Label: NEFAZODONE HYDROCHLORIDE tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 16590-166-30, 16590-166-60, 16590-166-90 - Packager: STAT RX USA LLC
- This is a repackaged label.
- Source NDC Code(s): 0093-1024
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated April 7, 2011
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
Suicidality and Antidepressant Drugs
Antidepressants increased the risk compared to placebo of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults in short-term studies of major depressive disorder (MDD) and other psychiatric disorders. Anyone considering the use of nefazodone hydrochloride tablets or any other antidepressant in a child, adolescent, or young adult must balance this risk with the clinical need. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction in risk with antidepressants compared to placebo in adults aged 65 and older. Depression and certain other psychiatric disorders are themselves associated with increases in the risk of suicide. Patients of all ages who are started on antidepressant therapy should be monitored appropriately and observed closely for clinical worsening, suicidality, or unusual changes in behavior. Families and caregivers should be advised of the need for close observation and communication with the prescriber. Nefazodone hydrochloride tablets are not approved for use in pediatric patients (see WARNINGS, Clinical Worsening and Suicide Risk; PRECAUTIONS, Information for Patients; and PRECAUTIONS, Pediatric Use).
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
Warning
Cases of life-threatening hepatic failure have been reported in patients treated with nefazodone hydrochloride tablets. The reported rate in the United States is about 1 case of liver failure resulting in death or transplant per 250,000 to 300,000 patient-years of nefazodone hydrochloride treatment. The total patient-years is a summation of each patient’s duration of exposure expressed in years. For example, 1 patient-year is equal to 2 patients each treated for 6 months, 3 patients each treated for 4 months, etc. (see WARNINGS).
Ordinarily, treatment with nefazodone hydrochloride tablets should not be initiated in individuals with active liver disease or with elevated baseline serum transaminases. There is no evidence that pre-existing liver disease increases the likelihood of developing liver failure, however, baseline abnormalities can complicate patient monitoring.
Patients should be advised to be alert for signs and symptoms of liver dysfunction (jaundice, anorexia, gastrointestinal complaints, malaise, etc.) and to report them to their doctor immediately if they occur.
Nefazodone hydrochloride tablets should be discontinued if clinical signs or symptoms suggest liver failure (see PRECAUTIONS, Information for Patients). Patients who develop evidence of hepatocellular injury such as increased serum AST or serum ALT levels ≥ 3 times the upper limit of NORMAL, while on nefazodone hydrochloride tablets should be withdrawn from the drug. These patients should be presumed to be at increased risk for liver injury if nefazodone hydrochloride is reintroduced. Accordingly, such patients should not be considered for re-treatment.
-
DESCRIPTION
Nefazodone hydrochloride tablets USP are an antidepressant for oral administration with a chemical structure unrelated to selective serotonin reuptake inhibitors, tricyclics, tetracyclics, or monoamine oxidase inhibitors (MAOI).
Nefazodone hydrochloride is a synthetically derived phenylpiperazine antidepressant. The chemical name for nefazodone hydrochloride is 2-[3-[4-(3-chlorophenyl)-1-piperazinyl]propyl]-5-ethyl-2,4-dihydro-4-(2-phenoxyethyl)-3H-1,2,4-triazol-3-one monohydrochloride. The structural formula is:
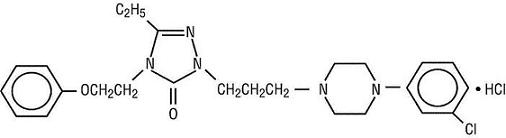
C25H32CIN5O2•HCl M.W. 506.5
Nefazodone hydrochloride is a nonhygroscopic, white crystalline solid. It is freely soluble in chloroform, soluble in propylene glycol, and slightly soluble in polyethylene glycol and water.
Nefazodone hydrochloride tablets USP are supplied as capsule-shaped tablets containing 50 mg, 100 mg, 150 mg, 200 mg, or 250 mg of nefazodone hydrochloride and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, sodium starch glycolate and povidone. Additionally, the 50 mg tablets include ferric oxide red as a colorant, the 150 mg tablets include ferric oxide red and yellow as colorants, and the 200 mg tablets include ferric oxide yellow as a colorant.
-
CLINICAL PHARMACOLOGY
Pharmacodynamics
The mechanism of action of nefazodone, as with other antidepressants, is unknown.
Preclinical studies have shown that nefazodone inhibits neuronal uptake of serotonin and norepinephrine.
Nefazodone occupies central 5-HT2 receptors at nanomolar concentrations, and acts as an antagonist at this receptor. Nefazodone was shown to antagonize alpha1-adrenergic receptors, a property which may be associated with postural hypotension. In vitro binding studies showed that nefazodone had no significant affinity for the following receptors: alpha2 and beta adrenergic, 5-HT1A, cholinergic, dopaminergic, or benzodiazepine.
Pharmacokinetics
Nefazodone is rapidly and completely absorbed but is subject to extensive metabolism, so that its absolute bioavailability is low, about 20%, and variable. Peak plasma concentrations occur at about one hour and the half-life of nefazodone is 2 to 4 hours.
Both nefazodone and its pharmacologically similar metabolite, hydroxynefazodone, exhibit nonlinear kinetics for both dose and time, with AUC and Cmax increasing more than proportionally with dose increases and more than expected upon multiple dosing over time, compared to single dosing. For example, in a multiple-dose study involving BID dosing with 50, 100, and 200 mg, the AUC for nefazodone and hydroxynefazodone increased by about 4 fold with an increase in dose from 200 to 400 mg per day; Cmax increased by about 3 fold with the same dose increase. In a multiple-dose study involving BID dosing with 25, 50, 100, and 150 mg, the accumulation ratios for nefazodone and hydroxynefazodone AUC, after 5 days of BID dosing relative to the first dose, ranged from approximately 3 to 4 at the lower doses (50 to 100 mg/day) and from 5 to 7 at the higher doses (200 to 300 mg/day); there were also approximately 2 to 4 fold increases in Cmax after 5 days of BID dosing relative to the first dose, suggesting extensive and greater than predicted accumulation of nefazodone and its hydroxy metabolite with multiple dosing. Steady-state plasma nefazodone and metabolite concentrations are attained within 4 to 5 days of initiation of BID dosing or upon dose increase or decrease.
Nefazodone is extensively metabolized after oral administration by n-dealkylation and aliphatic and aromatic hydroxylation, and less than 1% of administered nefazodone is excreted unchanged in urine. Attempts to characterize three metabolites identified in plasma, hydroxynefazodone (HO-NEF), meta-chlorophenylpiperazine (mCPP), and a triazole-dione metabolite, have been carried out. The AUC (expressed as a multiple of the AUC for nefazodone dosed at 100 mg BID) and elimination half-lives for these three metabolites were as follows:
AUC Multiples and T1/2 for Three Metabolites of Nefazodone (100 mg BID) Metabolite AUC Multiple T1/2 HO-NEF 0.4 1.5 to 4 h mCPP 0.07 4 to 8 h Triazole-dione 4.0 18 h HO-NEF possesses a pharmacological profile qualitatively and quantitatively similar to that of nefazodone. mCPP has some similarities to nefazodone, but also has agonist activity at some serotonergic receptor subtypes. The pharmacological profile of the triazole-dione metabolite has not yet been well characterized. In addition to the above compounds, several other metabolites were present in plasma but have not been tested for pharmacological activity.
After oral administration of radiolabeled nefazodone, the mean half-life of total label ranged between 11 and 24 hours. Approximately 55% of the administered radioactivity was detected in urine and about 20 to 30% in feces.
Distribution
Nefazodone is widely distributed in body tissues, including the central nervous system (CNS). In humans the volume of distribution of nefazodone ranges from 0.22 to 0.87 L/kg.
Protein Binding
At concentrations of 25 to 2500 ng/mL nefazodone is extensively (> 99%) bound to human plasma proteins in vitro. The administration of 200 mg BID of nefazodone for 1 week did not increase the fraction of unbound warfarin in subjects whose prothrombin times had been prolonged by warfarin therapy to 120 to 150% of the laboratory control (see PRECAUTIONS, Drug Interactions). While nefazodone did not alter the in vitro protein binding of chlorpromazine, desipramine, diazepam, diphenylhydantoin, lidocaine, prazosin, propranolol, or verapamil, it is unknown whether displacement of either nefazodone or these drugs occurs in vivo. There was a 5% decrease in the protein binding of haloperidol; this is probably of no clinical significance.
Effect of Food
Food delays the absorption of nefazodone and decreases the bioavailability of nefazodone by approximately 20%.
Renal Disease
In studies involving 29 renally impaired patients, renal impairment (creatinine clearances ranging from 7 to 60 mL/min/1.73 m2) had no effect on steady-state nefazodone plasma concentrations.
Liver Disease
In a multiple-dose study of patients with liver cirrhosis, the AUC values for nefazodone and HO-NEF at steady state were approximately 25% greater than those observed in normal volunteers.
Age/Gender Effects
After single doses of 300 mg to younger (18 to 45 years) and older patients (> 65 years), Cmax and AUC for nefazodone and hydroxynefazodone were up to twice as high in the older patients. With multiple doses, however, differences were much smaller, 10 to 20%. A similar result was seen for gender, with a higher Cmax and AUC in women after single doses but no difference after multiple doses.
