Label: SODIUM PHENYLBUTYRATE- sodium phenylbutyrate tablets, 500 mg tablet


- NDC Code(s): 49884-170-04
- Packager: Par Pharmaceutical, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated July 18, 2017
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
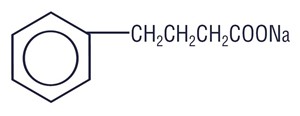
Sodium Phenylbutyrate Tablets for oral administration contain Sodium phenylbutyrate, USP. Sodium Phenylbutyrate, USP is an off-white crystalline substance which is soluble in water and has a strong salty taste. Sodium Phenylbutyrate, USP also is freely soluble in methanol and practically insoluble in acetone and diethyl ether. It is known chemically as 4-phenylbutyric acid, sodium salt with a molecular weight of 186 and the molecular formula C10H11O2Na.
Chemical Structure:
Each tablet of Sodium Phenylbutyrate Tablets contains 500 mg of Sodium Phenylbutyrate, USP and the inactive ingredients calcium stearate NF, colloidal silicon dioxide NF, magnesium stearate and microcrystalline cellulose.
-
CLINICAL PHARMACOLOGY
Sodium phenylbutyrate is a pro-drug and is rapidly metabolized to phenylacetate. Phenylacetate is a metabolically-active compound that conjugates with glutamine via acetylation to form phenylacetylglutamine. Phenylacetylglutamine then is excreted by the kidneys. On a molar basis, it is comparable to urea (each containing two moles of nitrogen). Therefore, phenylacetylglutamine provides an alternate vehicle for waste nitrogen excretion.
PHARMACOKINETICS
General:
Pharmacokinetic studies have not been conducted in the primary patient population (neonates, infants, and children), but pharmacokinetic data were obtained from normal adult subjects.
Absorption:
Peak plasma levels of phenylbutyrate occur within 1 hour after a single dose of 5 grams of sodium phenylbutyrate tablet with a Cmax of 218 mcg/mL under fasting conditions. The effect of food on phenylbutyrate's absorption is unknown.
Disposition:
The overall disposition of sodium phenylbutyrate and its metabolites has not been characterized fully. However, the drug is known to be metabolized to phenylacetate and subsequently to phenylacetylglutamine.
Following oral administration of 5 grams (tablets), measurable plasma levels of phenylbutyrate and phenylacetate were detected 15 and 30 minutes after dosing, respectively, and phenylacetylglutamine was detected shortly thereafter. The pharmacokinetic parameters for phenylbutyrate for Cmax (mcg/mL), Tmax (hours), and elimination half-life (hours) were 218, 1.35, and 0.77, respectively, and for phenylacetate were 48.5, 3.74, and 1.15, respectively.
The major sites for metabolism of sodium phenylbutyrate are the liver and kidney.
Excretion:
A majority of the administered compound (approximately 80 to 100%) was excreted by the kidneys within 24 hours as the conjugation product, phenylacetylglutamine. For each gram of sodium phenylbutyrate administered, it is estimated that between 0.12 to 0.15 grams of phenylacetylglutamine nitrogen are produced.
Pharmacodynamics:
In patients with urea cycle disorders, sodium phenylbutyrate decreases elevated plasma ammonia glutamine levels. It increases waste nitrogen excretion in the form of phenylacetylglutamine.
Special Populations
Gender:
Significant gender differences were found in the pharmacokinetics of phenylbutyrate and phenylacetate, but not for phenylacetylglutamine. The pharmacokinetic parameters (AUC and Cmax), for both plasma phenylbutyrate and phenylacetate were about 30 to 50 percent greater in females than in males.
Hepatic insufficiency:
In patients who did not have urea cycle disorders but had impaired hepatic function, the metabolism and excretion of sodium phenylbutyrate were not affected. However, this information was obtained from unvalidated, uncontrolled case studies.
-
INDICATIONS & USAGE
Sodium Phenylbutyrate Tablets is indicated as adjunctive therapy in the chronic management of patients with urea cycle disorders involving deficiencies of carbamylphosphate synthetase (CPS), ornithine transcarbamylase (OTC), or argininosuccinic acid synthetase (AS). It is indicated in all patients with neonatal-onset deficiency (complete enzymatic deficiency, presenting within the first 28 days of life). It is also indicated in patients with late-onset disease (partial enzymatic deficiency, presenting after the first month of life) who have a history of hyperammonemic encephalopathy. It is important that the diagnosis be made early and treatment initiated immediately to improve survival. Any episode of acute hyperammonemia should be treated as a life-threatening emergency.
