Label: DIFLUNISAL tablet, film coated

-
Contains inactivated NDC Code(s)
NDC Code(s): 68151-0848-0 - Packager: Carilion Materials Management
- This is a repackaged label.
- Source NDC Code(s): 0093-0755
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated July 6, 2017
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
Cardiovascular Thrombotic Events
- •
- Nonsteroidal anti-inflammatory drugs (NSAIDs) cause an increased risk of serious cardiovascular thrombotic events, including myocardial infarction and stroke, which can be fatal. This risk may occur early in treatment and may increase with duration of use (see WARNINGS and PRECAUTIONS).
- •
- Diflunisal tablets are contraindicated in the setting of coronary artery bypass graft (CABG) surgery (see CONTRAINDICATIONS and WARNINGS).
Gastrointestinal Risk
- •
- NSAIDs cause an increased risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. These events can occur at any time during use and without warning symptoms. Elderly patients are at greater risk for serious gastrointestinal events (see WARNINGS).
-
DESCRIPTION
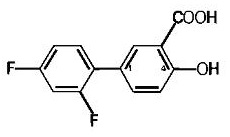
Diflunisal is 2',4'-difluoro-4-hydroxy-3-biphenylcarboxylic acid. Its structural formula is:
C13H8F2O3 M.W. 250.20
Diflunisal is a stable, white, crystalline compound with a melting point of 211° to 213°C. It is practically insoluble in water at neutral or acidic pH. Because it is an organic acid, it dissolves readily in dilute alkali to give a moderately stable solution at room temperature. It is soluble in most organic solvents including ethanol, methanol, and acetone.
Each tablet, for oral administration, contains 500 mg diflunisal, USP. In addition, each tablet contains the following inactive ingredients: croscarmellose sodium, FD&C Blue #2 aluminum lake, hypromellose, microcrystalline cellulose, pregelatinized starch, propylene glycol, sodium stearyl fumarate, and titanium dioxide.
-
CLINICAL PHARMACOLOGY
Action
Diflunisal is a non-steroidal drug with analgesic, anti-inflammatory and antipyretic properties. It is a peripherally-acting non-narcotic analgesic drug. Habituation, tolerance, and addiction have not been reported.
Diflunisal is a difluorophenyl derivative of salicylic acid. Chemically, diflunisal differs from aspirin (acetylsalicylic acid) in two respects. The first of these two is the presence of a difluorophenyl substituent at carbon 1. The second difference is the removal of the O-acetyl group from the carbon 4 position. Diflunisal is not metabolized to salicylic acid, and the fluorine atoms are not displaced from the difluorophenyl ring structure.
The precise mechanism of the analgesic and anti-inflammatory actions of diflunisal is not known. Diflunisal is a prostaglandin synthetase inhibitor. In animals, prostaglandins sensitize afferent nerves and potentiate the action of bradykinin in inducing pain. Since prostaglandins are known to be among the mediators of pain and inflammation, the mode of action of diflunisal may be due to a decrease of prostaglandins in peripheral tissues.
Pharmacokinetics and Metabolism
Diflunisal is rapidly and completely absorbed following oral administration with peak plasma concentrations occurring between 2 to 3 hours. The drug is excreted in the urine as two soluble glucuronide conjugates accounting for about 90% of the administered dose. Little or no diflunisal is excreted in the feces. Diflunisal appears in human milk in concentrations of 2 to 7% of those in plasma. More than 99% of diflunisal in plasma is bound to proteins.
As is the case with salicylic acid, concentration-dependent pharmacokinetics prevail when diflunisal is administered; a doubling of dosage produces a greater than doubling of drug accumulation. The effect becomes more apparent with repetitive doses. Following single doses, peak plasma concentrations of 41 ± 11 mcg/mL (mean ± S.D.) were observed following 250 mg doses, 87 ± 17 mcg/mL were observed following 500 mg and 124 ± 11 mcg/mL following single 1000 mg doses. However, following administration of 250 mg b.i.d., a mean peak level of 56 ± 14 mcg/mL was observed on day 8, while the mean peak level after 500 mg b.i.d. for 11 days was 190 ± 33 mcg/mL. In contrast to salicylic acid which has a plasma half-life of 2 1/2 hours, the plasma half-life of diflunisal is 3 to 4 times longer (8 to 12 hours), because of a difluorophenyl substituent at carbon 1. Because of its long half-life and nonlinear pharmacokinetics, several days are required for diflunisal plasma levels to reach steady state following multiple doses. For this reason, an initial loading dose is necessary to shorten the time to reach steady-state levels, and 2 to 3 days of observation are necessary for evaluating changes in treatment regimens if a loading dose is not used.
Studies in baboons to determine passage across the blood-brain barrier have shown that only small quantities of diflunisal, under normal or acidotic conditions are transported into the cerebrospinal fluid (CSF). The ratio of blood/CSF concentrations after intravenous doses of 50 mg/kg or oral doses of 100 mg/kg of diflunisal was 100:1. In contrast, oral doses of 500 mg/kg of aspirin resulted in a blood/CSF ratio of 5:1.
Mild to Moderate Pain
Diflunisal is a peripherally-acting analgesic agent with a long duration of action. Diflunisal produces significant analgesia within 1 hour and maximum analgesia within 2 to 3 hours.
