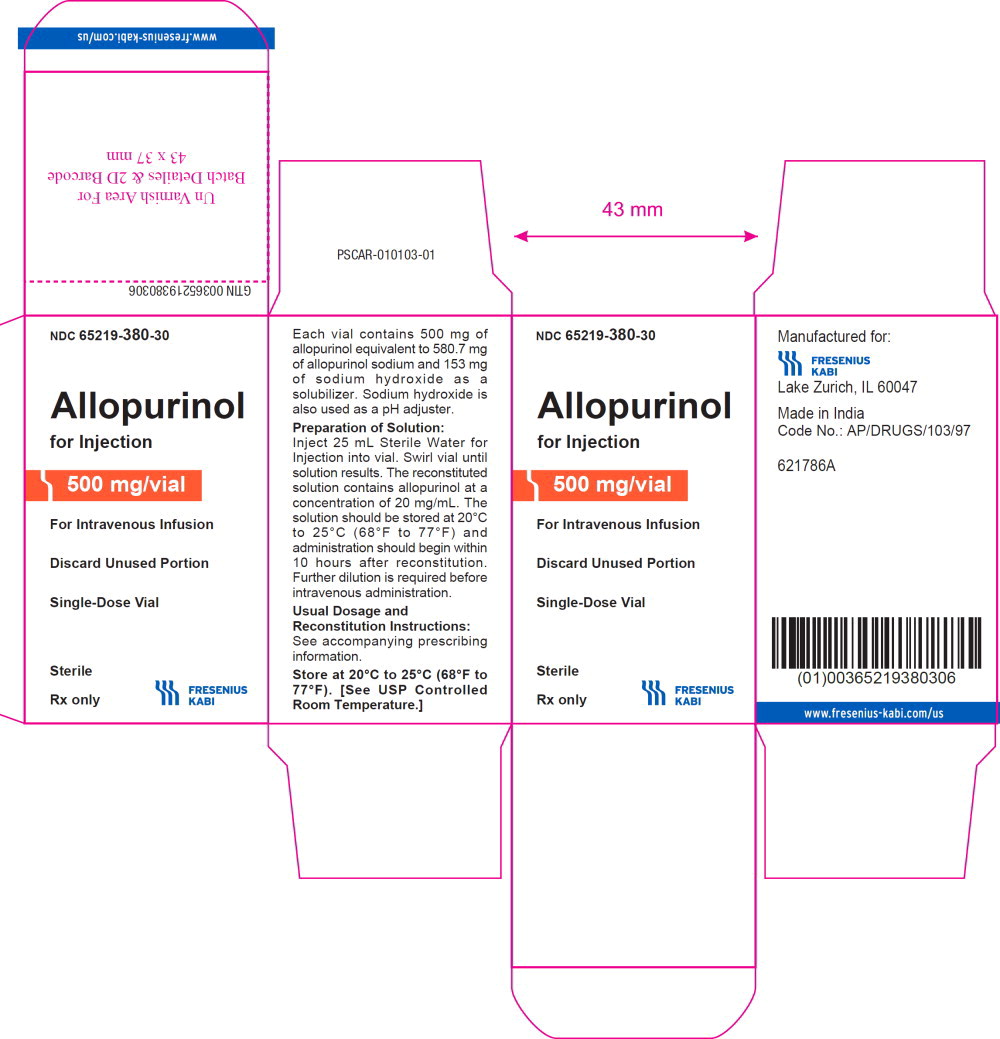
Label: ALLOPURINOL injection, powder, lyophilized, for solution


- NDC Code(s): 65219-380-30
- Packager: Fresenius Kabi USA, LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated January 24, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ALLOPURINOL FOR INJECTION safely and effectively. See full prescribing information for ALLOPURINOL FOR INJECTION.
