Label: BUSULFAN injection


- NDC Code(s): 72606-559-01, 72606-559-02
- Packager: CELLTRION USA, INC.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated December 1, 2019
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BUSULFAN INJECTION, safely and effectively. See full prescribing information for BUSULFAN INJECTION.
BUSULFAN injection, for intravenous use
Initial U.S. Approval: 1999INDICATIONS AND USAGE
Busulfan injection is an alkylating drug indicated for:
- Use in combination with cyclophosphamide as a conditioning regimen prior to allogeneic hematopoietic progenitor cell transplantation for chronic myelogenous leukemia (CML) (1)
DOSAGE AND ADMINISTRATION
- Pre-medicate with anticonvulsants (e.g. benzodiazepines, phenytoin, valproic acid or levetiracetam) and antiemetic ( 2.1, 5.2)
- Dilute and administer as intravenous infusion. Do not administer as intravenous push or bolus ( 2.1, 2.3)
- Recommended adult dose: 0.8 mg per kg of ideal body weight or actual body weight, whichever is lower, administered intravenously via a central venous catheter as a two-hour infusion every six hours for four consecutive days for a total of 16 doses (2.1)
DOSAGE FORMS AND STRENGTHS
- 60 mg per 10 mL (6 mg per mL) single-dose vial (3)
CONTRAINDICATIONS
- Busulfan injection is contraindicated in patients with a history of hypersensitivity to any of its components (4)
WARNINGS AND PRECAUTIONS
- Seizures: Initiate anticonvulsant prophylactic therapy prior to treatment with busulfan injection. Monitor patients with history of seizure disorder, head trauma or receiving epileptogenic drugs (5.2)
- Hepatic Veno-Occlusive Disease (HVOD): Increased risk of developing HVOD at AUC greater than 1,500 μM•min. Monitor serum transaminases, alkaline phosphatase and bilirubin daily (5.3)
- Embryo-fetal Toxicity: Can cause fetal harm. Advise of potential risk to a fetus and use of effective contraception (5.4, 8.1, 8.3)
- Cardiac tamponade has been reported in pediatric patients with thalassemia who received high doses of oral busulfan and cyclophosphamide. Abdominal pain and vomiting precede the tamponade in most patients (5.5)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 60%) were: myelosuppression, nausea, stomatitis, vomiting, anorexia, diarrhea, insomnia, fever, hypomagnesemia, abdominal pain, anxiety, headache, hyperglycemia and hypokalemia (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Shilpa Medicare Limited at 1-888-557-1212 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: MYLOSUPPRESSION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Initial Dosing Information
2.2 Preparation and Administration Precautions
2.3 Preparation for Intravenous Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
5.2 Seizures
5.3 Hepatic Veno-Occlusive Disease (HVOD)
5.4 Embryo-fetal Toxicity
5.5 Cardiac Tamponade
5.6 Bronchopulmonary Dysplasia
5.7 Cellular Dysplasia
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
6.3 Oral Busulfan Literature Review
7 DRUG INTERACTIONS
7.1 Drugs that Decrease Busulfan Clearance
7.2 Drugs that Increase Busulfan Clearance
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: MYLOSUPPRESSION
Busulfan Injection causes severe and prolonged myelosuppression at the recommended dosage. Hematopoietic progenitor cell transplantation is required to prevent potentially fatal complications of the prolonged myelosuppression. [See Warnings and Precautions (5.1)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Initial Dosing Information
- Administer busulfan injection in combination with cyclophosphamide as a conditioning regimen prior to bone marrow or peripheral blood progenitor cell replacement. For patients weighing more than 12 kg, the recommended doses are:
- Busulfan injection 0.8 mg per kg (ideal body weight or actual body weight, whichever is lower) intravenously via a central venous catheter as a two-hour infusion every six hours for four consecutive days for a total of 16 doses (Days -7, -6, -5 and -4).
- Cyclophosphamide 60 mg per kg intravenously as a one-hour infusion on each of two days beginning no sooner than six hours following the 16th dose of busulfan injection (Days -3 and -2).
- Administer hematopoietic progenitor cells on Day 0.
- Premedicate patients with anticonvulsants (e.g., benzodiazepines, phenytoin, valproic acid or levetiracetam) to prevent seizures reported with the use of high dose busulfan injection. Administer anticonvulsants 12 hours prior to busulfan injection to 24 hours after the last dose of busulfan injection [see Warnings and Precautions (5.2)].
- Administer antiemetics prior to the first dose of busulfan injection and continue on a fixed schedule through busulfan injection administration.
- Busulfan injection clearance is best predicted when the busulfan injection dose is administered based on adjusted ideal body weight. Dosing busulfan injection based on actual body weight, ideal body weight or other factors can produce significant differences in busulfan injection clearance among lean, normal and obese patients.
- Calculate ideal body weight (IBW) as follows (height in cm, and weight in kg):
Women: IBW (kg)=45+0.91x (height in cm -152)
- For obese or severely obese patients, base busulfan injection dosing on adjusted ideal body weight (AIBW):
2.2 Preparation and Administration Precautions
Busulfan injection is incompatible with polycarbonate. Do not use any infusion components (syringes, filter needles, intravenous tubing, etc.) containing polycarbonate with busulfan injection.
Use an administration set with minimal residual hold-up volume (2mL - 5mL) for product administration.
Busulfan injection is a cytotoxic drug. Follow applicable special handling and disposal procedures. Skin reactions may occur with accidental exposure. Use gloves when preparing busulfan injection. If busulfan or diluted busulfan solution contacts the skin or mucosa, wash the skin or mucosa thoroughly with water.
