatment
CONTRAVE is not approved for smoking cessation treatment, but serious neuropsychiatric adverse events have been reported in patients taking bupropion for smoking cessation. These postmarketing reports have included changes in mood (including depression and mania), psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, agitation, anxiety, and panic, as well as suicidal ideation, suicide attempt, and completed suicide [see Warnings and Precautions (5.1)]. Some patients who stopped smoking may have been experiencing symptoms of nicotine withdrawal, including depressed mood. Depression, rarely including suicidal ideation, has been reported in smokers undergoing a smoking cessation attempt without medication. However, some of these adverse events occurred in patients taking bupropion who continued to smoke.
Neuropsychiatric adverse events occurred in patients without and with pre-existing psychiatric disease; some patients experienced worsening of their psychiatric illnesses. Observe patients for the occurrence of neuropsychiatric adverse events. Advise patients and caregivers that the patient should stop taking CONTRAVE and contact a healthcare provider immediately if agitation, depressed mood, or changes in behavior or thinking that are not typical for the patient are observed, or if the patient develops suicidal ideation or suicidal behavior. In many postmarketing cases, resolution of symptoms after discontinuation of bupropion was reported. However, the symptoms persisted in some cases, therefore, ongoing monitoring and supportive care should be provided until symptoms resolve.
Depression, suicide, attempted suicide and suicidal ideation have been reported in the postmarketing experience with naltrexone used in the treatment of opioid dependence. No causal relationship has been demonstrated.
5.3 Seizures
Bupropion, a component of CONTRAVE, can cause seizures. The risk of seizure is dose- related. The incidence of seizure in patients receiving CONTRAVE in clinical trials was approximately 0.1% vs 0% on placebo. CONTRAVE should be discontinued and not restarted in patients who experience a seizure while being treated with CONTRAVE.
The risk of seizures is also related to patient factors, clinical situations, and concomitant medications that lower the seizure threshold. Consider these risks before initiating treatment with CONTRAVE. CONTRAVE is contraindicated in patients with a seizure disorder, current or prior diagnosis of anorexia nervosa or bulimia, or undergoing abrupt discontinuation of alcohol, benzodiazepines, barbiturates, and antiepileptic drugs. Caution should be used when prescribing CONTRAVE to patients with predisposing factors that may increase the risk of seizure including:
- history of head trauma or prior seizure, severe stroke, arteriovenous malformation, central nervous system tumor or infection, or metabolic disorders (e.g., hypoglycemia, hyponatremia, severe hepatic impairment, and hypoxia)
- excessive use of alcohol or sedatives, addiction to cocaine or stimulants, or withdrawal from sedatives
- patients with diabetes treated with insulin and/or oral diabetic medications (sulfonylureas and meglitinides) that may cause hypoglycemia
- concomitant administration of medications that may lower the seizure threshold, including other bupropion products, antipsychotics, tricyclic antidepressants, theophylline, systemic steroids
Recommendations for Reducing the Risk of Seizure: Clinical experience with bupropion suggests that the risk of seizure may be minimized by adhering to the recommended dosing recommendations [see Dosage and Administration (2)], in particular:
- the total daily dose of CONTRAVE does not exceed 360 mg of the bupropion component (i.e., four tablets per day)
- the daily dose is administered in divided doses (twice daily)
- the dose is escalated gradually
- no more than two tablets are taken at one time
- coadministration of CONTRAVE with high-fat meals is avoided [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)]
- if a dose is missed, a patient should wait until the next scheduled dose to resume the regular dosing schedule
5.4 Patients Receiving Opioid Analgesics
Vulnerability to Opioid Overdose: CONTRAVE should not be administered to patients receiving chronic opioids, due to the naltrexone component, which is an opioid receptor antagonist [see Contraindications (4)]. If chronic opiate therapy is required, CONTRAVE treatment should be stopped. In patients requiring intermittent opiate treatment, CONTRAVE therapy should be temporarily discontinued and lower doses of opioids may be needed. Patients should be alerted that they may be more sensitive to opioids, even at lower doses, after CONTRAVE treatment is discontinued.
An attempt by a patient to overcome any naltrexone opioid blockade by administering large amounts of exogenous opioids is especially dangerous and may lead to a fatal overdose or life-threatening opioid intoxication (e.g., respiratory arrest, circulatory collapse). Patients should be told of the serious consequences of trying to overcome the opioid blockade.
Precipitated Opioid Withdrawal: The symptoms of spontaneous opioid withdrawal, which are associated with the discontinuation of opioid in a dependent individual, are uncomfortable, but they are not generally believed to be severe or necessitate hospitalization. However, when withdrawal is precipitated abruptly, the resulting withdrawal syndrome can be severe enough to require hospitalization. To prevent occurrence of either precipitated withdrawal in patients dependent on opioids or exacerbation of a pre-existing subclinical withdrawal symptoms, opioid-dependent patients, including those being treated for alcohol dependence, should be opioid-free (including tramadol) before starting CONTRAVE treatment. An opioid-free interval of a minimum of 7 to 10 days is recommended for patients previously dependent on short- acting opioids, and those patients transitioning from buprenorphine or methadone may need as long as two weeks. Patients should be made aware of the risks associated with precipitated withdrawal and encouraged to give an accurate account of last opioid use.
5.5 Increase in Blood Pressure and Heart Rate
CONTRAVE can cause an increase in systolic and/or diastolic blood pressure as well as an increase in resting heart rate. In clinical practice with other bupropion-containing products, hypertension, in some cases severe and requiring acute treatment, has been reported. The clinical significance of the increases in blood pressure and heart rate observed with CONTRAVE treatment is unclear, especially for patients with cardiac and cerebrovascular disease, since patients with a history of myocardial infarction or stroke in the previous 6 months, life-threatening arrhythmias, or congestive heart failure were excluded from CONTRAVE clinical trials. Blood pressure and pulse should be measured prior to starting therapy with CONTRAVE and should be monitored at regular intervals consistent with usual clinical practice, particularly among patients with controlled hypertension prior to treatment [see Dosage and Administration (2.1)]. CONTRAVE should not be given to patients with uncontrolled hypertension [see Contraindications (4)].
Among patients treated with CONTRAVE in placebo-controlled clinical trials, mean systolic and diastolic blood pressure was approximately 1 mmHg higher than baseline at Weeks 4 and 8, similar to baseline at Week 12, and approximately 1 mmHg below baseline between Weeks 24 and 56. In contrast, among patients treated with placebo, mean blood pressure was approximately 2 to 3 mmHg below baseline throughout the same time points, yielding statistically significant differences between the groups at every assessment during this period. The largest mean differences between the groups were observed during the first 12 weeks (treatment difference +1.8 to +2.4 mmHg systolic, all p<0.001; +1.7 to +2.1 mmHg diastolic, all p<0.001).
For heart rate, at both Weeks 4 and 8, mean heart rate was statistically significantly higher (2.1 bpm) in the CONTRAVE group compared with the placebo group; at Week 52, the difference between groups was +1.7 bpm (p<0.001).
In an ambulatory blood pressure monitoring substudy of 182 patients, the mean change from baseline in systolic blood pressure after 52 weeks of treatment was -0.2 mmHg for the CONTRAVE group and -2.8 mmHg for the placebo group (treatment difference, +2.6 mmHg, p=0.08); the mean change in diastolic blood pressure was +0.8 mmHg for the CONTRAVE group and -2.1 mmHg for the placebo group (treatment difference, +2.9 mmHg, p=0.004).
A greater percentage of subjects had adverse reactions related to blood pressure or heart rate in the CONTRAVE group compared to the placebo group (6.3% vs 4.2%, respectively), primarily attributable to adverse reactions of Hypertension/Blood Pressure Increased (5.9% vs 4.0%, respectively). These events were observed in both patients with and without evidence of preexisting hypertension. In a trial that enrolled individuals with diabetes, 12.0% of patients in the CONTRAVE group and 6.5% in the placebo group had a blood pressure-related adverse reaction.
5.6 Allergic Reactions
Anaphylactoid/anaphylactic reactions characterized by symptoms such as pruritus, urticaria, angioedema, and dyspnea requiring medical treatment have been reported in clinical trials with bupropion. In addition, there have been rare spontaneous postmarketing reports of erythema multiforme, Stevens-Johnson syndrome, and anaphylactic shock associated with bupropion. Instruct patients to discontinue CONTRAVE and consult a healthcare provider if they develop an allergic or anaphylactoid/anaphylactic reaction (e.g., skin rash, pruritus, hives, chest pain, edema, or shortness of breath) during treatment.
Arthralgia, myalgia, fever with rash, and other symptoms suggestive of delayed hypersensitivity have been reported in association with bupropion. These symptoms may resemble serum sickness.
5.7 Hepatotoxicity
Cases of hepatitis and clinically significant liver dysfunction were observed in association with naltrexone exposure during naltrexone clinical trials and in postmarketing reports for patients using naltrexone. Transient, asymptomatic hepatic transaminase elevations were also observed. When patients presented with elevated transaminases, there were often other potential causative or contributory etiologies identified, including pre-existing alcoholic liver disease, hepatitis B and/or C infection, and concomitant usage of other potentially hepatotoxic drugs. Although clinically significant liver dysfunction is not typically recognized as a manifestation of opioid withdrawal, opioid withdrawal that is precipitated abruptly may lead to systemic sequelae, including acute liver injury.
Patients should be warned of the risk of hepatic injury and advised to seek medical attention if they experience symptoms of acute hepatitis. Use of CONTRAVE should be discontinued in the event of symptoms and/or signs of acute hepatitis.
In CONTRAVE clinical trials, there were no cases of elevated transaminases greater than three times the upper limit of normal (ULN) in conjunction with an increase in bilirubin greater than two times ULN.
5.8 Activation of Mania
Bupropion, a component of CONTRAVE, is a drug used for the treatment of depression. Antidepressant treatment can precipitate a manic, mixed, or hypomanic episode. The risk appears to be increased in patients with bipolar disorder or who have risk factors for bipolar disorder. Prior to initiating CONTRAVE, screen patients for a history of bipolar disorder and the presence of risk factors for bipolar disorder (e.g., family history of bipolar disorder, suicide, or depression). CONTRAVE is not approved for use in treating bipolar depression. No activation of mania or hypomania was reported in the clinical trials evaluating effects of CONTRAVE in obese patients; however, patients receiving antidepressant medications and patients with a history of bipolar disorder or recent hospitalization because of psychiatric illness were excluded from CONTRAVE clinical trials.
5.9 Angle-Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs including bupropion, a component of CONTRAVE, may trigger an angle-closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy.
