Label: DURYSTA- bimatoprost implant

- NDC Code(s): 0023-9652-01, 0023-9652-03
- Packager: Allergan, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated November 16, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DURYSTA safely and effectively. See full prescribing information for DURYSTA.
DURYSTA® (bimatoprost intracameral implant), for intracameral administration
Initial U.S. Approval: 2001
INDICATIONS AND USAGE
DURYSTA is a prostaglandin analog indicated for the reduction of intraocular pressure (IOP) in patients with open angle glaucoma (OAG) or ocular hypertension (OHT). (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Intracameral implant containing bimatoprost 10 mcg, in a drug delivery system. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
-
Endothelial cell loss: Due to possible corneal endothelial cell loss, administration of DURYSTA should be limited to a single implant per eye without retreatment. (5.1)
-
Corneal Adverse Reactions: DURYSTA has been associated with corneal adverse reactions and risks are increased with multiple implants. Use caution in patients with limited corneal endothelial cell reserve. (5.1)
- Iridocorneal Angle: DURYSTA should be used with caution in patients with narrow angles or anatomical angle obstruction. (5.2)
ADVERSE REACTIONS
In controlled studies, the most common ocular adverse reaction reported by 27% of patients was conjunctival hyperemia. Other common adverse reactions reported in 5-10% of patients were foreign body sensation, eye pain, photophobia, conjunctival hemorrhage, dry eye, eye irritation, intraocular pressure increased, corneal endothelial cell loss, vision blurred, iritis, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2023
-
Endothelial cell loss: Due to possible corneal endothelial cell loss, administration of DURYSTA should be limited to a single implant per eye without retreatment. (5.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Information
2.2 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
4.2 Corneal Endothelial Cell Dystrophy
4.3 Prior Corneal Transplantation
4.4 Absent or Ruptured Posterior Lens Capsule
4.5 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Corneal Adverse Reactions
5.2 Iridocorneal Angle
5.3 Macular Edema
5.4 Intraocular Inflammation
5.5 Pigmentation
5.6 Endophthalmitis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2
DOSAGE AND ADMINISTRATION
2.1 General Information
DURYSTA is an ophthalmic drug delivery system for a single intracameral administration of a biodegradable implant. DURYSTA should not be readministered to an eye that received a prior DURYSTA.
2.2 Administration
The intracameral injection procedure must be performed under magnification that allows clear visualization of the anterior chamber structures and should be carried out using standard aseptic conditions for intracameral procedures, with the patient’s head in a stabilized position. The eye should not be dilated prior to the procedure.
Remove the foil pouch from the carton and examine for damage. Then, open the foil pouch over a sterile field and gently drop the applicator on a sterile tray. Once the foil pouch is opened, use promptly.
Figure 1
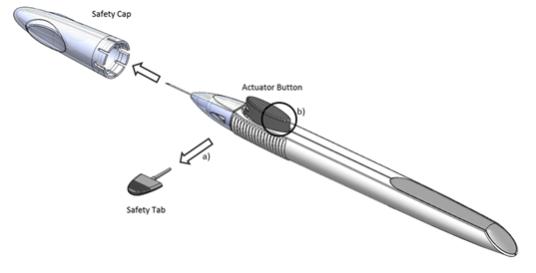
Perform a detailed visual inspection of the applicator, including ensuring that the actuator button has not been depressed, and the safety tab is in place. Carefully remove the plastic safety cap taking care to avoid contacting the needle tip. Inspect the needle tip for damage under magnification prior to use; the implant retention plug may be visible in the bevel and should not be removed.
Prior to use, remove the safety tab by pulling it out perpendicular to the long axis of the applicator (refer to Figure 1a above). Do not twist or bend the tab.
Stabilize the eye as the needle is advanced through the cornea. Enter the anterior chamber with the needle bevel visible through clear cornea. Enter parallel to the iris plane, adjacent to the limbus through clear cornea in the superotemporal quadrant.
The needle should be inserted approximately 2 bevel lengths with the bevel completely within the anterior chamber; avoid positioning the needle bevel directly over the pupil. Ensure the needle is not bent before depressing the actuator button. See Figure 2.
Figure 2
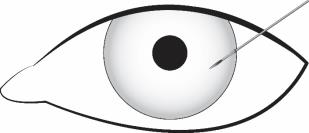
Depress the back half of the actuator button (refer to Figure 1b above) firmly until an audible and/or palpable click is noted.
Following the release of the implant, remove the needle via the same track in which it was inserted and tamponade the opening. The implant should not be left in the corneal injection track.
Check the injection site for leaks; make sure that it is self-sealing and the anterior chamber is formed.
After injection, do not recap the needle. Dispose of the used applicator in a sharps disposal container and in accordance with local requirements.
Instruct the patient to remain upright for at least 1 hour after the procedure so the implant can settle.