Visually inspect parenteral drug products for particulate matter and discoloration prior to administration whenever the solution and container permit. Do not use if particulate matter is seen in the busulfan injection vial.
2.3 Preparation for Intravenous Administration
Busulfan injection must be diluted prior to intravenous infusion with either 0.9% Sodium Chloride Injection, USP (normal saline) or 5% Dextrose Injection, USP (D5W). The diluent quantity should be 10 times the volume of busulfan injection, so that the final concentration of busulfan is approximately 0.5 mg per mL. Calculation of the dose for a 70 kg patient would be performed as follows:
(70 kg patient) x (0.8 mg per kg) ÷ (6 mg per mL) =9.3 mL busulfan injection (56 mg total dose).To prepare the final solution for infusion, add 9.3 mL of busulfan injection to 93 mL of diluent (normal saline or D5W) as calculated below:
(9.3 mL busulfan injection) x (10) =93 mL of either diluent plus the 9.3 mL of busulfan injection to yield a final concentration of busulfan of 0.54 mg per mL (9.3 mL x 6 mg per mL ÷ 102.3 mL =0.54 mg per mL).All transfer procedures require strict adherence to aseptic techniques, preferably employing a vertical laminar flow safety hood while wearing gloves and protective clothing.
DO NOT put the busulfan injection into an intravenous bag or large-volume syringe that does not contain normal saline or D5W. Always add the busulfan injection to the diluent, not the diluent to the busulfan injection. Mix thoroughly by inverting several times.
Infusion pumps should be used to administer the diluted busulfan injection solution. Set the flow rate of the pump to deliver the entire prescribed busulfan injection dose over two hours. Prior to and following each infusion, flush the indwelling catheter line with approximately 5 mL of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP. DO NOT infuse concomitantly with another intravenous solution of unknown compatibility. WARNING: RAPID INFUSION OF BUSULFAN INJECTION HAS NOT BEEN TESTED AND IS NOT RECOMMENDED.
- Administer busulfan injection in combination with cyclophosphamide as a conditioning regimen prior to bone marrow or peripheral blood progenitor cell replacement. For patients weighing more than 12 kg, the recommended doses are:
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
The most frequent serious consequence of treatment with busulfan at the recommended dose and schedule is prolonged myelosuppression, occurring in all patients (100%). Severe granulocytopenia, thrombocytopenia, anemia, or any combination thereof may develop. Hematopoietic progenitor cell transplantation is required to prevent potentially fatal complications of the prolonged myelosuppression. Monitor complete blood counts, including white blood cell differentials, and quantitative platelet counts daily during treatment and until engraftment is demonstrated. Absolute neutrophil counts dropped below 0.5x109/L at a median of 4 days post-transplant in 100% of patients treated in the busulfan clinical trial. The absolute neutrophil count recovered at a median of 13 days following allogeneic transplantation when prophylactic filgrastim was used in the majority of patients. Thrombocytopenia (less than 25,000/mm3 or requiring platelet transfusion) occurred at a median of 5-6 days in 98% of patients. Anemia (hemoglobin less than 8.0 g/dL) occurred in 69% of patients. Use antibiotic therapy and platelet and red blood cell support when medically indicated.
5.2 Seizures
Seizures have been reported in patients receiving high-dose oral busulfan at doses producing plasma drug levels similar to those achieved following the recommended dosage of busulfan. Despite prophylactic therapy with phenytoin, one seizure (1/42 patients) was reported during an autologous transplantation clinical trial of busulfan. This episode occurred during the cyclophosphamide portion of the conditioning regimen, 36 hours after the last busulfan dose. Initiate phenytoin therapy or any other alternative anti-convulsant prophylactic therapy (e.g., benzodiazepines, valproic acid or levetiracetam) prior to busulfan treatment [See Dosage and Administration (2.1)]. Use caution when administering the recommended dose of busulfan to patients with a history of a seizure disorder or head trauma or who are receiving other potentially epileptogenic drugs.
5.3 Hepatic Veno-Occlusive Disease (HVOD)
Current literature suggests that high busulfan area under the plasma concentration verses time curve (AUC) values (greater than 1,500 μM•min) may be associated with an increased risk of developing HVOD. Patients who have received prior radiation therapy, greater than or equal to three cycles of chemotherapy, or a prior progenitor cell transplant may be at an increased risk of developing HVOD with the recommended busulfan dose and regimen. Based on clinical examination and laboratory findings, HVOD was diagnosed in 8% (5/61) of patients treated with busulfan in the setting of allogeneic transplantation, was fatal in 2/5 cases (40%), and yielded an overall mortality from HVOD in the entire study population of 2/61 (3%). Three of the five patients diagnosed with HVOD were retrospectively found to meet the Jones' criteria. The incidence of HVOD reported in the literature from the randomized, controlled trials was 7.7%-12% [See Clinical Studies (14)]. Monitor serum transaminases, alkaline phosphatase, and bilirubin daily through BMT Day +28 to detect hepatotoxicity, which may herald the onset of HVOD.
5.4 Embryo-fetal Toxicity
Busulfan can cause fetal harm when administered to a pregnant woman based on animal data. Busulfan was teratogenic in mice, rats, and rabbits. The solvent, DMA, may also cause fetal harm when administered to a pregnant woman based on findings in animals. Advise pregnant women of the potential risk to a fetus. Advise females and males of reproductive potential to use effective contraception during and after treatment with busulfan [see Use in Specific Populations (8.1, 8.3)].