Treatment with nefazodone should be initiated at half the usual dose in elderly patients, especially women (see DOSAGE AND ADMINISTRATION), but the therapeutic dose range is similar in younger and older patients.
Clinical Efficacy Trial Results
Studies in Outpatients With Depression
During its premarketing development, the efficacy of nefazodone was evaluated at doses within the therapeutic range in five well-controlled, short-term (6 to 8 weeks) clinical investigations. These trials enrolled outpatients meeting DSM-III or DSM-IIIR criteria for major depression. Among these trials, two demonstrated the effectiveness of nefazodone, and two provided additional support for that conclusion.
One trial was a 6 week dose-titration study comparing nefazodone in two dose ranges (up to 300 mg/day and up to 600 mg/day [mean modal dose for this group was about 400 mg/day], on a BID schedule) and placebo. The second trial was an 8 week dose-titration study comparing nefazodone (up to 600 mg/day; mean modal dose was 375 mg/day), imipramine (up to 300 mg/day), and placebo, all on a BID schedule. Both studies demonstrated nefazodone, at doses titrated between 300 mg to 600 mg/day (therapeutic dose range), to be superior to placebo on at least three of the following four measures: 17 Item Hamilton Depression Rating Scale or HDRS (total score), Hamilton Depressed Mood item, Clinical Global Impressions (CGI) Severity score, and CGI Improvement score. Significant differences were also found for certain factors of the HDRS (e.g., anxiety factor, sleep disturbance factor, and retardation factor). In the two supportive studies, nefazodone was titrated up to 500 or 600 mg/day (mean modal doses of 462 mg/day and 363 mg/day). In the fifth study, the differentiation in response rates between nefazodone and placebo was not statistically significant. Three additional trials were conducted using subtherapeutic doses of nefazodone.
Overall, approximately two thirds of patients in these trials were women, and an analysis of the effects of gender on outcome did not suggest any differential responsiveness on the basis of sex. There were too few elderly patients in these trials to reveal possible age-related differences in response.
Since its initial marketing as an antidepressant drug product, additional clinical investigations of nefazodone have been conducted. These studies explored nefazodone’s use under conditions not evaluated fully at the time initial marketing approval was granted.
Studies in “Inpatients”
Two studies were conducted to evaluate nefazodone’s effectiveness in hospitalized depressed patients. These were 6 week, dose-titration trials comparing nefazodone (up to 600 mg/day) and placebo, on a BID schedule. In one study, nefazodone was superior to placebo. In this study, the mean modal dose of nefazodone was 503 mg/day, and 85% of these inpatients were melancholic; at baseline, patients were distributed at the higher end of the 7 point CGI Severity scale, as follows: 4 = moderately ill (17%); 5 = markedly ill (48%); 6 = severely ill (32%). In the other study, the differentiation in response rates between nefazodone and placebo was not statistically significant. This result may be explained by the “high” rate of spontaneous improvement among the patients randomized to placebo.
Studies of “Relapse Prevention in Patients Recently Recovered (Clinically) From Depression”
Two studies were conducted to assess nefazodone’s capacity to maintain a clinical remission in acutely depressed patients who were judged to have responded adequately (HDRS total score ≤ 10) after a 16 week period of open treatment with nefazodone (titration up to 600 mg/day). In one study, nefazodone was superior to placebo. In this study, patients (n = 131) were randomized to continuation on nefazodone or placebo for an additional 36 weeks (1 year total). This study demonstrated a significantly lower relapse rate (HDRS total score ≥ 18) for patients taking nefazodone compared to those on placebo. The second study was of appropriate design and power, but the sample of patients admitted for evaluation did not suffer relapses at a high enough incidence to provide a meaningful test of nefazodone’s efficacy for this use.
Comparisons of Clinical Trial Results
Highly variable results have been seen in the clinical development of all antidepressant drugs. Furthermore, in those circumstances when the drugs have not been studied in the same controlled clinical trial(s), comparisons among the findings of studies evaluating the effectiveness of different antidepressant drug products are inherently unreliable. Because conditions of testing (e.g., patient samples, investigators, doses of the treatments administered and compared, outcome measures, etc.) vary among trials, it is virtually impossible to distinguish a difference in drug effect from a difference due to one or more of the confounding factors just enumerated.
-
INDICATIONS AND USAGE
Nefazodone hydrochloride tablets are indicated for the treatment of depression. When deciding among the alternative treatments available for this condition, the prescriber should consider the risk of hepatic failure associated with nefazodone hydrochloride treatment (see WARNINGS). In many cases, this would lead to the conclusion that other drugs should be tried first.
The efficacy of nefazodone in the treatment of depression was established in 6 to 8 week controlled trials of outpatients and in a 6 week controlled trial of depressed inpatients whose diagnoses corresponded most closely to the DSM-III or DSM-IIIR category of major depressive disorder (see CLINICAL PHARMACOLOGY).
A major depressive episode implies a prominent and relatively persistent depressed or dysphoric mood that usually interferes with daily functioning (nearly every day for at least 2 weeks). It must include either depressed mood or loss of interest or pleasure and at least five of the following nine symptoms: depressed mood, loss of interest in usual activities, significant change in weight and/or appetite, insomnia or hypersomnia, psychomotor agitation or retardation, increased fatigue, feelings of guilt or worthlessness, slowed thinking or impaired concentration, a suicide attempt or suicidal ideation.
The efficacy of nefazodone in reducing relapse in patients with major depression who were judged to have had a satisfactory clinical response to 16 weeks of open-label nefazodone treatment for an acute depressive episode has been demonstrated in a randomized placebo-controlled trial (see CLINICAL PHARMACOLOGY). Although remitted patients were followed for as long as 36 weeks in the study cited (i.e., 52 weeks total), the physician who elects to use nefazodone for extended periods should periodically reevaluate the long-term usefulness of the drug for the individual patient.
-
CONTRAINDICATIONS
Coadministration of terfenadine, astemizole, cisapride, pimozide, or carbamazepine with nefazodone hydrochloride is contraindicated (see WARNINGS and PRECAUTIONS).
Nefazodone hydrochloride tablets are contraindicated in patients who were withdrawn from nefazodone because of evidence of liver injury (see BOXED WARNING). Nefazodone hydrochloride tablets are also contraindicated in patients who have demonstrated hypersensitivity to nefazodone hydrochloride, its inactive ingredients, or other phenylpiperazine antidepressants.
The coadministration of triazolam and nefazodone causes a significant increase in the plasma level of triazolam (see WARNINGS and PRECAUTIONS), and a 75% reduction in the initial triazolam dosage is recommended if the two drugs are to be given together. Because not all commercially available dosage forms of triazolam permit a sufficient dosage reduction, the coadministration of triazolam and nefazodone should be avoided for most patients, including the elderly.
-
WARNINGS
Clinical Worsening and Suicide Risk
Patients with major depressive disorder (MDD), both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a long-standing concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phases of treatment. Pooled analyses of short-term placebo-controlled trials of antidepressant drugs (SSRIs and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18 to 24) with major depressive disorder (MDD) and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction with antidepressants compared to placebo in adults aged 65 and older.
The pooled analyses of placebo-controlled trials in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term trials of 9 antidepressant drugs in over 4400 patients. The pooled analyses of placebo-controlled trials in adults with MDD or other psychiatric disorders included a total of 295 short-term trials (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk differences (drug vs placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1000 patients treated) are provided in Table1.
Table 1 Age Range Drug-Placebo Difference in Number of Cases of Suicidality per 1000 Patients Treated Increases Compared to Placebo < 18 14 additional cases 18 to 24 5 additional cases Decreases Compared to Placebo 25 to 64 1 fewer case ≥ 65 6 fewer cases No suicides occurred in any of the pediatric trials. There were suicides in the adult trials, but the number was not sufficient to reach any conclusion about drug effect on suicide.
It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with depression that the use of antidepressants can delay the recurrence of depression.
All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases.
The following symptoms, anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality.
Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient's presenting symptoms.
Families and caregivers of patients being treated with antidepressants for major depressive disorder or other indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to health care providers.Such monitoring should include daily observation by families and caregivers. Prescriptions for nefazodone hydrochloride tablets should be written for the smallest quantity of tablets consistent with good patient management, in order to reduce the risk of overdose.
Screening Patients for Bipolar Disorder
A major depressive episode may be the initial presentation of bipolar disorder. It is generally believed (though not established in controlled trials) that treating such an episode with an antidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder. Whether any of the symptoms described above represent such a conversion is unknown. However, prior to initiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression. It should be noted that nefazodone hydrochloride tablets are not approved for use in treating bipolar depression.
Hepatotoxicity
(See BOXED WARNING.)
Cases of life-threatening hepatic failure have been reported in patients treated with nefazodone hydrochloride tablets.
The reported rate in the United States is about 1 case of liver failure resulting in death or transplant per 250,000 to 300,000 patient-years of nefazodone treatment. This represents a rate of about 3 to 4 times the estimated background rate of liver failure. This rate is an underestimate because of under reporting, and the true risk could be considerably greater than this. A large cohort study of antidepressant users found no cases of liver failure leading to death or transplant among nefazodone users in about 30,000 patient-years of exposure. The spontaneous report data and the cohort study results provide estimates of the upper and lower limits of the risk of liver failure in nefazodone-treated patients, but are not capable of providing a precise risk estimate.
