Label: TEPEZZA- teprotumumab injection, powder, lyophilized, for solution

- NDC Code(s): 75987-130-15
- Packager: Horizon Therapeutics USA, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated November 10, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TEPEZZA® safely and effectively. See full prescribing information for TEPEZZA.
TEPEZZA (teprotumumab-trbw) for injection, for intravenous use
Initial U.S. Approval: 2020RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TEPEZZA is an insulin-like growth factor-1 receptor inhibitor indicated for the treatment of Thyroid Eye Disease (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
For Injection: 500 mg lyophilized powder in a single-dose vial for reconstitution (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Infusion Reactions: If an infusion reaction occurs, interrupt or slow the rate of infusion and use appropriate medical management (5.1)
- Inflammatory Bowel Disease (IBD): Monitor patients; for signs and symptoms of disease, including patients without a history of IBD; discontinue TEPEZZA if IBD exacerbation is suspected (5.2)
- Hyperglycemia: Assess patients for elevated blood glucose and symptoms of hyperglycemia prior to infusion and continue to monitor while on treatment with TEPEZZA. Ensure patients with hyperglycemia or preexisting diabetes are under appropriate glycemic control before and while receiving TEPEZZA (5.3)
- Hearing Impairment Including Hearing Loss: TEPEZZA may cause severe hearing impairment including hearing loss, which in some cases may be permanent. Assess patients' hearing before, during, and after treatment with TEPEZZA and consider the benefit-risk of treatment with patients (5.4)
ADVERSE REACTIONS
Most common adverse reactions (incidence greater than 5%) are muscle spasm, nausea, alopecia, diarrhea, fatigue, hyperglycemia, hearing impairment, dry skin, dysgeusia, headache, weight decreased, and nail disorder (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Females of Reproductive Potential: Appropriate forms of contraception should be implemented prior to initiation, during treatment and for 6 months following the last dose of TEPEZZA (8.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Reconstitution and Preparation
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infusion Reactions
5.2 Inflammatory Bowel Disease
5.3 Hyperglycemia
5.4 Hearing Impairment Including Hearing Loss
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dose of TEPEZZA is an intravenous infusion of 10 mg/kg for the initial dose followed by an intravenous infusion of 20 mg/kg every three weeks for 7 additional infusions.
2.2 Reconstitution and Preparation
Step 1: Calculate the dose (mg) and determine the number of vials needed for the 10 or 20 mg/kg dosage based on patient weight. Each TEPEZZA vial contains 500 mg of the teprotumumab antibody.
Step 2: Using appropriate aseptic technique, reconstitute each TEPEZZA vial with 10 mL of Sterile Water for Injection, USP. Ensure that the stream of diluent is not directed onto the lyophilized powder, which has a cake-like appearance. Do not shake, but gently swirl the solution by rotating the vial until the lyophilized powder is dissolved. The reconstituted solution has a total volume of 10.5 mL. Withdraw 10.5 mL of reconstituted solution to obtain 500 mg. After reconstitution, the final concentration is 47.6 mg/mL.
Step 3: The reconstituted TEPEZZA solution must be further diluted in 0.9% Sodium Chloride Injection, USP prior to infusion. To prepare the diluted solution, use 100 mL infusion bags for a dose less than 1800 mg, and 250 mL infusion bags for a dose equal to or greater than 1800 mg. To maintain a constant volume in the infusion bag, a sterile syringe and needle should be used to remove the volume equivalent to the amount of the reconstituted TEPEZZA solution to be placed into the infusion bag. Discard the volume of 0.9% Sodium Chloride Injection, USP withdrawn.
Step 4: Withdraw the required volume from the reconstituted TEPEZZA vial(s) based on the patient's weight (in kg) and transfer into the intravenous bag containing 0.9% Sodium Chloride Injection, USP. Mix diluted solution by gentle inversion. Do not shake. If refrigerated prior to administration, allow the diluted solution to reach room temperature prior to infusion.