5.5 Cardiac Tamponade
Cardiac tamponade has been reported in pediatric patients with thalassemia (8/400 or 2% in one series) who received high doses of oral busulfan and cyclophosphamide as the preparatory regimen for hematopoietic progenitor cell transplantation. Six of the eight children died and two were saved by rapid pericardiocentesis. Abdominal pain and vomiting preceded the tamponade in most patients. Monitor for signs and symptoms, promptly evaluate and treat if cardiac tamponade is suspected.
5.6 Bronchopulmonary Dysplasia
Bronchopulmonary dysplasia with pulmonary fibrosis is a rare but serious complication following chronic busulfan therapy. The average onset of symptoms is 4 years after therapy (range 4 months to 10 years).
5.7 Cellular Dysplasia
Busulfan may cause cellular dysplasia in many organs. Cytologic abnormalities characterized by giant, hyperchromatic nuclei have been reported in lymph nodes, pancreas, thyroid, adrenal glands, liver, lungs and bone marrow. This cytologic dysplasia may be severe enough to cause difficulty in the interpretation of exfoliative cytologic examinations of the lungs, bladder, breast and the uterine cervix.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Myelosuppression [See Warnings and Precautions (5.1)]
- Seizures [See Warnings and Precautions (5.2)]
- Hepatic Veno-Occlusive Disease (HVOD) [See Warnings and Precautions (5.3)]
- Embryo-fetal Toxicity [See Warnings and Precautions (5.4)]
- Cardiac Tamponade [See Warnings and Precautions (5.5)]
- Bronchopulmonary Dysplasia [See Warnings and Precautions (5.6)]
- Cellular Dysplasia [See Warnings and Precautions (5.7)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reaction information is primarily derived from the clinical study (N=61) of busulfan and the data obtained for high-dose oral busulfan conditioning in the setting of randomized, controlled trials identified through a literature review.
In the busulfan Injection allogeneic stem cell transplantation clinical trial, all patients were treated with busulfan 0.8 mg per kg as a two-hour infusion every six hours for 16 doses over four days, combined with cyclophosphamide 60 mg per kg x2 days. Ninety-three percent (93%) of evaluable patients receiving this dose of busulfan maintained an AUC less than 1,500 μM•min for dose 9, which has generally been considered the level that minimizes the risk of HVOD.
Table 1 lists the non-hematologic adverse reactions events through Bone Marrow Transplantation (BMT) Day +28 at a rate greater than or equal to 20% in patients treated with busulfan prior to allogeneic hematopoietic cell transplantation.
Table 1: Summary of the Incidence (greater than or equal to 20%) of Non-Hematologic Adverse Reactions through BMT Day +28 in Patients who Received Busulfan injection Prior to Allogeneic Hematopoietic Progenitor Cell Transplantation Non-Hematological Adverse Reactions1 Percent Incidence BODY AS A WHOLE Fever 80 Headache 69 Asthenia 51 Chills 46 Pain 44 Edema General 28 Allergic Reaction 26 Chest Pain 26 Inflammation at Injection Site 25 Back Pain 23 CARDIOVASCULAR SYSTEM Tachycardia 44 Hypertension 36 Thrombosis 33 Vasodilation 25 DIGESTIVE SYSTEM Nausea 98 Stomatitis (Mucositis) 97 Vomiting 95 Anorexia 85 Diarrhea 84 Abdominal Pain 72 Dyspepsia 44 Constipation 38 Dry Mouth 26 Rectal Disorder 25 Abdominal Enlargement 23 METABOLIC AND NUTRITIONAL SYSTEM Hypomagnesemia 77 Hyperglycemia 66 Hypokalemia 64 Hypocalcemia 49 Hyperbilirubinemia 49 Edema 36 SGPT Elevation 31 Creatinine Increased 21 NERVOUS SYSTEM Insomnia 84 Anxiety 72 Dizziness 30 Depression 23 RESPIRATORY SYSTEM Rhinitis 44 Lung Disorder 34 Cough 28 Epistaxis 25 Dyspnea 25 SKIN AND APPENDAGES Rash 57 Pruritus 28 1. Includes all reported adverse reactions regardless of severity (toxicity grades 1-4) Additional Adverse Reactions by Body System
Hematologic: Prolonged prothrombin time
Gastrointestinal: Esophagitis, ileus, hematemesis, pancreatitis, rectal discomfort
Hepatic: Alkaline phosphatase increases, jaundice, hepatomegaly
Graft-versus-host disease: Graft-versus-host disease. There were 3 deaths (5%) attributed to GVHD.
Edema: Hypervolemia, or documented weight increase
Infection: Infection, pneumonia (fatal in one patient and life-threatening in 3% of patients)
Cardiovascular: Arrhythmia, atrial fibrillation, ventricular extrasystoles, third degree heart block, thrombosis (all episodes were associated with the central venous catheter), hypotension, flushing and hot flashes, cardiomegaly, ECG abnormality, left-sided heart failure, and pericardial effusion
Pulmonary: Hyperventilation, alveolar hemorrhage (fatal in 3%), pharyngitis, hiccup, asthma, atelectasis, pleural effusion, hypoxia, hemoptysis, sinusitis, and interstitial fibrosis (fatal in a single case)
Neurologic: Cerebral hemorrhage, coma, delirium, agitation, encephalopathy, confusion, hallucinations, lethargy, somnolence
Renal: BUN increased, dysuria, oliguria, hematuria, hemorrhagic cystitis
Skin: Alopecia, vesicular rash, maculopapular rash, vesiculo-bullous rash, exfoliative dermatitis, erythema nodosum, acne, skin discoloration
Metabolic: Hypophosphatemia, hyponatremia
Other Events: Injection site pain, myalgia, arthralgia, ear disorder
6.2 Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following adverse reactions have been identified during post-approval use of Busulfan Injection:
Blood and Lymphatic System Disorders: febrile neutropenia
Gastrointestinal Disorders: tooth hypoplasia
Metabolism and Nutrition Disorders: tumor lysis syndrome
Vascular Disorders: thrombotic microangiopathy (TMA)
Infections and Infestations: severe bacterial, viral (e.g., cytomegalovirus viremia) and fungal infections; and sepsis.