Sodium Phenylbutyrate Tablets must be combined with dietary protein restriction and, in some cases, essential amino acid supplementation. (See Nutritional Supplementation subsection of the DOSAGE AND ADMINISTRATION section.)
Previously, neonatal-onset disease was almost universally fatal within the first year of life, even when treated with peritoneal dialysis and essential amino acids or their nitrogen-free analogs. However, with hemodialysis, use of alternative waste nitrogen excretion pathways (sodium phenylbutyrate, sodium benzoate, and sodium phenylacetate), dietary protein restriction, and, in some cases, essential amino acid supplementation, the survival rate in newborns diagnosed after birth but within the first month of life is almost 80%. Most deaths have occurred during an episode of acute hyperammonemic encephalopathy. Patients with neonatal-onset disease have a high incidence of mental retardation. Those who had IQ tests administered had an incidence of mental retardation as follows: ornithine transcarbamylase deficiency, 100% (14/14 patients tested); argininosuccinic acid synthetase deficiency, 88% (15/17 patients tested); and carbamylphosphate synthetase deficiency, 57% (4/7 patients tested). Retardation was severe in the majority of the retarded patients.
In patients diagnosed during gestation and treated prior to any episode of hyperammonemic encephalopathy, survival is 100%, but even in these patients, most subsequently demonstrate cognitive impairment or other neurologic deficits.
In late-onset deficiency patients, including females heterozygous for ornithine transcarbamylase deficiency, who recover from hyperammonemic encephalopathy and are then treated chronically with Sodium Phenylbutyrate Tablets and dietary protein restriction, the survival rate is 98%. The two deaths in this group of patients occurred during episodes of hyperammonemic encephalopathy. However, compliance with the therapeutic regimen has not been adequately documented to allow evaluation of the potential for Sodium Phenylbutyrate Tablets and dietary protein restriction to prevent mental deterioration and recurrence of hyperammonemic encephalopathy if carefully adhered to. The majority of these patients tested (30/46 or 65%) have IQ's in the average to low average/borderline mentally retarded range. Reversal of pre-existing neurologic impairment is not likely to occur with treatment and neurologic deterioration may continue in some patients.
Even on therapy, acute hyperammonemic encephalopathy recurred in the majority of patients for whom the drug is indicated.
Sodium Phenylbutyrate Tablets may be required life-long unless orthotopic liver transplantation is elected.
(See CLINICAL PHARMACOLOGY, Pharmacodynamics subsection for the biochemical effects of Sodium Phenylbutyrate Tablets).
- CONTRAINDICATIONS
-
WARNINGS
Each sodium phenylbutyrate tablet contains 62 mg of sodium (9.2 % w/w) (corresponding to 124 mg of sodium per gram of sodium phenylbutyrate [12.4% w/w]). Sodium phenylbutyrate should be used with great care, if at all, in patients with congestive heart failure or severe renal insufficiency, and in clinical states in which there is sodium retention with edema.
Because sodium phenylbutyrate is metabolized in the liver and kidney, and phenylacetylglutamine is primarily excreted by the kidney, use caution when administering the drug to patients with hepatic or renal insufficiency or inborn errors of beta oxidation. Probenecid is known to inhibit the renal transport of many organic compounds, including hippuric acid, and may affect renal excretion of the conjugated product of sodium phenylbutyrate as well as its metabolite.
Use of corticosteroids may cause the breakdown of body protein and increase plasma ammonia levels.
-
PRECAUTIONS
General:
Sodium phenylbutyrate should not be administered to patients with known hypersensitivity to sodium phenylbutyrate or any component of this preparation.
There have been published reports of hyperammonemia being induced by haloperidol and by valproic acid.
Neurotoxicity of phenylacetate in animals:
When given subcutaneously to rat pups, 190 to 474 mg/kg phenylacetate caused decreased proliferation and increased loss of neurons, and it reduced CNS myelin. Cerebral synapse maturation was retarded, and the number of functioning nerve terminals in the cerebrum was reduced, which resulted in impaired brain growth. Prenatal exposure of rat pups to phenylacetate produced lesions in layer 5 of the cortical pyramidal cells; dendritic spines were longer and thinner than normal and reduced in number.
Information for Patients:
The full text of the separate insert of information for patients is reprinted at the end of the labeling.
Laboratory Tests:
Plasma levels of ammonia, arginine, branched-chain amino acids, and serum proteins should be maintained within normal limits, and plasma glutamine should be maintained at levels less than 1,000 µmol/L. Serum drug levels of phenylbutyrate and its metabolites, phenylacetate and phenylacetylglutamine, should be monitored periodically.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
Carcinogenicity, mutagenicity, and fertility studies of sodium phenylbutyrate have not been conducted.