The product does not contain any preservative. The combined storage time of reconstituted TEPEZZA solution in the vial and the diluted solution in the infusion bag containing 0.9% Sodium Chloride Injection, USP is a total of 4 hours at room temperature 20°C to 25°C (68°F to 77°F) or up to 48 hours under refrigerated conditions 2°C to 8°C (36°F to 46°F) protected from light.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Upon reconstitution, TEPEZZA is a nearly colorless or slightly brown, clear to opalescent solution which is free of foreign particulate matter. Discard the solution if any particulate matter or discoloration are observed.
Do not freeze the reconstituted or diluted solution.
Discard vial(s) and all unused contents.
No incompatibilities between TEPEZZA and polyethylene (PE), polyvinyl chloride (PVC), polyurethane (PUR) or polyolefin (PO) bags and intravenous administration sets have been observed.
2.3 Administration
Administer the diluted solution intravenously over at least 90 minutes for the first two infusions. If well tolerated, the minimum time for subsequent infusions can be reduced to 60 minutes. If not well tolerated, the minimum time for subsequent infusions should remain at 90 minutes, and pre-medication is recommended for subsequent infusions.
Do not administer as an intravenous push or bolus. TEPEZZA should not be infused concomitantly with other agents.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Infusion Reactions
TEPEZZA may cause infusion reactions. Infusion reactions have been reported in approximately 4% of patients treated with TEPEZZA. Signs and symptoms of infusion-related reactions include transient increases in blood pressure, feeling hot, tachycardia, dyspnea, headache and muscular pain. Infusion reactions may occur during any of the infusions or within 1.5 hours after an infusion. Reported infusion reactions are usually mild or moderate in severity and can usually be successfully managed with corticosteroids and antihistamines. In patients who experience an infusion reaction, consideration should be given to pre-medicating with an antihistamine, antipyretic, corticosteroid and/or administering all subsequent infusions at a slower infusion rate.
5.2 Inflammatory Bowel Disease
TEPEZZA may cause an exacerbation of inflammatory bowel disease (IBD). IBD has been reported in some patients without a prior diagnosis of IBD [see Adverse Reactions (6.2)]. Monitor patients for signs and symptoms of IBD. If IBD exacerbation is suspected, discontinue use of TEPEZZA.
5.3 Hyperglycemia
Hyperglycemia or increased blood glucose may occur in patients treated with TEPEZZA. In the premarketing clinical trials, 10% of patients (two-thirds of whom had preexisting diabetes or impaired glucose tolerance) experienced hyperglycemia [see Adverse Reactions (6.1)]. Hyperglycemic events should be controlled with medications for glycemic control, if necessary.
Assess patients for elevated blood glucose and symptoms of hyperglycemia prior to infusion and continue to monitor while on treatment with TEPEZZA. Ensure patients with hyperglycemia or preexisting diabetes are under appropriate glycemic control before and while receiving TEPEZZA.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Infusion Reactions [see Warnings and Precautions (5.1)]
- Inflammatory Bowel Disease [see Warnings and Precautions (5.2)]
- Hyperglycemia [see Warnings and Precautions (5.3)]
- Hearing Impairment Including Hearing Loss [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of TEPEZZA was evaluated in two premarketing randomized, double-masked, placebo-controlled clinical trials (Study 1 [NCT01868997] and Study 2 [NCT03298867]) consisting of 170 patients with Thyroid Eye Disease (84 received TEPEZZA and 86 received placebo). Patients were treated with TEPEZZA (10 mg/kg for first infusion and 20 mg/kg for the remaining 7 infusions) or placebo given as an intravenous infusion every 3 weeks for a total of 8 infusions. The majority of patients completed 8 infusions (89% of TEPEZZA patients and 93% of placebo patients).
The most common adverse reactions (≥ 5%) that occurred at greater incidence in the TEPEZZA group than in the control group during the treatment period of Studies 1 and 2 are summarized in Table 1.