5.10 Potential Risk of Hypoglycemia in Patients with Type 2 Diabetes Mellitus on Antidiabetic Therapy
Weight loss may increase the risk of hypoglycemia in patients with type 2 diabetes mellitus treated with insulin and/or insulin secretagogues (e.g., sulfonylureas). Measurement of blood glucose levels prior to starting CONTRAVE and during CONTRAVE treatment is recommended in patients with type 2 diabetes. Decreases in medication doses for antidiabetic medications which are non-glucose-dependent should be considered to mitigate the risk of hypoglycemia. If a patient develops hypoglycemia after starting CONTRAVE, appropriate changes should be made to the antidiabetic drug regimen.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Suicidal Behavior and Ideation [see Boxed Warning, Warnings and Precautions (5.1)]
- Neuropsychiatric Adverse Events [see Warnings and Precautions (5.2)]
- Seizures [see Contraindications (4), Warnings and Precautions (5.3)]
- Increase in Blood Pressure and Heart Rate [see Warnings and Precautions (5.5)]
- Allergic Reactions [see Warnings and Precautions (5.6)]
- Angle-Closure Glaucoma [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
CONTRAVE was evaluated for safety in five double-blind placebo-controlled trials in 4,754 overweight or obese patients (3,239 patients treated with CONTRAVE and 1,515 patients treated with placebo) for a treatment period up to 56 weeks. The majority of patients were treated with CONTRAVE 32 mg/360 mg total daily dose. In addition, some patients were treated with other combination daily doses including naltrexone up to 50 mg and bupropion up to 400 mg. All subjects received study drug in addition to diet and exercise counseling. One trial (N=793) evaluated patients participating in an intensive behavioral modification program and another trial (N= 505) evaluated patients with type 2 diabetes. In these randomized, placebo-controlled trials, 2,545 patients received CONTRAVE 32 mg/360 mg for a mean treatment duration of 36 weeks (median, 56 weeks). Baseline patient characteristics included a mean age of 46 years, 82% women, 78% white, 25% with hypertension, 13% with type 2 diabetes, 56% with dyslipidemia, 25% with BMI greater than 40 kg/m2, and less than 2% with coronary artery disease. Dosing was initiated and increased weekly to reach the maintenance dose within 4 weeks.
In CONTRAVE clinical trials, 24% of subjects receiving CONTRAVE and 12% of subjects receiving placebo discontinued treatment because of an adverse event. The most frequent adverse reactions leading to discontinuation with CONTRAVE were nausea (6.3%), headache (1.7%) and vomiting (1.1%).
Common Adverse Reactions
Adverse reactions that were reported by greater than or equal to 2% of patients and were more frequently reported by patients treated with CONTRAVE compared to placebo, are summarized in Table 3.
Table 3. Adverse Reactions Reported by Obese or Overweight Patients With an Incidence (%) of at Least 2% Among Patients Treated with CONTRAVE and More Common than with Placebo Adverse Reaction CONTRAVE
32 mg/360 mg
N=2545
%Placebo
N=1515
%Nausea 32.5 6.7 Constipation 19.2 7.2 Headache 17.6 10.4 Vomiting 10.7 2.9 Dizziness 9.9 3.4 Insomnia 9.2 5.9 Dry mouth 8.1 2.3 Diarrhea 7.1 5.2 Anxiety 4.2 2.8 Hot flush 4.2 1.2 Fatigue 4.0 3.4 Tremor 4.0 0.7 Upper abdominal pain 3.5 1.3 Viral gastroenteritis 3.5 2.6 Influenza 3.4 3.2 Tinnitus 3.3 0.6 Urinary tract infection 3.3 2.8 Hypertension 3.2 2.2 Abdominal pain 2.8 1.4 Hyperhidrosis 2.6 0.6 Irritability 2.6 1.8 Blood pressure increased 2.4 1.5 Dysgeusia 2.4 0.7 Rash 2.4 2.0 Muscle strain 2.2 1.7 Palpitations 2.1 0.9 Other Adverse Reactions
The following additional adverse reactions were reported in less than 2% of patients treated with CONTRAVE but with an incidence at least twice that of placebo:
Cardiac Disorders: tachycardia, myocardial infarction
Ear and Labyrinth Disorders: vertigo, motion sickness
Gastrointestinal Disorders: lower abdominal pain, eructation, lip swelling, hematochezia, hernia
General Disorders and Administration Site Conditions: feeling jittery, feeling abnormal, asthenia, thirst, feeling hot
Hepatobiliary Disorders: cholecystitis
Infections and Infestations: pneumonia, staphylococcal infection, kidney infection
Investigations: increased blood creatinine, increased hepatic enzymes, decreased hematocrit
Metabolism and Nutrition Disorders: dehydration
Musculoskeletal and Connective Tissue Disorders: intervertebral disc protrusion, jaw pain
Nervous System Disorders: disturbance in attention, lethargy, intention tremor, balance
disorder, memory impairment, amnesia, mental impairment, presyncope
Psychiatric Disorders: abnormal dreams, nervousness, dissociation (feeling spacey), tension, agitation, mood swings
Renal and Urinary Disorders: micturition urgency
Reproductive System and Breast Disorders: vaginal hemorrhage, irregular menstruation, erectile dysfunction, vulvovaginal dryness
Skin and Subcutaneous Tissue Disorders: alopeciaPsychiatric and Sleep Disorders
In the one-year controlled trials of CONTRAVE, the proportion of patients reporting one or more adverse reactions related to psychiatric and sleep disorders was higher in the CONTRAVE 32/360 mg group than the placebo group (22.2% and 15.5%, respectively). These events were further categorized into sleep disorders (13.8% CONTRAVE, 8.4% placebo), depression (6.3% CONTRAVE, 5.9% placebo), and anxiety (6.1% CONTRAVE, 4.4% placebo). Patients who were 65 years or older experienced more psychiatric and sleep disorder adverse reactions in the CONTRAVE group (28.6%) compared to placebo (6.3%), although the sample size in this subgroup was small (56 CONTRAVE, 32 placebo); the majority of these events were insomnia (10.7% CONTRAVE, 3.1% placebo) and depression (7.1% CONTRAVE, 3.1% placebo).
Neurocognitive Adverse Reactions
Adverse reactions involving attention, dizziness, and syncope occurred more often in individuals randomized to CONTRAVE 32/360 mg group compared to placebo (15.0% and 5.5%, respectively). The most common cognitive-related adverse reactions were attention disorders (2.5% CONTRAVE, 0.6% placebo). Adverse reactions involving dizziness and syncope were more common in patients treated with CONTRAVE (10.6%) than in placebo-treated patients (3.6%); dizziness accounted for almost all of these reported events (10.4% CONTRAVE, 3.4% placebo). Dizziness was the primary reason for discontinuation for 0.9% and 0.3% of patients in the CONTRAVE and placebo groups, respectively.
Increases in Serum Creatinine
In the one-year controlled trials of CONTRAVE, larger mean increases in serum creatinine from baseline to trial endpoint were observed in the CONTRAVE group compared with the placebo group (0.07 mg/dL and 0.01 mg/dL, respectively) as well as from baseline to the maximum value during follow-up (0.15 mg/dL and 0.07 mg/dL, respectively). Increases in serum creatinine that exceeded the upper limit of normal and were also greater than or equal to 50% higher than baseline occurred in 0.6% of subjects receiving CONTRAVE compared to 0.1% receiving placebo. The observed increase in serum creatinine may be the result of OCT2 inhibition [see Clinical Pharmacology (12.3)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of CONTRAVE, naltrexone, or bupropion. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Loss of consciousness, malaise
- Brugada pattern/syndrome
- Drug reaction with eosinophilia and systemic symptoms (DRESS)
- Acute generalized exanthematous pustulosis (AGEP)
- Aseptic meningitis
-
7 DRUG INTERACTIONS
7.1 Monoamine Oxidase Inhibitors (MAOI)
Concomitant use of MAOIs and bupropion is contraindicated. Bupropion inhibits the re-uptake of dopamine and norepinephrine and can increase the risk for hypertensive reactions when used concomitantly with drugs that also inhibit the re-uptake of dopamine or norepinephrine, including MAOIs. Studies in animals demonstrate that the acute toxicity of bupropion is enhanced by the MAOI phenelzine. At least 14 days should elapse between discontinuation of an MAOI and initiation of treatment with CONTRAVE. Conversely, at least 14 days should be allowed after stopping CONTRAVE before starting an MAOI [see Contraindications (4)].
7.2 Opioid Analgesics
Patients taking CONTRAVE may not fully benefit from treatment with opioid-containing medicines, such as cough and cold remedies, antidiarrheal preparations, and opioid analgesics. In patients requiring intermittent opiate treatment, CONTRAVE therapy should be temporarily discontinued, and opiate dose should not be increased above the standard dose. CONTRAVE may be used with caution after chronic opioid use has been stopped for 7 to 10 days in order to prevent precipitation of withdrawal [see Contraindications (4) and Warnings and Precautions (5.4)].
During CONTRAVE clinical studies, the use of concomitant opioid or opioid-like medications, including analgesics or antitussives, were excluded.
7.3 Potential for CONTRAVE to Affect Other Drugs
Metabolized by CYP2D6
In a clinical study, CONTRAVE (32 mg naltrexone/360 mg bupropion) daily was coadministered with a 50 mg dose of metoprolol (a CYP2D6 substrate). CONTRAVE increased metoprolol AUC and Cmax by approximately 4- and 2-fold, respectively, relative to metoprolol alone. Similar clinical drug interactions resulting in increased pharmacokinetic exposure of CYP2D6 substrates have also been observed with bupropion as a single agent with desipramine or venlafaxine.
Coadministration of CONTRAVE with drugs that are metabolized by CYP2D6 isozyme including certain antidepressants (SSRIs and many tricyclics), antipsychotics (e.g., haloperidol, risperidone and thioridazine), beta-blockers (e.g., metoprolol) and Type 1C antiarrhythmics (e.g., propafenone and flecainide), should be approached with caution and should be initiated at the lower end of the dose range of the concomitant medication. If CONTRAVE is added to the treatment regimen of a patient already receiving a drug metabolized by CYP2D6, the need to decrease the dose of the original medication should be considered, particularly for those concomitant medications with a narrow therapeutic index [see Clinical Pharmacology (12.3)].
Digoxin
Coadministration of CONTRAVE with digoxin may decrease plasma digoxin levels. Monitor plasma digoxin levels in patients treated concomitantly with CONTRAVE and digoxin [see Clinical Pharmacology (12.3)].
7.4 Potential for Other Drugs to Affect CONTRAVE
Bupropion is primarily metabolized to hydroxybupropion by CYP2B6. Therefore, the potential exists for drug interactions between CONTRAVE and drugs that are inhibitors or inducers of CYP2B6.