The time to liver injury for the reported liver failure cases resulting in death or transplant generally ranged from 2 weeks to 6 months on nefazodone therapy. Although some reports described dark urine and nonspecific prodromal symptoms (e.g., anorexia, malaise, and gastrointestinal symptoms), other reports did not describe the onset of clear prodromal symptoms prior to the onset of jaundice.
The physician may consider the value of liver function testing. Periodic serum transaminase testing has not been proven to prevent serious injury but it is generally believed that early detection of drug-induced hepatic injury along with immediate withdrawal of the suspect drug enhances the likelihood for recovery.
Patients should be advised to be alert for signs and symptoms of liver dysfunction (jaundice, anorexia, gastrointestinal complaints, malaise, etc.) and to report them to their doctor immediately if they occur. Ongoing clinical assessment of patients should govern physician interventions, including diagnostic evaluations and treatment.
Nefazodone should be discontinued if clinical signs or symptoms suggest liver failure (see PRECAUTIONS, Information for Patients). Patients who develop evidence of hepatocellular injury such as increased serum AST or serum ALT levels ≥ 3 times the upper limit of NORMAL, while on nefazodone should be withdrawn from the drug. These patients should be presumed to be at increased risk for liver injury if nefazodone is reintroduced. Accordingly, such patients should not be considered for re-treatment.
Potential for Interaction With Monoamine Oxidase Inhibitors
In patients receiving antidepressants with pharmacological properties similar to nefazodone in combination with a monoamine oxidase inhibitor (MAOI), there have been reports of serious, sometimes fatal, reactions. For a selective serotonin reuptake inhibitor (SSRI), these reactions have included hyperthermia, rigidity, myoclonus, autonomic instability with possible rapid fluctuations of vital signs, and mental status changes that include extreme agitation progressing to delirium and coma. These reactions have also been reported in patients who have recently discontinued that drug and have been started on an MAOI. Some cases presented with features resembling neuroleptic malignant syndrome. Severe hyperthermia and seizures, sometimes fatal, have been reported in association with the combined use of tricyclic antidepressants and MAOIs. These reactions have also been reported in patients who have recently discontinued these drugs and have been started on an MAOI.
Although the effects of combined use of nefazodone and MAOI have not been evaluated in humans or animals, because nefazodone is an inhibitor of both serotonin and norepinephrine reuptake, it is recommended that nefazodone not be used in combination with an MAOI, or within 14 days of discontinuing treatment with an MAOI. At least 1 week should be allowed after stopping nefazodone before starting an MAOI.
Interaction With Triazolobenzodiazepines
Interaction studies of nefazodone with two triazolobenzodiazepines, i.e., triazolam and alprazolam, metabolized by cytochrome P450 3A4, have revealed substantial and clinically important increases in plasma concentrations of these compounds when administered concomitantly with nefazodone.
Triazolam
When a single oral 0.25 mg dose of triazolam was coadministered with nefazodone (200 mg BID) at steady state, triazolam half-life and AUC increased 4 fold and peak concentrations increased 1.7 fold. Nefazodone plasma concentrations were unaffected by triazolam. Coadministration of nefazodone potentiated the effects of triazolam on psychomotor performance tests. If triazolam is coadministered with nefazodone, a 75% reduction in the initial triazolam dosage is recommended. Because not all commercially available dosage forms of triazolam permit sufficient dosage reduction, coadministration of triazolam with nefazodone should be avoided for most patients, including the elderly. In the exceptional case where coadministration of triazolam with nefazodone may be considered appropriate, only the lowest possible dose of triazolam should be used (see CONTRAINDICATIONS and PRECAUTIONS).
Alprazolam
When alprazolam (1 mg BID) and nefazodone (200 mg BID) were coadministered, steady-state peak concentrations, AUC and half-life values for alprazolam increased by approximately 2 fold. Nefazodone plasma concentrations were unaffected by alprazolam. If alprazolam is coadministered with nefazodone, a 50% reduction in the initial alprazolam dosage is recommended. No dosage adjustment is required for nefazodone.
Potential Terfenadine, Astemizole, Cisapride, and Pimozide Interactions
Terfenadine, astemizole, cisapride, and pimozide are all metabolized by the cytochrome P450 3A4 (CYP3A4) isozyme, and it has been demonstrated that ketoconazole, erythromycin, and other inhibitors of CYP3A4 can block the metabolism of these drugs, which can result in increased plasma concentrations of parent drug. Increased plasma concentrations of terfenadine, astemizole, cisapride, and pimozide are associated with QT prolongation and with rare cases of serious cardiovascular adverse events, including death, due principally to ventricular tachycardia of the torsade de pointes type. Nefazodone has been shown in vitro to be an inhibitor of CYP3A4. Consequently, it is recommended that nefazodone not be used in combination with either terfenadine, astemizole, cisapride, or pimozide (see CONTRAINDICATIONS and PRECAUTIONS).
Interaction With Carbamazepine
The coadministration of carbamazepine 200 mg BID with nefazodone 200 mg BID, at steady state for both drugs, resulted in almost 95% reductions in AUCs for nefazodone and hydroxynefazodone, likely resulting in insufficient plasma nefazodone and hydroxynefazodone concentrations for achieving an antidepressant effect for nefazodone. Consequently, it is recommended that nefazodone not be used in combination with carbamazepine (see CONTRAINDICATIONS and PRECAUTIONS).
-
PRECAUTIONS
General
Postural Hypotension
A pooled analysis of the vital signs monitored during placebo-controlled premarketing studies revealed that 5.1% of nefazodone patients compared to 2.5% of placebo patients (p ≤ 0.01) met criteria for a potentially important decrease in blood pressure at some time during treatment (systolic blood pressure ≤ 90 mmHg and a change from baseline of ≥ 20 mmHg). While there was no difference in the proportion of nefazodone and placebo patients having adverse events characterized as ‘syncope’ (nefazodone, 0.2%; placebo, 0.3%), the rates for adverse events characterized as ‘postural hypotension’ were as follows: nefazodone (2.8%), tricyclic antidepressants (10.9%), SSRI (1.1%), and placebo (0.8%). Thus, the prescriber should be aware that there is some risk of postural hypotension in association with nefazodone use. Nefazodone should be used with caution in patients with known cardiovascular or cerebrovascular disease that could be exacerbated by hypotension (history of myocardial infarction, angina, or ischemic stroke) and conditions that would predispose patients to hypotension (dehydration, hypovolemia, and treatment with antihypertensive medication).
Activation of Mania/Hypomania
During premarketing testing, hypomania or mania occurred in 0.3% of nefazodone-treated unipolar patients, compared to 0.3% of tricyclic- and 0.4% of placebo-treated patients. In patients classified as bipolar the rate of manic episodes was 1.6% for nefazodone, 5.1% for the combined tricyclic-treated groups, and 0% for placebo-treated patients. Activation of mania/hypomania is a known risk in a small proportion of patients with major affective disorder treated with other marketed antidepressants. As with all antidepressants, nefazodone should be used cautiously in patients with a history of mania.
Seizures
During premarketing testing, a recurrence of a petit mal seizure was observed in a patient receiving nefazodone who had a history of such seizures. In addition, one nonstudy participant reportedly experienced a convulsion (type not documented) following a multiple-drug overdose (see OVERDOSAGE). Rare occurrences of convulsions (including grand mal seizures) following nefazodone administration have been reported since market introduction. A causal relationship to nefazodone has not been established (see ADVERSE REACTIONS).
Priapism
While priapism did not occur during premarketing experience with nefazodone, rare reports of priapism have been received since market introduction. A causal relationship to nefazodone has not been established (see ADVERSE REACTIONS). If patients present with prolonged or inappropriate erections, they should discontinue therapy immediately and consult their physicians. If the condition persists for more than 24 hours, a urologist should be consulted to determine appropriate management.
Use in Patients With Concomitant Illness
Nefazodone has not been evaluated or used to any appreciable extent in patients with a recent history of myocardial infarction or unstable heart disease. Patients with these diagnoses were systematically excluded from clinical studies during the product’s premarketing testing. Evaluation of electrocardiograms of 1153 patients who received nefazodone in 6 to 8 week, double-blind, placebo-controlled trials did not indicate that nefazodone is associated with the development of clinically important ECG abnormalities. However, sinus bradycardia, defined as heart rate ≤ 50 bpm and a decrease of at least 15 bpm from baseline, was observed in 1.5% of nefazodone-treated patients compared to 0.4% of placebo-treated patients (p ≤ 0.05). Because patients with a recent history of myocardial infarction or unstable heart disease were excluded from clinical trials, such patients should be treated with caution.
In patients with cirrhosis of the liver, AUC values of nefazodone and HO-NEF were increased by approximately 25%.