In addition, menstrual disorders (amenorrhea, metrorrhagia, dysmenorrhea) were reported in approximately 23% (5 of 22 patients) of menstruating women treated with TEPEZZA compared to 4% (1 of 25 patients) treated with placebo in the premarketing clinical trials.
Table 1. Adverse Reactions Occurring in 5% or More of Patients Treated with TEPEZZA and Greater Incidence than Placebo Adverse Reactions TEPEZZA
N = 84
N (%)Placebo
N = 86
N (%)- *
- Fatigue includes asthenia
- †
- Hyperglycemia includes blood glucose increase
- ‡
- Hearing impairment including hearing loss (deafness, including sensorineural deafness, eustachian tube dysfunction, hyperacusis, hypoacusis, autophony and tinnitus)
- §
- Nail disorder (includes nail discoloration, nail disorder and onychoclasis)
Muscle spasms 21 (25%) 6 (7%) Nausea 14 (17%) 8 (9%) Alopecia 11 (13%) 7 (8%) Diarrhea 10 (12%) 7 (8%) Fatigue* 10 (12%) 6 (7%) Hyperglycemia† 8 (10%) 1 (1%) Hearing impairment‡ 8 (10%) 0 Dysgeusia 7 (8%) 0 Headache 7 (8%) 6 (7%) Dry skin 7 (8%) 0 Weight decreased 5 (6%) 0 Nail disorder§ 4 (5%) 0 In clinical trials, gastrointestinal complaints including exacerbation of inflammatory bowel disease were reported.
In two postapproval trials, the following were the adverse reactions occurring ≥ 5% of participants treated with TEPEZZA during the double-masked treatment period: muscle spasms (29%), hearing impairment (19%), hyperglycemia (18%) diarrhea (16%), fatigue (13%), headache (13%), alopecia (10%), dry skin (10%), dysgeusia (9%), ear discomfort (9%), and nail disorder (6%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of TEPEZZA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal Disorders: bowel perforation, exacerbation of inflammatory bowel disease (IBD), including patients without a prior diagnosis of IBD
Metabolism and Nutrition Disorders: diabetic ketoacidosis, hyperosmolar hyperglycemic state (HHS)
Otologic: severe hearing impairment including hearing loss, which in some cases may be permanent
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animals and its mechanism of action inhibiting insulin-like growth factor-1 receptor (IGF-1R) signaling, TEPEZZA may cause fetal harm when administered to a pregnant woman. Adequate and well-controlled studies with TEPEZZA have not been conducted in pregnant women. There are insufficient data with TEPEZZA use in pregnant women to inform any drug associated risks for adverse developmental outcomes. Cynomolgus monkeys dosed once weekly with teprotumumab during pregnancy resulted in placental changes, decreased fetal growth during pregnancy, decreased fetal size and weight, and multiple external and skeletal abnormalities in offspring. Teprotumumab exposure may lead to an increase in fetal loss [see Data]. Therefore, TEPEZZA should not be used in pregnancy, and appropriate forms of contraception should be implemented prior to initiation, during treatment and for 6 months following the last dose of TEPEZZA. Women of childbearing age should have a pregnancy test performed by their doctor before starting treatment with TEPEZZA. If the patient becomes pregnant during treatment, TEPEZZA should be discontinued and the patient advised of the potential risk to the fetus.
The background rate of major birth defects and miscarriage is unknown for the indicated population. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2-4% and 15-20%, respectively.
Data
Animal Data
In an abridged pilot embryo-fetal development study, seven pregnant cynomolgus monkeys were dosed intravenously at one dose level of teprotumumab, 75 mg/kg (2.8-fold the maximum recommended human dose (MRHD) based on AUC) once weekly from gestation day 20 through the end of gestation. The incidence of abortion was higher for the teprotumumab treated group compared to the control group. Teprotumumab caused decreased fetal growth during pregnancy, decreased fetal size and weight at caesarean section, decreased placental weight and size, and decreased amniotic fluid volume. Multiple external and skeletal abnormalities were observed in each exposed fetus, including: misshapen cranium, closely set eyes, micrognathia, pointing and narrowing of the nose, and ossification abnormalities of skull bones, sternebrae, carpals, tarsals and teeth. The test dose, 75 mg/kg of teprotumumab, was the maternal no observed adverse effect level (NOAEL).