ALLOPURINOL for injection, for intravenous use
Initial U.S. Approval: 1966
INDICATIONS AND USAGE
Allopurinol for injection is a xanthine oxidase inhibitor indicated for the management of adult and pediatric patients with leukemia, lymphoma, and solid tumor malignancies who are receiving cancer therapy which causes elevations of serum and urinary uric acid levels and who cannot tolerate oral therapy. (1)
DOSAGE AND ADMINISTRATION
- Recommended Dosage (2.2)
Adult Patients 200 mg/m2/day to 400 mg/m2/day Maximum 600 mg/day Pediatric Patients Starting Dose 200 mg/m2/day Maximum 400 mg/day Creatinine Clearance Recommended Daily
Dose10 to 20 mL/min 200 mg/day Less than 10 mL/min 100 mg/day On dialysis 50 mg every 12 hours, or
100 mg every 24 hoursDOSAGE FORMS AND STRENGTHS
For injection: 500 mg as a white lyophilized powder or cake in a single-dose vial for reconstitution (3)
CONTRAINDICATIONS
Known hypersensitivity to allopurinol. (4)
WARNINGS AND PRECAUTIONS
- Skin Rash and Hypersensitivity: Discontinue allopurinol at the first appearance of skin rash or other signs which may indicate a hypersensitivity reaction. Allopurinol has been associated with serious and sometimes fatal dermatologic reactions. (5.1)
- Renal Function Impairment: Patients with decreased renal function require lower doses of allopurinol. (5.2)
- Hepatotoxicity: If signs and symptoms of hepatotoxicity develop, liver function evaluation should be performed. (5.3)
- Myelosuppression: Bone marrow suppression has been reported with allopurinol. (5.4)
- Drowsiness: Drowsiness has been reported in patients taking allopurinol. (5.5)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 1%) are skin rash, nausea, vomiting, and renal failure/insufficiency. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Fresenius Kabi USA, LLC at 1-800-551-7176 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Capecitabine: Avoid the concomitant use of allopurinol (7.2).
- Pegloticase: Discontinue and do not institute allopurinol therapy during treatment with pegloticase (7.2).
- Mercaptopurine or Azathioprine: Reduce mercaptopurine or azathioprine dose as recommended in the respective prescribing information (7.2).
- See full prescribing information for complete list of significant drug interactions (7).
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modifications in Patients with Renal Impairment
2.3 Preparation Instructions
2.4 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Skin Rash and Hypersensitivity
5.2 Renal Function Impairment
5.3 Hepatotoxicity
5.4 Myelosuppression
5.5 Drowsiness
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Drugs Known to Affect the Occurrence of Skin Rash and Hypersensitivity
7.2 Other Drugs Known to Have Clinically Important Drug Interactions with Allopurinol
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Initiate therapy with allopurinol for injection 24 to 48 hours before the start of chemotherapy known to cause tumor cell lysis. Additionally, administer fluids sufficient to yield a daily urinary output of at least two liters in adults with a neutral or, preferably, slightly alkaline urine.
The recommended daily dose of allopurinol for injection is shown in Table 1. Administer the daily dose as single infusion or in equally divided infusions at 6-, 8-, or 12-hour intervals at a rate appropriate for the volume of infusate.
Table 1: Recommended Daily Dose of Allopurinol for Injection Adult Patients 200 mg/m2/day to 400 mg/m2/day intravenously Maximum 600 mg/day Pediatric Patients Starting Dose 200 mg/m2/day intravenously
Maximum 400 mg/dayThe dosage of allopurinol for injection to lower serum uric acid to normal or near-normal varies with the severity of the disease. Monitor serum uric acid levels at least daily and administer allopurinol for injection at a dose and frequency to maintain the serum uric acid within the normal range. Discontinue allopurinol for injection when the patient is able to take oral therapy or when the risk of tumor lysis has abated.
2.2 Dosage Modifications in Patients with Renal Impairment
Reduce the dose of allopurinol for injection in patients with impaired renal function [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. The recommended dosage reductions of allopurinol for injection in adult patients with renal impairment are shown in Table 2.