Information for Patients (see Patient Information)
Prescribers or other health professionals should inform patients, their families, and their caregivers about the benefits and risks associated with treatment with nefazodone hydrochloride tablets and should counsel them in its appropriate use. A patient Medication Guide about “Antidepressant Medicines, Depression and other Serious Mental Illnesses, and Suicidal Thoughts or Actions” is available for nefazodone hydrochloride tablets. The prescriber or health professional should instruct patients, their families, and their caregivers to read the Medication Guide and should assist them in understanding its contents. Patients should be given the opportunity to discuss the contents of the Medication Guide and to obtain answers to any questions they may have. The complete text of the Medication Guide is reprinted at the end of this document.
Patients should be advised of the following issues and asked to alert their prescriber if these occur while taking nefazodone hydrochloride tablets.
Clinical Worsening and Suicide Risk
Patients, their families, and their caregivers should be encouraged to be alert to the emergence of anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, mania, other unusual changes in behavior, worsening of depression, and suicidal ideation, especially early during antidepressant treatment and when the dose is adjusted up or down. Families and caregivers of patients should be advised to look for the emergence of such symptoms on a day-to-day basis, since changes may be abrupt. Such symptoms should be reported to the patient's prescriber or health professional, especially if they are severe, abrupt in onset, or were not part of the patient's presenting symptoms. Symptoms such as these may be associated with an increased risk for suicidal thinking and behavior and indicate a need for very close monitoring and possibly changes in the medication.
Hepatotoxicity
Patients should be informed that nefazodone therapy has been associated with liver abnormalities ranging from asymptomatic reversible serum transaminase increases to cases of liver failure resulting in transplant and/or death. At present, there is no way to predict who is likely to develop liver failure. Ordinarily, patients with active liver disease should not be treated with nefazodone. Patients should be advised to be alert for signs of liver dysfunction (jaundice, anorexia, gastrointestinal complaints, malaise, etc.) and to report them to their doctor immediately if they occur.
Time to Response/Continuation
As with all antidepressants, several weeks on treatment may be required to obtain the full antidepressant effect. Once improvement is noted, it is important for patients to continue drug treatment as directed by their physician.
Interference With Cognitive and Motor Performance
Since any psychoactive drug may impair judgment, thinking, or motor skills, patients should be cautioned about operating hazardous machinery, including automobiles, until they are reasonably certain that nefazodone therapy does not adversely affect their ability to engage in such activities.
Pregnancy
Patients should be advised to notify their physician if they become pregnant or intend to become pregnant during therapy.
Nursing
Patients should be advised to notify their physician if they are breast-feeding an infant (see PRECAUTIONS, Nursing Mothers).
Concomitant Medication
Patients should be advised to inform their physicians if they are taking, or plan to take, any prescription or over-the-counter drugs, since there is a potential for interactions. Significant caution is indicated if nefazodone is to be used in combination with XANAX®1 (alprazolam), concomitant use with HALCION®1 (triazolam) should be avoided for most patients including the elderly, and concomitant use with SELDANE®2 (terfenadine), HISMANAL®3 (astemizole), PROPULSID®3 (cisapride), ORAP®4 (pimozide), or TEGRETOL®5 (carbamazepine) is contraindicated (see CONTRAINDICATIONS and WARNINGS).
Drug Interactions
Drugs Highly Bound to Plasma Protein
Because nefazodone is highly bound to plasma protein (see CLINICAL PHARMACOLOGY, Pharmacokinetics), administration of nefazodone to a patient taking another drug that is highly protein bound may cause increased free concentrations of the other drug, potentially resulting in adverse events. Conversely, adverse effects could result from displacement of nefazodone by other highly bound drugs.
Warfarin – There were no effects on the prothrombin or bleeding times or upon the pharmacokinetics of R-warfarin when nefazodone (200 mg BID) was administered for 1 week to subjects who had been pretreated for 2 weeks with warfarin. Although the coadministration of nefazodone did decrease the subjects’ exposure to S-warfarin by 12%, the lack of effects on the prothrombin and bleeding times indicates this modest change is not clinically significant. Although these results suggest no adjustments in warfarin dosage are required when nefazodone is administered to patients stabilized on warfarin, such patients should be monitored as required by standard medical practices.
CNS-Active Drugs
Monoamine Oxidase Inhibitors – See WARNINGS.
Haloperidol – When a single oral 5 mg dose of haloperidol was coadministered with nefazodone (200 mg BID) at steady state, haloperidol apparent clearance decreased by 35% with no significant increase in peak haloperidol plasma concentrations or time of peak. This change is of unknown clinical significance. Pharmacodynamic effects of haloperidol were generally not altered significantly. There were no changes in the pharmacokinetic parameters for nefazodone. Dosage adjustment of haloperidol may be necessary when coadministered with nefazodone.
Lorazepam – When lorazepam (2 mg BID) and nefazodone (200 mg BID) were coadministered to steady state, there was no change in any pharmacokinetic parameter for either drug compared to each drug administered alone. Therefore, dosage adjustment is not necessary for either drug when coadministered.
Triazolam/Alprazolam – See CONTRAINDICATIONS and WARNINGS.
Alcohol – Although nefazodone did not potentiate the cognitive and psychomotor effects of alcohol in experiments with normal subjects, the concomitant use of nefazodone and alcohol in depressed patients is not advised.
Buspirone – In a study of steady-state pharmacokinetics in healthy volunteers, coadministration of buspirone (2.5 or 5 mg BID) with nefazodone (250 mg BID) resulted in marked increases in plasma buspirone concentrations (increases up to 20 fold in Cmax and up to 50 fold in AUC) and statistically significant decreases (about 50%) in plasma concentrations of the buspirone metabolite 1-pyrimidinylpiperazine. With 5 mg BID doses of buspirone, slight increases in AUC were observed for nefazodone (23%) and its metabolites hydroxynefazodone (17%) and mCPP (9%). Subjects receiving nefazodone 250 mg BID and buspirone 5 mg BID experienced lightheadedness, asthenia, dizziness, and somnolence, adverse events also observed with either drug alone. If the two drugs are to be used in combination, a low dose of buspirone (e.g., 2.5 mg QD) is recommended. Subsequent dose adjustment of either drug should be based on clinical assessment.
Pimozide – See CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS, Pharmacokinetics of Nefazodone in ‘Poor Metabolizers’ and Potential Interaction With Drugs That Inhibit and/or Are Metabolized by Cytochrome P450 Isozymes.
Fluoxetine – When fluoxetine (20 mg QD) and nefazodone (200 mg BID) were administered at steady state there were no changes in the pharmacokinetic parameters for fluoxetine or its metabolite, norfluoxetine. Similarly, there were no changes in the pharmacokinetic parameters of nefazodone or HO-NEF; however, the mean AUC levels of the nefazodone metabolites mCPP and triazole-dione increased by 3 to 6 fold and 1.3 fold, respectively. When a 200 mg dose of nefazodone was administered to subjects who had been receiving fluoxetine for 1 week, there was an increased incidence of transient adverse events such as headache, lightheadedness, nausea, or paresthesia, possibly due to the elevated mCPP levels. Patients who are switched from fluoxetine to nefazodone without an adequate washout period may experience similar transient adverse events. The possibility of this happening can be minimized by allowing a washout period before initiating nefazodone therapy and by reducing the initial dose of nefazodone. Because of the long half-life of fluoxetine and its metabolites, this washout period may range from one to several weeks depending on the dose of fluoxetine and other individual patient variables.
Phenytoin – Pretreatment for 7 days with 200 mg BID of nefazodone had no effect on the pharmacokinetics of a single 300 mg oral dose of phenytoin. However, due to the nonlinear pharmacokinetics of phenytoin, the failure to observe a significant effect on the single-dose pharmacokinetics of phenytoin does not preclude the possibility of a clinically significant interaction with nefazodone when phenytoin is dosed chronically. However, no change in the initial dosage of phenytoin is considered necessary and any subsequent adjustment of phenytoin dosage should be guided by usual clinical practices.
Desipramine – When nefazodone (150 mg BID) and desipramine (75 mg QD) were administered together there were no changes in the pharmacokinetics of desipramine or its metabolite, 2-hydroxy desipramine. There were also no changes in the pharmacokinetics of nefazodone or its triazole-dione metabolite, but the AUC and Cmax of mCPP increased by 44% and 48%, respectively, while the AUC of HO-NEF decreased by 19%. No changes in doses of either nefazodone or desipramine are necessary when the two drugs are given concomitantly. Subsequent dose adjustments should be made on the basis of clinical response.
Lithium – In 13 healthy subjects the coadministration of nefazodone (200 mg BID) with lithium (500 mg BID) for 5 days (steady-state conditions) was found to be well tolerated. When the two drugs were coadministered, there were no changes in the steady-state pharmacokinetics of either lithium, nefazodone, or its metabolite HO-NEF; however, there were small decreases in the steady-state plasma concentrations of two nefazodone metabolites, mCPP and triazole-dione, which are considered not to be of clinical significance. Therefore, no dosage adjustment of either lithium or nefazodone is required when they are coadministered.