6.3 Oral Busulfan Literature Review
A literature review identified four randomized, controlled trials that evaluated a high-dose oral busulfan-containing conditioning regimen for allogeneic bone marrow transplantation in the setting of CML [see Clinical Studies (14)]. The safety outcomes reported in those trials are summarized in Table 2 below for a mixed population of hematological malignancies (AML, CML, and ALL).
Table 2: Summary of safety analyses from the randomized, controlled trials utilizing a high dose oral busulfan-containing conditioning regimen that were identified in a literature review. Clift
CML Chronic PhaseTRM1 VOD2 GVHD3 Pulmonary Hemorrhagic Cystitis Seizure Death ≤100d=4.1% (3/73) No Report Acute ≥Grade 2 =35%
Chronic=41% (30/73)1 death from Idiopathic Interstitial Pneumonitis And 1 death from Pulmonary Fibrosis No Report No Report Devergie
CML Chronic PhaseTRM VOD GVHD Pulmonary Hemorrhagic Cystitis Seizure 38% 7.7% (5/65)
Deaths=4.6%
(3/65)Acute ≥Grade 2 =41% (24/59 at risk) Interstitial Pneumonitis=16.9% (11/65) 10.8% (7/65) No report Ringden
CML, AML, ALLTRM VOD GVHD Pulmonary Hemorrhagic Cystitis Seizure 28% 12% Acute ≥Grade 2 GVHD=26%
Chronic GVHD=45%Interstitial Pneumonitis=14% 24% 6% Blume
CML, AML, ALLTRM VOD GVHD Pulmonary Hemorrhagic Cystitis Seizure No Report Deaths=4.9% Acute ≥Grade 2 GVHD=22% (13/58 at risk)
Chronic GVHD=31% (14/45 at risk)No Report No Report No Report 1. TRM = Transplantation Related Mortality
2. VOD = Veno-Occlusive Diseases of the Liver
3. GVHD = Graft versus Host Disease
-
7 DRUG INTERACTIONS
7.1 Drugs that Decrease Busulfan Clearance
Itraconazole decreases busulfan clearance by up to 25%. Metronidazole decreases the clearance of busulfan to a greater extent than does itraconazole; metronidazole coadministration has been associated with increased busulfan toxicity. Fluconazole (200 mg) has been used with busulfan.
Decreased clearance of busulfan was observed with concomitant use with deferasirox. The mechanism of this interaction is not fully elucidated. Discontinue iron chelating agents well in advance of administration of busulfan to avoid increased exposure to busulfan.
Because busulfan is eliminated from the body via conjugation with glutathione, use of acetaminophen prior to (less than 72 hours) or concurrent with busulfan may result in reduced busulfan clearance based upon the known property of acetaminophen to decrease glutathione levels in the blood and tissues.
7.2 Drugs that Increase Busulfan Clearance
Phenytoin increases the clearance of busulfan by 15% or more, possibly due to the induction of glutathione-S-transferase. Since the pharmacokinetics of busulfan was studied in patients treated with phenytoin, the clearance of busulfan at the recommended dose may be lower and exposure (AUC) higher in patients not treated with phenytoin.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Busulfan can cause fetal harm when administered to a pregnant woman based on animal data. Busulfan was teratogenic in mice, rats, and rabbits following administration during organogenesis. The solvent, DMA, may also cause fetal harm when administered to a pregnant woman. In rats, DMA doses of approximately 40% of the daily dose of DMA in the busulfan dose on a mg/m² basis given during organogenesis caused significant developmental anomalies [see Data]. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated populations are unknown. However, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.
Animal Data
Following administration during organogenesis in animals, busulfan caused malformations and anomalies, including significant alterations in the musculoskeletal system, body weight gain, and size. In pregnant rats, busulfan produced sterility in both male and female offspring due to the absence of germinal cells in the testes and ovaries. The solvent, N,N-dimethylacetamide (DMA), administered to rats at doses of 400 mg/kg/day (about 40% of the daily dose of DMA in the busulfan dose on a mg/m² basis) during organogenesis caused significant developmental anomalies. The most striking abnormalities included anasarca, cleft palate, vertebral anomalies, rib anomalies, and serious anomalies of the vessels of the heart.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Busulfan can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with busulfan and for 6 months following cessation of therapy.
Males
Busulfan may damage spermatozoa and testicular tissue, resulting in possible genetic fetal abnormalities. Males with female sexual partners of reproductive potential should use effective contraception during treatment with busulfan and for 3 months after cessation of therapy [see Nonclinical Toxicology (13.1)].
Infertility
Females
Ovarian suppression and amenorrhea commonly occur in premenopausal women undergoing chronic, low-dose busulfan therapy for chronic myelogenous leukemia. Busulfan may cause temporary or permanent infertility in prepubertal girls or in females of child-bearing potential treated with high-dose busulfan in the conditioning regimen prior to allogeneic hematopoietic progenitor cell transplantation.