Table 2: Recommended Daily Dose of Allopurinol for Injection in Adult Patients with Renal Impairment Creatinine Clearance Recommended Daily Dose 10 to 20 mL/min 200 mg/day Less than 10 mL/min 100 mg/day On dialysis 50 mg every 12 hours, or
100 mg every 24 hoursTreatment with allopurinol for injection has not been studied in pediatric patients with severe renal impairment or on dialysis. For pediatric patients with severe renal impairment or on dialysis, consider the risks and potential benefits before initiating treatment with allopurinol for injection [see Warnings and Precautions (5.2) and Use in Specific Populations (8.6)].
2.3 Preparation Instructions
Reconstitute and further dilute allopurinol for injection prior to intravenous infusion.
Reconstitution
- Reconstitute each vial of allopurinol for injection with 25 mL of Sterile Water for Injection, USP to obtain a concentration of 20 mg/mL of allopurinol.
- Inspect the reconstituted solution for discoloration and particulate matter. The reconstituted solution should appear as a clear, almost colorless solution with no more than a slight opalescence. Do not use if the reconstituted solution contains particulate matter or discoloration is present.
Dilution
- Dilute with 0.9% Sodium Chloride Injection, USP or 5% Dextrose for Injection, USP to obtain a final concentration of less than 6 mg/mL.
- Inspect the diluted solution for particulate matter or discoloration and discard if present.
- If not used immediately, the diluted allopurinol for injection solution can be stored at 20° to 25°C (68° to 77°F) for up to 10 hours after initial reconstitution. The storage includes time for infusion. Do not refrigerate the reconstituted and/or diluted product.
- If stored, the administration should be completed within 10 hours after reconstitution.
- Discard unused portion.
2.4 Administration Instructions
Do not mix allopurinol for injection with or administer it through the same intravenous port as agents which are incompatible in solution with allopurinol for injection. The following table lists drugs that are known to be physically incompatible in solution with allopurinol for injection.
Table 3: Drugs That Are Physically Incompatible in Solution with Allopurinol for Injection Amikacin sulfate Hydroxyzine HCl Amphotericin B Idarubicin HCl Carmustine Imipenem-cilastatin sodium Cefotaxime sodium Mechlorethamine HCl Chlorpromazine HCl Meperidine HCl Cimetidine HCl Metoclopramide HCl Clindamycin phosphate Methylprednisolone sodium succinate Cytarabine Minocycline HCl Dacarbazine Nalbuphine HCl Daunorubicin HCl Ondansetron HCl Diphenhydramine HCl Prochlorperazine edisylate Doxorubicin HCl Promethazine HCl Doxycycline hyclate Sodium bicarbonate Droperidol Streptozocin Floxuridine Tobramycin sulfate Gentamicin sulfate Vinorelbine tartrate Haloperidol lactate - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Skin Rash and Hypersensitivity
Serious and sometimes fatal dermatologic reactions, including toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome (SJS), and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported in patients taking allopurinol [see Adverse Reactions (6.1)]. These reactions occur in approximately 5 in 10,000 (0.05%) patients taking allopurinol. Other serious hypersensitivity reactions that have been reported include exfoliative, urticarial and purpuric lesions; generalized vasculitis; and irreversible hepatotoxicity. Discontinue allopurinol at the first appearance of skin rash or other signs which may indicate a hypersensitivity reaction.
The HLA-B*58:01 allele is a genetic marker for severe skin reactions indicative of hypersensitivity to allopurinol. Patients who carry the HLA-B*58:01 allele are at a higher risk of allopurinol hypersensitivity syndrome (AHS), but hypersensitivity reactions have been reported in patients who do not carry this allele. The frequency of this allele is higher in individuals of African, Asian (e.g., Han Chinese, Korean, Thai), and Native Hawaiian/Pacific Islander ancestry [see Clinical Pharmacology (12.5)]. The use of allopurinol is not recommended in HLA-B*58:01 positive patients unless the benefits clearly outweigh the risks.