Carbamazepine – The coadministration of nefazodone (200 mg BID) for 5 days to 12 healthy subjects on carbamazepine who had achieved steady state (200 mg BID) was found to be well tolerated. Steady-state conditions for carbamazepine, nefazodone, and several of their metabolites were achieved by day 5 of coadministration. With coadministration of the two drugs there were significant increases in the steady-state Cmax and AUC of carbamazepine (23% and 23%, respectively), while the steady-state Cmax and the AUC of the carbamazepine metabolite, 10,11 epoxycarbamazepine, decreased by 21% and 20%, respectively. The coadministration of the two drugs significantly reduced the steady-state Cmax and AUC of nefazodone by 86% and 93%, respectively. Similar reductions in the Cmax and AUC of HO-NEF were also observed (85% and 94%), while the reductions in Cmax and AUC of mCPP and triazole-dione were more modest (13% and 44% for the former and 28% and 57% for the latter). Due to the potential for coadministration of carbamazepine to result in insufficient plasma nefazodone and hydroxynefazodone concentrations for achieving an antidepressant effect for nefazodone, it is recommended that nefazodone not be used in combination with carbamazepine (see CONTRAINDICATIONS and WARNINGS).
General Anesthetics – Little is known about the potential for interaction between nefazodone and general anesthetics; therefore, prior to elective surgery, nefazodone hydrochloride should be discontinued for as long as clinically feasible.
Other CNS-Active Drugs – The use of nefazodone in combination with other CNS-active drugs has not been systematically evaluated. Consequently, caution is advised if concomitant administration of nefazodone and such drugs is required.
Cimetidine
When nefazodone (200 mg BID) and cimetidine (300 mg QID) were coadministered for one week, no change in the steady-state pharmacokinetics of either nefazodone or cimetidine was observed compared to each dosed alone. Therefore, dosage adjustment is not necessary for either drug when coadministered.
Theophylline
When nefazodone (200 mg BID) was given to patients being treated with theophylline (600 to 1200 mg/day) for chronic obstructive pulmonary disease, there was no change in the steady-state pharmacokinetics of either nefazodone or theophylline. FEV1 measurements taken when theophylline and nefazodone were coadministered did not differ from baseline dosage (i.e., when theophylline was administered alone). Therefore, dosage adjustment is not necessary for either drug when coadministered.
Cardiovascular-Active Drugs
Digoxin – When nefazodone (200 mg BID) and digoxin (0.2 mg QD) were coadministered for 9 days to healthy male volunteers (n = 18) who were phenotyped as CYP2D6 extensive metabolizers, Cmax, Cmin, and AUC of digoxin were increased by 29%, 27%, and 15%, respectively. Digoxin had no effects on the pharmacokinetics of nefazodone and its active metabolites. Because of the narrow therapeutic index of digoxin, caution should be exercised when nefazodone and digoxin are coadministered; plasma level monitoring for digoxin is recommended.
Propranolol – The coadministration of nefazodone (200 mg BID) and propranolol (40 mg BID) for 5.5 days to healthy male volunteers (n = 18), including 3 poor and 15 extensive CYP2D6 metabolizers, resulted in 30% and 14% reductions in Cmax and AUC of propranolol, respectively, and a 14% reduction in Cmax for the metabolite, 4-hydroxypropranolol. The kinetics of nefazodone, hydroxynefazodone, and triazole-dione were not affected by coadministration of propranolol. However, Cmax, Cmin, and AUC of m-chlorophenylpiperazine were increased by 23%, 54%, and 28%, respectively. No change in initial dose of either drug is necessary and dose adjustments should be made on the basis of clinical response.
HMG-CoA Reductase Inhibitors – When single 40 mg doses of simvastatin or atorvastatin, both substrates of CYP3A4, were given to healthy adult volunteers who had received nefazodone hydrochloride, 200 mg BID for 6 days, approximately 20 fold increases in plasma concentrations of simvastatin and simvastatin acid and 3 to 4 fold increases in plasma concentrations of atorvastatin and atorvastatin lactone were seen. These effects appear to be due to the inhibition of CYP3A4 by nefazodone because, in the same study, nefazodone had no significant effect on the plasma concentrations of pravastatin, which is not metabolized by CYP3A4 to a clinically significant extent.
There have been rare reports of rhabdomyolysis involving patients receiving the combination of nefazodone and either simvastatin or lovastatin, also a substrate of CYP3A4 (see ADVERSE REACTIONS, Postintroduction Clinical Experience). Rhabdomyolysis has been observed in patients receiving HMG-CoA reductase inhibitors administered alone (at recommended dosages) and in particular, for certain drugs in this class, when given in combination with inhibitors of the CYP3A4 isozyme.
Caution should be used if nefazodone is administered in combination with HMG-CoA reductase inhibitors that are metabolized by CYP3A4, such as simvastatin, atorvastatin, and lovastatin, and dosage adjustments of these HMG-CoA reductase inhibitors are recommended. Since metabolic interactions are unlikely between nefazodone and HMG-CoA reductase inhibitors that undergo little or no metabolism by the CYP3A4 isozyme, such as pravastatin or fluvastatin, dosage adjustments should not be necessary.
Immunosuppressive Agents
There have been reports of increased blood concentrations of cyclosporine and tacrolimus into toxic ranges when patients received these drugs concomitantly with nefazodone. Both cyclosporine and tacrolimus are substrates of CYP3A4, and nefazodone is known to inhibit this enzyme. If either cyclosporine or tacrolimus is administered with nefazodone, blood concentrations of the immunosuppressive agent should be monitored and dosage adjusted accordingly.
Pharmacokinetics of Nefazodone in ‘Poor Metabolizers’ and Potential Interaction With Drugs That Inhibit and/or Are Metabolized by Cytochrome P450 Isozymes
CYP3A4 Isozyme – Nefazodone has been shown in vitro to be an inhibitor of CYP3A4. This is consistent with the interactions observed between nefazodone and triazolam, alprazolam, buspirone, atorvastatin, and simvastatin, drugs metabolized by this isozyme. Consequently, caution is indicated in the combined use of nefazodone with any drugs known to be metabolized by CYP3A4. In particular, the combined use of nefazodone with triazolam should be avoided for most patients, including the elderly. The combined use of nefazodone with terfenadine, astemizole, cisapride, or pimozide is contraindicated (see CONTRAINDICATIONS and WARNINGS).
CYP2D6 Isozyme – A subset (3% to 10%) of the population has reduced activity of the drug-metabolizing enzyme CYP2D6. Such individuals are referred to commonly as “poor metabolizers” of drugs such as debrisoquin, dextromethorphan, and the tricyclic antidepressants. The pharmacokinetics of nefazodone and its major metabolites are not altered in these “poor metabolizers.” Plasma concentrations of one minor metabolite (mCPP) are increased in this population; the adjustment of nefazodone dosage is not required when administered to “poor metabolizers.” Nefazodone and its metabolites have been shown in vitro to be extremely weak inhibitors of CYP2D6. Thus, it is not likely that nefazodone will decrease the metabolic clearance of drugs metabolized by this isozyme.
CYP1A2 Isozyme – Nefazodone and its metabolites have been shown in vitro not to inhibit CYP1A2. Thus, metabolic interactions between nefazodone and drugs metabolized by this isozyme are unlikely.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
There is no evidence of carcinogenicity with nefazodone. The dietary administration of nefazodone to rats and mice for 2 years at daily doses of up to 200 mg/kg and 800 mg/kg, respectively, which are approximately 3 and 6 times, respectively, the maximum human daily dose on a mg/m2 basis, produced no increase in tumors.
Mutagenesis
Nefazodone has been shown to have no genotoxic effects based on the following assays: bacterial mutation assays, a DNA repair assay in cultured rat hepatocytes, a mammalian mutation assay in Chinese hamster ovary cells, an in vivo cytogenetics assay in rat bone marrow cells, and a rat dominant lethal study.
Pregnancy
Teratogenic Effects
Pregnancy category C
Reproduction studies have been performed in pregnant rabbits and rats at daily doses up to 200 and 300 mg/kg, respectively (approximately 6 and 5 times, respectively, the maximum human daily dose on a mg/m2 basis). No malformations were observed in the offspring as a result of nefazodone treatment. However, increased early pup mortality was seen in rats at a dose approximately five times the maximum human dose, and decreased pup weights were seen at this and lower doses, when dosing began during pregnancy and continued until weaning. The cause of these deaths is not known. The no-effect dose for rat pup mortality was 1.3 times the human dose on a mg/m2 basis. There are no adequate and well-controlled studies in pregnant women. Nefazodone should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether nefazodone or its metabolites are excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when nefazodone is administered to a nursing woman.
Pediatric Use
Safety and effectiveness in the pediatric population have not been established (see BOXED WARNING and WARNINGS, Clinical Worsening and Suicide Risk). Two placebo-controlled trials in 286 pediatric patients with MDD have been conducted with nefazodone, and the data were not sufficient to support a claim for use in pediatric patients. Anyone considering the use of nefazodone hydrochloride tablets in a child or adolescent must balance the potential risks with the clinical need.