Some degree of eye redness and discomfort is expected following administration. However, it is recommended to instruct patients that if the eye becomes progressively red, sensitive to light, painful, or develops a change in vision, they should immediately contact the physician.
- 3 DOSAGE FORMS AND STRENGTHS
-
4
CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
DURYSTA is contraindicated in patients with active or suspected ocular or periocular infections.
4.2 Corneal Endothelial Cell Dystrophy
DURYSTA is contraindicated in patients with corneal endothelial cell dystrophy (e.g., Fuchs’ Dystrophy) [see Warnings and Precautions (5.1)].
4.3 Prior Corneal Transplantation
DURYSTA is contraindicated in patients with prior corneal transplantation, or endothelial cell transplants [e.g., Descemet’s Stripping Automated Endothelial Keratoplasty (DSAEK)].
4.4 Absent or Ruptured Posterior Lens Capsule
DURYSTA is contraindicated in patients whose posterior lens capsule is absent or ruptured, due to the risk of implant migration into the posterior segment. Laser posterior capsulotomy in pseudophakic patients is not a contraindication for DURYSTA use if the intraocular lens fully covers the opening in the posterior capsule.
4.5 Hypersensitivity
DURYSTA is contraindicated in patients with hypersensitivity to bimatoprost or to any other components of the product [see Adverse Reactions (6.1)].
-
5
WARNINGS AND PRECAUTIONS
5.1 Corneal Adverse Reactions
The presence of DURYSTA implants has been associated with corneal adverse reactions and increased risk of corneal endothelial cell loss. Administration of DURYSTA should be limited to a single implant per eye without retreatment. Caution should be used when prescribing DURYSTA in patients with limited corneal endothelial cell reserve.
5.2 Iridocorneal Angle
Following administration with DURYSTA, the intracameral implant is intended to settle within the inferior angle. DURYSTA should be used with caution in patients with narrow iridocorneal angles (Shaffer grade ˂ 3) or anatomical obstruction (e.g., scarring) that may prohibit settling in the inferior angle.
5.3 Macular Edema
Macular edema, including cystoid macular edema, has been reported during treatment with ophthalmic bimatoprost, including DURYSTA intracameral implant. DURYSTA should be used with caution in aphakic patients, in pseudophakic patients with a torn posterior lens capsule, or in patients with known risk factors for macular edema.
5.4 Intraocular Inflammation
Prostaglandin analogs, including DURYSTA, have been reported to cause intraocular inflammation. DURYSTA should be used with caution in patients with active intraocular inflammation (e.g., uveitis) because the inflammation may be exacerbated.
5.5 Pigmentation
Ophthalmic bimatoprost, including DURYSTA intracameral implant, has been reported to cause changes to pigmented tissues, such as increased pigmentation of the iris. Pigmentation of the iris is likely to be permanent. Patients who receive treatment should be informed of the possibility of increased pigmentation. The pigmentation change is due to increased melanin content in the melanocytes rather than to an increase in the number of melanocytes. While treatment with DURYSTA can be continued in patients who develop noticeably increased iris pigmentation, these patients should be examined regularly.
-
6
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Implant migration [see Contraindications (4.4)]
- Hypersensitivity [see Contraindications (4.5)]
- Corneal adverse reactions [see Warnings and Precautions (5.1)]
- Macular edema [see Warnings and Precautions (5.3)]
- Intraocular inflammation [see Warnings and Precautions (5.4)]
- Pigmentation [see Warnings and Precautions (5.5)]
- Endophthalmitis [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common ocular adverse reaction observed in two randomized, active-controlled clinical trials with DURYSTA in patients with OAG or OHT was conjunctival hyperemia, which was reported in 27% of patients. Other common ocular adverse reactions reported in 5-10% of patients were foreign body sensation, eye pain, photophobia, conjunctival hemorrhage, dry eye, eye irritation, intraocular pressure increased, corneal endothelial cell loss, vision blurred, and iritis. Ocular adverse reactions occurring in 1-5% of patients were anterior chamber cell, lacrimation increased, corneal edema, aqueous humor leakage, iris adhesions, ocular discomfort, corneal touch, iris hyperpigmentation, anterior chamber flare, anterior chamber inflammation, and macular edema. The following additional adverse drug reactions occurred in less than 1% of patients: hyphema, iridocyclitis, uveitis, corneal opacity, product administered at inappropriate site, corneal decompensation, cystoid macular edema, and drug hypersensitivity.
The most common nonocular adverse reaction was headache, which was observed in 5% of patients.