Consistent with its long half-life, clinical effects of diflunisal mirror its pharmacokinetic behavior, which is the basis for recommending a loading dose when instituting therapy. Patients treated with diflunisal, on the first dose, tend to have a slower onset of pain relief when compared with drugs achieving comparable peak effects. However, diflunisal produces longer lasting responses than the comparative agents.
Comparative single dose clinical studies have established the analgesic efficacy of diflunisal at various dose levels relative to other analgesics. Analgesic effect measurements were derived from hourly evaluations by patients during eight and twelve hour postdosing observation periods. The following information may serve as a guide for prescribing diflunisal.
Diflunisal 500 mg was comparable in analgesic efficacy to aspirin 650 mg, acetaminophen 600 mg or 650 mg, and acetaminophen 650 mg with propoxyphene napsylate 100 mg. Patients treated with diflunisal had longer lasting responses than the patients treated with the comparative analgesics.
Diflunisal 1000 mg was comparable in analgesic efficacy to acetaminophen 600 mg with codeine 60 mg. Patients treated with diflunisal had longer lasting responses than the patients who received acetaminophen with codeine.
A loading dose of 1000 mg provides faster onset of pain relief, shorter time to peak analgesic effect, and greater peak analgesic effect than an initial 500 mg dose.
In contrast to the comparative analgesics, a significantly greater proportion of patients treated with diflunisal did not remedicate and continued to have a good analgesic effect eight to twelve hours after dosing. Seventy-five percent (75%) of patients treated with diflunisal continued to have a good analgesic response at four hours. When patients having a good analgesic response at four hours were followed, 78% of these patients continued to have a good analgesic response at eight hours and 64% at twelve hours.
Chronic Anti-Inflammatory Therapy in Osteoarthritis and Rheumatoid Arthritis
In the controlled, double-blind clinical trials in which diflunisal (500 mg to 1000 mg a day) was compared with anti-inflammatory doses of aspirin (2 to 4 grams a day), patients treated with diflunisal had a significantly lower incidence of tinnitus and of adverse effects involving the gastrointestinal system than patients treated with aspirin (see also Effect on Fecal Blood Loss).
Osteoarthritis
The effectiveness of diflunisal for the treatment of osteoarthritis was studied in patients with osteoarthritis of the hip and/or knee. The activity of diflunisal was demonstrated by clinical improvement in the signs and symptoms of disease activity.
In a double-blind multicenter study of 12 weeks' duration in which dosages were adjusted according to patient response, diflunisal 500 or 750 mg daily was shown to be comparable in effectiveness to aspirin 2000 or 3000 mg daily. In open-label extensions of this study to 24 or 48 weeks, diflunisal continued to show similar effectiveness and generally was well tolerated.
Rheumatoid Arthritis
In controlled clinical trials, the effectiveness of diflunisal was established for both acute exacerbations and long-term management of rheumatoid arthritis. The activity of diflunisal was demonstrated by clinical improvement in the signs and symptoms of disease activity.
In a double-blind multicenter study of 12 weeks' duration in which dosages were adjusted according to patient response, diflunisal 500 or 750 mg daily was comparable in effectiveness to aspirin 2600 or 3900 mg daily. In open-label extensions of this study to 52 weeks, diflunisal continued to be effective and was generally well tolerated.
Diflunisal 500, 750, or 1000 mg daily was compared with aspirin 2000, 3000, or 4000 mg daily in a multicenter study of 8 weeks' duration in which dosages were adjusted according to patient response. In this study, diflunisal was comparable in efficacy to aspirin.
In a double-blind multicenter study of 12 weeks' duration in which dosages were adjusted according to patient needs, diflunisal 500 or 750 mg daily and ibuprofen 1600 or 2400 mg daily were comparable in effectiveness and tolerability.
In a double-blind multicenter study of 12 weeks' duration, diflunisal 750 mg daily was comparable in efficacy to naproxen 750 mg daily. The incidence of gastrointestinal adverse effects and tinnitus was comparable for both drugs. This study was extended to 48 weeks on an open-label basis. Diflunisal continued to be effective and generally well tolerated.
In patients with rheumatoid arthritis, diflunisal and gold salts may be used in combination at their usual dosage levels. In clinical studies, diflunisal added to the regimen of gold salts usually resulted in additional symptomatic relief but did not alter the course of the underlying disease.
Antipyretic Activity
Diflunisal tablets are not recommended for use as an antipyretic agent. In single 250 mg, 500 mg, or 750 mg doses, diflunisal produced measurable but not clinically useful decreases in temperature in patients with fever; however, the possibility that it may mask fever in some patients, particularly with chronic or high doses, should be considered.
Uricosuric Effect
In normal volunteers, an increase in the renal clearance of uric acid and a decrease in serum uric acid was observed when diflunisal was administered at 500 mg or 750 mg daily in divided doses. Patients on long-term therapy taking diflunisal at 500 mg to 1000 mg daily in divided doses showed a prompt and consistent reduction across studies in mean serum uric acid levels, which were lowered as much as 1.4 mg%. It is not known whether diflunisal interferes with the activity of other uricosuric agents.