8.4 Pediatric Use
The effectiveness of busulfan in the treatment of CML has not been specifically studied in pediatric patients. An open-label, uncontrolled study evaluated the pharmacokinetics of busulfan in 24 pediatric patients receiving busulfan as part of a conditioning regimen administered prior to hematopoietic progenitor cell transplantation for a variety of malignant hematologic (N=15) or non-malignant diseases (N=9). Patients ranged in age from 5 months to 16 years (median 3 years). Busulfan dosing was targeted to achieve an area under the plasma concentration curve (AUC) of 900-1350 μM•min with an initial dose of 0.8 mg per kg or 1.0 mg per kg (based on Actual Body Weight (ABW)) if the patient was greater than 4 or less than or equal to 4 years, respectively. The dose was adjusted based on plasma concentration after completion of dose 1.
Patients received busulfan doses every six hours as a two-hour infusion over four days for a total of 16 doses, followed by cyclophosphamide 50 mg per kg once daily for four days. After one rest day, hematopoietic progenitor cells were infused. All patients received phenytoin as seizure prophylaxis. The target AUC (900-1350±5% μM•min) for busulfan was achieved at dose 1 in 71% (17/24) of patients. Steady state pharmacokinetic testing was performed at dose 9 and 13. Busulfan levels were within the target range for 21 of 23 evaluable patients.
All 24 patients experienced neutropenia (absolute neutrophil count (ANC) less than 0.5×109/L) and thrombocytopenia (platelet transfusions or platelet count less than 20,000/mm3). Seventy-nine percent (19/24) of patients experienced lymphopenia (absolute lymphocyte count less than 0.1×109). In 23 patients, the ANC recovered to greater than 0.5×109/L (median time to recovery = BMT day +13; range = BMT day +9 to +22). One patient who died on day +20 had not recovered to an ANC >0.5×109/L.
Four (17%) patients died during the study. Two patients died within 28 days of transplant; one with pneumonia and capillary leak syndrome, and the other with pneumonia and veno-occlusive disease. Two patients died prior to day 100; one due to progressive disease and one due to multi-organ failure.
Adverse reactions were reported in all 24 patients during the study period (BMT day -10 through BMT day +28) or post-study surveillance period (day +29 through +100). These included vomiting (100%), nausea (83%), stomatitis (79%), HVOD (21%), graft-versus host disease (GVHD) (25%), and pneumonia (21%).
Based on the results of this 24-patient clinical trial, a suggested dosing regimen of busulfan in pediatric patients is shown in the following dosing nomogram:
Busulfan Dosing Nomogram Patient's Actual Body Weight (ABW) Busulfan Dosage less than or equal to12 kg 1.1 (mg per kg) greater than 12 kg 0.8 (mg per kg) Simulations based on a pediatric population pharmacokinetic model indicate that approximately 60% of pediatric patients will achieve a target busulfan exposure (AUC) between 900 to 1350 μM•min with the first dose of busulfan using this dosing nomogram. Therapeutic drug monitoring and dose adjustment following the first dose of busulfan is recommended.
Dose Adjustment Based on Therapeutic Drug Monitoring
Instructions for measuring the AUC of busulfan at dose 1 (see Blood Sample Collection for AUC Determination) and the formula for adjustment of subsequent doses to achieve the desired target AUC (1125 µM•min), are provided below.
Adjusted dose (mg) = Actual Dose (mg) x Target AUC (μM•min)/Actual AUC (μM•min)
For example, if a patient received a dose of 11 mg busulfan and if the corresponding AUC measured was 800 µM•min, for a target AUC of 1125 µM•min, the target mg dose would be:
Mg dose =11 mg x 1125 μM•min /800 μM•min =15.5 mg
Busulfan dose adjustment may be made using this formula and instructions below.
Blood Sample Collection for AUC Determination
Calculate the AUC (µM•min) based on blood samples collected at the following time points:
For dose 1:2 hr (end of infusion), 4 hr and 6 hr (immediately prior to the next scheduled busulfan administration). Actual sampling times should be recorded.
For doses other than dose 1: Pre-infusion (baseline), 2 hr (end of infusion), 4 hr and 6 hr (immediately prior to the next scheduled busulfan administration).
AUC calculations based on fewer than the three specified samples may result in inaccurate AUC determinations.
For each scheduled blood sample, collect one to three mL of blood into heparinized (Na or Li heparin) Vacutainer® tubes. The blood samples should be placed on wet ice immediately after collection and should be centrifuged (at 4°C) within one hour. The plasma, harvested into appropriate cryovial storage tubes, is to be frozen immediately at -20°C. All plasma samples are to be sent in a frozen state (i.e., on dry ice) to the assay laboratory for the determination of plasma busulfan concentrations.
Calculation of AUC
Busulfan AUC calculations may be made using the following instructions and appropriate standard pharmacokinetic formula:
Dose 1 AUCinfinity Calculation: AUCinfinity = AUC0–6hr +AUCextrapolated, where AUC0–6hr is to be estimated using the linear trapezoidal rule and AUC extrapolated can be computed by taking the ratio of the busulfan concentration at Hour 6 and the terminal elimination rate constant, λz. The λz must be calculated from the terminal elimination phase of the busulfan concentration vs. time curve. A "0" pre-dose busulfan concentration should be assumed, and used in the calculation of AUC.
If the AUC is assessed subsequent to Dose 1, steady-state AUCss (AUC0–6hr) is to be estimated from the trough, 2 hr, 4 hr and 6 hr concentrations using the linear trapezoidal rule.