- Implant migration [see Contraindications (4.4)]
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies of DURYSTA (bimatoprost intracameral implant) administration in pregnant women to inform a drug associated risk. Oral administration of bimatoprost to pregnant rats and mice throughout organogenesis did not produce adverse maternal or fetal effects at clinically relevant exposures. Oral administration of bimatoprost to rats from the start of organogenesis to the end of lactation did not produce adverse maternal, fetal or neonatal effects at clinically relevant exposures [see Animal Data].
Data
Animal Data
In an embryofetal development rat study, oral administration of bimatoprost to pregnant rats during organogenesis produced abortion at 0.6 mg/kg/day (1770-times the human systemic exposure to bimatoprost from DURYSTA, based on Cmax and a blood-to plasma partition ratio of 0.858). The No Observed Adverse Effect Level (NOAEL) for abortion was 0.3 mg/kg/day (estimated at 470-times the human systemic exposure to bimatoprost from DURYSTA, based on Cmax). No fetal abnormalities were observed at doses up to 0.6 mg/kg/day.
In an embryofetal development mouse study, oral administration of bimatoprost to pregnant mice during organogenesis produced abortion and early delivery at 0.3 mg/kg/day (2240-times the human systemic exposure to bimatoprost from DURYSTA, based on plasma Cmax level; blood-to plasma partition ratio of 0.858). The NOAEL for abortion and early delivery was 0.1 mg/kg/day (400-times the human systemic exposure to bimatoprost from DURYSTA, based on Cmax). No fetal abnormalities were observed at doses up to 0.6 mg/kg/day (5200-times the human systemic exposure to bimatoprost from DURYSTA, based on Cmax).
In a pre/postnatal development study, oral administration of bimatoprost to pregnant rats from gestation day 7 through lactation resulted in reduced gestation length, increased late resorptions, fetal deaths, and postnatal pup mortality, and reduced pup body weight at 0.3 mg/kg/day (estimated 470-times the human systemic exposure to bimatoprost from DURYSTA, based plasma Cmax and a blood-to plasma partition ratio of 0.858). No adverse effects were observed in rat offspring at 0.1 mg/kg/day (estimated 350-times the human systemic exposure to bimatoprost from DURYSTA, based on plasma Cmax).
8.2 Lactation
Risk Summary
There is no information regarding the presence of bimatoprost in human milk, the effects on the breastfed infants, or the effects on milk production. In animal studies, topical bimatoprost has been shown to be excreted in breast milk. Because many drugs are excreted in human milk, caution should be exercised when DURYSTA is administered to a nursing woman.
The developmental and health benefits of breastfeeding should be considered, along with the mother's clinical need for DURYSTA and any potential adverse effects on the breastfed child from DURYSTA.
-
11
DESCRIPTION
DURYSTA is a sterile intracameral implant containing 10 mcg of bimatoprost, a prostaglandin analog, in a solid polymer sustained-release drug delivery system (DDS). The drug delivery system consists of poly (D,L-lactide), poly (D,L-lactide-co-glycolide), poly (D,L-lactide) acid end, and polyethylene glycol 3350. DURYSTA is preloaded into a single-use, DDS applicator to facilitate injection of the rod-shaped implant directly into the anterior chamber of the eye. The chemical name for bimatoprost is (Z)-7-[(1R,2R,3R,5S)-3,5-dihydroxy-2-[(1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-N-ethyl-5-heptenamide, and its molecular weight is 415.57. Its molecular formula is C25H37NO4. Its structural formula is:
![The chemical name for bimatoprost is (Z)-7-[(1R,2R,3R,5S)-3,5-dihydroxy-2-[(1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-N-ethyl-5-heptenamide, and its molecular weight is 415.57. Its molecular formula is C25H37NO4.](/dailymed/image.cfm?name=durysta-03.jpg&setid=3f59da84-0bcc-4c84-b3e2-e215681ef341)
Bimatoprost is a white to off-white powder, soluble in ethyl alcohol and methyl alcohol and slightly soluble in water. The polymer matrix slowly degrades to lactic acid and glycolic acid.
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bimatoprost, a prostaglandin analog, is a synthetic structural analog of prostaglandin with ocular hypotensive activity. Bimatoprost is believed to lower IOP in humans by increasing outflow of aqueous humor through both the trabecular meshwork (conventional) and uveoscleral routes (unconventional). Elevated IOP presents a major risk factor for glaucomatous field loss. The higher the level of IOP, the greater the likelihood of optic nerve damage and visual field loss.
12.3 Pharmacokinetics
After a single administration of DURYSTA, bimatoprost concentrations were below the lower limit of quantitation (0.001 ng/mL) in the majority (approximately 92%) of patients. The maximum bimatoprost concentration observed in any patient was 0.00224 ng/mL. Bimatoprost acid concentrations were also below the lower limit of quantitation (0.01 ng/mL) in almost all (approximately 99%) of patients.