Geriatric Use
Of the approximately 7000 patients in clinical studies who received nefazodone for the treatment of depression, 18% were 65 years and older, while 5% were 75 years and older. Based on monitoring of adverse events, vital signs, electrocardiograms, and results of laboratory tests, no overall differences in safety between elderly and younger patients were observed in clinical studies. Efficacy in the elderly has not been demonstrated in placebo-controlled trials. Other reported clinical experience has not identified differences in responses between elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Due to the increased systemic exposure to nefazodone seen in single-dose studies in elderly patients (see CLINICAL PHARMACOLOGY, Pharmacokinetics), treatment should be initiated at half the usual dose, but titration upward should take place over the same range as in younger patients (see DOSAGE AND ADMINISTRATION). The usual precautions should be observed in elderly patients who have concomitant medical illnesses or who are receiving concomitant drugs.
-
ADVERSE REACTIONS
Associated With Discontinuation of Treatment
Approximately 16% of the 3496 patients who received nefazodone in worldwide premarketing clinical trials discontinued treatment due to an adverse experience. The more common (≥ 1%) events in clinical trials associated with discontinuation and considered to be drug related (i.e., those events associated with dropout at a rate approximately twice or greater for nefazodone compared to placebo) included: nausea (3.5%), dizziness (1.9%), insomnia (1.5%), asthenia (1.3%), and agitation (1.2%).
Incidence in Controlled Trials
Commonly Observed Adverse Events in Controlled Clinical Trials
The most commonly observed adverse events associated with the use of nefazodone (incidence of 5% or greater) and not seen at an equivalent incidence among placebo-treated patients (i.e., significantly higher incidence for nefazodone compared to placebo, p ≤ 0.05), derived from the table below, were: somnolence, dry mouth, nausea, dizziness, constipation, asthenia, lightheadedness, blurred vision, confusion, and abnormal vision.
Adverse Events Occurring at an Incidence of 1% or More Among Nefazodone-Treated Patients
The table that follows enumerates adverse events that occurred at an incidence of 1% or more, and were more frequent than in the placebo group, among nefazodone-treated patients who participated in short-term (6 to 8 week) placebo-controlled trials in which patients were dosed with nefazodone to ranges of 300 to 600 mg/day. This table shows the percentage of patients in each group who had at least one episode of an event at some time during their treatment. Reported adverse events were classified using standard COSTART-based Dictionary terminology.
The prescriber should be aware that these figures cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contribution of drug and nondrug factors to the side-effect incidence rate in the population studied.
Treatment-Emergent Adverse Experience Incidence in 6 to 8 Week Placebo-Controlled Clinical Trials1, Nefazodone 300 to 600 mg/day Dose Range Percent of Patients Body System Preferred Term Nefazodone (n = 393) Placebo (n = 394) Body as a Whole Headache 36 33 Asthenia 11 5 Infection 8 6 Flu syndrome 3 2 Chills 2 1 Fever 2 1 Neck rigidity 1 0 Cardiovascular Postural hypotension 4 1 Hypotension 2 1 Dermatological Pruritus 2 1 Rash 2 1 Gastrointestinal Dry mouth 25 13 Nausea 22 12 Constipation 14 8 Dyspepsia 9 7 Diarrhea 8 7 Increased appetite 5 3 Nausea & vomiting 2 1 Metabolic Peripheral edema 3 2 Thirst 1 < 1 Musculoskeletal Arthralgia 1 < 1 Nervous Somnolence 25 14 Dizziness 17 5 Insomnia 11 9 Lightheadedness 10 3 Confusion 7 2 Memory impairment 4 2 Paresthesia 4 2 Vasodilatation2 4 2 Abnormal dreams 3 2 Concentration decreased 3 1 Ataxia 2 0 Incoordination 2 1 Psychomotor retardation 2 1 Tremor 2 1 Hypertonia 1 0 Libido decreased 1 < 1 Respiratory Pharyngitis 6 5 Cough increased 3 1 Special Senses Blurred vision 9 3 Abnormal vision3 7 1 Tinnitus 2 1 Taste perversion 2 1 Visual field defect 2 0 Urogenital Urinary frequency 2 1 Urinary tract infection 2 1 Urinary retention 2 1 Vaginitis4 2 1 Breast pain4 1 < 1 1 Events reported by at least 1% of patients treated with nefazodone and more frequent than the placebo group are included; incidence is rounded to the nearest 1% (< 1% indicates an incidence less than 0.5%). Events for which the nefazodone incidence was equal to or less than placebo are not listed in the table, but included the following: abdominal pain, pain, back pain, accidental injury, chest pain, neck pain, palpitation, migraine, sweating, flatulence, vomiting, anorexia, tooth disorder, weight gain, edema, myalgia, cramp, agitation, anxiety, depression, hypesthesia, CNS stimulation, dysphoria, emotional lability, sinusitis, rhinitis, dysmenorrhea4, dysuria.
2 Vasodilatation – flushing, feeling warm.
3 Abnormal vision – scotoma, visual trails.
4 Incidence adjusted for gender.
Dose Dependency of Adverse Events
The table that follows enumerates adverse events that were more frequent in the nefazodone dose range of 300 to 600 mg/day than in the nefazodone dose range of up to 300 mg/day. This table shows only those adverse events for which there was a statistically significant difference (p ≤ 0.05) in incidence between the nefazodone dose ranges as well as a difference between the high dose range and placebo.
Dose Dependency of Adverse Events in Placebo-Controlled Trials Percent of Patients Body System Preferred Term Nefazodone 300 to 600 mg/day (n = 209) Nefazodone ≤ 300 mg/day (n = 211) Placebo (n = 212) Gastrointestinal Nausea 23 14 12 Constipation 17 10 9 Nervous Somnolence 28 16 13 Dizziness 22 11 4 Confusion 8 2 1 Special Senses Abnormal Vision 10 0 2 Blurred Vision 9 3 2 Tinnitus 3 0 1 Visual Disturbances
In controlled clinical trials, blurred vision occurred in 9% of nefazodone-treated patients compared to 3% of placebo-treated patients. In these same trials abnormal vision, including scotomata and visual trails, occurred in 7% of nefazodone-treated patients compared to 1% of placebo-treated (see Treatment-Emergent Adverse Experience table, above). Dose-dependency was observed for these events in these trials, with none of the scotomata and visual trails at doses below 300 mg/day. However, scotomata and visual trails observed at doses below 300 mg/day have been reported in postmarketing experience with nefazodone (see PRECAUTIONS, Information for Patients).
Weight Changes
In a pooled analysis of placebo-controlled premarketing studies, there were no differences between nefazodone and placebo groups in the proportions of patients meeting criteria for potentially important increases or decreases in body weight (a change of ≥ 7%).
Laboratory Changes
Of the serum chemistry, serum hematology, and urinalysis parameters monitored during placebo-controlled premarketing studies with nefazodone, a pooled analysis revealed a statistical trend between nefazodone and placebo for hematocrit, i.e., 2.8% of nefazodone patients met criteria for a potentially important decrease in hematocrit (≤ 37% male or ≤ 32% female) compared to 1.5% of placebo patients (0.05 < p ≤ 0.10). Decreases in hematocrit, presumably dilutional, have been reported with many other drugs that block alpha1-adrenergic receptors. There was no apparent clinical significance of the observed changes in the few patients meeting these criteria.
ECG Changes
Of the ECG parameters monitored during placebo-controlled premarketing studies with nefazodone, a pooled analysis revealed a statistically significant difference between nefazodone and placebo for sinus bradycardia, i.e., 1.5% of nefazodone patients met criteria for a potentially important decrease in heart rate (≤ 50 bpm and a decrease of ≥ 15 bpm) compared to 0.4% of placebo patients (p < 0.05). There was no obvious clinical significance of the observed changes in the few patients meeting these criteria.
Other Events Observed During the Premarketing Evaluation of Nefazodone
During its premarketing assessment, multiple doses of nefazodone were administered to 3496 patients in clinical studies, including more than 250 patients treated for at least one year. The conditions and duration of exposure to nefazodone varied greatly, and included (in overlapping categories) open and double-blind studies, uncontrolled and controlled studies, inpatient and outpatient studies, fixed-dose and titration studies. Untoward events associated with this exposure were recorded by clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse events without first grouping similar types of untoward events into a smaller number of standardized event categories.
In the tabulations that follow, reported adverse events were classified using standard COSTART-based Dictionary terminology. The frequencies presented, therefore, represent the proportion of the 3496 patients exposed to multiple doses of nefazodone who experienced an event of the type cited on at least one occasion while receiving nefazodone. All reported events are included except those already listed in the Treatment-Emergent Adverse Experience Incidence table, those events listed in other safety-related sections of this insert, those adverse experiences subsumed under COSTART terms that are either overly general or excessively specific so as to be uninformative, those events for which a drug cause was very remote, and those events which were not serious and occurred in fewer than two patients.
It is important to emphasize that, although the events reported occurred during treatment with nefazodone, they were not necessarily caused by it.
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse events are those occurring on one or more occasions in at least 1/100 patients (only those not already listed in the tabulated results from placebo-controlled trials appear in this listing); infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare events are those occurring in fewer than 1/1000 patients.
Body as a whole – Infrequent: allergic reaction, malaise, photosensitivity reaction, face edema, hangover effect, abdomen enlarged, hernia, pelvic pain, and halitosis. Rare: cellulitis.
Cardiovascular system – Infrequent: tachycardia, hypertension, syncope, ventricular extrasystoles, and angina pectoris. Rare: AV block, congestive heart failure, hemorrhage, pallor, and varicose vein.