Based on mechanism of action inhibiting IGF-1R, postnatal exposure to teprotumumab may cause harm.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Based on its mechanism of action inhibiting IGF-1R, TEPEZZA may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception prior to initiation, during treatment with TEPEZZA and for 6 months after the last dose of TEPEZZA.
8.5 Geriatric Use
Of the 171 patients in the two randomized trials, 15% were 65 years of age or older; the number of patients 65 years or older was similar between treatment groups. No overall differences in efficacy or safety were observed between patients 65 years or older and younger patients (less than 65 years of age).
- 10 OVERDOSAGE
-
11 DESCRIPTION
Teprotumumab-trbw, an insulin-like growth factor-1 receptor inhibitor (IGF-1R), is a fully human IgG1 monoclonal antibody produced in Chinese hamster ovary (CHO-DG44) cells. It has a molecular weight of approximately 148 kilodaltons.
TEPEZZA (teprotumumab-trbw) for injection is supplied as a sterile, preservative-free, white to off-white, lyophilized powder for intravenous infusion. Each single-dose vial delivers 500 mg of teprotumumab-trbw, histidine (7.82 mg), L-histidine hydrochloride monohydrate (33.43 mg), polysorbate 20 (1.05 mg), and trehalose (898.43 mg). After reconstitution with 10 mL of Sterile Water for Injection, USP, the final concentration is 47.6 mg/mL with a pH of 5.5.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Teprotumumab-trbw's mechanism of action in patients with Thyroid Eye Disease has not been fully characterized. Teprotumumab-trbw binds to IGF-1R and blocks its activation and signaling.
12.3 Pharmacokinetics
The pharmacokinetics of teprotumumab-trbw was described by a two compartment population PK model based on data from 40 patients with Thyroid Eye Disease receiving an initial intravenous infusion of 10 mg/kg, followed by infusions of 20 mg/kg TEPEZZA every 3 weeks in one clinical trial. Following this regimen, the mean (± standard deviation) estimates for steady-state area under the concentration curve (AUC), peak (Cmax), and trough (Ctrough) concentrations of teprotumumab-trbw were 138 (± 34) mg∙hr/mL, 632 (± 139) mcg/mL, and 176 (± 56) mcg/mL, respectively.
Distribution
Following the recommended TEPEZZA dosing regimen, the population PK estimated mean (± standard deviation) for central and peripheral volume of distribution of teprotumumab-trbw were 3.26 (± 0.87) L and 4.32 (± 0.67) L, respectively. The mean (± standard deviation) estimated inter-compartment clearance was 0.74 (± 0.16) L/day.
Elimination
Following the recommended TEPEZZA dosing regimen, the population PK estimated mean (± standard deviation) for the clearance of teprotumumab-trbw was 0.27 (± 0.08) L/day and for the elimination half-life was 20 (± 5) days.
Specific Populations
No clinically significant differences in the pharmacokinetics of teprotumumab-trbw were observed following administration of TEPEZZA based on patient's age (18-80 years), gender, race/ethnicity (103 White, 10 Black, and 3 Asian), weight (46-169 kg), mild to moderate renal impairment (creatinine clearance 30 to 89 mL/min estimated by Cockcroft-Gault Equation), bilirubin levels (2.7-24.3 mcmol/L), aspartate aminotransferase (AST) levels (11-221 U/L), or alanine aminotransferase (ALT) levels (7-174 U/L). The effect of hepatic impairment on the pharmacokinetics of teprotumumab-trbw is unknown.
12.6 Immunogenicity
The observed incidence of anti-drug antibody is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibody in the studies described below with the incidence of anti-drug antibodies in other studies, including those of TEPEZZA.