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Bimatoprost was not carcinogenic in either mice or rats when administered by oral gavage at doses up to 2 mg/kg/day and 1 mg/kg/day respectively for 104 weeks (approximately 3100 and 1700 times, respectively, the maximum human exposure [based on plasma Cmax levels; blood-to-plasma partition ratio of 0.858]).
Mutagenesis
Bimatoprost was not mutagenic or clastogenic in the Ames test, in the mouse lymphoma test, or in the in vivo mouse micronucleus tests.
Impairment of Fertility
Bimatoprost did not impair fertility in male or female rats up to doses of 0.6 mg/kg/day (1770-times the maximum human exposure, based on plasma Cmax; blood-to-plasma partition ratio of 0.858).
-
14
CLINICAL STUDIES
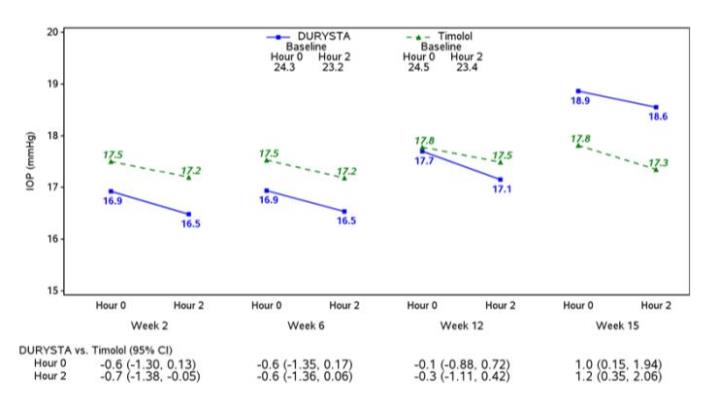
Efficacy was evaluated in two multicenter, randomized, parallel-group, controlled 20-month (including 8-month extended follow-up) studies of DURYSTA compared to twice daily topical timolol 0.5% drops, in patients with OAG or OHT. DURYSTA demonstrated an IOP reduction of approximately 5-8 mmHg in patients with a mean baseline IOP of 24.5 mmHg (see Figures 3 and 4).
Figure 3: Study 1 Mean IOP (mmHg) by Treatment Group and Treatment Difference in Mean IOP
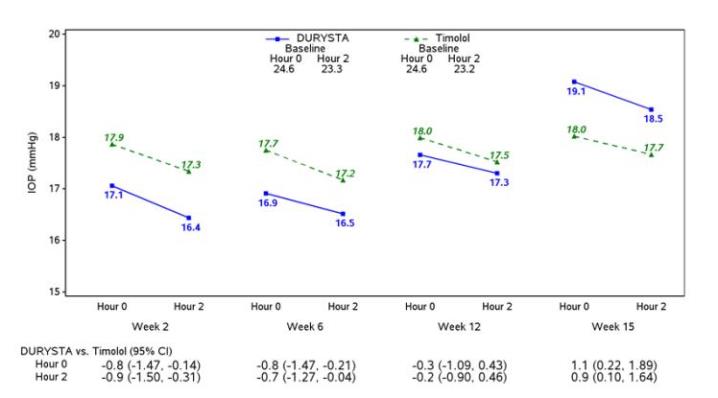
Figure 4: Study 2 Mean IOP (mmHg) by Treatment Group and Treatment Difference in Mean IOP
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17
PATIENT COUNSELING INFORMATION
Treatment-related Effects
Advise patients about the potential risk for complications including, but not limited to, the development of corneal adverse events, intraocular inflammation or endophthalmitis [see Warnings and Precautions (5.1, 5.4, 5.6)].
Potential for Pigmentation
Advise patients about the potential for increased brown pigmentation of the iris, which may be permanent [see Warnings and Precautions (5.5)].
When to Seek Physician Advice
Advise patients that if the eye becomes red, sensitive to light, painful, or develops a change in vision, they should seek immediate care from an ophthalmologist [see Warnings and Precautions (5.6)].
Distributed by: AbbVie Inc.
North Chicago, IL 60064© 2024 AbbVie. All rights reserved.
DURYSTA and its design are trademarks of Allergan, Inc., an AbbVie company.
v1.2USPI9652
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
DURYSTA
bimatoprost implantProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0023-9652 Route of Administration INTRACAMERAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BIMATOPROST (UNII: QXS94885MZ) (BIMATOPROST - UNII:QXS94885MZ) BIMATOPROST 10 ug Inactive Ingredients Ingredient Name Strength POLYLACTIDE (UNII: 459TN2L5F5) POLY(DL-LACTIC-CO-GLYCOLIC ACID), (50:50; 12000 MW) (UNII: WE369X5600) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0023-9652-01 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 03/04/2020 2 NDC:0023-9652-03 1 in 1 POUCH; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 03/04/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211911 03/04/2020 Labeler - Allergan, Inc. (144796497)