Inhibitors of CYP2B6: Ticlopidine and Clopidogrel: Concomitant treatment with these drugs can increase bupropion exposure but decrease hydroxybupropion exposure. During concomitant use with CYP2B6 inhibitors (e.g., ticlopidine or clopidogrel), the CONTRAVE daily dose should not exceed two tablets (one tablet each morning and evening) [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
Inducers of CYP2B6: Ritonavir, Lopinavir, and Efavirenz: Concomitant treatment with these drugs can decrease bupropion and hydroxybupropion exposure and may reduce efficacy. Avoiding concomitant use with ritonavir, lopinavir, or efavirenz is recommended [see Clinical Pharmacology (12.3)].
7.5 Drugs That Lower Seizure Threshold
Use extreme caution when coadministering CONTRAVE with other drugs that lower seizure threshold (e.g., antipsychotics, antidepressants, theophylline, or systemic corticosteroids). Use low initial doses and increase the dose gradually. Concomitant use of other bupropion-containing products is contraindicated [see Contraindications (4) and Warnings and Precautions (5.3)].
7.6 Dopaminergic Drugs (Levodopa and Amantadine)
Bupropion, levodopa, and amantadine have dopamine agonist effects. CNS toxicity has been reported when bupropion was coadministered with levodopa or amantadine. Adverse reactions have included restlessness, agitation, tremor, ataxia, gait disturbance, vertigo, and dizziness. It is presumed that the toxicity results from cumulative dopamine agonist effects. Use caution and monitor for such adverse reactions when administering CONTRAVE concomitantly with these drugs.
7.7 Use with Alcohol
In postmarketing experience, there have been rare reports of adverse neuropsychiatric events or reduced alcohol tolerance in patients who were drinking alcohol during treatment with bupropion. The consumption of alcohol during treatment with CONTRAVE should be minimized or avoided.
7.8 Drug-Laboratory Test Interactions
False-positive urine immunoassay screening tests for amphetamines have been reported in patients taking bupropion. This is due to lack of specificity of some screening tests. False-positive test results may result even following discontinuation of bupropion therapy. Confirmatory tests, such as gas chromatography/mass spectrometry, will distinguish bupropion from amphetamines.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Weight loss offers no benefit to a pregnant patient and may cause fetal harm. When a pregnancy is recognized, advise the pregnant patient of the risk to the fetus, and discontinue CONTRAVE (see Clinical Considerations). Available pharmacovigilance data and data from clinical trials with the individual components of CONTRAVE use in pregnant patients have not demonstrated a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes.
Bupropion
Data from epidemiological studies of pregnant patients exposed to bupropion in the first trimester have not identified an increased risk of congenital malformations overall (see Data). When bupropion was administered to pregnant rats during organogenesis, there was no evidence of fetal malformations at doses up to approximately 20 times the maximum recommended human dose (MRHD) of 360 mg/day. When given to pregnant rabbits during organogenesis, non-dose–related increases in incidence of fetal malformations, and skeletal variations were observed at doses approximately twice the MRHD and greater. Decreased fetal weights were seen at doses 5 times the MRHD and greater (see Data).
Naltrexone
Limited case report data of pregnant patients exposed to naltrexone in the first trimester have not identified an increased risk of congenital malformations overall. Daily oral administration of naltrexone during the period of organogenesis has been shown to increase the incidence of early fetal loss in rats and rabbits at doses ≥ 15 times and ≥ 60 times the MRHD of 32 mg/day, respectively. There was no evidence of fetal malformations in rats and rabbits at doses up to approximately 100 and 200 times the MHRD, respectively. (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Human Data
In clinical studies, 21 (0.7%) of 3,024 women became pregnant while taking CONTRAVE: 11 carried to term and gave birth to a healthy infant, three had elective abortions, four had spontaneous abortions, and the outcome of three pregnancies were unknown.
Data from the international bupropion Pregnancy Registry (675 first trimester exposures) and a retrospective cohort study using the United Healthcare database (1,213 first trimester exposures) did not show an increased risk for malformations overall.
No increased risk for cardiovascular malformations overall has been observed after bupropion exposure during the first trimester. The prospectively observed rate of cardiovascular malformations in pregnancies with exposure to bupropion in the first trimester from the international Pregnancy Registry was 1.3% (9 cardiovascular malformations out of 675 first-trimester maternal bupropion exposures), which is similar to the background rate of cardiovascular malformations (approximately 1%). Data from the United Healthcare database and a case-control study (6,853 infants with cardiovascular malformations and 5,763 with non-cardiovascular malformations) from the National Birth Defects Prevention Study (NBDPS) did not show an increased risk for cardiovascular malformations overall after bupropion exposure during the first trimester.
Study findings on bupropion exposure during the first trimester and risk for left ventricular outflow tract obstruction (LVOTO) are inconsistent and do not allow conclusions regarding a possible association. The United Healthcare database lacked sufficient power to evaluate this association; the NBDPS found increased risk for LVOTO (n = 10; adjusted odds ratio [OR] = 2.6; 95% CI: 1.2, 5.7), and the Slone Epidemiology case control study did not find increased risk for LVOTO.
Study findings on bupropion exposure during the first trimester and risk for ventricular septal defect (VSD) are inconsistent and do not allow conclusions regarding a possible association. The Slone Epidemiology Study found an increased risk for VSD following first trimester maternal bupropion exposure (n = 17; adjusted OR = 2.5; 95% CI: 1.3, 5.0) but did not find increased risk for any other cardiovascular malformations studied (including LVOTO as above). The NBDPS and United Healthcare database study did not find an association between first trimester maternal bupropion exposure and VSD.
For the findings of LVOTO and VSD, the studies were limited by the small number of exposed cases, inconsistent findings among studies, and the potential for chance findings from multiple comparisons in case control studies.
Animal Data
Reproduction and developmental studies have not been conducted for the combined products naltrexone and bupropion in CONTRAVE. Separate studies with bupropion and naltrexone have been conducted in pregnant rats and rabbits. Safety margins were estimated using body surface area exposure (mg/m2) based on a body weight of 100 kg.
Daily oral administration of naltrexone has been shown to increase the incidence of early fetal loss when given to rats at doses ≥30 mg/kg/day (15 times the MHRD on a mg/m2 basis) and to rabbits at oral doses ≥60 mg/kg/day (60 times the MHRD on a mg/m2 basis).
Daily oral administration of naltrexone to rats and rabbits during the period of organogenesis did not induce malformations at doses up to 200 mg/kg/day (approximately 100 and 200 times the MHRD, respectively, on a mg/m2 basis).
Rats do not form appreciable quantities of the major human metabolite, 6-beta-naltrexol; therefore, the potential reproductive toxicity of the metabolite in rats is not known. In studies conducted in pregnant rats and rabbits, bupropion was administered orally during the period of organogenesis at doses of up to 450 and 150 mg/kg/day, respectively (approximately 20 and 14 times the MRHD, respectively, on a mg/m2 basis). There was no evidence of fetal malformations in rats. When given to pregnant rabbits during organogenesis, non-dose-related increases in incidence of fetal malformations and skeletal variations were observed at the lowest dose tested (25 mg/kg/day, approximately 2 times the MRHD on a mg/m2 basis) and greater. Decreased fetal weights were observed at doses of 50 mg/kg/day (approximately 5 times the MRHD on a mg/m2 basis) and greater. No maternal toxicity was evident at doses of 50 mg/kg/day or less.
In a pre- and postnatal development study, bupropion administered orally to pregnant rats at doses of up to 150 mg/kg/day (approximately 7 times the MRHD on a mg/m2 basis) from embryonic implantation through lactation had no effect on pup growth or development.
8.2 Lactation
Risk Summary
Data from published literature report the presence of bupropion and its metabolites in human milk. Limited data from postmarketing reports with bupropion use during lactation have not identified a clear association of adverse effects on a breastfed infant (see Data). Naltrexone and its major metabolite, 6β-naltrexol, are present in human milk. There are no data on bupropion, naltrexone, or their metabolites on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CONTRAVE and any potential adverse effects on the breastfed infant from CONTRAVE or from the mother's underlying condition.
Data
In a lactation study of ten women, levels of orally dosed bupropion and its active metabolites were measured in expressed milk. The average daily infant exposure (assuming 150 mL/kg daily consumption) to bupropion and its active metabolites was 2% of the maternal weight-adjusted dose. Postmarketing reports have described seizures in breastfed infants. The relationship of bupropion exposure and these seizures is unclear.
8.4 Pediatric Use
The safety and effectiveness of CONTRAVE in pediatric patients below the age of 18 have not been established and the use of CONTRAVE is not recommended in pediatric patients.
8.5 Geriatric Use
Of the 3,239 subjects who participated in clinical trials with CONTRAVE, 62 (2%) were 65 years and older and none were 75 years and older. Clinical studies of CONTRAVE did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Older individuals may be more sensitive to the central nervous system adverse effects of CONTRAVE. Naltrexone and bupropion are known to be substantially excreted by the kidney, and the risk of adverse reactions to CONTRAVE may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. CONTRAVE should be used with caution in patients over 65 years of age.
8.6 Renal Impairment
In a pharmacokinetic study conducted for CONTRAVE in subjects with renal impairment (mild, moderate and severe), exposure to naltrexone metabolite, 6-beta naltrexol, and bupropion metabolites, threohydrobupropion, and erythrohydrobupropion was increased. Therefore, the maximum recommended daily maintenance dose for CONTRAVE is two tablets (one tablet each morning and evening) in patients with moderate or severe renal impairment. CONTRAVE is not recommended for use in patients with end-stage renal disease [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
In a pharmacokinetic study conducted for CONTRAVE in subjects with hepatic impairment (mild, moderate, and severe), exposure to naltrexone, bupropion, and their metabolites were increased. Therefore, the maximum recommended daily maintenance dose of CONTRAVE is two tablets (one tablet each morning and evening) in patients with moderate hepatic impairment. CONTRAVE is not recommended for use in patients with severe hepatic impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
-
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
Humans
CONTRAVE has not been systematically studied in humans for its potential for abuse, tolerance, or physical dependence. However, in outpatient clinical studies of up to 56 weeks in duration, there was no evidence of euphoric drug intoxication, physical dependence, diversion, or abuse. There was no evidence of an abstinence syndrome following abrupt or tapered drug discontinuation after 56 weeks of double-blind, placebo-controlled, randomized treatment.
Naltrexone is a pure opioid antagonist. It does not lead to physical or psychological dependence. Tolerance to the opioid antagonistic effect is not known to occur.