Dermatological system – Infrequent: dry skin, acne, alopecia, urticaria, maculopapular rash, vesiculobullous rash, and eczema.
Gastrointestinal system – Frequent: gastroenteritis. Infrequent: eructation, periodontal abscess, abnormal liver function tests, gingivitis, colitis, gastritis, mouth ulceration, stomatitis, esophagitis, peptic ulcer, and rectal hemorrhage. Rare: glossitis, hepatitis, dysphagia, gastrointestinal hemorrhage, oral moniliasis, and ulcerative colitis.
Hemic and lymphatic system – Infrequent: ecchymosis, anemia, leukopenia, and lymphadenopathy .
Metabolic and nutritional system – Infrequent: weight loss, gout, dehydration, lactic dehydrogenase increased, SGOT increased, and SGPT increased. Rare: hypercholesteremia and hypoglycemia.
Musculoskeletal system – Infrequent: arthritis, tenosynovitis, muscle stiffness, and bursitis. Rare: tendinous contracture.
Nervous system – Infrequent: vertigo, twitching, depersonalization, hallucinations, suicide attempt, apathy, euphoria, hostility, suicidal thoughts, abnormal gait, thinking abnormal, attention decreased, derealization, neuralgia, paranoid reaction, dysarthria, increased libido, suicide, and myoclonus. Rare: hyperkinesia, increased salivation, cerebrovascular accident, hyperesthesia, hypotonia, ptosis, and neuroleptic malignant syndrome.
Respiratory system – Frequent: dyspnea and bronchitis. Infrequent: asthma, pneumonia, laryngitis, voice alteration, epistaxis, hiccup. Rare: hyperventilation and yawn.
Special senses – Frequent: eye pain. Infrequent: dry eye, ear pain, abnormality of accommodation, diplopia, conjunctivitis, mydriasis, keratoconjunctivitis, hyperacusis, and photophobia. Rare: deafness, glaucoma, night blindness, and taste loss.
Urogenital system – Frequent: impotencea. Infrequent: cystitis, urinary urgency, metrorrhagiaa, amenorrheaa, polyuria, vaginal hemorrhagea, breast enlargementa, menorrhagiaa, urinary incontinence, abnormal ejaculationa, hematuria, nocturia, and kidney calculus. Rare: uterine fibroids enlargeda, uterine hemorrhagea, anorgasmia, and oliguria.
a Adjusted for gender.
Postintroduction Clinical Experience
Postmarketing experience with nefazodone has shown an adverse experience profile similar to that seen during the premarketing evaluation of nefazodone. Voluntary reports of adverse events temporally associated with nefazodone have been received since market introduction that are not listed above and for which a causal relationship has not been established. These include:
Anaphylactic reactions; angioedema; convulsions (including grand mal seizures); galactorrhea; gynecomastia (male); hyponatremia; liver necrosis and liver failure, in some cases leading to liver transplantation and/or death (see WARNINGS); priapism (see PRECAUTIONS); prolactin increased; rhabdomyolysis involving patients receiving the combination of nefazodone and lovastatin or simvastatin (see PRECAUTIONS); serotonin syndrome; and Stevens-Johnson syndrome; and thrombocytopenia.
-
DRUG ABUSE AND DEPENDENCE
Physical and Psychological Dependence
In animal studies, nefazodone did not act as a reinforcer for intravenous self-administration in monkeys trained to self-administer cocaine, suggesting no abuse liability. In a controlled study of abuse liability in human subjects, nefazodone showed no potential for abuse.
Nefazodone has not been systematically studied in humans for its potential for tolerance, physical dependence, or withdrawal. While the premarketing clinical experience with nefazodone did not reveal any tendency for a withdrawal syndrome or any drug-seeking behavior, it is not possible to predict on the basis of this limited experience the extent to which a CNS-active drug will be misused, diverted, and/or abused once marketed. Consequently, physicians should carefully evaluate patients for a history of drug abuse and follow such patients closely, observing them for signs of misuse or abuse of nefazodone (e.g., development of tolerance, dose escalation, drug-seeking behavior).
-
OVERDOSAGE
Human Experience
In premarketing clinical studies, there were seven reports of nefazodone overdose alone or in combination with other pharmacological agents. The amount of nefazodone ingested ranged from 1000 mg to 11,200 mg. Commonly reported symptoms from overdose of nefazodone included nausea, vomiting, and somnolence. One nonstudy participant took 2000 to 3000 mg of nefazodone with methocarbamol and alcohol; this person reportedly experienced a convulsion (type not documented). None of these patients died.
In postmarketing experience, overdose with nefazodone alone and in combination with alcohol and/or other substances has been reported. Commonly reported symptoms were similar to those reported from overdose in premarketing experience. While there have been rare reports of fatalities in patients taking overdoses of nefazodone, predominantly in combination with alcohol and/or other substances, no causal relationship to nefazodone has been established.
Overdosage Management
Treatment should consist of those general measures employed in the management of overdosage with any antidepressant.
Ensure an adequate airway, oxygenation, and ventilation. Monitor cardiac rhythm and vital signs. General supportive and symptomatic measures are also recommended. Induction of emesis is not recommended. Gastric lavage with a large-bore orogastric tube with appropriate airway protection, if needed, may be indicated if performed soon after ingestion, or in symptomatic patients.
Activated charcoal should be administered. Due to the wide distribution of nefazodone in body tissues, forced diuresis, dialysis, hemoperfusion, and exchange transfusion are unlikely to be of benefit. No specific antidotes for nefazodone are known.
In managing overdosage, consider the possibility of multiple drug involvement. The physician should consider contacting a poison control center for additional information on the treatment of any overdose. Telephone numbers for certified poison control centers are listed in the Physicians’ Desk Reference (PDR).
-
DOSAGE AND ADMINISTRATION
When deciding among the alternative treatments available for depression, the prescriber should consider the risk of hepatic failure associated with nefazodone hydrochloride treatment (see WARNINGS).
Initial Treatment
The recommended starting dose for nefazodone hydrochloride tablets USP is 200 mg/day, administered in two divided doses (BID). In the controlled clinical trials establishing the antidepressant efficacy of nefazodone, the effective dose range was generally 300 to 600 mg/day. Consequently, most patients, depending on tolerability and the need for further clinical effect, should have their dose increased. Dose increases should occur in increments of 100 mg/day to 200 mg/day, again on a BID schedule, at intervals of no less than 1 week. As with all antidepressants, several weeks on treatment may be required to obtain a full antidepressant response.
Dosage for Elderly or Debilitated Patients
The recommended initial dose for elderly or debilitated patients is 100 mg/day, administered in two divided doses (BID). These patients often have reduced nefazodone clearance and/or increased sensitivity to the side effects of CNS-active drugs. It may also be appropriate to modify the rate of subsequent dose titration. As steady-state plasma levels do not change with age, the final target dose based on a careful assessment of the patient’s clinical response may be similar in healthy younger and older patients.
Maintenance/Continuation/Extended Treatment
There is no body of evidence available from controlled trials to indicate how long the depressed patient should be treated with nefazodone. It is generally agreed, however, that pharmacological treatment for acute episodes of depression should continue for up to 6 months or longer. Whether the dose of antidepressant needed to induce remission is identical to the dose needed to maintain euthymia is unknown. Systematic evaluation of the efficacy of nefazodone has shown that efficacy is maintained for periods of up to 36 weeks following 16 weeks of open-label acute treatment (treated for 52 weeks total) at dosages that averaged 438 mg/day. For most patients, their maintenance dose was that associated with response during acute treatment (see CLINICAL PHARMACOLOGY). The safety of nefazodone in long-term use is supported by data from both double-blind and open-label trials involving more than 250 patients treated for at least one year.
-
HOW SUPPLIED
Nefazodone hydrochloride tablets USP, 50 mg, are light-pink to pink (mottled), capsule-shaped, beveled-edged tablets, debossed “7178” on one side and debossed “93” on the other side. They are available in bottles of 100.
Nefazodone hydrochloride tablets USP, 100 mg, are white to off-white, capsule-shaped tablets, debossed “1024” on one side and scored on the other side with a debossed “93” on one side of the score. They are available in bottles of 60.
Nefazodone hydrochloride tablets USP, 150 mg, are peach (mottled), capsule-shaped tablets, debossed “7113” on one side and scored on the other side with a debossed “93” on one side of the score. They are available in bottles of 60.
Nefazodone hydrochloride tablets USP, 200 mg, are light-yellow to yellow (mottled), capsule-shaped tablets, debossed “1025” on one side and debossed “93” on the other side. They are available in bottles of 60.
Nefazodone hydrochloride tablets USP, 250 mg, are white to off-white, capsule-shaped tablets, debossed “1026” on one side and debossed “93” on the other side. They are available in bottles of 60.
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
-
REFERENCES
1 HALCION® and XANAX® are registered trademarks of Pharmacia & Upjohn.
2 SELDANE® is a registered trademark of Hoechst Marion Roussel Inc. (now Aventis Pharmaceuticals).
3 HISMANAL® and PROPULSID® are registered trademarks of Janssen Pharmaceutica Products, L.P.
4 ORAP® is a registered trademark of Gate Pharmaceuticals, a division of Teva Pharmaceuticals USA.
5 TEGRETOL® is a registered trademark of Novartis Pharmaceuticals Corporation.
Manufactured In Israel By:
TEVA PHARMACEUTICAL IND. LTD.