Effect on Platelet Function
As an inhibitor of prostaglandin synthetase, diflunisal has a dose-related effect on platelet function and bleeding time. In normal volunteers, 250 mg b.i.d. for 8 days had no effect on platelet function, and 500 mg b.i.d., the usual recommended dose, had a slight effect. At 1000 mg b.i.d., which exceeds the maximum recommended dosage, however, diflunisal inhibited platelet function. In contrast to aspirin, these effects of diflunisal were reversible, because of the absence of the chemically labile and biologically reactive O-acetyl group at the carbon 4 position. Bleeding time was not altered by a dose of 250 mg b.i.d., and was only slightly increased at 500 mg b.i.d. At 1000 mg b.i.d., a greater increase occurred, but was not statistically significantly different from the change in the placebo group.
Effect on Fecal Blood Loss
When diflunisal was given to normal volunteers at the usual recommended dose of 500 mg twice daily, fecal blood loss was not significantly different from placebo. Aspirin at 1000 mg four times daily produced the expected increase in fecal blood loss. Diflunisal at 1000 mg twice daily (NOTE: exceeds the recommended dosage) caused a statistically significant increase in fecal blood loss, but this increase was only one-half as large as that associated with aspirin 1300 mg twice daily.
-
INDICATIONS AND USAGE
Carefully consider the potential benefits and risks of diflunisal tablets and other treatment options before deciding to use diflunisal tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
Diflunisal tablets are indicated for acute or long-term use for symptomatic treatment of the following:
- 1.
- Mild to moderate pain
- 2.
- Osteoarthritis
- 3.
- Rheumatoid arthritis
-
CONTRAINDICATIONS
Diflunisal tablets are contraindicated in patients with known hypersensitivity to diflunisal or the excipients (see DESCRIPTION).
Diflunisal tablets should not be given to patients who have experienced asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal, anaphylactic/analphylactoid reactions to NSAIDs have been reported in such patients (see WARNINGS, Anaphylactic/Anaphylactoid Reactions and PRECAUTIONS, Preexisting Asthma).
Diflunisal tablets are contraindicated in the setting of coronary artery bypass graft (CABG) surgery (see WARNINGS).
-
WARNINGS
Cardiovascular Effects
Cardiovascular Thrombotic Events
Clinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, including myocardial infarction (MI) and stroke, which can be fatal. Based on available data, it is unclear that the risk for CV thrombotic events is similar for all NSAIDs. The relative increase in serious CV thrombotic events over baseline conferred by NSAID use appears to be similar in those with and without known CV disease or risk factors for CV disease. However, patients with known CV disease or risk factors had a higher absolute incidence of excess serious CV thrombotic events, due to their increased baseline rate. Some observational studies found that this increased risk of serious CV thrombotic events began as early as the first weeks of treatment. The increase in CV thrombotic risk has been observed most consistently at higher doses.
To minimize the potential risk for an adverse CV event in NSAID-treated patients, use the lowest effective dose for the shortest duration possible. Physicians and patients should remain alert for the development of such events, throughout the entire treatment course, even in the absence of previous CV symptoms. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID, such as diflunisal, increases the risk of serious gastrointestinal (GI) events (see WARNINGS).
Status Post Coronary Artery Bypass Graft (CABG) Surgery
Two large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10 to 14 days following CABG surgery found an increased incidence of myocardial infarction and stroke. NSAIDs are contraindicated in the setting of CABG (see CONTRAINDICATIONS).
Post-MI Patients
Observational studies conducted in the Danish National Registry have demonstrated that patients treated with NSAIDs in the post-MI period were at increased risk of reinfarction, CV-related death, and all-cause mortality beginning in the first week of treatment. In this same cohort, the incidence of death in the first year post MI was 20 per 100 person years in NSAID-treated patients compared to 12 per 100 person years in non-NSAID exposed patients. Although the absolute rate of death declined somewhat after the first year post-MI, the increased relative risk of death in NSAID users persisted over at least the next four years of follow-up.
Avoid the use of diflunisal tablets in patients with a recent MI unless the benefits are expected to outweigh the risk of recurrent CV thrombotic events. If diflunisal tablets are used in patients with a recent MI, monitor patients for signs of cardiac ischemia.
Hypertension
NSAIDs, including diflunisal tablets, can lead to onset of new hypertension or worsening of preexisting hypertension, either of which may contribute to the increased incidence of CV events. Patients taking thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs. NSAIDs, including diflunisal tablets, should be used with caution in patients with hypertension. Blood pressure (BP) should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy.
Heart Failure and Edema
The Coxib and traditional NSAID Trialists’ Collaboration meta-analysis of randomized controlled trials demonstrated an approximately two-fold increase in hospitalizations for heart failure in COX-2 selective-treated patients and nonselective NSAID-treated patients compared to placebo-treated patients. In a Danish National Registry study of patients with heart failure, NSAID use increased the risk of MI, hospitalization for heart failure, and death.
Additionally, fluid retention and edema have been observed in some patients treated with NSAIDs. Use of diflunisal may blunt the CV effects of several therapeutic agents used to treat these medical conditions [e.g., diuretics, ACE inhibitors, or angiotensin receptor blockers (ARBs)] (see DRUG INTERACTIONS).