Controlled clinical trials of bupropion (immediate-release formulation) conducted in normal volunteers, in subjects with a history of multiple drug abuse, and in depressed subjects showed some increase in motor activity and agitation/excitement. In a population of individuals experienced with drugs of abuse, a single dose of 400 mg of bupropion produced mild amphetamine-like activity as compared with placebo on the Morphine-Benzedrine Subscale of the Addiction Research Center Inventories (ARCI) and a score intermediate between placebo and amphetamine on the Liking Scale of the ARCI. These scales measure general feelings of euphoria and drug desirability.
Findings in clinical trials, however, are not known to reliably predict the abuse potential of drugs. Nonetheless, evidence from single-dose studies does suggest that the recommended daily dosage of bupropion when administered in divided doses is not likely to be significantly reinforcing to amphetamine or CNS stimulant abusers.
The inhalation of crushed tablets or injection of dissolved bupropion has been reported. Seizures and/or cases of death have been reported when bupropion has been administered intranasally or by parenteral injection. CONTRAVE (naltrexone HCl and bupropion HCl) extended-release tablets are intended for oral use only.
Animals
Studies in rodents and primates have shown that bupropion exhibits some pharmacologic actions common to psychostimulants. In rodents, it has been shown to increase locomotor activity, elicit a mild stereotyped behavioral response, and increased rates of responding in several schedule-controlled behavior paradigms. In primate models assessing the positive reinforcing effects of psychoactive drugs, bupropion was self-administered intravenously. In rats, bupropion produced amphetamine-like and cocaine-like discriminative stimulus effects in drug discrimination paradigms used to characterize the subjective effects of psychoactive drugs.
-
10 OVERDOSAGE
Human Experience
Overdoses of up to 30 grams or more of bupropion (equivalent of up to 83 times the recommended daily dose of CONTRAVE 32 mg/360 mg) have been reported. Seizure was reported in approximately one third of all cases. Other serious reactions reported with overdoses of bupropion alone included hallucinations, loss of consciousness, mental status changes, sinus tachycardia, ECG changes such as conduction disturbances (including QRS prolongation) or arrhythmias, clonus, myoclonus, and hyperreflexia. Fever, muscle rigidity, rhabdomyolysis, hypotension, stupor, coma, and respiratory failure have been reported mainly when bupropion was part of multiple drug overdoses.
Although most patients recovered without sequelae, deaths associated with overdoses of bupropion alone have been reported in patients ingesting large doses of the drug. Multiple uncontrolled seizures, bradycardia, cardiac failure, and cardiac arrest prior to death were reported in these patients.
Animal Experience
In the mouse, rat, and guinea pig, the oral LD50s for naltrexone were 1,100 to 1,550 mg/kg; 1,450 mg/kg; and 1,490 mg/kg; respectively. High doses of naltrexone (generally greater than or equal to 1,000 mg/kg) produced salivation, depression/reduced activity, tremors, and convulsions. Mortality in animals due to high-dose naltrexone administration usually was due to clonic-tonic convulsions and/or respiratory failure.
Overdosage Management
If over-exposure occurs, call your poison control center at 1-800-222-1222. There are no known antidotes for CONTRAVE. In case of an overdose, provide supportive care, including close medical supervision and monitoring. Consider the possibility of multiple drug overdose. Ensure an adequate airway, oxygenation, and ventilation. Monitor cardiac rhythm and vital signs. Induction of emesis is not recommended.
-
11 DESCRIPTION
CONTRAVE extended-release tablets contain naltrexone hydrochloride (HCl) and bupropion HCl.
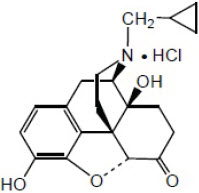
Naltrexone HCl, USP, an opioid antagonist, is a synthetic congener of oxymorphone with no opioid agonist properties. Naltrexone differs in structure from oxymorphone in that the methyl group on the nitrogen atom is replaced by a cyclopropylmethyl group. Naltrexone HCl is also related to the potent opioid antagonist, naloxone, or n-allylnoroxymorphone.
Naltrexone HCl has the chemical name of morphinan-6-one, 17-(cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxy-, hydrochloride, (5α)-. The empirical formula is C20H23NO4∙HCl and the molecular weight is 377.86. The structural formula is:
Naltrexone HCl is a white to yellowish, crystalline compound. It is soluble in water to the extent of about 100 mg/mL.
Bupropion HCl is an antidepressant of the aminoketone class. Bupropion HCl closely resembles the structure of diethylpropion. It is designated as (±)-1-(3 chlorophenyl)-2-[(1,1-dimethylethyl)amino]-1-propranone hydrochloride. It is related to phenylethylamines. The empirical formula is C13H18ClNO∙HCl and the molecular weight is 276.2. The structural formula is:
Bupropion HCl powder is white, crystalline, and highly soluble in water.
CONTRAVE is available for oral administration as a round, bi-convex, film-coated, extended-release tablet. Each tablet has a trilayer core composed of two drug layers, containing the drug and excipients, separated by a more rapidly dissolving inert layer. Each tablet contains 8 mg of naltrexone HCl and 90 mg of bupropion HCl. Tablets are blue and are debossed with NB-890 on one side. Each tablet contains the following inactive ingredients: microcrystalline cellulose, hydroxypropyl cellulose, lactose anhydrous, L-cysteine hydrochloride, crospovidone, magnesium stearate, hypromellose, edetate disodium, lactose monohydrate, colloidal silicon dioxide, Opadry II Blue and FD&C Blue #2 aluminum lake.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CONTRAVE has two components: naltrexone, an opioid antagonist, and bupropion, a relatively weak inhibitor of the neuronal reuptake of dopamine and norepinephrine. Nonclinical studies suggest that naltrexone and bupropion have effects on two separate areas of the brain involved in the regulation of food intake: the hypothalamus (appetite regulatory center) and the mesolimbic dopamine circuit (reward system). The exact neurochemical effects of CONTRAVE leading to weight loss are not fully understood.
12.2 Pharmacodynamics
Combined, bupropion and naltrexone increased the firing rate of hypothalamic pro-opiomelanocortin (POMC) neurons in vitro, which are associated with regulation of appetite. The combination of bupropion and naltrexone also reduced food intake when injected directly into the ventral tegmental area of the mesolimbic circuit in mice, an area associated with regulation of reward pathways.
12.3 Pharmacokinetics
Absorption
Food Effect on Absorption
When CONTRAVE was administered with a high-fat meal, the AUC and Cmax for naltrexone increased 2.1-fold and 3.7-fold, respectively, and the AUC and Cmax for bupropion increased 1.4-fold and 1.8-fold, respectively. At steady state, the food effect increased AUC and Cmax for naltrexone by 1.7-fold and 1.9-fold, respectively, and increased AUC and Cmax for bupropion by 1.1-fold and 1.3-fold, respectively. Thus, CONTRAVE should not be taken with high-fat meals because of the resulting significant increases in bupropion and naltrexone systemic exposure.
Distribution
Metabolism and Excretion
Naltrexone
The major metabolite of naltrexone is 6-beta-naltrexol. The activity of naltrexone is believed to be the result of both the parent and the 6-beta-naltrexol metabolite. Though less potent, 6-beta-naltrexol is eliminated more slowly and thus circulates at much higher concentrations than naltrexone. Naltrexone and 6-beta-naltrexol are not metabolized by cytochrome P450 enzymes and in vitro studies indicate that there is no potential for inhibition or induction of important isozymes.
Naltrexone and its metabolites are excreted primarily by the kidney (53% to 79% of the dose). Urinary excretion of unchanged naltrexone accounts for less than 2% of an oral dose. Urinary excretion of unchanged and conjugated 6-beta-naltrexol accounts for 43% of an oral dose. The renal clearance for naltrexone ranges from 30 to 127 mL/min, suggesting that renal elimination is primarily by glomerular filtration. The renal clearance for 6-beta-naltrexol ranges from 230 to 369 mL/min suggesting an additional renal tubular secretory mechanism. Fecal excretion is a minor elimination pathway.
Following single oral administration of CONTRAVE tablets to healthy subjects, mean elimination half-life (T1/2) was approximately 5 hours for naltrexone. Following twice daily administration of CONTRAVE, naltrexone did not accumulate and its kinetics appeared linear. However, in comparison to naltrexone, 6-beta-naltrexol accumulates to a larger extent (accumulation ratio ~3).
Bupropion
Bupropion is extensively metabolized with three active metabolites: hydroxybupropion, threohydrobupropion and erythrohydrobupropion. The metabolites have longer elimination half-lives than bupropion and accumulate to a greater extent. Following bupropion administration, more than 90% of the exposure is a result of metabolites. In vitro findings suggest that CYP2B6 is the principal isozyme involved in the formation of hydroxybupropion whereas cytochrome P450 isozymes are not involved in the formation of the other active metabolites. Bupropion and its metabolites inhibit CYP2D6. Plasma protein binding of hydroxybupropion is similar to that of bupropion (84%) whereas the other two metabolites have approximately half the binding.
Following oral administration of 200 mg of 14C-bupropion in humans, 87% and 10% of the radioactive dose were recovered in the urine and feces, respectively. The fraction of the oral dose of bupropion excreted unchanged was 0.5%, a finding consistent with the extensive metabolism of bupropion.
Following single oral administration of CONTRAVE tablets to healthy subjects, mean elimination half-life (T½) was approximately 21 hours for bupropion. Following twice daily administration of CONTRAVE, metabolites of bupropion, and to a lesser extent unchanged bupropion, accumulate and reach steady-state concentrations in approximately one week.
Specific Populations
Gender
Pooled analysis of CONTRAVE data suggested no clinically meaningful differences in the pharmacokinetic parameters of bupropion or naltrexone based on gender.
Race
Pooled analysis of CONTRAVE data suggested no clinically meaningful differences in the pharmacokinetic parameters of bupropion or naltrexone based on race.
Elderly
The pharmacokinetics of CONTRAVE have not been evaluated in the geriatric population. The effects of age on the pharmacokinetics of naltrexone or bupropion and their metabolites have not been fully characterized. An exploration of steady-state bupropion concentrations from several depression efficacy studies involving patients dosed in a range of 300 to 750 mg/day, on a three times daily schedule, revealed no relationship between age (18 to 83 years) and plasma concentration of bupropion. A single-dose pharmacokinetic study demonstrated that the disposition of bupropion and its metabolites in elderly subjects was similar to that of younger subjects. These data suggest there is no prominent effect of age on bupropion concentration; however, another pharmacokinetic study, single and multiple dose, has suggested that the elderly are at increased risk for accumulation of bupropion and its metabolites [see Use in Specific Populations (8.5)].