Prior to starting allopurinol, consider testing for the HLA-B*58:01 allele in genetically at-risk populations. Screening is generally not recommended in patients from populations in which the prevalence of HLA-B*58:01 is low, or in current allopurinol users, as the risk of SJS/TEN/DRESS is largely confined to the first few months of therapy, regardless of HLA-B*58:01 status.
Hypersensitivity reactions to allopurinol may be increased in patients with decreased renal function receiving thiazide diuretics and allopurinol concurrently. In addition, concomitant use of the following drugs may increase the risk of skin rash, which may be severe: bendamustine, thiazide diuretics, ampicillin and amoxicillin [see Drug Interactions (7.1)]. Patients should stop allopurinol and seek medical attention if they develop a rash.
5.2 Renal Function Impairment
Treatment with allopurinol may result in renal impairment due to formation of xanthine calculi or due to precipitation of urates in patients receiving concomitant uricosuric agents. Patients with pre-existing renal disease, including renal impairment or history of kidney stones, may be at increased risk for worsening renal impairment due to xanthine calculi or precipitation of urates while receiving treatment with allopurinol.
Monitor serum creatinine at least daily during the early stages of allopurinol administration. Maintain fluid intake sufficient to yield a urinary output of at least 2 liters per day in adults. In patients with severely impaired renal function or increase in uric acid concentration associated with decreased urate clearance, reduce the dosage of allopurinol [see Use in Specific Populations (8.6) and Dosage and Administration (2.1, 2.2)].
5.3 Hepatotoxicity
Cases of reversible clinical hepatotoxicity have been noted in patients taking oral allopurinol. In some patients, asymptomatic rises in serum alkaline phosphatase or serum transaminase have been observed. If anorexia, weight loss, or pruritus develop in patients on allopurinol, evaluate liver enzymes. In patients with pre-existing liver disease, monitor liver enzymes periodically during the early stages of therapy. Discontinue allopurinol in patients with elevated liver enzymes.
5.4 Myelosuppression
Myelosuppression, manifested by anemia, leukopenia or thrombocytopenia, has been reported in patients receiving allopurinol [see Adverse Reactions (6.1)]. The cytopenias have occurred from as early as 6 weeks to as late as 6 years after the initiation of allopurinol therapy. Discontinue use of allopurinol in patients with unexplained cytopenias. Concomitant use with allopurinol with cytotoxic drugs associated with myelosuppression may increase the risk of myelosuppression. Monitor blood counts more frequently [see Drug Interactions (7)].
Concomitant use with allopurinol increases the exposure of either mercaptopurine or azathioprine which may increase the risk of myelosuppression. Reduce the dosage of mercaptopurine or azathioprine as recommended in the respective prescribing information when used concomitantly with allopurinol [see Drug Interactions (7)].
5.5 Drowsiness
Drowsiness has been reported in patients taking allopurinol [see Adverse Reactions (6.1)]. Advise patients to avoid operation of automobiles or other dangerous machinery and activities made hazardous by decreased alertness when starting allopurinol or increasing the dose until they know how the drug affects them. Advise patients that the central nervous system depressant effects of allopurinol may be additive to those of alcohol and other CNS depressants.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Skin Rash and Hypersensitivity [see Warnings and Precautions (5.1)]
- Renal Function Impairment [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Myelosuppression [see Warnings and Precautions (5.4)]
- Drowsiness [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of allopurinol was evaluated in an uncontrolled compassionate use study of 1,378 patients with advanced malignancies requiring treatment with cytotoxic chemotherapy and in patients with other serious conditions.
Adverse reactions were reported in 9% (125/1378) of the patients treated with allopurinol. The most common adverse reaction was skin rash. Two patients experience serious adverse reactions (decreased renal function and generalized seizure) and one patient experienced severe diarrhea. Approximately 1.1% of patients experienced allergic adverse reactions (including rash, eosinophilia, local injection site reaction).