Pregnancy:
Pregnancy Category C. Animal reproduction studies have not been conducted with sodium phenylbutyrate. It is also not known whether sodium phenylbutyrate can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity.
Sodium phenylbutyrate should be given to a pregnant woman only if clearly needed.
Nursing Mothers:
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when sodium phenylbutyrate is administered to a nursing woman.
Pediatric Use:
The use of tablets for neonates, infants and children to the weight of 20 kg is not recommended. (See DOSAGE AND ADMINISTRATION).
-
ADVERSE REACTIONS
The assessment of clinical adverse events came from 206 patients treated with sodium phenylbutyrate. Adverse events (both clinical and laboratory) were not collected systematically in these patients, but were obtained from patient-visit reports by the 65 co-investigators. Causality of adverse effects is sometimes difficult to determine in this patient population because they may result from either the underlying disease, the patient's restricted diet, intercurrent illness, or sodium phenylbutyrate. Furthermore, the rates may be under-estimated because they were reported primarily by parent or guardian and not the patient.
CLINICAL ADVERSE EVENTS
In female patients, the most common clinical adverse event reported was amenorrhea/menstrual dysfunction (irregular menstrual cycles), which occurred in 23% of the menstruating patients.
Decreased appetite occurred in 4% of all patients. Body odor (probably caused by the metabolite, phenylacetate) and bad taste or taste aversion were each reported in 3% of patients.
Other adverse events reported in 2% or fewer patients were:
Gastrointestinal: abdominal pain, gastritis, nausea and vomiting; constipation, rectal bleeding, peptic ulcer disease, and pancreatitis each occurred in one patient.
Hematologic: aplastic anemia and ecchymoses each occurred in one patient.
Cardiovascular: arrhythmia and edema each occurred in one patient.
Renal: renal tubular acidosis
Psychiatric: depression
Skin: rash
Miscellaneous: headache, syncope, and weight gain
Neurotoxicity was reported in cancer patients receiving intravenous phenylacetate, 250 to 300 mg/kg/day for 14 days, repeated at 4-week intervals. Manifestations were predominately somnolence, fatigue, and lightheadedness; with less frequent headache, dysgeusia, hypoacusis, disorientation, impaired memory, and exacerbation of a pre-existing neuropathy. These adverse events were mainly mild in severity. The acute onset and reversibility when the phenylacetate infusion was discontinued suggest a drug effect.
LABORATORY ADVERSE EVENTS
In patients with urea cycle disorders, the frequency of laboratory adverse events by body system were:
Metabolic: acidosis (14%), alkalosis and hyperchloremia (each 7%), hypophosphatemia (6%), hyperuricemia and hyperphosphatemia (each 2%), and hypernatremia and hypokalemia (each 1%).
Nutritional: hypoalbuminemia (11%) and decreased total protein (3%).
Hepatic: increased alkaline phosphatase (6%), increased liver transaminases (4%), and hyperbilirubinemia (1%).
Hematologic: anemia (9%), leukopenia and leukocytosis (each 4%), thrombocytopenia (3%), and thrombocytosis (1%).
The clinician is advised to routinely perform urinalysis, blood chemistry profiles, and hematologic tests.
- OVERDOSAGE
-
DOSAGE & ADMINISTRATION
For oral use only.
The use of Sodium Phenylbutyrate Tablets is indicated for children weighing more than 20 kg and for adults.
The usual total daily dose of Sodium Phenylbutyrate Tablets for patients with urea cycle disorders is 450 to 600 mg/kg/day in patients weighing less than 20 kg, or 9.9 to 13 g/m2/day in larger patients. The tablets is to be taken in equally divided amounts with each meal or feeding (i.e., three to six times per day).
The safety or efficacy of doses in excess of 20 grams per day has not been established.
NUTRITIONAL MANAGEMENT
To promote growth and development, plasma levels of ammonia, arginine, branched-chain amino acids, and serum protein should be maintained within normal limits while plasma glutamine is maintained at levels less than 1,000 µmol/L. Minimum daily protein intake for a patient of a particular age should be taken from, for example, "Recommended Dietary Allowances", 10th ed., Food and Nutrition Board, National Academy of Sciences, 1989. The allocation of dietary nitrogen into natural protein and essential amino acids is a function of age, residual urea-cycle enzyme activity, and the dose of sodium phenylbutyrate.