Smokers
Pooled analysis of CONTRAVE data revealed no meaningful differences in the plasma concentrations of bupropion or naltrexone in smokers compared with nonsmokers. The effects of cigarette smoking on the pharmacokinetics of bupropion were studied in 34 healthy male and female volunteers; 17 were chronic cigarette smokers and 17 were nonsmokers. Following oral administration of a single 150 mg dose of bupropion, there was no statistically significant difference in Cmax, half-life, Tmax, AUC, or clearance of bupropion or its active metabolites between smokers and nonsmokers.
Hepatic Impairment
A single dose pharmacokinetic study conducted for CONTRAVE, comparing patients with mild (n=8), moderate (n=8) and severe (n=7) hepatic impairment based on CHILD-PUGH classification system to subjects with normal hepatic function (n=13), showed that hepatic impairment had a significant effect on the PK parameters of the parent drugs naltrexone and bupropion. Systemic exposure to some metabolites was also increased in patients with impaired hepatic function [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)].
Following a single dose of naltrexone/bupropion, AUCinf of naltrexone was approximately 2.8-, 6.1-, and 34-fold higher in patients with mild, moderate, and severe hepatic impairment, respectively. In patients with moderate and severe hepatic impairment, AUCinf of bupropion was approximately 2.0- and 3.6-fold higher, respectively, compared to subjects with normal hepatic function. There was no effect of mild hepatic impairment on bupropion exposure. Exposure to bupropion metabolite, threohydrobupropion, was increased by 1.9-, 3.4-, and 2.5-fold in patients with mild, moderate, and severe hepatic impairment, respectively [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)].
Renal Impairment
A single-dose pharmacokinetic study conducted for CONTRAVE, comparing patients with mild (n=8), moderate (n=8) and severe (n=7) renal impairment to subjects with normal renal function (n=13), showed that renal impairment had no significant effect on the PK parameters of the parent drugs naltrexone and bupropion. However, systemic exposure (AUCinf) of some metabolites was increased in patients with impairment of renal function [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Following a single-dose of 16 mg naltrexone /180 mg bupropion, AUCinf of 6-beta-naltrexol was approximately1.5-, 1.7-, and 2.2-fold higher in patients with mild, moderate, and severe renal impairment, respectively. In patients with mild, moderate, and severe renal impairment, AUC of bupropion metabolites threohydrobupropion and erythrohydrobupropion increased approximately 1.3-, 1.9-, and 1.7-fold and 1.2-, 1.8-, and 1.5-fold, respectively. No studies have been conducted for CONTRAVE in patients with end stage renal disease.
The following information is available for individual components.
In a study of seven patients with end-stage renal disease requiring dialysis, peak plasma concentrations of naltrexone were elevated at least 6-fold compared to healthy subjects. An inter-trial comparison between normal subjects and patients with end-stage renal failure demonstrated that the bupropion Cmax and AUC values were comparable in the two groups, whereas the hydroxybupropion and threohydrobupropion metabolites had a 2.3- and 2.8-fold increase, respectively, in AUC for patients with end-stage renal failure.
Drug Interactions
In Vitro Assessment of Drug Interactions
At therapeutically relevant concentrations, naltrexone and 6-beta-naltrexol are not major inhibitors of CYP isoforms CYP1A2, CYP2B6, CYP2C8, CYP2E1, CYP2C9, CYP2C19, CYP2D6 or CYP3A4. Both naltrexone and 6-beta-naltrexol are not major inducers of CYP isoforms CYP1A2, CYP2B6, or CYP3A4.
Bupropion and its metabolites (hydroxybupropion, erythrohydrobupropion, threohydrobupropion) are inhibitors of CYP2D6.
In vitro studies suggest that paroxetine, sertraline, norfluoxetine, fluvoxamine, and nelfinavir inhibit the hydroxylation of bupropion.
Bupropion (IC50 9.3 mcM) and its metabolites, hydroxybupropion (IC50 82 mcM) and threohydrobupropion and erythrohydrobupropion (1:1 mixture; IC50 7.8 mcM), inhibited the renal organic transporter OCT2 to a clinically relevant level.
Effects of Naltrexone/Bupropion on the Pharmacokinetics of Other Drugs
Drug interaction between CONTRAVE and CYP2D6 substrates (metoprolol) or other drugs (atorvastatin, glyburide, lisinopril, nifedipine, valsartan) has been evaluated. In addition, drug interaction between bupropion, a component of CONTRAVE, and CYP2D6 substrates (desipramine) or other drugs (citalopram, lamotrigine) has also been evaluated.
Table 4. Effect of Naltrexone/Bupropion Coadministration on Systemic Exposure of Other Drugs Naltrexone/Bupropion Dosage Coadministered Drug Name and Dose Regimens Change in Systemic Exposure Initiate the following drugs at the lower end of the dose range during concomitant use with CONTRAVE [see Drug Interactions 7]: Bupropion
150 mg twice daily for 10 daysDesipramine
50 mg single dose↑5-fold AUC, ↑2-fold Cmax Bupropion
300 mg (as XL) once daily for 14 daysCitalopram
40 mg once daily for 14 days↑40% AUC, ↑30% Cmax Naltrexone/Bupropion
16 mg/180 mg twice daily for 7 daysMetoprolol
50 mg single dose↑4-fold AUC, ↑2-fold Cmax No dose adjustment needed for the following drugs during concomitant use with CONTRAVE: Naltrexone/Bupropion
16 mg/180 mg single doseAtorvastatin
80 mg single doseNo Effect Naltrexone/Bupropion
16 mg/180 mg single doseGlyburide
6 mg single doseNo Effect Naltrexone/Bupropion
16 mg/180 mg single doseLisinopril
40 mg single doseNo Effect Naltrexone/Bupropion
16 mg/180 mg twice dailyMetformin
850 mg single dose↑23% AUC;
No effect on CmaxNaltrexone/Bupropion
16 mg/180 mg single doseNifedipine
90 mg single doseNo Effect Naltrexone/Bupropion
16 mg/180 mg single doseValsartan
320 mg single doseNo Effect Bupropion
150 mg twice daily for 12 daysLamotrigine
100 mg single doseNo Effect Effects of Other Drugs on the Pharmacokinetics of Naltrexone/Bupropion
Drug interactions between CYP2B6 inhibitors (ticlopidine, clopidogrel, prasugrel), CYP2B6 inducers (ritonavir, lopinavir) and bupropion (one of the CONTRAVE components), or between other drugs (atorvastatin, glyburide, metoprolol, lisinopril, nifedipine, valsartan) and CONTRAVE have been evaluated. While not systematically studied, carbamazepine, phenobarbital, or phenytoin may induce the metabolism of bupropion.
Table 5. Effect of Coadministered Drugs on Systemic Exposure of Naltrexone/Bupropion Name and Dose Regimens Coadministered Drug CONTRAVE Components Change in Systemic Exposure - *
- Results were confounded by the food-effect due to oral glucose coadministered with the treatment.
Do not exceed one tablet twice daily dose of CONTRAVE with the following drugs: Ticlopidine
250 mg twice daily for 4 daysBupropion
Hydroxybupropion↑85% AUC, ↑38% Cmax
↓84% AUC, ↓78% CmaxClopidogrel
75 mg once daily for 4 daysBupropion
Hydroxybupropion↑60% AUC, ↑40% Cmax
↓52% AUC, ↓50% CmaxNo dose adjustment needed for CONTRAVE with the following drugs: Atorvastatin
80 mg single doseNaltrexone
6-beta naltrexol
Bupropion
Hydroxybupropion
Threohydrobupropion
ErythrohydrobupropionNo Effect
No Effect
No Effect
No Effect
No Effect
No EffectLisinopril
40 mg single doseNaltrexone
6-beta naltrexol
Bupropion
Hydroxybupropion
Threohydrobupropion
ErythrohydrobupropionNo Effect
No Effect
No Effect
No Effect
No Effect
No EffectValsartan
320 mg single doseNaltrexone
6-beta naltrexol
Bupropion
Hydroxybupropion
Threohydrobupropion
ErythrohydrobupropionNo Effect
No Effect
No Effect
↓14% AUC, No Effect on Cmax
No Effect
No EffectCimetidine
800 mg single doseBupropion
Hydroxybupropion
Threo/ErythrohydrobupropionNo Effect
No Effect
↑16% AUC, ↑32% CmaxCitalopram
40 mg once daily for 14 daysBupropion
Hydroxybupropion
Threohydrobupropion
ErythrohydrobupropionNo Effect
No Effect
No Effect
No EffectMetoprolol
50 mg single doseNaltrexone
6-beta naltrexol
Bupropion
Hydroxybupropion
Threohydrobupropion
Erythrohydrobupropion↓25% AUC, ↓29% Cmax
No Effect
No Effect
No Effect
No Effect
No EffectMetformin
850 mg single doseBupropion
Hydroxybupropion
Threohydrobupropion
Erythrohydrobupropion
Naltrexone
6-beta naltrexolNo Effect
No Effect
No Effect
No Effect
No Effect
No EffectNifedipine
90 mg single doseNaltrexone
6-beta naltrexol
Bupropion
Hydroxybupropion
Threohydrobupropion
Erythrohydrobupropion↑24% AUC, ↑58% Cmax
No Effect
No Effect on AUC, ↑22% Cmax
No Effect
No Effect
No EffectPrasugrel
10 mg once daily for 6 daysBupropion
Hydroxybupropion↑18% AUC, ↑14% Cmax
↓24%AUC, ↓32% CmaxUse CONTRAVE with caution with the following drugs: Glyburide
6 mg single dose*Naltrexone
6-beta naltrexol
Bupropion
Hydroxybupropion
Threohydrobupropion
Erythrohydrobupropion↑2-fold AUC, ↑2-fold Cmax
No Effect
↑36% AUC, ↑18% Cmax
↑22% AUC, ↑21% Cmax
No Effect on AUC, ↑15% Cmax
No EffectAvoid concomitant use of CONTRAVE with following drugs: Ritonavir
100 mg twice daily for 17 daysBupropion
Hydroxybupropion
Threohydrobupropion
Erythrohydrobupropion↓22% AUC, ↓21 % Cmax
↓23% AUC, No Effect on Cmax
↓38% AUC, ↓39 % Cmax
↓48% AUC, ↓28 % Cmax600 mg twice daily for 8 days Bupropion
Hydroxybupropion
Threohydrobupropion
Erythrohydrobupropion↓66% AUC, ↓62% Cmax
↓78% AUC, ↓42 % Cmax
↓50% AUC, ↓58% Cmax
↓68% AUC, ↓48 % CmaxLopinavir/Ritonavir
400 mg/100 mg twice daily for 14 daysBupropion
Hydroxybupropion↓57% AUC, ↓57% Cmax
↓50% AUC, ↓31% CmaxEfavirenz
600 mg once daily for 2 weeksBupropion
Hydroxybupropion↓55% AUC, ↓34% Cmax
No Effect on AUC, ↑50% Cmax -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies to evaluate carcinogenesis, mutagenesis, or impairment of fertility with the combined products in CONTRAVE have not been conducted. The following findings are from studies performed individually with naltrexone and bupropion. The potential carcinogenic, mutagenic and fertility effects of the metabolite 6-beta-naltrexol are unknown. Safety margins were estimated using body surface area exposure (mg/m2) based on a body weight of 100 kg.