A listing of the adverse reactions reported from clinical trials follows:
Incidence Greater Than 1%:
Cutaneous/Dermatologic: rash (1.5%)
Genitourinary: renal failure/insufficiency (1.2%)
Gastrointestinal: nausea (1.3%), vomiting (1.2%)
Incidence Less Than 1%:
Body as a Whole: fever, pain, chills, alopecia, infection, sepsis, enlarged abdomen, mucositis/pharyngitis, blast crisis, cellulitis, hypervolemia
Cardiovascular: heart failure, cardiorespiratory arrest, hypertension, pulmonary embolus, hypotension, decreased venous pressure, flushing, headache, stroke, septic shock, cardiovascular disorder, ECG abnormality, hemorrhage, bradycardia, thrombophlebitis, ventricular fibrillation
Cutaneous/Dermatologic: urticaria, pruritus, local injection site reaction
Gastrointestinal: diarrhea, gastrointestinal bleeding, hyperbilirubinemia, splenomegaly, hepatomegaly, intestinal obstruction, jaundice, flatulence, constipation, liver failure, proctitis
Genitourinary: hematuria, increased creatinine, oliguria, kidney function abnormality, urinary tract infection
Hematologic: leukopenia, marrow aplasia, thrombocytopenia, eosinophilia, neutropenia, anemia, pancytopenia, ecchymosis, bone marrow suppression, disseminated intravascular coagulation
Metabolic: hypocalcemia, hyperphosphatemia, hypokalemia, hyperuricemia, electrolyte abnormality, hypercalcemia, hyperglycemia, hypernatremia, hyponatremia, metabolic acidosis, edema, glycosuria, hyperkalemia, lactic acidosis, water intoxication, hypomagnesemia
Neurologic: seizure, status epilepticus, myoclonus, twitching, agitation, mental status changes, cerebral infarction, coma, dystonia, paralysis, tremor
Pulmonary: respiratory failure/insufficiency, ARDS, increased respiration rate, apnea
Musculoskeletal: arthralgia
Other: hypotonia, diaphoresis, tumor lysis syndrome
-
7 DRUG INTERACTIONS
Clinically important interactions with the drugs listed below were observed in patients undergoing treatment with an oral allopurinol formulation.
7.1 Drugs Known to Affect the Occurrence of Skin Rash and Hypersensitivity
Concomitant use of the following drugs may increase the risk of skin rash, which may be severe: bendamustine, thiazide diuretics, ampicillin and amoxicillin. Renal impairment may further increase risk with concomitant use of thiazide diuretics [see Warnings and Precautions (5.1) (5.2) and Clinical Pharmacology (12.2)].
Monitor renal function and reduce the dose of allopurinol in patients with concomitant thiazide diuretic use and impaired renal function [see Dosage and Administration (2.2)].
Discontinue allopurinol at the first appearance of skin rash or other signs which may indicate a hypersensitivity reaction when use concomitantly with these drugs.