In a randomized placebo-controlled study, HZNP-TEP-301, where active TED patients were administered placebo or TEPEZZA over a 24-week period, 2.4% (1 of 42) of placebo and 0% (0 of 41) of TEPEZZA treated patients tested positive for anti-teprotumumab antibodies at post-baseline visits.
Because of the low occurrence of anti-teprotumumab antibodies, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of teprotumumab products is unknown.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
TEPEZZA was evaluated in 2 randomized, double-masked, placebo-controlled studies in 171 patients with Thyroid Eye Disease: Study 1 (NCT01868997) and Study 2 (NCT03298867). Patients were randomized to receive TEPEZZA or placebo in a 1:1 ratio. Patients were given intravenous infusions (10 mg/kg for first infusion and 20 mg/kg for the remaining 7 infusions) every 3 weeks for a total of 8 infusions. Patients had a clinical diagnosis of Thyroid Eye Disease with symptoms and were euthyroid or had thyroxine and free triiodothyronine levels less than 50% above or below normal limits. Prior surgical treatment for Thyroid Eye Disease was not permitted. Proptosis ranged from 16 to 33 mm and 125 patients (73%) had diplopia at baseline.
A total of 84 patients were randomized to TEPEZZA and 87 patients were randomized to placebo. The median age was 52 years (range 20 to 79 years), 86% were White, 9% were Black or African-American, 4% were Asian and 1% identified as Other. The majority (73%) were female. At baseline, 27% of patients were smokers.
The proptosis responder rate at week 24 was defined as the percentage of patients with ≥ 2 mm reduction in proptosis in the study eye from baseline, without deterioration in the non-study eye (≥ 2 mm increase) in proptosis. Additional evaluations included signs and symptoms of Thyroid Eye Disease including pain, gaze evoked orbital pain, swelling, eyelid erythema, redness, chemosis, inflammation, clinical activity score and assessments of functional vision and patient appearance. Results for proptosis are found in Table 2.
Table 2. Efficacy Results in Patients with Thyroid Eye Disease in Study 1 and 2 Study 1 Study 2 Teprotumumab
(N = 42)Placebo
(N = 45)Difference
(95% CI)Teprotumumab
(N = 41)Placebo
(N = 42)Difference
(95% CI)- *
- Difference and its corresponding 95% Confidence Interval (CI) is based on a weighted average of the difference within each randomization stratum (tobacco user, tobacco non-use) using CMH weights.
- †
- Results were obtained from an MMRM with an unstructured covariance matrix and including treatment, smoking status, baseline value, visit, treatment by visit, and visit by baseline value interaction as fixed effects. A change from baseline of 0 was imputed at the first post-baseline visit for any subject without a post baseline value.
Proptosis responder rate at week 24, % (n) * 71% (30) 20% (9) 51%
(33, 69)83% (34) 10% (4) 73%
(59, 88)Proptosis (mm) average change from baseline through week 24, LS Mean (SE)† -2.5 (0.2) -0.2 (0.2) -2.3
(-2.8, -1.8)-2.8 (0.2) -0.5 (0.2) -2.3
(-2.8, -1.8)In Study 2, improvement of proptosis as measured by mean change from Baseline was observed as early as 6 weeks and continued to improve through week 24 as shown in Figure 1. Similar results were seen in Study 1.