Instructions for Drug Administration and Blood Sample Collection for Therapeutic Drug Monitoring
Use an administration set with minimal residual hold up (priming) volume (1- to 3 mL) for drug infusion to ensure accurate delivery of the entire prescribed dose and to ensure accurate collection of blood samples for therapeutic drug monitoring and dose adjustment.
Prime the administration set tubing with drug solution to allow accurate documentation of the start time of busulfan infusion. Collect the blood sample from a peripheral IV line to avoid contamination with infusing drug. If the blood sample is taken directly from the existing central venous catheter (CVC), DO NOT COLLECT THE BLOOD SAMPLE WHILE THE DRUG IS INFUSING to ensure that the end of infusion sample is not contaminated with any residual drug. At the end of infusion (2 hr), disconnect the administration tubing and flush the CVC line with 5 ml of normal saline prior to the collection of the end of infusion sample from the CVC port. Collect the blood samples from a different port than that used for the busulfan infusion. When recording the busulfan infusion stop time, do not include the time required to flush the indwelling catheter line. Discard the administration tubing at the end of the two-hour infusion [see Dosage and Administration (2.3)].
-
10 OVERDOSAGE
There is no known antidote to busulfan injection other than hematopoietic progenitor cell transplantation. In the absence of hematopoietic progenitor cell transplantation, the recommended dosage for busulfan would constitute an overdose of busulfan. The principal toxic effect is profound bone marrow hypoplasia/aplasia and pancytopenia, but the central nervous system, liver, lungs, and gastrointestinal tract may be affected. Monitor hematologic status closely and institute vigorous supportive measures as medically indicated. Survival after a single 140 mg dose of Myleran® Tablets in an 18 kg, 4-year old child has been reported. Inadvertent administration of a greater than normal dose of oral busulfan (2.1 mg per kg; total dose of 23.3 mg per kg) occurred in a 2-year old child prior to a scheduled bone marrow transplant without sequelae. An acute dose of 2.4 g was fatal in a 10-year old boy. There is one report that busulfan is dialyzable, thus dialysis should be considered in the case of overdose.
-
11 DESCRIPTION
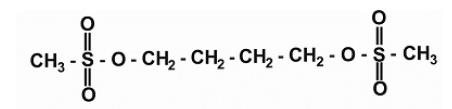
Busulfan is a bifunctional alkylating agent known chemically as 1,4-butanediol, dimethanesulfonate. Busulfan Injection is intended for intravenous administration. It is supplied as a clear, colorless, sterile, solution in 10 mL single-dose vials. Each vial of busulfan injection contains 60 mg (6 mg/mL) of busulfan, the active ingredient, a white crystalline powder with a molecular formula of CH3SO2O(CH2)4OSO2CH3 and a molecular weight of 246 g/mole. Busulfan has the following chemical structure:
Busulfan is dissolved in N,N-dimethylacetamide (DMA), 3.3 mL and Polyethylene Glycol 400, NF 6.7 mL. The solubility of busulfan in water is 0.1 g per L and the pH of busulfan diluted to approximately 0.5 mg per mL busulfan in 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP as recommended for infusion reflects the pH of the diluent used and ranges from 3.4 to 3.9.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Busulfan is a bifunctional alkylating agent in which two labile methanesulfonate groups are attached to opposite ends of a four-carbon alkyl chain. In aqueous media, busulfan hydrolyzes to release the methanesulfonate groups. This produces reactive carbonium ions that can alkylate DNA. DNA damage is thought to be responsible for much of the cytotoxicity of busulfan.
12.3 Pharmacokinetics
The pharmacokinetics of busulfan was studied in 59 patients participating in a prospective trial of a busulfan cyclophosphamide preparatory regimen prior to allogeneic hematopoietic progenitor stem cell transplantation. Patients received 0.8 mg/kg busulfan every six hours, for a total of 16 doses over four days. Fifty-five of fifty-nine patients (93%) administered busulfan maintained AUC values below the target value (less than1500 μM•min).
Table 3: Steady State Pharmacokinetic Parameters Following Busulfan (0.8 mg per kg; N=59) Mean CV (%) Range Cmax (ng per mL) 1222 18 496 - 1684 AUC (μM•min) 1167 20 556 - 1673 CL (mL per min per kg)1 2.52 25 1.49 - 4.31 1 Clearance normalized to actual body weight for all patients. Busulfan pharmacokinetics showed consistency between dose 9 and dose 13 as demonstrated by reproducibility of steady state Cmax and a low coefficient of variation for this parameter.
Distribution: Busulfan achieves concentrations in the cerebrospinal fluid approximately equal to those in plasma. Busulfan primarily binds to albumin (Mean ± standard deviation=32.4 ± 2.2%).
Metabolism: Busulfan is predominantly metabolized by conjugation with glutathione, both spontaneously and by glutathione S-transferase (GST) catalysis. This conjugate undergoes extensive oxidative metabolism in the liver.
Excretion: Following administration of 14C-labeled busulfan to humans, approximately 30% of the radioactivity was excreted into the urine over 48 hours; negligible amounts were recovered in feces.