In a two-year carcinogenicity study in rats with naltrexone, there were small increases in the numbers of testicular mesotheliomas in males and tumors of vascular origin in males and females. The incidence of mesothelioma in males given naltrexone at a dietary dose of 100 mg/kg/day (approximately 50 times the recommended therapeutic dose on a mg/m2 basis for the naltrexone maintenance dose for CONTRAVE) was 6%, compared with a maximum historical incidence of 4%. The incidence of vascular tumors in males and females given dietary doses of 100 mg/kg/day was 4%, but only the incidence in females was increased compared with a maximum historical control incidence of 2%. There was no evidence of carcinogenicity in a two-year dietary study with naltrexone in male and female mice.
Lifetime carcinogenicity studies of bupropion were performed in rats and mice at doses up to 300 and 150 mg/kg/day, respectively. These doses are approximately 14 and 3 times the maximum recommended human dose (MRHD) of the bupropion component in CONTRAVE, respectively, on a mg/m2 basis. In the rat study there was an increase in nodular proliferative lesions of the liver at doses of 100 to 300 mg/kg/day (approximately 5 to 14 times the MRHD of the bupropion component in CONTRAVE on a mg/m2 basis); lower doses were not tested. The question of whether or not such lesions may be precursors of neoplasms of the liver is currently unresolved. Similar liver lesions were not seen in the mouse study, and no increase in malignant tumors of the liver and other organs was seen in either study.
There was limited evidence of a weak genotoxic effect of naltrexone in one gene mutation assay in a mammalian cell line, in the Drosophila recessive lethal assay, and in non-specific DNA repair tests with E. coli. However, no evidence of genotoxic potential was observed in a range of other in vitro tests, including assays for gene mutation in bacteria, yeast, or in a second mammalian cell line, a chromosomal aberration assay, and an assay for DNA damage in human cells. Naltrexone did not exhibit clastogenicity in an in vivo mouse micronucleus assay.
Bupropion produced a positive response (two to three times control mutation rate) in two of five strains in the Ames bacterial mutagenicity test and an increase in chromosomal aberrations in one of three in vivo rat bone marrow cytogenetic studies.
Naltrexone administered orally to rats caused a significant increase in pseudopregnancy and a decrease in pregnancy rates in rats at 100 mg/kg/day (approximately 50 times the MRHD of the naltrexone component in CONTRAVE on a mg/m2 basis). There was no effect on male fertility at this dose level. The relevance of these observations to human fertility is not known.
A fertility study of bupropion in rats at doses up to 300 mg/kg/day (approximately 14 times the MRHD of the bupropion component in CONTRAVE on a mg/m2 basis) revealed no evidence of impaired fertility.
-
14 CLINICAL STUDIES
The effects of CONTRAVE on weight reduction and long-term maintenance in conjunction with reduced caloric intake and increased physical activity was studied in double-blind, placebo-controlled trials (BMI range 27 to 45 kg/m2) with study durations of 16 to 56 weeks randomized to naltrexone and/or bupropion or placebo.
Effect on Weight Reduction and Long-Term Maintenance
Four 56-week multicenter, double-blind, placebo-controlled obesity trials (CONTRAVE Obesity Research, or COR-I, COR-II, COR-BMOD, and COR-Diabetes) were conducted to evaluate the effect of CONTRAVE on weight reduction in conjunction with lifestyle modification in 4,536 patients randomized to CONTRAVE or placebo. The COR-I, COR-II, and COR-BMOD trials enrolled patients with obesity (BMI 30 kg/m2 or greater) or overweight (BMI 27 kg/m2 or greater) and at least one comorbidity (hypertension or dyslipidemia). The COR-Diabetes trial enrolled patients with BMI greater than 27 kg/m2 with type 2 diabetes with or without hypertension and/or dyslipidemia.
Treatment was initiated with a three-week dose-escalation period followed by approximately 1 year of continued therapy. Patients were instructed to take CONTRAVE with food. COR-I and COR-II included a program consisting of a reduced-calorie diet resulting in an approximate 500 kcal/day decrease in caloric intake, behavioral counseling, and increased physical activity. COR-BMOD included an intensive behavioral modification program consisting of 28 group counseling sessions over 56 weeks as well as a prescribed diet and exercise regimen. COR-Diabetes evaluated patients with type 2 diabetes not achieving glycemic goal of a HbA1c less than 7% either with oral antidiabetic agents or with diet and exercise alone. Of the overall population from these four trials, 24% had hypertension, 54% had dyslipidemia at study entry, and 10% had type 2 diabetes.
Apart from COR-Diabetes, which only enrolled patients with type 2 diabetes, the demographic characteristics of patients were similar across all four trials. For the four trial populations combined, the mean age was 46 years, 83% were female, 77% were Caucasian, 18% were black, and 5% were other races. At baseline, mean BMI was 36 kg/m2 and mean waist circumference was 110 cm.
A substantial percentage of randomized patients withdrew from the trials prior to Week 56: 45% for the placebo group and 46% for the CONTRAVE group. The majority of these patients discontinued within the first 12 weeks of treatment. Approximately 24% of patients treated with CONTRAVE and 12% of patients treated with placebo discontinued treatment because of an adverse reaction [see Adverse Reactions (6.1)].
The co-primary endpoints were percent change from baseline body weight and the proportion of patients achieving at least a 5% reduction in body weight. In the 56-week COR-I trial, the mean change in body weight was -5.4% among patients assigned to CONTRAVE 32 mg/360 mg compared with -1.3% among patients assigned to placebo (Intent-To-Treat [ITT] population), as shown in Table 6 and Figure 1. In this trial, the achievement of at least a 5% reduction in body weight from baseline occurred more frequently for patients treated with CONTRAVE 32 mg/360 mg compared with placebo (42% vs 17%; Table 6). Results from COR-BMOD and COR-Diabetes are shown in Table 6 and Figures 2 and 3.
Table 6. Changes in Weight in 56-Week Trials with CONTRAVE (ITT/LOCF*) COR-I COR-BMOD COR-Diabetes CONTRAVE
32 mg/360 mgPlacebo CONTRAVE
32 mg/360 mgPlacebo CONTRAVE
32 mg/360 mgPlacebo N 538 536 565 196 321 166 Type 1 error was controlled across all 3 endpoints - *
- Based on last observation carried forward (LOCF) in all randomized subjects who had a baseline body weight measurement and at least one post baseline body weight measurement during the defined treatment phase. All available body weight data during the double-blind treatment phase are included in the analysis, including data collected from subjects who discontinued study drug.
- †
- Difference from placebo, p<0.001
- ‡
- Difference from placebo, p<0.01
Weight (kg) Baseline mean (SD) 99.8
(16.1)99.5
(14.4)100.3
(15.5)101.8
(15.0)104.2
(19.1)105.3
(16.9)LS Mean % Change From Baseline (SE) -5.4
(0.3)-1.3
(0.3)-8.1
(0.4)-4.9
(0.6)-3.7
(0.3)-1.7
(0.4)Difference from placebo (95% CI) -4.1†
(-4.9, -3.3)-3.2†
(-4.5, -1.8)-2.0†
(-3.0, -1.0)Percentage of patients losing greater than or equal to 5% body weight 42 17 57 43 36 18 Risk difference vs placebo (95% CI) 25†
(19, 30)14†
(6, 22)18†
(9, 25)Percentage of patients losing greater than or equal to 10% body weight 21 7 35 21 15 5 Risk difference vs placebo (95% CI) 14†
(10, 18)14†
(7, 21)10‡
(4, 15)The percentages of patients who achieved at least 5% or at least 10% body weight loss from baseline were greater among those assigned to CONTRAVE, compared with placebo, in all four obesity trials (Table 6).
Figure 1. Weight Loss Over Time in Completer Population: COR-I Trial Figure 2. Weight Loss Over Time in Completer Population: COR-BMOD Trial 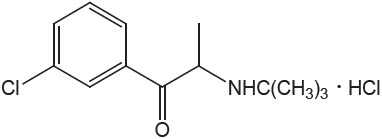
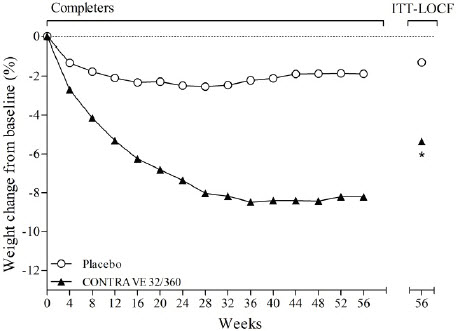
*p<0.001 vs placebo
COR-I trial: 50.1% in the placebo group and 49.2% in the CONTRAVE group discontinued study drug.*p<0.001 vs placebo COR-BMOD trial: 41.6% in the placebo group and 42.1% in the CONTRAVE group discontinued study drug. Figure 3. Weight Loss Over Time in Completer Population: COR-Diabetes Trial 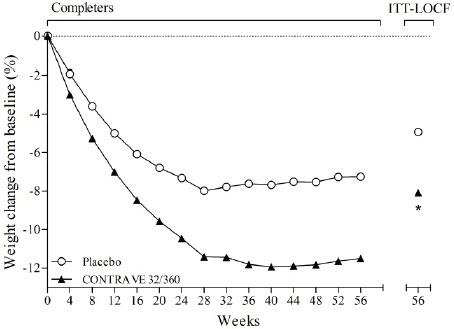
*p<0.001 vs placebo
COR-Diabetes trial: 41.2% in the placebo group and 47.8% in the CONTRAVE group discontinued study drug.Effect on Cardiovascular and Metabolic Parameters
Changes in cardiovascular and metabolic parameters associated with obesity are presented for COR-I and COR-BMOD (Table 7). Changes in mean blood pressure and heart rate are further described elsewhere [see Warnings and Precautions (5.5)].