Avoid the use of diflunisal tablets in patients with severe heart failure unless the benefits are expected to outweigh the risk of worsening heart failure. If diflunisal tablets are used in patients with severe heart failure, monitor patients for signs of worsening heart failure.
Gastrointestinal Effects – Risk of Ulceration, Bleeding and Perforation
NSAIDs, including diflunisal tablets, can cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs. Only one in five patients, who develop a serious upper GI adverse event on NSAID therapy, is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occur in approximately 1% of patients treated for 3 to 6 months, and in about 2 to 4% of patients treated for one year. These trends continue with longer duration of use, increasing the likelihood of developing a serious GI event at some time during the course of therapy. However, even short-term therapy is not without risk.
NSAIDs should be prescribed with extreme caution in those with a prior history of ulcer disease or gastrointestinal bleeding. Patients with a prior history of peptic ulcer disease and/or gastrointestinal bleeding who use NSAIDs have a greater than 10 fold increased risk for developing a GI bleed compared to patients with neither of these risk factors. Other factors that increase the risk for GI bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore, special care should be taken in treating this population.
To minimize the potential risk for an adverse GI event in patients treated with an NSAID, the lowest effective dose should be used for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of GI ulceration and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious GI adverse event is suspected. This should include discontinuation of the NSAID until a serious GI adverse event is ruled out. For high risk patients, alternate therapies that do not involve NSAIDs should be considered.
Renal Effects
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of a non-steroidal anti-inflammatory drug may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, patients who are volume depleted, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
Advanced Renal Disease
No information is available from controlled clinical studies regarding the use of diflunisal tablets in patients with advanced renal disease. Therefore, treatment with diflunisal tablets is not recommended in these patients with advanced renal disease. If diflunisal tablet therapy must be initiated, close monitoring of the patient's renal function is advisable.
Anaphylactic/Anaphylactoid Reactions
As with other NSAIDs, anaphylactic/anaphylactoid reactions may occur in patients without known prior exposure to diflunisal tablets. Diflunisal tablets should not be given to patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs (see CONTRAINDICATIONS and PRECAUTIONS, Preexisting Asthma). Emergency help should be sought in cases where an anaphylactic/ anaphylactoid reaction occurs.
Skin Reactions
NSAIDs, including diflunisal tablets, can cause serious skin adverse events such as exfoliative dermatitis, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Patients should be informed about the signs and symptoms of serious skin manifestations and use of the drug should be discontinued at the first appearance of skin rash or any other sign of hypersensitivity.
Hypersensitivity Syndrome
A potentially life-threatening, apparent hypersensitivity syndrome has been reported. This multisystem syndrome includes constitutional symptoms (fever, chills), and cutaneous findings (see ADVERSE REACTIONS, Dermatologic). It may also include involvement of major organs (changes in liver function, jaundice, leukopenia, thrombocytopenia, eosinophilia, disseminated intravascular coagulation, renal impairment, including renal failure), and less specific findings (adenitis, arthralgia, myalgia, arthritis, malaise, anorexia, disorientation). If evidence of hypersensitivity occurs, therapy with diflunisal tablets should be discontinued.
-
PRECAUTIONS
General
Diflunisal tablets cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to disease exacerbation. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
The pharmacological activity of diflunisal tablets in reducing fever and inflammation may diminish the utility of these diagnostic signs in detecting complications of presumed noninfectious, painful conditions.
Hepatic Effects
Borderline elevations of one or more liver tests may occur in up to 15% of patients taking NSAIDs including diflunisal tablets. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continuing therapy. Notable elevations of ALT or AST (approximately three or more times the upper limit of normal) have been reported in approximately 1% of patients in clinical trials with NSAIDs. In addition, rare cases of severe hepatic reactions, including jaundice and fatal fulminant hepatitis, liver necrosis and hepatic failure, some of them with fatal outcomes have been reported.
A patient with symptoms and/or signs suggesting liver dysfunction, or in whom an abnormal liver test has occurred, should be evaluated for evidence of the development of a more severe hepatic reaction while on therapy with diflunisal tablets. If clinical signs and symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, etc.), diflunisal tablets should be discontinued.
Hematological Effects
Anemia is sometimes seen in patients receiving NSAIDs, including diflunisal tablets. This may be due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. Patients on long-term treatment with NSAIDs, including diflunisal tablets, should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia.
NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible. Patients receiving diflunisal tablets who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants, should be carefully monitored.
Preexisting Asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm which can be fatal. Since cross reactivity, including bronchospasm, between aspirin and other non-steroidal anti-inflammatory drugs has been reported in such aspirin-sensitive patients, diflunisal tablets should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with preexisting asthma.
Ocular Effects
Because of reports of adverse eye findings with agents of this class, it is recommended that patients who develop eye complaints during treatment with diflunisal tablets have ophthalmologic studies.
Reye’s Syndrome
Acetylsalicylic acid has been associated with Reye’s syndrome. Because diflunisal is a derivative of salicylic acid, the possibility of its association with Reye’s syndrome cannot be excluded.
Information for Patients
Patients should be informed of the following information before initiating therapy with an NSAID and periodically during the course of ongoing therapy. Patients should also be encouraged to read the NSAID Medication Guide that accompanies each prescription dispensed.