7.2 Other Drugs Known to Have Clinically Important Drug Interactions with Allopurinol
Table 4: Interventions for Clinically Important Drug Interactions with Allopurinol Capecitabine Clinical Impact Concomitant use with allopurinol may decrease concentration of capecitabine's active metabolites, which may decrease capecitabine efficacy. Intervention Avoid the use of allopurinol during treatment with capecitabine. Cyclosporine Clinical Impact Concomitant use of allopurinol increases cyclosporine concentrations which may increase the risk of adverse reactions. Intervention Increase frequency of monitoring cyclosporine concentrations as reflected
in the prescribing information when used concomitantly with allopurinol.Cytotoxic Agents Clinical Impact Concomitant use of allopurinol with cytotoxic agents increases bone marrow suppression among patients with neoplastic disease, except leukemia [see Warnings and Precautions (5.4) and Clinical Pharmacology
(12.2)].Intervention Blood count monitoring and regular physician follow-up recommended. Fluorouracil Clinical Impact Based on non-clinical data, allopurinol may decrease anti-tumor activity
due to suppression of phosphorylation of 5-fluorouracil.Intervention Concomitant administration with fluorouracil should be avoided. Mercaptopurine or Azathioprine Clinical Impact Allopurinol inhibits xanthine oxidase mediated metabolism of mercaptopurine and azathioprine. Concomitant use of allopurinol increases the exposure of either mercaptopurine or azathioprine which may increase the risk of their adverse reactions including myelosuppression [see Warnings and Precautions (5.4)]. Intervention Reduce the dosage of mercaptopurine or azathioprine as recommended in
the respective prescribing information.Pegloticase Clinical Impact Concomitant use of allopurinol and pegloticase may potentially blunt the rise of serum uric acid levels and increase the risk of pegloticase related anaphylaxis in patients whose uric acid level increase to above 6 mg/dL. Intervention Discontinue and do not institute allopurinol therapy during treatment with
pegloticase.Theophylline Clinical Impact Concomitant use of allopurinol doses greater than or equal to 600 mg/day
may decrease the clearance of theophylline.Intervention Monitor and adjust theophylline doses as reflected in the prescribing
information.Uricosuric Agents Clinical Impact Uricosuric agents increase the excretion of the active allopurinol metabolite oxypurinol. Concomitant use with uricosuric agents decreases oxypurinol exposure which may reduce the inhibition of xanthine oxidase by oxypurinol and increases the urinary excretion of uric acid. Intervention Monitor uric acid levels due to the increased chance of hypouricemic
effects.Warfarin Clinical Impact Allopurinol may inhibit the metabolism of warfarin, possibly enhancing its anticoagulant effect. Intervention Patients on concomitant therapy should be monitored for excessive anticoagulation. The INR should be checked frequently and warfarin dosage adjusted accordingly when allopurinol is added to warfarin therapy. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animals, allopurinol may cause fetal harm when administered to a pregnant woman. Adverse developmental outcomes have been described in exposed animals (see Data). Allopurinol and its metabolite oxypurinol have been shown to cross the placenta following administration of maternal allopurinol.
Available limited published data on allopurinol use in pregnant women do not demonstrate a clear pattern or increase in frequency of adverse developmental outcomes. Among approximately 50 pregnancies described in published literature, 2 infants with major congenital malformations have been reported with following maternal allopurinol exposure. Advise pregnant women of the potential risk to a fetus.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
There was no evidence of fetotoxicity or teratogenicity in rats or rabbits treated during the period of organogenesis with oral allopurinol at doses up to 200 mg/kg/day and up to 100 mg/kg/day, respectively (about three times the human dose on a mg/m2 basis). However, there is a published report in pregnant mice that single intraperitoneal doses of 50 or 100 mg/kg (about 1/3 or 3/4 the human dose on a mg/m2 basis) of allopurinol on gestation days 10 or 13 produced significant increases in fetal deaths and teratogenic effects (cleft palate, harelip, and digital defects). It is uncertain whether these findings represented a fetal effect or an effect secondary to maternal toxicity. In another published study with no reported maternal toxicity, allopurinol administered orally at 15 or 45 mg/kg to pregnant rats during organogenesis caused embryonic resorptions, growth retardation, decreased fetal weight, and skeletal, liver, kidney, and brain abnormalities. In rats, maternal treatment with allopurinol in normoxic pregnancy has been shown to increase the cardiac protein levels of sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2a) in the adult male offspring. The mechanism underlying this effect is not understood. However, this effect was not matched by an increase in left ventricular end diastolic pressure or sympathetic dominance in hearts of adult male offspring of normoxic pregnancy treated with allopurinol.