At the recommended dose of sodium phenylbutyrate, it is suggested that infants with neonatal-onset CPS and OTC deficiencies initially receive a daily dietary protein intake limited to approximately 1.6 g/kg/day for the first 4 months of life. If tolerated, the daily protein intake may be increased to 1.9 g/kg/day during this period. Protein tolerance will decrease as the growth rate decreases, requiring a reduction in dietary nitrogen intake. From 4 months to 1 year of age, it is recommended that the infant receive at least 1.4 g/kg/day, but 1.7 g/kg/day is advisable. From 1 to 3 years of age, the protein intake should not be less than 1.2 g/kg/day; 1.4 g/kg/day is advisable during this period. For neonatal-onset patients with carbamylphosphate synthetase deficiency or ornithine transcarbamylase deficiency who are at least 6 months of age, it is recommended that the daily protein intake be equally divided between natural protein and supplemental essential amino acids.
Patients with argininosuccinic acid synthetase deficiency and those with late-onset disease (partial deficiencies, including females heterozygous for ornithine transcarbamylase), initially may receive a diet containing the age-determined minimal daily natural protein allowance. The protein intake may be increased as tolerated and determined by plasma glutamine and other amino acid levels. However, many patients with partial deficiencies avoid dietary protein.
Citrulline supplementation is required and recommended for patients diagnosed with neonatal-onset deficiency of carbamylphosphate synthetase or ornithine transcarbamylase; citrulline daily intake is recommended at 0.17 g/kg/day or 3.8 g/m2/day.
The free-base form of arginine may be used instead of citrulline in patients with milder forms of carbamylphosphate synthetase and ornithine transcarbamylase deficiency (daily intake is recommended at 0.17 g/kg/day or 3.8 g/m2/day).
Arginine supplementation is needed for patients diagnosed with deficiency of argininosuccinic acid synthetase; arginine (free base) daily intake is recommended at 0.4 to 0.7 g/kg/day or 8.8 to 15.4 g/m2/day.
If caloric supplementation is indicated, a protein-free product is recommended. Caloric intake should be based upon the "Recommended Dietary Allowances", 10th ed., Food and Nutrition Board, National Research Council, National Academy of Sciences, 1989.
-
HOW SUPPLIED
Sodium Phenylbutyrate Tablets are available as 500 mg in bottles of 250 Sodium Phenylbutyrate Tablets (NDC 49884-170-04). The bottles are equipped with child-resistant caps. White to off-white, oval shaped tablet debossed 'EP 340' on one side and plain on the other side. Each tablet contains 500 mg of sodium phenylbutyrate, USP.
Store at 20º to 25ºC (68º to 77ºF); [see USP Controlled Temperature]; Excursions permitted to 15º to 30ºC (59º to 86ºF).
After opening, keep bottle tightly closed.
-
PATIENT PACKAGE INSERT
Sodium Phenylbutyrate Tablets
SOE-dee-um fen-il-BUE-ti-rate.
What is the most important information I should know about sodium phenylbutyrate tablets?
Sodium phenylbutyrate tablets is prescribed along with changes in diet for long-term treatment of urea cycle disorders. Sodium phenylbutyrate tablets can only be obtained with a prescription from your doctor.
Sodium phenylbutyrate tablets must be taken exactly as the doctor prescribes; do not increase or decrease the dose of this medication without the doctor's approval.
What are urea cycle disorders?
Urea cycle disorders include a group of diseases, each having a specific liver enzyme deficiency. Because they are inherited, other family members may be affected. These disorders vary in severity and may be first detected at various ages, from newborn infants to adults. They lead to increased amounts of ammonia in the blood, which may cause disturbed brain function and severe brain damage. Typical signs of the disease are decreased mental awareness, vomiting, combativeness, slurred speech, unstable gait, and unconsciousness. The diagnosis of urea cycle disorders requires special laboratory tests. These typical signs of the disease may recur after the diagnosis is made if the condition is not under control. If they do, the doctor should be notified immediately because this is a medical emergency. An infection can cause the condition to go out of control. Therefore, if a fever develops, the doctor should be seen immediately.
A patient or carrier of these disorders should wear a Medic Alert tag stating the diagnosis. In the event that the patient has a sudden, rapid accumulation of ammonia in the blood, and, therefore, in the brain, leading to unconsciousness, the doctor will be alerted to treat the disease properly.
Periodically, depending upon the severity of a particular patient's urea cycle disorder, it will be necessary to perform blood tests. These include plasma ammonia, plasma amino acid levels, and other more routine blood tests to evaluate nutritional status.
What is sodium phenylbutyrate tablets?