- 1.
-
Cardiovascular Thrombotic Events
Advise patients to be alert for the symptoms of cardiovascular thrombotic events, including chest pain, shortness of breath, weakness, or slurring of speech, and to report any of these symptoms to their health care provider immediately (see WARNINGS).
- 2.
- Diflunisal tablets, like other NSAIDs, can cause GI discomfort and, rarely, serious GI side effects, such as ulcers and bleeding, which may result in hospitalization and even death. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding, and should ask for medical advice when observing any indicative signs or symptoms including epigastric pain, dyspepsia, melena, and hematemesis. Patients should be apprised of the importance of this follow-up (see WARNINGS, Gastrointestinal Effects - Risk of Ulceration, Bleeding, and Perforation).
- 3.
- Diflunisal tablets, like other NSAIDs, can cause serious skin side effects such as exfoliative dermatitis, SJS, and TEN, which may result in hospitalization and even death. Although serious skin reactions may occur without warning, patients should be alert for the signs and symptoms of skin rash and blisters, fever, or other signs of hypersensitivity such as itching, and should ask for medical advice when observing any indicative signs or symptoms. Patients should be advised to stop the drug immediately if they develop any type of rash and contact their physicians as soon as possible.
- 4.
-
Heart Failure And Edema
Advise patients to be alert for the symptoms of congestive heart failure including shortness of breath, unexplained weight gain, or edema and to contact their healthcare provider if such symptoms occur (see WARNINGS).
- 5.
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness, and "flu-like" symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical therapy.
- 6.
- Patients should be informed of the signs of an anaphylactic/anaphylactoid reaction (e.g., difficulty breathing, swelling of the face or throat). If these occur, patients should be instructed to seek immediate emergency help (see WARNINGS).
- 7.
- In late pregnancy, as with other NSAIDs, diflunisal tablets should be avoided because they may cause premature closure of the ductus arteriosus.
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of GI bleeding. Patients on long-term treatment with NSAIDs, should have their CBC and a chemistry profile checked periodically. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash, etc.) or if abnormal liver tests persist or worsen, diflunisal tablets should be discontinued.
Drug Interactions
ACE-inhibitors and Angiotensin II Anagonists
Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE-inhibitors and angiotensin II antagonists. These interactions should be given consideration in patients taking NSAIDs concomitantly with ACE-inhibitors or angiotensin II antagonists. In some patients with compromised renal function, the coadministration of an NSAID and an ACE-inhibitor or an angiotensin II antagonist may result in further deterioration of renal function, including possible acute renal failure, which is usually reversible.
Acetaminophen
In normal volunteers, concomitant administration of diflunisal and acetaminophen resulted in an approximate 50% increase in plasma levels of acetaminophen. Acetaminophen had no effect on plasma levels of diflunisal. Since acetaminophen in high doses has been associated with hepatotoxicity, concomitant administration of diflunisal tablets and acetaminophen should be used cautiously, with careful monitoring of patients.
Concomitant administration of diflunisal and acetaminophen in dogs, but not in rats, at approximately 2 times the recommended maximum human therapeutic dose of each (40 to 52 mg/kg/day of diflunisal/acetaminophen), resulted in greater gastrointestinal toxicity than when either drug was administered alone. The clinical significance of these findings has not been established.
Antacids
Concomitant administration of antacids may reduce plasma levels of diflunisal. This effect is small with occasional doses of antacids, but may be clinically significant when antacids are used on a continuous schedule.
Aspirin
When diflunisal is administered with aspirin, its protein binding is reduced, although the clearance of free diflunisal is not altered. The clinical significance of this interaction is not known; however, as with other NSAIDs, concomitant administration of diflunisal tablets and aspirin is not generally recommended because of the potential of increased adverse effects.
In normal volunteers, a small decrease in diflunisal levels was observed when multiple doses of diflunisal and aspirin were administered concomitantly.
Cyclosporine
Administration of non-steroidal anti-inflammatory drugs concomitantly with cyclosporine has been associated with an increase in cyclosporine-induced toxicity, possibly due to decreased synthesis of renal prostacyclin. NSAIDs should be used with caution in patients taking cyclosporine, and renal function should be carefully monitored.
Diuretics
Clinical studies, as well as postmarketing observations, have shown that diflunisal can reduce the natriuretic effect of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis.
In normal volunteers, concomitant administration of diflunisal and hydrochlorothiazide resulted in significantly increased plasma levels of hydrochlorothiazide. Diflunisal decreased the hyperuricemic effect of hydrochlorothiazide. During concomitant therapy with NSAIDs, the patient should be observed closely for signs of renal failure (see WARNINGS, Renal Effects), as well as to assure diuretic efficacy.
Lithium
NSAIDs have produced an elevation of plasma lithium levels and a reduction in renal lithium clearance. The mean minimum lithium concentration increased 15% and the renal clearance was decreased by approximately 20%. These effects have been attributed to inhibition of renal prostaglandin synthesis by the NSAID. Thus, when NSAIDs and lithium are administered concurrently, subjects should be observed carefully for signs of lithium toxicity.