Table 7. Change in Markers of Cardiovascular and Metabolic Parameters from Baseline in 56 Week Trials with CONTRAVE 32 mg/360 mg (COR-I and COR-BMOD)* Parameter COR-I COR-BMOD CONTRAVE
32 mg/360 mg
N=471Placebo
N=511CONTRAVE minus Placebo
(LS Mean)CONTRAVE
32 mg/360 mg
N=482Placebo
N=193CONTRAVE minus Placebo
(LS Mean)Q1: first quartile; Q3: third quartile Triglycerides, mg/dL Baseline median (Q1, Q3) 113
(86, 158)112
(78, 157)-10.7† 110
(78, 162)103
(76, 144)-9.9† Median % change -11.6 1.7 -17.8 -7.4 HDL-C, mg/dL Baseline mean (SD) 51.9
(13.6)52.0
(13.6)7.2 53.6
(13.5)55.3
(12.9)6.6 LS Mean % change (SE) 8.0
(0.9)0.8
(0.9)9.4
(1.0)2.8
(1.6)LDL-C, mg/dL Baseline mean (SD) 118.8
(32.6)119.7
(34.8)-1.5 109.5
(27.5)109.2
(27.3)-2.9 LS Mean % change (SE) -2.0
(1.0)-0.5
(1.1)7.1
(1.4)10.0
(2.2)Waist circumference, cm Baseline mean (SD) 108.8
(11.3)110.0
(12.2)-3.8‡ 109.3
(11.4)109.0
(11.8)-3.2‡ LS Mean change (SE) -6.2
(0.4)-2.5
(0.4)-10.0
(0.5)-6.8
(0.8)Heart rate, bpm Baseline mean (SD) 72.1
(8.7)71.8
(8.0)1.2 70.7
(8.3)70.4
(9.0)0.9 LS Mean change (SE) 1.0
(0.3)-0.2
(0.3)1.1
(0.4)0.2
(0.5)Systolic blood pressure, mmHg Baseline mean (SD) 118.9
(9.8)119.0
(9.8)1.8 116.9
(9.9)116.7
(10.9)2.6 LS Mean change (SE) -0.1
(0.4)-1.9
(0.4)-1.3
(0.5)-3.9
(0.7)Diastolic blood pressure, mmHg Baseline mean (SD) 77.1
(7.2)77.3
(6.6)0.9 78.2
(7.2)77.2
(7.4)1.4 LS Mean change (SE) 0.0
(0.3)-0.9
(0.3)-1.4
(0.3)-2.8
(0.5)Effect of CONTRAVE on Cardiometabolic Parameters and Anthropometry in Patients with Type 2 Diabetes Mellitus
Changes in glycemic control observed from baseline to Week 56 among patients with type 2 diabetes and obesity assigned to either CONTRAVE 32 mg/360 mg or placebo are shown in Table 8.
Table 8. Changes in Cardiometabolic Parameters and Waist Circumference in Patients with Type 2 Diabetes Mellitus in a 56 Week Trial with CONTRAVE 32 mg/360 mg (COR-Diabetes) CONTRAVE
32 mg/360 mg
N=265Placebo
N=159Based on last observation carried forward (LOCF) while on study drug Baseline Change from Baseline
(LS Mean)Baseline Change from Baseline
(LS Mean)CONTRAVE minus Placebo
(LS Mean)HbA1c (%) 8.0 -0.6 8.0 -0.1 -0.5* Fasting Glucose (mg/dL) 160.0 -11.9 163.9 -4.0 -7.9 Waist Circumference (cm) 115.6 -5.0 114.3 -2.9 -2.1 Systolic blood pressure (mmHg) 125.0 0.0 124.5 -1.1 1.2 Diastolic blood pressure (mmHg) 77.5 -1.1 77.4 -1.5 0.4 Heart rate (bpm) 72.9 0.7 73.1 -0.2 0.9 Baseline % Change from Baseline
(LS Mean)Baseline % Change from Baseline
(LS Mean)CONTRAVE minus Placebo
(LS Mean)Triglycerides (mg/dL)† 147 (98, 200) -7.7 168 (114, 236) -8.6 -3.3 HDL Cholesterol (mg/dL) 46.2 7.4 46.1 -0.2 7.6 LDL Cholesterol (mg/dL) 100.2 2.4 101.0 4.2 -1.9 Effect on Body Composition
In a subset of 124 patients (79 CONTRAVE, 45 placebo), body composition was measured using dual energy X-ray absorptiometry (DEXA). The DEXA assessment showed that mean total body fat mass decreased by 4.7 kg (11.7%) in the CONTRAVE group vs 1.4 kg (4.3%) in the placebo group at Week 52/LOCF (treatment difference, -3.3 kg [-7.4%], p<0.01).
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
Patient information is printed at the end of this insert. This information and the instructions provided in the Medication Guide should be discussed with patients.
Patients should be advised to take CONTRAVE exactly as prescribed. Patients should be instructed to follow the dose escalation schedule and not to take more than the recommended dose of CONTRAVE.
Patients should be made aware that CONTRAVE contains the same active ingredient (bupropion) found in certain antidepressants and smoking cessation products (including, but not limited to, WELLBUTRIN, WELLBUTRIN SR, WELLBUTRIN XL, APLENZIN and ZYBAN) and that CONTRAVE should not be used in combination with any other medications that contain bupropion.
Patients should be advised that some patients have experienced changes in mood (including depression and mania), psychosis, hallucinations, paranoia, delusions, homicidal ideation, aggression, hostility, agitation, anxiety, and panic, as well as suicidal ideation, suicide attempt, and completed suicide when attempting to quit smoking while taking bupropion. Instruct patients to discontinue CONTRAVE and contact a healthcare professional if they experience such symptoms.
Patients should be advised of the potential serious risks associated with the use of CONTRAVE, including suicidality, seizures, and increases in blood pressure or heart rate.
Patients should be advised to call their healthcare provider to report new or sudden changes in mood, behavior, thoughts, or feelings.
Patients should be advised that taking CONTRAVE can cause mild pupillary dilation, which in susceptible individuals, can lead to an episode of angle-closure glaucoma. Pre-existing glaucoma is almost always open-angle glaucoma because angle-closure glaucoma, when diagnosed, can be treated definitively with iridectomy. Open-angle glaucoma is not a risk factor for angle-closure glaucoma. Patients may wish to be examined to determine whether they are susceptible to angle closure, and have a prophylactic procedure (e.g., iridectomy), if they are susceptible.
Patients should be educated on the symptoms of hypersensitivity and to discontinue CONTRAVE if they have a severe allergic reaction to CONTRAVE.
Patients should be told that CONTRAVE should be discontinued and not restarted if they experience a seizure while on treatment.
Patients should be advised that the excessive use or abrupt discontinuation of alcohol, benzodiazepines, antiepileptic drugs, or sedatives/hypnotics can increase the risk of seizure. Patients should be advised to minimize or avoid use of alcohol.
Patients should be advised that if they previously used opioids, they may be more sensitive to lower doses of opioids and at risk of accidental overdose should they use opioids after CONTRAVE treatment is discontinued or temporarily interrupted.
Patients should be advised that because naltrexone, a component of CONTRAVE, can block the effects of opioids, they will not perceive any effect if they attempt to self-administer any opioid drug in small doses while on CONTRAVE. Further advise patients that the attempt to administer large doses of any opioid or to bypass the blockade while on CONTRAVE may lead to serious injury, coma, or death.
Patients should be off all opioids for a minimum of 7 to 10 days before starting CONTRAVE in order to avoid precipitation of withdrawal. Advise patients they should not take CONTRAVE if they have any symptoms of opioid withdrawal.
Patients should be advised to call their healthcare provider if they experience increased blood pressure or heart rate.
Patients should be advised to notify their healthcare provider if they are taking, or plan to take, any prescription or over-the-counter drugs. Concern is warranted because CONTRAVE and other drugs may affect each other's metabolism.
Pregnancy
Advise pregnant patients and all patients who can become pregnant of the potential risk to a fetus. Advise patients to inform their healthcare provider of a known or suspected pregnancy to discuss if CONTRAVE should be discontinued [see Use in Specific Populations (8.1)].
Patients with type 2 diabetes mellitus on antidiabetic therapy should be advised to monitor their blood glucose levels and report symptoms of hypoglycemia to their healthcare provider(s).
Patients should be advised to swallow CONTRAVE tablets whole so that the release rate is not altered. Do not chew, divide, or crush tablets.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: 11/2025 MEDICATION GUIDE
CONTRAVE® (CON-trayv)
(naltrexone HCl and bupropion HCl) extended-release tabletsWhat is the most important information I should know about CONTRAVE? CONTRAVE can cause serious side effects, including: -
Suicidal thoughts or actions. One of the ingredients in CONTRAVE is bupropion. Bupropion has caused some people to have suicidal thoughts or actions or unusual changes in behavior, whether or not they are taking medicines used to treat depression.
Bupropion may increase suicidal thoughts or actions in some children, teenagers, and young adults within the first few months of treatment.
If you already have depression or other mental illnesses, taking bupropion may cause it to get worse, especially within the first few months of treatment.
Stop taking CONTRAVE and call a healthcare provider right away if you, or your family member, have any of the following symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling very agitated or restless
- panic attacks
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
- trouble sleeping (insomnia)
While taking CONTRAVE, you or your family members should: - Pay close attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings. This is very important when you start taking CONTRAVE or when your dose changes.
- Keep all follow-up visits with your healthcare provider as scheduled. Call your healthcare provider between visits as needed, especially if you have concerns about symptoms.
CONTRAVE has not been studied in and is not approved for use in children under the age of 18. What is CONTRAVE? CONTRAVE is a prescription medicine which contains 2 medicines (naltrexone and bupropion) that may help adults with obesity, or some adults with overweight who also have weight-related medical problems, to lose excess body weight and keep the weight off. CONTRAVE should be used with a reduced calorie-diet and increased physical activity. It is not known if CONTRAVE changes your risk of heart problems or stroke or of death due to heart problems or stroke. It is not known if CONTRAVE is safe and effective in children under 18 years of age. CONTRAVE contains naltrexone and bupropion and should not be used with other products that contain naltrexone or bupropion. Bupropion is the same ingredient in some other medicines used to treat depression and to help people quit smoking. CONTRAVE is not approved to treat depression or other mental illnesses, or to help people quit smoking (smoking cessation) Do not take CONTRAVE if you: - have uncontrolled hypertension.
- have or have had seizures.
- use other medicines that contain bupropion such as WELLBUTRIN, WELLBUTRIN SR, WELLBUTRIN XL, APLENZIN and ZYBAN.
- have or have had an eating disorder called anorexia (eating very little) or bulimia (eating too much and vomiting to avoid gaining weight).
- are dependent on opioid pain medicines or use medicines to help stop taking opioids such as methadone or buprenorphine, or are in opiate withdrawal.