Jerusalem, 91010, Israel
Manufactured For:
TEVA PHARMACEUTICALS USA
Sellersville, PA 18960
Rev. J 8/2008
-
PATIENT INFORMATION
NEFAZODONE HYDROCHLORIDE TABLETS USP
Rx only
Read this information completely before using nefazodone. Read the information each time you get more medicine. There may be new information. This leaflet provides a summary about nefazodone and does not include everything there is to know about your medicine. This information is not meant to take the place of talking with your doctor.
What is the most important information that I should know about nefazodone?
Rarely, people who take nefazodone can develop serious liver problems. If you get any of the following symptoms while taking nefazodone, call your doctor right away because you may be developing a liver problem:
- Yellowing of the skin or whites of eyes (jaundice)
- Unusually dark urine
- Loss of appetite that lasts several days or longer
- Nausea
- Abdominal (lower stomach) pain
People who currently have liver problems should not take nefazodone.
What is nefazodone?
Nefazodone is a medicine used to treat depression. Nefazodone is thought to treat depression by correcting an imbalance in the amounts of certain natural chemicals, such as serotonin and norepinephrine, which are in your brain.
Who should not take nefazodone?
Do not take nefazodone if you
- are allergic to nefazodone or the related medicine Desyrel® (trazodone).
- are taking Seldane® (terfenadine), an antihistamine; Hismanal® (astemizole), an antihistamine; Propulsid® (cisapride), used for heartburn; Halcion® (triazolam), used for insomnia; Orap® (pimozide), used to treat Tourette’s syndrome; or Tegretol® (carbamazepine), used to control seizures.
- currently have liver problems.
- are taking or have taken within the last 14 days one of the medicines for depression known as monoamine oxidase inhibitors (MAOIs), such as Nardil® or Parnate®.
Be sure to tell your doctor if you
- have ever had liver problems;
- are taking any other medicine, vitamin supplement, or herbal remedy, including those sold without a prescription (over-the-counter);
- have heart problems or have had a heart attack or stroke;
- have had manic episodes (extreme agitation or excitability);
- have ever attempted suicide;
- have had convulsions (seizures);
- are pregnant or breast-feeding.
How should I take nefazodone?
- Take nefazodone at the same time every day exactly as prescribed by your doctor. You may take nefazodone with or without food.
- It may take a while for you to feel that nefazodone is working. You may not feel the full effect for several weeks. Once you feel better, it is important to keep taking nefazodone as directed by your doctor.
- If you miss a dose of nefazodone, skip that dose and continue with your regular schedule. Never take 2 doses at the same time.
- If you think that you have taken more nefazodone than prescribed, contact your doctor, local poison control center, or emergency room right away.
What should I avoid while taking nefazodone?
- Do not drive or operate possibly dangerous machinery (such as an automobile, power mower, or power tool) or participate in any hazardous activity that requires full mental alertness until you know how nefazodone affects you.
- Before taking nefazodone, tell your doctor about any medicines you are taking, including vitamin supplements, herbal remedies, and any non-prescription (over-the-counter) medicines. Some of these medicines may affect how nefazodone works and should not be used in combination without talking to your doctor.
- Do not drink alcoholic beverages while taking nefazodone.
- Tell your doctor if you are pregnant, planning to become pregnant, or become pregnant while taking nefazodone. It is not known whether nefazodone can harm your unborn baby.
- Talk with your doctor before taking nefazodone if you are breast-feeding. It is not known whether nefazodone can pass through your breast milk to the baby.
What are the possible side effects of nefazodone?
The most common side effects of nefazodone are sleepiness, dry mouth, nausea, dizziness, constipation, weakness, lightheadedness, problems with vision, and confusion.
Call your doctor right away if you have any of the following side effects:
- Yellowing of the skin or whites of eyes (jaundice)
- Unusually dark urine
- Loss of appetite that lasts several days or longer
- Severe nausea
- Abdominal (lower stomach) pain
- Rash or hives
- Seizure (convulsion)
- Fainting
- Erection that lasts too long
Tell your doctor right away about any side effects that you have or discomfort that you experience. Do not change your dose or stop taking nefazodone without talking with your doctor first.
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Your doctor has prescribed nefazodone for you and you alone. Do not give nefazodone to other people even if they have the same condition. It may harm them.
This leaflet provides a summary of the most important information about nefazodone. If you would like more information, talk with your doctor or pharmacist. You can ask for information about nefazodone that is written for healthcare professionals.
Seldane® is a registered trademark of Hoechst Marion Roussel Inc. (now Aventis Pharmaceuticals).
Hismanal® and Propulsid® are registered trademarks of Janssen Pharmaceutica Products, L.P.
Nardil® is a registered trademark of Parke-Davis.
Parnate® is a registered trademark of SmithKline Beecham Pharmaceuticals.
Halcion® is a registered trademark of Pharmacia & Upjohn.
Orap® is a registered trademark of Gate Pharmaceuticals, a division of TEVA Pharmaceuticals USA.
Tegretol® is a registered trademark of Novartis Pharmaceuticals Corporation.
Manufactured In Israel By:
TEVA PHARMACEUTICAL IND. LTD.
Jerusalem, 91010, Israel
Manufactured For:
TEVA PHARMACEUTICALS USA
Sellersville, PA 18960
Rev. A 8/2008
This Patient Information Leaflet has been approved by the U.S. Food and Drug Administration.
-
MEDICATION GUIDE
Medication Guide
Antidepressant Medicines, Depression and other Serious Mental Illnesses, and Suicidal Thoughts or Actions
Rx only
Read the Medication Guide that comes with your or your family member’s antidepressant medicine. This Medication Guide is only about the risk of suicidal thoughts and actions with antidepressant medicines. Talk to your, or your family member’s, healthcare provider about:
- all risks and benefits of treatment with antidepressant medicines
- all treatment choices for depression or other serious mental illness
What is the most important information I should know about antidepressant medicines, depression and other serious mental illnesses, and suicidal thoughts or actions?
1. Antidepressant medicines may increase suicidal thoughts or actions in some children, teenagers, and young adults within the first few months of treatment.
2. Depression and other serious mental illnesses are the most important causes of suicidal thoughts and actions. Some people may have a particularly high risk of having suicidal thoughts or actions. These include people who have (or have a family history of) bipolar illness (also called manic-depressive illness) or suicidal thoughts or actions.
3. How can I watch for and try to prevent suicidal thoughts and actions in myself or a family member?
- Pay close attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings. This is very important when an antidepressant medicine is started or when the dose is changed.
- Call the healthcare provider right away to report new or sudden changes in mood, behavior, thoughts, or feelings.
- Keep all follow-up visits with the healthcare provider as scheduled. Call the healthcare provider between visits as needed, especially if you have concerns about symptoms.
Call a healthcare provider right away if you or your family member has any of the following symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling very agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
What else do I need to know about antidepressant medicines?
- Never stop an antidepressant medicine without first talking to a healthcare provider. Stopping an antidepressant medicine suddenly can cause other symptoms.
- Antidepressants are medicines used to treat depression and other illnesses. It is important to discuss all the risks of treating depression and also the risks of not treating it. Patients and their families or other caregivers should discuss all treatment choices with the healthcare provider, not just the use of antidepressants.
- Antidepressant medicines have other side effects. Talk to the healthcare provider about the side effects of the medicine prescribed for you or your family member.
- Antidepressant medicines can interact with other medicines. Know all of the medicines that you or your family member takes. Keep a list of all medicines to show the healthcare provider. Do not start new medicines without first checking with your healthcare provider.
- Not all antidepressant medicines prescribed for children are FDA approved for use in children. Talk to your child’s healthcare provider for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
This Medication Guide has been approved by the U.S. Food and Drug Administration for all antidepressants.
Manufactured In Israel By:
TEVA PHARMACEUTICAL IND. LTD.
Jerusalem, 91010, Israel
Manufactured For:
TEVA PHARMACEUTICALS USA
Sellersville, PA 18960
Rev. E 5/2008
- PACKAGE LABEL - NEFAZODONE - 100MG TABS
-
INGREDIENTS AND APPEARANCE
NEFAZODONE HYDROCHLORIDE
nefazodone hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:16590-166(NDC:0093-1024) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength NEFAZODONE HYDROCHLORIDE (UNII: 27X63J94GR) (NEFAZODONE - UNII:59H4FCV1TF) NEFAZODONE HYDROCHLORIDE 100 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) COLLOIDAL SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE (UNII: FZ989GH94E) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) Product Characteristics Color white (white to off-white) Score 2 pieces Shape OVAL (capsule-shaped) Size 12mm Flavor Imprint Code 1024;93 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:16590-166-30 30 in 1 BOTTLE 2 NDC:16590-166-60 60 in 1 BOTTLE 3 NDC:16590-166-90 90 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA076037 08/18/2010 Labeler - STAT RX USA LLC (786036330) Establishment Name Address ID/FEI Business Operations STAT RX USA LLC 786036330 relabel, repack