Specific Populations
Pediatric Patients: In a pharmacokinetic study of busulfan injection in 24 pediatric patients, the population pharmacokinetic (PPK) estimates of busulfan injection for clearance (CL) and volume of distribution (V) were determined. For actual body weight, PPK estimates of CL and V were 4.04 L/hr per 20 kg (3.37 mL per min per kg; interpatient variability 23%); and 12.8 L per 20 kg (0.64 L per kg; interpatient variability 11%).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Busulfan is a mutagen and a clastogen. In in vitro tests it caused mutations in Salmonella typhimurium and Drosophila melanogaster. Chromosomal aberrations induced by busulfan have been reported in vivo (rats, mice, hamsters, and humans) and in vitro (rodent and human cells). The intravenous administration of busulfan (48 mg/kg given as biweekly doses of 12 mg/kg, or 30% of the total busulfan dose on a mg/m² basis) has been shown to increase the incidence of thymic and ovarian tumors in mice.
Busulfan depleted oocytes of female rats and induced sterility in male rats and hamsters. The solvent DMA may also impair fertility. A DMA daily dose of 0.45 g/kg/day given to rats for nine days (equivalent to 44% of the daily dose of DMA contained in the recommended dose of busulfan on a mg/m² basis) significantly decreased spermatogenesis in rats. A single subcutaneous dose of 2.2 g/kg (27% of the total DMA dose contained in busulfan on a mg/m² basis) four days after insemination terminated pregnancy in 100% of tested hamsters [see Use in Specific Populations (8.3)].
-
14 CLINICAL STUDIES
Documentation of the safety and efficacy of busulfan as a component of a conditioning regimen prior to allogeneic hematopoietic progenitor cell reconstitution is derived from two sources:
i) analysis of a prospective clinical trial of busulfan that involved 61 patients diagnosed with various hematologic malignancies, and
ii) the published reports of randomized, controlled trials that employed high-dose oral busulfan as a component of a conditioning regimen for transplantation, which were identified in a literature review of five established commercial databases.
Prospective Clinical Trial of Busulfan: The prospective trial was a single-arm, open-label study in 61 patients who received busulfan as part of a conditioning regimen for allogeneic hematopoietic stem cell transplantation. The study included patients with acute leukemia past first remission (first or subsequent relapse), with high-risk first remission, or with induction failure; chronic myelogenous leukemia (CML) in chronic phase, accelerated phase, or blast crisis; primary refractory or resistant relapsed Hodgkin's disease or non-Hodgkin's lymphoma; and myelodysplastic syndrome. Forty-eight percent of patients (29/61) were heavily pretreated, defined as having at least one of the following: prior radiation, greater than or equal to 3 prior chemotherapeutic regimens, or prior hematopoietic stem cell transplant. Seventy-five percent of patients (46/61) were transplanted with active disease.
Patients received 16 busulfan doses of 0.8 mg per kg every 6 hours as a two-hour infusion for 4 days, followed by cyclophosphamide 60 mg per kg once per day for two days (BuCy2 regimen). All patients received 100% of their scheduled busulfan regimen. No dose adjustments were made. After one rest day, allogeneic hematopoietic progenitor cells were infused. The efficacy parameters in this study were myeloablation (defined as one or more of the following: absolute neutrophil count [ANC] less than 0.5×109/L, absolute lymphocytes count [ALC] less than 0.1×109/L, thrombocytopenia defined as a platelet count less than 20,000/mm3 or a platelet transfusion requirement) and engraftment (ANC greater than or equal to 0.5×109/L).
All patients (61/61) experienced myeloablation. The median time to neutropenia was 4 days. All evaluable patients (60/60) engrafted at a median of 13 days post-transplant (range 9 to 29 days); one patient was considered non-evaluable because he died of a fungal pneumonia 20 days after BMT and before engraftment occurred. All but 13 of the patients were treated with prophylactic G-CSF. Evidence of donor cell engraftment and chimerism was documented in all patients who had a chromosomal sex marker or leukemic marker (43/43), and no patient with chimeric evidence of allogeneic engraftment suffered a later loss of the allogeneic graft. There were no reports of graft failure in the overall study population. The median number of platelet transfusions per patient was 6, and the median number of red blood cell transfusions per patient was 4.
Twenty-three patients (38%) relapsed at a median of 183 days post-transplant (range 36 to 406 days). Sixty-two percent of patients (38/61) were free from disease with a median follow-up of 269 days post-transplant (range 20 to 583 days). Forty-three patients (70%) were alive with a median follow up of 288 days post-transplant (range 51 to 583 days). There were two deaths before BMT Day +28 and six additional patients died by BMT Day +100. Ten patients (16%) died after BMT Day +100, at a median of 199 days post-transplant (range 113 to 275 days).
Oral Busulfan Literature Review: Four publications of randomized, controlled trials that evaluated a high-dose oral busulfan-containing conditioning regimen (busulfan 4 mg/kg/d x4 days + cyclophosphamide 60 mg/kg/d x2 days) for allogeneic transplantation in the setting of CML were identified. Two of the studies (Clift and Devergie) had populations confined to CML in chronic phase that were randomized between conditioning with busulfan/cyclophosphamide (BU/CY) and cyclophosphamide/total body irradiation (CY/TBI). A total of 138 patients were treated with BU/CY in these studies. The populations of the two remaining studies (Ringden and Blume) included patients with CML, acute lymphoblastic leukemia (ALL), and acute myelogenous leukemia (AML). In the Nordic BMT Group study published by Ringden, et al., 57 patients had CML, and of those, 30 were treated with BU/CY. Patients with CML in chronic phase, accelerated phase, and blast crisis were eligible for this study. The participants with CML (34/122 patients) in a SWOG study published by Blume, et al., had disease beyond first chronic phase. Twenty of those CML patients were treated with BU/CY, and the TBI comparator arm utilized etoposide instead of cyclophosphamide.