Figure 1. Change from Baseline in Proptosis over 24 Weeks in Study 2
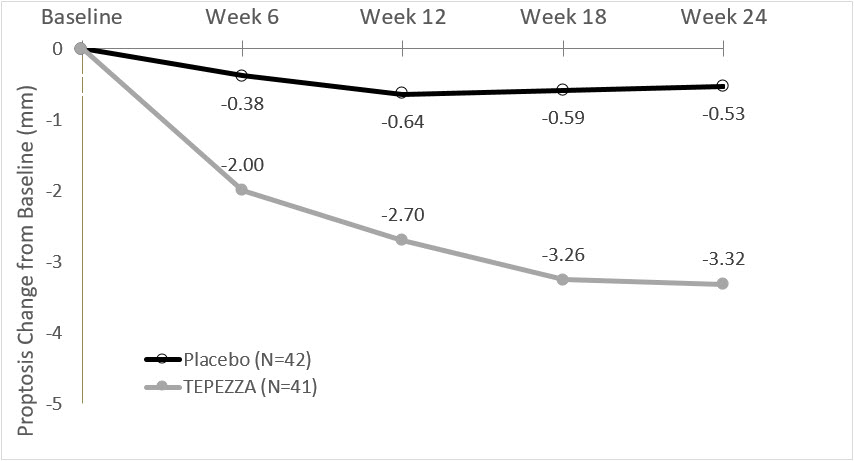
P < 0.01 at each timepoint
TEPEZZA also led to improvement in the less severely impacted "fellow" eye.
Diplopia (double vision) was evaluated in a subgroup of patients that had diplopia at baseline in Study 1 and 2. Results are shown in Table 3.
Table 3. Diplopia in Patients with Thyroid Eye Disease in Study 1 and 2 Parameter TEPEZZA
(n = 66)Placebo
(n = 59)P < 0.01 - *
- Diplopia was evaluated on a 4-point scale where scores ranged from 0 for no diplopia to 3 for constant diplopia. A diplopia responder was defined as a patient with baseline diplopia > 0 and a score of 0 at week 24.
Diplopia Responder rate* at week 24, % (n) 53% (35) 25% (15) Following discontinuation of treatment in Study 1, 53% of patients (16 of 30 patients) who were proptosis responders at week 24 maintained proptosis response 51 weeks after the last TEPEZZA infusion. 67% of patients (12 of 18) who were diplopia responders at week 24 maintained diplopia response 51 weeks after the last TEPEZZA infusion.
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Embryo-Fetal Toxicity
- Advise females of reproductive potential that TEPEZZA can cause harm to a fetus and to inform their healthcare provider of a known or suspected pregnancy.
- Educate and counsel females of reproductive potential about the need to use effective contraception prior to initiation, during treatment with TEPEZZA and for 6 months after the last dose of TEPEZZA.
Infusion-related reactions
- Advise patients that TEPEZZA may cause infusion reactions that can occur at any time. Instruct patients to recognize the signs and symptoms of infusion reaction and to contact their healthcare provider immediately for signs or symptoms of potential infusion-related reactions.
Inflammatory Bowel Disease
- Advise patients on the risk of inflammatory bowel disease (IBD) including patients with or without a prior diagnosis of IBD. Advise patients to seek medical advice immediately if they experience diarrhea, with or without blood or rectal bleeding, associated with abdominal pain or cramping/colic, fecal urgency, tenesmus or incontinence.
Hyperglycemia
- Advise patients on the risk of hyperglycemia and, if diabetic, discuss with healthcare provider to adjust glycemic control measures including medications as appropriate. Encourage compliance with glycemic control.
Hearing Impairment Including Hearing Loss
- Advise patients that TEPEZZA may cause severe hearing impairment including hearing loss, which in some cases may be permanent. Instruct patients to contact their healthcare provider if they experience any signs or symptoms of hearing impairment or any changes in hearing.
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 500 mg Vial Carton
-
INGREDIENTS AND APPEARANCE
TEPEZZA
teprotumumab injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:75987-130 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TEPROTUMUMAB (UNII: Y64GQ0KC0A) (TEPROTUMUMAB - UNII:Y64GQ0KC0A) TEPROTUMUMAB 500 mg Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 7.45 mg HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) POLYSORBATE 20 (UNII: 7T1F30V5YH) TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) Product Characteristics Color WHITE (white to off-white) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:75987-130-15 1 in 1 CARTON 01/21/2020 1 1 in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761143 01/21/2020 Labeler - Horizon Therapeutics USA, Inc. (033470838) Registrant - Catalent Indiana, LLC (172209277)