- drink a lot of alcohol and abruptly stop drinking, or use medicines called sedatives (these make you sleepy), benzodiazepines, or anti-seizure medicines and you stop using them all of a sudden.
- are taking medicines called monoamine oxidase inhibitors (MAOIs). Ask your healthcare provider or pharmacist if you are not sure if you take an MAOI, including linezolid. Do not start CONTRAVE until you have stopped taking your MAOI for at least 14 days.
- are allergic to naltrexone or bupropion or any of the ingredients in CONTRAVE. See the end of this Medication Guide for a complete list of ingredients in CONTRAVE.
Before taking CONTRAVE, tell your healthcare provider about all of your medical conditions, including if you: - have or have had depression or other mental illnesses (such as bipolar disorder)
- have attempted suicide in the past
- have or have had seizures
- have had a head injury
- have had a tumor or infection of your brain or spine (central nervous system)
- have had a problem with low blood sugar (hypoglycemia) or low levels of sodium in your blood (hyponatremia)
- have or have had liver problems
- have high blood pressure
- have or have had a heart attack, heart problems, or have had a stroke
- have kidney problems
- are diabetic taking insulin or other medicines to control your blood sugar
- have or have had an eating disorder
- drink a lot of alcohol
- abuse prescription medicines or street drugs
- are over the age of 65
- are pregnant or plan to become pregnant. Losing weight while pregnant may harm your unborn baby. Stop taking CONTRAVE if you become pregnant. Tell your healthcare provider if you become pregnant or you think you may be pregnant during treatment with CONTRAVE.
- are breastfeeding or plan to breastfeed. CONTRAVE can pass into your breast milk and may harm your baby. You and your healthcare provider should decide if you should take CONTRAVE or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. CONTRAVE may affect the way other medicines work and other medicines may affect the way CONTRAVE works causing side effects. Ask your healthcare provider for a list of these medicines if you are not sure. Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine. How should I take CONTRAVE? How to take CONTRAVE Morning Dose Evening Dose Starting: Week 1 1 tablet None Week 2 1 tablet 1 tablet Week 3 2 tablets 1 tablet Week 4 Onward 2 tablets 2 tablets - Take CONTRAVE exactly as your healthcare provider tells you to.
- If you have kidney or liver problems, your final dose may be lower (for example, one tablet in the morning and one tablet in the evening or one tablet in the morning).
- Your healthcare provider will change your dose if needed.
- Do not change your CONTRAVE dose without talking with your healthcare provider.
- Your healthcare provider should tell you to stop taking CONTRAVE if you have not lost a certain amount of weight after 16 weeks of treatment.
- Swallow CONTRAVE tablets whole. Do not cut, chew, or crush CONTRAVE tablets. Tell your healthcare provider if you cannot swallow CONTRAVE tablets whole.
- Do not take more than 2 tablets in the morning and 2 tablets in the evening.
- Do not take more than 2 tablets at the same time or more than 4 tablets in 1 day.
- Do not take CONTRAVE with high-fat meals. It may increase your risk of seizures.
- If you miss a dose of CONTRAVE, wait until your next regular time to take it. Do not take more than 1 dose of CONTRAVE at a time.
- If you take too much CONTRAVE, call your healthcare provider or your poison control center at 1-800-222-1222 right away, or go to the nearest emergency room.
What should I avoid while taking CONTRAVE? - Do not drink a lot of alcohol while taking CONTRAVE. If you drink a lot of alcohol, talk with your healthcare provider before suddenly stopping. If you suddenly stop drinking alcohol, you may increase your chance of having a seizure.
What are the possible side effects of CONTRAVE? CONTRAVE may cause serious side effects, including: - See "What is the most important information I should know about CONTRAVE?"
-
Seizures. There is a risk of having a seizure when you take CONTRAVE. The risk of seizure is higher in people who:
- take higher doses of CONTRAVE
- have certain medical conditions
- take CONTRAVE with certain other medicines
If you have a seizure while taking CONTRAVE, stop taking CONTRAVE and call your healthcare provider right away.
You should not take CONTRAVE again if you have a seizure. -
Risk of opioid overdose. One of the ingredients in CONTRAVE (naltrexone) can increase your chance of having an opioid overdose if you take opioid medicines while taking CONTRAVE.
You can accidentally overdose in 2 ways:- Naltrexone blocks the effects of opioids, such as heroin, methadone or opioid pain medicines. Do not take large amounts of opioids, including opioid-containing medicines, such as heroin or prescription pain pills, to try to overcome the opioid-blocking effects of naltrexone. This can lead to serious injury, coma, or death.
- After you take naltrexone, its blocking effect slowly decreases and completely goes away over time. If you have used opioid street drugs or opioid-containing medicines in the past, using opioids in amounts that you used before treatment with naltrexone can lead to overdose and death. You may also be more sensitive to the effects of lower amounts of opioids:
- after you have gone through detoxification
- when your next dose of CONTRAVE is due
- if you miss a dose of CONTRAVE
- after you stop CONTRAVE treatment
You or someone close to you should get emergency medical help right away if you:
- have trouble breathing
- become very drowsy with slowed breathing
- have slow, shallow breathing (little chest movement with breathing)
- feel faint, very dizzy, confused, or have unusual symptoms
- Sudden opioid withdrawal. People who take CONTRAVE must not use any type of opioid (must be opioid-free) including street drugs, prescription pain medicines (including tramadol), cough, cold, or diarrhea medicines that contain opioids, or opioid dependence treatments, buprenorphine or methadone, for at least 7 to 10 days before starting CONTRAVE. Using opioids in the 7 to 10 days before you start taking CONTRAVE may cause you to suddenly have symptoms of opioid withdrawal when you take it. Sudden opioid withdrawal can be severe, and you may need to go to the hospital. Tell your healthcare provider you are taking CONTRAVE before a medical procedure or surgery.
- Severe allergic reactions. Some people have had a severe allergic reaction to bupropion, one of the ingredients in CONTRAVE. Stop taking CONTRAVE and call your healthcare provider or go to the nearest hospital emergency room right away if you have any of the following signs and symptoms of an allergic reaction:
- rash
- itching
- hives
- fever
- swollen lymph glands
- painful sores in your mouth or around your eyes
- swelling of your lips or tongue
- chest pain
- trouble breathing
- Increases in blood pressure or heart rate. Some people may get high blood pressure or have a higher heart rate when taking CONTRAVE. Your healthcare provider should check your blood pressure and heart rate before you start taking, and while you take CONTRAVE.
-
Liver damage or hepatitis. One of the ingredients in CONTRAVE, naltrexone can cause liver damage or hepatitis. Stop taking CONTRAVE and tell your healthcare provider if you have any of the following symptoms of liver problems:
- stomach area pain lasting more than a few days
- dark urine
- yellowing of the whites of your eyes
- tiredness
- Manic episodes. One of the ingredients in CONTRAVE, bupropion can cause some people who were manic or depressed in the past to become manic or depressed again.
-
Visual problems (angle-closure glaucoma). One of the ingredients in CONTRAVE, bupropion, can cause some people to have visual problems (angle-closure glaucoma). Signs and symptoms of angle-closure glaucoma may include:
- eye pain
- changes in vision
- swelling or redness in or around the eye
- Increased risk of low blood sugar (hypoglycemia) in people with type 2 diabetes mellitus who also take medicines to treat their diabetes. Weight loss can cause low blood sugar in people with type 2 diabetes mellitus who also take medicines used to treat type 2 diabetes mellitus (such as insulin or sulfonylureas). You should check your blood sugar before you start taking CONTRAVE and while you take CONTRAVE.
The most common side effects of CONTRAVE include: - nausea
- constipation
- headache
- vomiting
- dizziness
- trouble sleeping
- dry mouth
- diarrhea
These are not all of the possible side effects of CONTRAVE. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store CONTRAVE? Store CONTRAVE at room temperature between 68°F to 77°F (20°C to 25°C). Keep CONTRAVE and all medicines out of the reach of children. General information about the safe and effective use of CONTRAVE. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use CONTRAVE for a condition for which it was not prescribed. Do not give CONTRAVE to other people, even if they have the same symptoms that you have. It may harm them. If you take a urine drug screening test, CONTRAVE may make the test result positive for amphetamines. If you tell the person giving you the drug screening test that you are taking CONTRAVE, they can do a more specific drug screening test that should not have this problem. You can ask your pharmacist or healthcare provider for information about CONTRAVE that is written for health professionals. What are the ingredients in CONTRAVE? Active ingredient: naltrexone hydrochloride and bupropion hydrochloride Inactive ingredients: microcrystalline cellulose, hydroxypropyl cellulose, lactose anhydrous, L-cysteine hydrochloride, crospovidone, magnesium stearate, hypromellose, edetate disodium, lactose monohydrate, colloidal silicon dioxide, Opadry II Blue and FD&C Blue #2 aluminum lake Distributed by: Currax™ Pharmaceuticals LLC. Brentwood, TN 37027
CONTRAVE® is a registered trademark of Nalpropion Pharmaceuticals LLC
All other trademarks are the property of their respective owners.
© 2025 Nalpropion Pharmaceuticals LLC
For more information, go to www.contrave.com or call 1-800-793-2145.
-
Suicidal thoughts or actions. One of the ingredients in CONTRAVE is bupropion. Bupropion has caused some people to have suicidal thoughts or actions or unusual changes in behavior, whether or not they are taking medicines used to treat depression.
- PRINCIPAL DISPLAY PANEL - 8 mg/90 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
CONTRAVE EXTENDED-RELEASE
naltrexone hydrochloride and bupropion hydrochloride tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:51267-890 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NALTREXONE HYDROCHLORIDE (UNII: Z6375YW9SF) (NALTREXONE - UNII:5S6W795CQM) NALTREXONE HYDROCHLORIDE 8 mg BUPROPION HYDROCHLORIDE (UNII: ZG7E5POY8O) (BUPROPION - UNII:01ZG3TPX31) BUPROPION HYDROCHLORIDE 90 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) CYSTEINE HYDROCHLORIDE (UNII: ZT934N0X4W) CROSPOVIDONE (120 .MU.M) (UNII: 68401960MK) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) EDETATE DISODIUM (UNII: 7FLD91C86K) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Product Characteristics Color BLUE Score no score Shape ROUND (bi-convex) Size 12mm Flavor Imprint Code NB;890 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:51267-890-99 120 in 1 BOTTLE; Type 0: Not a Combination Product 10/22/2014 2 NDC:51267-890-07 7 in 1 BOTTLE; Type 0: Not a Combination Product 01/14/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA200063 10/22/2014 Labeler - Nalpropion Pharmaceuticals LLC (081341086)