Methotrexate
NSAIDs have been reported to competitively inhibit methotrexate accumulation in rabbit kidney slices. This may indicate that they could enhance the toxicity of methotrexate. Caution should be used when NSAIDs are administered concomitantly with methotrexate.
NSAIDs
The administration of diflunisal to normal volunteers receiving indomethacin decreased the renal clearance and significantly increased the plasma levels of indomethacin. In some patients the combined use of indomethacin and diflunisal has been associated with fatal gastrointestinal hemorrhage. Therefore, indomethacin and diflunisal tablets should not be used concomitantly.
The concomitant use of diflunisal tablets and other NSAIDs is not recommended due to the increased possibility of gastrointestinal toxicity, with little or no increase in efficacy. The following information was obtained from studies in normal volunteers.
Oral Anticoagulants
In some normal volunteers, the concomitant administration of diflunisal and warfarin, acenocoumarol, or phenprocoumon resulted in prolongation of prothrombin time. This may occur because diflunisal competitively displaces coumarins from protein binding sites. Accordingly, when diflunisal tablets are administered with oral anticoagulants, the prothrombin time should be closely monitored during and for several days after concomitant drug administration. Adjustment of dosage of oral anticoagulants may be required. The effects of warfarin and NSAIDs on GI bleeding are synergistic, such that users of both drugs together have a risk of serious GI bleeding higher than users of either drug alone.
Tolbutamide
In diabetic patients receiving diflunisal and tolbutamide, no significant effects were seen on tolbutamide plasma levels or fasting blood glucose.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Diflunisal did not affect the type or incidence of neoplasia in a 105 week study in the rat given doses up to 40 mg/kg/day (equivalent to approximately 1.3 times the maximum recommended human dose), or in long-term carcinogenic studies in mice given diflunisal at doses up to 80 mg/kg/day (equivalent to approximately 2.7 times the maximum recommended human dose). It was concluded that there was no carcinogenic potential for diflunisal.
Diflunisal passes the placental barrier to a minor degree in the rat. Diflunisal had no mutagenic activity after oral administration in the dominant lethal assay, in the Ames microbial mutagen test or in the V-79 Chinese hamster lung cell assay.
No evidence of impaired fertility was found in reproduction studies in rats at doses up to 50 mg/kg/day.
Pregnancy
Teratogenic Effects
Pregnancy category C
A dose of 60 mg/kg/day of diflunisal (equivalent to two times the maximum human dose) was maternotoxic, embryotoxic, and teratogenic in rabbits. In three of six studies in rabbits, evidence of teratogenicity was observed at doses ranging from 40 to 50 mg/kg/day. Teratology studies in mice, at doses up to 45 mg/kg/day, and in rats at doses up to 100 mg/kg/day, revealed no harm to the fetus due to diflunisal. Aspirin and other salicylates have been shown to be teratogenic in a wide variety of species, including the rat and rabbit, at doses ranging from 50 to 400 mg/kg/day (approximately one to eight times the human dose). Animal reproduction studies are not always predictive of human response. There are no adequate and well controlled studies with diflunisal in pregnant women. Diflunisal tablets should be used in pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nonteratogenic Effects
Because of the known effects of non-steroidal anti-inflammatory drugs on the fetal cardiovascular system (closure of ductus arteriosus), use during pregnancy (particularly late pregnancy) should be avoided.
The known effects of drugs of this class on the human fetus during the third trimester of pregnancy include: constriction of the ductus arteriosus prenatally, tricuspid incompetence, and pulmonary hypertension; non-closure of the ductus arteriosus postnatally which may be resistant to medical management; myocardial degenerative changes, platelet dysfunction with resultant bleeding, intracranial bleeding, renal dysfunction or failure, renal injury/dysgenesis which may result in prolonged or permanent renal failure, oligohydramnios, gastrointestinal bleeding or perforation, and increased risk of necrotizing enterocolitis.
In rats at a dose of one and one-half times the maximum human dose, there was an increase in the average length of gestation. Similar increases in the length of gestation have been observed with aspirin, indomethacin, and phenylbutazone, and may be related to inhibition of prostaglandin synthetase.
Labor and Delivery
In rat studies with NSAIDs, as with other drugs known to inhibit prostaglandin synthesis, an increased incidence of dystocia, delayed parturition, and decreased pup survival occurred. The effects of diflunisal tablets on labor and delivery in pregnant women are unknown.
Nursing Mothers
Diflunisal is excreted in human milk in concentrations of 2 to 7% of those in plasma. Because of the potential for serious adverse reactions in nursing infants from diflunisal, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness of diflunisal in pediatric patients below the age of 12 have not been established. Use of diflunisal tablets in pediatric patients below the age of 12 is not recommended.
The adverse effects observed following diflunisal administration to neonatal animals appear to be species, age, and dose-dependent. At dose levels approximately 3 times the usual human therapeutic dose, both aspirin (200 to 400 mg/kg/day) and diflunisal (80 mg/kg/day) resulted in death, leukocytosis, weight loss, and bilateral cataracts in neonatal (4 to 5-day-old) beagle puppies after 2 to 10 doses. Administration of an 80 mg/kg/day dose of diflunisal to 25-day-old puppies resulted in lower mortality, and did not produce cataracts. In newborn rats, a 400 mg/kg/day dose of aspirin resulted in increased mortality and some cataracts, whereas the effects of diflunisal administration at doses up to 140 mg/kg/day were limited to a decrease in average body weight gain.