Table 4 summarizes the efficacy analyses reported from these 4 studies.
Table 4: Summary of efficacy analyses from the randomized, controlled trials utilizing a high dose oral busulfan-containing conditioning regimen identified in a literature review. Clift, 1994
CML Chronic Phase;3 year Overall
Survival3 year DFS
(p=0.43)Relapse Time to Engraftment
(ANC greater than or equal to 500)BU/CY CY/TBI BU/CY CY/TBI BU/CY CY/TBI BU/CY CY/TBI 80% 80% 71% 68% 13% 13% 22.6 days 22.3 days Devergie, 1995
CML Chronic Phase;5 year Overall Survival
(p=0.5)5 year DFS (p=0.75) Relapse (Relative Risk analysis BU/CY:CY/TBI)
(p=0.04)Time to Engraftment
(ANC greater than or equal to 500)BU/CY CY/TBI BU/CY CY/TBI BU/CY CY/TBI BU/CY CY/TBI 60.6%
±11.7%65.8%
±12.5%59.1%
±11.8%51.0%
±14%4.10
(95%CI =1.00-20.28)None
GivenNone
GivenRingden, 1994
CML, AML, ALL;3 year Overall
Survival
(p<0.03)3 year Relapse
Free Survival
(p=0.065)Relapse
(p=0.9)Time to Engraftment
(ANC greater than 500)BU/CY CY/TBI BU/CY CY/TBI BU/CY CY/TBI BU/CY CY/TBI 62% 76% 56% 67% 22% 26% 20 days 20 days Blume, 19931
CML, AML, ALL; Relative Risk Analysis BU/CY: Etoposide/TBIRR of Mortality DFS RR of Relapse
(Relative Risk analysis BU/CY:Eto/TBI)Time to Engraftment BU/CY Eto/TBI BU/CY Eto/TBI BU/CY Eto/TBI BU/CY Eto/TBI 0.97
(95% CI=0.64-1.48)Not Given 1.02
(95% CI=0.56-1.86)Not Given 1.Eto=etoposide. TBI was combined with etoposide in the comparator arm of this study.
BU = Busulfan
CY = Cyclophosphamide
TBI = Total Body Irradiation
DFS = Disease Free Survival
ANC = Absolute Neutrophil Count
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Busulfan injection is packaged as a sterile solution in 10 mL single-dose clear glass vials each containing 60 mg of busulfan at a concentration of 6 mg per mL for intravenous use, NDC 72606-559-01.
Busulfan injection is distributed as a unit carton of eight vials NDC 72606-559-02
16.2 Storage and Handling
Unopened vials of busulfan injection must be stored under refrigerated conditions between 2°C to 8°C (36°F to 46°F).
Busulfan injection diluted in 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP is stable at room temperature (25°C) for up to 8 hours but the infusion must be completed within that time.
Busulfan injection diluted in 0.9% Sodium Chloride Injection, USP is stable at refrigerated conditions (2°C to 8°C) for up to 12 hours but the infusion must be completed within that time.
Busulfan injection is a cytotoxic drug. Follow applicable special handling and disposal procedures1.
-
17 PATIENT COUNSELING INFORMATION
Myelosuppression
Advise patients of the possibility of developing low blood cell counts and the need for hematopoietic progenitor cell infusion. Instruct patients to immediately report to their healthcare provider if fever develops [see Warnings and Precautions (5.1)].
Seizures
Advise patients of the possibility of seizures and that they will be given medication to prevent them. Patients should be asked to report a history of seizure or head trauma [see Warnings and Precautions (5.2)].
Hepatic Veno-Occlusive Disease (HVOD)
Advise patients of the risks associated with the use of busulfan injection as well as the plan for regular blood monitoring during therapy. Specifically inform patients of the following: The risk of veno-occlusive liver disease [see Warnings and Precautions (5.3)].
Embryo-fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider with a known or suspected pregnancy [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)].
Females of Reproductive Potential
Advise females of reproductive potential to use effective contraception during treatment with busulfan injection and for 6 months following cessation of therapy [see Use in Specific Populations (8.3)].
Males of Reproductive Potential
Advise males with female sexual partners of reproductive potential to use effective contraception during treatment with busulfan injection and for 3 months following cessation of therapy [see Use in Specific Populations (8.3)].
Lactation
Advise females to discontinue breastfeeding during treatment with busulfan injection [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that busulfan injection may cause temporary or permanent infertility [see Use in Specific Populations (8.3)].
Cardiac Tamponade
Advise patients of the risk of cardiac tamponade. Instruct patients to report to their healthcare provider symptoms of abdominal pain and vomiting [see Warnings and Precautions (5.5)].
Bronchopulmonary Dysplasia
Advise patients of the possibility of bronchopulmonary dysplasia with pulmonary fibrosis with chronic busulfan injection therapy. Instruct patients to report symptoms of shortness of breath and cough to their healthcare provider. These symptoms could occur several months or years after therapy with busulfan injection [see Warnings and Precautions (5.6)].
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BUSULFAN
busulfan injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72606-559 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BUSULFAN (UNII: G1LN9045DK) (BUSULFAN - UNII:G1LN9045DK) BUSULFAN 6 mg in 1 mL Inactive Ingredients Ingredient Name Strength N,N-DIMETHYLACETAMIDE (UNII: JCV5VDB3HY) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72606-559-02 8 in 1 CARTON 12/01/2019 1 NDC:72606-559-01 1 in 1 CARTON 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA210931 12/01/2019 Labeler - CELLTRION USA, INC. (116587378)