Sodium phenylbutyrate tablets is a drug that helps to prevent ammonia from accumulating in the blood. Sodium phenylbutyrate tablets aids the body in eliminating substances that produce ammonia. However, despite drug treatment, blood ammonia levels may become elevated periodically and there may be episodes of altered brain function in association with these ammonia elevations. Patients who have disease onset as newborns have a high incidence of mental retardation. Medical attention should be obtained as soon as signs appear (see above under "What are urea cycle disorders?"). Sodium phenylbutyrate tablets may be used as life-long therapy or as a temporary measure until liver transplantation is performed.
What diet should I or my child follow?
In addition to taking sodium phenylbutyrate tablets, it is equally important that a prescribed diet be followed. Because there is great variability in the severity of urea cycle disorders, each patient's diet should be custom designed by a physician and a nutritionist. Because the diet is so important, it is recommended that the prescribed diet be discussed with a nutritionist who is familiar with urea cycle disorders.
Who should not take sodium phenylbutyrate tablets?
Sodium phenylbutyrate tablets is prescribed only for patients with urea cycle disorders. It is not to be used for any other reason. Keep the medication in a safe place where children cannot reach it.
What other medical conditions may also be present that could increase the risk of taking sodium phenylbutyrate tablets?
Heart failure or decreased kidney function may lead to retention of the sodium content of sodium phenylbutyrate tablets with potentially serious consequences such as worsening heart failure, high blood pressure, and swelling. If these medical conditions are present, the doctor will determine if your child should take sodium phenylbutyrate tablets.
How should I or my child take sodium phenylbutyrate tablets?
The dose of sodium phenylbutyrate tablets prescribed for adults and children is based upon the patient's weight or size. It is very important that the full amount prescribed for any 24-hour period be taken. If a dose is missed it should be administered as soon as possible that same day. The total daily dose should be administered in equally divided amounts with meals.
What medications should I or my child avoid or be cautious of taking while on sodium phenylbutyrate tablets?
Patients with urea cycle disorders usually should not take Depakene® (valproic acid), a drug sometimes prescribed for seizure disorders, or Haldol® (haloperidol), a drug used to treat certain types of psychiatric or neurologic disorders. Both of these drugs have been reported to increase blood ammonia levels. Steroids may break down body protein, thereby increasing blood ammonia levels. The doctor should be consulted before administering medications containing steroids.
What medications may affect the way the body breaks down the drug?
Probenecid, a medication used to treat gout, may affect the way the kidneys excrete sodium phenylbutyrate tablets (consult the doctor for details).
What are the most common side effects of sodium phenylbutyrate tablets?
The most common side effect reported in premenopausal women taking sodium phenylbutyrate tablets was absent or irregular menstrual periods. Decreased appetite was reported in 4% of all people treated. Body odor and bad taste were each reported in 3% of all patients treated.
A breakdown product of sodium phenylbutyrate tablets has been associated mainly with sleepiness and light-headedness. Because these symptoms may also be due to the urea cycle going out of control, a doctor should see the patient immediately if these symptoms occur, so the cause can be determined. Blood tests should be performed periodically for adverse effects and for levels of medication and its breakdown products.
Call your doctor for medical advice about the side effects. You may report side effects to FDA at 1-800-FDA-1088
How should sodium phenylbutyrate tablets be stored?
Sodium phenylbutyrate tablets should be stored in a tightly closed bottle at room temperature.
This leaflet provides a brief summary of the information available on sodium phenylbutyrate tablets. The information here is incomplete and is not designed to take the place of your doctor's instructions. For more complete information, consult your physician or call or write Par Pharmaceutical Companies, Inc. One Ram Ridge Road, Chestnut Ridge, New York 10977. Call toll free 1-800-828-9393.
Distributed by:
Par Pharmaceutical Companies, Inc.
Chestnut Ridge, NY 10977 U.S.A.
Manufactured by:
Par Formulations Private Limited,
9/215, Pudupakkam, Kelambakkam – 603 103
Made in India
Mfg. Lic. No.: TN00002121
OS170-01-74-01
Issued: 06/2017
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SODIUM PHENYLBUTYRATE
sodium phenylbutyrate tablets, 500 mg tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:49884-170 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SODIUM PHENYLBUTYRATE (UNII: NT6K61736T) (PHENYLBUTYRIC ACID - UNII:7WY7YBI87E) SODIUM PHENYLBUTYRATE 500 mg Inactive Ingredients Ingredient Name Strength CALCIUM STEARATE (UNII: 776XM7047L) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color WHITE Score no score Shape OVAL Size 16mm Flavor Imprint Code EP340 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:49884-170-04 1 in 1 CARTON 04/29/2016 1 250 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA204395 04/29/2016 Labeler - Par Pharmaceutical, Inc. (092733690)