Geriatric Use
As with any NSAID, caution should be exercised in treating the elderly (65 years and older) since advancing age appears to increase the possibility of adverse reactions. Elderly patients seem to tolerate ulceration or bleeding less well than other individuals and many spontaneous reports of fatal GI events are in this population (see WARNINGS, Gastrointestinal Effects – Risk of Ulceration, Bleeding, and Perforation).
This drug is known to be substantially excreted by the kidney and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection and it may be useful to monitor renal function (see WARNINGS, Renal Effects).
-
ADVERSE REACTIONS
The adverse reactions observed in controlled clinical trials encompass observations in 2,427 patients.
Listed below are the adverse reactions reported in the 1,314 of these patients who received treatment in studies of two weeks or longer. Five hundred thirteen patients were treated for at least 24 weeks, 255 patients were treated for at least 48 weeks, and 46 patients were treated for 96 weeks. In general, the adverse reactions listed below were 2 to 14 times less frequent in the 1,113 patients who received short-term treatment for mild to moderate pain.
Incidence Greater Than 1%
Incidence Less Than 1 in 100
The following adverse reactions, occurring less frequently than 1 in 100, were reported in clinical trials or since the drug was marketed. The probability exists of a causal relationship between diflunisal and these adverse reactions.
Dermatologic
Erythema multiforme, exfoliative dermatitis, Stevens-Johnson syndrome, toxic epidermal necrolysis, urticaria, pruritus, sweating, dry mucous membranes, stomatitis, photosensitivity.
Gastrointestinal
Peptic ulcer, gastrointestinal bleeding, anorexia, eructation, gastrointestinal perforation, gastritis. Liver function abnormalities; jaundice, sometimes with fever; cholestasis; hepatitis.
Genitourinary
Dysuria; renal impairment, including renal failure; interstitial nephritis; hematuria; proteinuria.
Causal Relationship Unknown
Other reactions have been reported in clinical trials or since the drug was marketed, but occurred under circumstances where a causal relationship could not be established. However, in these rarely reported events, that possibility cannot be excluded. Therefore, these observations are listed to serve as alerting information to physicians.
Miscellaneous
Chest pain.
A rare occurrence of fulminant necrotizing fasciitis, particularly in association with Group A β-hemolytic streptococcus, has been described in persons treated with non-steroidal anti-inflammatory agents, including diflunisal, sometimes with fatal outcome (see also PRECAUTIONS, General).
-
OVERDOSAGE
Cases of overdosage have occurred and deaths have been reported. Most patients recovered without evidence of permanent sequelae. The most common signs and symptoms observed with overdosage were drowsiness, vomiting, nausea, diarrhea, hyperventilation, tachycardia, sweating, tinnitus, disorientation, stupor, and coma. Diminished urine output and cardiorespiratory arrest have also been reported. The lowest dosage of diflunisal at which a death has been reported was 15 grams without the presence of other drugs. In a mixed drug overdose, ingestion of 7.5 grams of diflunisal resulted in death.
In the event of overdosage, the stomach should be emptied by inducing vomiting or by gastric lavage, and the patient carefully observed and given symptomatic and supportive treatment. Because of the high degree of protein binding, hemodialysis may not be effective.
The oral LD50 of the drug is 500 mg/kg and 826 mg/kg in female mice and female rats, respectively.
-
DOSAGE AND ADMINISTRATION
Carefully consider the potential benefits and risks of diflunisal tablets and other treatment options before deciding to use diflunisal tablets. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS).
After observing the response to initial therapy with diflunisal tablets, the dose and frequency should be adjusted to suit an individual patient's needs.
Concentration-dependent pharmacokinetics prevail when diflunisal is administered; a doubling of dosage produces a greater than doubling of drug accumulation. The effect becomes more apparent with repetitive doses.
For mild to moderate pain, an initial dose of 1000 mg followed by 500 mg every 12 hours is recommended for most patients. Following the initial dose, some patients may require 500 mg every 8 hours.
A lower dosage may be appropriate depending on such factors as pain severity, patient response, weight, or advanced age; for example, 500 mg initially, followed by 250 mg every 8 to 12 hours.
For osteoarthritis and rheumatoid arthritis, the suggested dosage range is 500 mg to 1000 mg daily in two divided doses. The dosage of diflunisal may be increased or decreased according to patient response.
Maintenance doses higher than 1500 mg a day are not recommended.
Tablets should be swallowed whole, not crushed or chewed.
- HOW SUPPLIED
-
MEDICATION GUIDE
Medication Guide for Nonsteroidal Anti-inflammatory Drugs (NSAIDs)
What is the most important information I should know about medicines called Nonsteroidal Anti-inflammatory Drugs (NSAIDs)?
NSAIDs can cause serious side effects, including:
- •
- Increased risk of a heart attack or stroke that can lead to death. This risk may happen early in treatment and may increase:
- o
- with increasing doses of NSAIDs
- o
- with longer use of NSAIDs
- Do not take NSAIDs right before or after a heart surgery called a “coronary artery bypass graft (CABG).”
- Avoid taking NSAIDs after a recent heart attack, unless your healthcare provider tells you to. You may have an increased risk of another heart attack if you take NSAIDs after a recent heart attack.
- •
- Increased risk of bleeding, ulcers, and tears (perforation) of the esophagus (tube leading from the mouth to the stomach), stomach and intestines:
- o
- anytime during use
- o
- without warning symptoms
- o
- that may cause death
- The risk of getting an ulcer or bleeding increases with:
- o
- past history of stomach ulcers, or stomach or intestinal bleeding with use of NSAIDs
- o
- taking medicines called “corticosteroids”, “anticoagulants”, “SSRIs”, or “SNRIs”
- o
- increasing doses of NSAIDs
- o
- older age
- o
- longer use of NSAIDs
- o
- poor health
- o
- smoking
- o
- advanced liver disease
- o
- drinking alcohol
- o
- bleeding problems
NSAIDs should only be used:
- o
- exactly as prescribed
- o
- at the lowest dose possible for your treatment
- o
- for the shortest time needed
What are NSAIDs?
NSAIDs are used to treat pain and redness, swelling, and heat (inflammation) from medical conditions such as different types of arthritis, menstrual cramps, and other types of short-term pain.
Who should not take NSAIDs?
Do not take NSAIDs:
- •
- if you have had an asthma attack, hives, or other allergic reaction with aspirin or any other NSAIDs.
- •
- right before or after heart bypass surgery.
Before taking NSAIDs, tell your healthcare provider about all of your medical conditions, including if you:
- •
- have liver or kidney problems
- •
- have high blood pressure
- •
- have asthma
- •
- are pregnant or plan to become pregnant. Talk to your healthcare provider if you are considering taking NSAIDs during pregnancy. You should not take NSAIDs after 29 weeks of pregnancy.
- •
- are breastfeeding or plan to breast feed.
Tell your healthcare provider about all of the medicines you take, including prescription or over-the-counter medicines, vitamins or herbal supplements. NSAIDs and some other medicines can interact with each other and cause serious side effects. Do not start taking any new medicine without talking to your healthcare provider first.
What are the possible side effects of NSAIDs?
NSAIDs can cause serious side effects, including:
See “What is the most important information I should know about medicines called Nonsteroidal Anti-inflammatory Drugs (NSAIDs)?”
- •
- new or worse high blood pressure
- •
- heart failure
- •
- liver problems including liver failure
- •
- kidney problems including kidney failure
- •
- low red blood cells (anemia)
- •
- life-threatening skin reactions
- •
- life-threatening allergic reactions
- •
- Other side effects of NSAIDs include: stomach pain, constipation, diarrhea, gas, heartburn, nausea, vomiting, and dizziness.
Get emergency help right away if you get any of the following symptoms:
- •
- shortness of breath or trouble breathing
- •
- slurred speech
- •
- chest pain
- •
- swelling of the face or throat
- •
- weakness in one part or side of your body
Stop taking your NSAID and call your healthcare provider right away if you get any of the following symptoms:
- •
- nausea
- •
- vomit blood
- •
- more tired or weaker than usual
- •
- there is blood in your bowel movement or it
- •
- diarrhea
is black and sticky like tar
- •
- itching
- •
- unusual weight gain
- •
- your skin or eyes look yellow
- •
- skin rash or blisters with fever
- •
- indigestion or stomach pain
- •
- swelling of the arms, legs, hands and feet
- •
- flu-like symptoms
If you take too much of your NSAID, call your healthcare provider or get medical help right away.
These are not all the possible side effects of NSAIDs. For more information, ask your healthcare provider or pharmacist about NSAIDs.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
Other information about NSAIDs
- •
- Aspirin is an NSAID but it does not increase the chance of a heart attack. Aspirin can cause bleeding in the brain, stomach, and intestines. Aspirin can also cause ulcers in the stomach and intestines.
- •
- Some NSAIDs are sold in lower doses without a prescription (over-the-counter). Talk to your healthcare provider before using over-the-counter NSAIDs for more than 10 days.
General information about the safe and effective use of NSAIDs
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use NSAIDs for a condition for which it was not prescribed. Do not give NSAIDs to other people, even if they have the same symptoms that you have. It may harm them.
If you would like more information about NSAIDs, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about NSAIDs that is written for health professionals.
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised July 2015
- Diflunisal 500 MG TAB
-
INGREDIENTS AND APPEARANCE
DIFLUNISAL
diflunisal tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:68151-0848(NDC:0093-0755) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DIFLUNISAL (UNII: 7C546U4DEN) (DIFLUNISAL - UNII:7C546U4DEN) DIFLUNISAL 500 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) INDIGOTINDISULFONATE SODIUM (UNII: D3741U8K7L) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color BLUE (blue-purple) Score no score Shape OVAL (capsule-shaped) Size 19mm Flavor Imprint Code 755;93 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:68151-0848-0 1 in 1 PACKAGE; Type 0: Not a Combination Product 11/01/1992 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA073673 11/01/1992 Labeler - Carilion Materials Management (079239644) Establishment Name Address ID/FEI Business Operations Carilion Materials Management 079239644 REPACK(68151-0848)