8.2 Lactation
Risk Summary
Allopurinol and oxypurinol are present in human milk. Based on information from a single case report, allopurinol and its active metabolite, oxypurinol, were detected in the milk of a mother at five weeks postpartum at an estimated relative infant dose of 0.14 and 0.2 mg/kg of allopurinol and between 7.2 to 8 mg/kg of oxypurinol daily. There was no report of effects of allopurinol on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatments with allopurinol and for one week after the last dose.
8.4 Pediatric Use
The safety and effectiveness of allopurinol have been established in approximately 200 pediatric patients. The efficacy and safety profile observed in this patient population were similar to that observed in adults.
8.5 Geriatric Use
Clinical studies of allopurinol did not include sufficient numbers of patients 65 years and older to determine whether they respond differently than younger patients.
8.6 Renal Impairment
Allopurinol and its primary active metabolite, oxypurinol, are eliminated by the kidneys [see Clinical Pharmacology (12.3)]. Therefore, changes in renal function will likely increase allopurinal and oxypurinol exposure. In patients with decreased renal function, or who have concurrent illnesses that can affect renal function such as hypertension and diabetes mellitus, perform periodic laboratory parameters of renal function, particularly BUN and serum creatinine or creatinine clearance, should be performed.
In patients with severely impaired renal function or decreased urate clearance, the half-life of oxypurinol in the plasma is greatly prolonged. Reduce the dose of allopurinol in patients with creatinine clearance ≤ 20 mL/min [see Dosage and Administration (2.2)]. Patients should be treated with the lowest effective dose, in order to minimize possible side effects.
- 10 OVERDOSAGE
-
11 DESCRIPTION
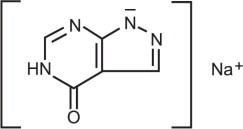
Allopurinol for Injection, a xanthine oxidase inhibitor, is a sterile, white, lyophilized powder or cake, in a single-dose vial for reconstitution. Each vial contains 500 mg of allopurinol equivalent to 580.7 mg of allopurinol sodium and 153 mg of sodium hydroxide as a solubilizer. Sodium hydroxide is also used as a pH adjuster. Allopurinol for Injection contains no preservatives.
Allopurinol is a xanthine oxidase inhibitor. The chemical name for allopurinol sodium is 1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one monosodium salt. It is a white amorphous mass with a molecular weight of 158.09 and molecular formula C5H3N4NaO. The structural formula is:
The pKa of allopurinol sodium is 9.31.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Allopurinol is a structural analogue of the natural purine base, hypoxanthine. Allopurinol and its oxypurinol metabolite inhibitor xanthine oxidase, the enzyme responsible for the conversion of hypoxanthine to xanthine and of xanthine to uric acid, the end product of purine metabolism in humans. Allopurinol does not disrupt the biosynthesis of purines.
The action of oral allopurinol differs from that of uricosuric agents, which lower the serum uric acid level by increasing urinary excretion of uric acid. Allopurinol reduces both the serum and urinary uric acid levels by inhibiting the formation of uric acid. The use of allopurinol to block the formation of urates avoids the hazard of increased renal excretion of uric acid posed by uricosuric drugs.
12.2 Pharmacodynamics
Allopurinol reduces the production of uric acid by inhibiting the biochemical reactions immediately preceding its formation in a dose dependent manner. The pharmacological action of allopurinol is generally believed to be mediated by its oxypurinol metabolite.
Effect on Hypoxanthine and Xanthine
Reutilization of both hypoxanthine and xanthine for nucleotide and nucleic acid synthesis is markedly enhanced when their oxidations are inhibited by allopurinol and oxypurinol. This reutilization does not disrupt normal nucleic acid anabolism because feedback inhibition is an integral part of purine biosynthesis. As a result of xanthine oxidase inhibition, the serum concentration of hypoxanthine plus xanthine in patients receiving allopurinol for treatment of hyperuricemia is usually in the range of 0.3 to 0.4 mg/dL compared to a normal level of approximately 0.15 mg/dL. A maximum of 0.9 mg/dL of these oxypurines has been reported when the serum urate was lowered to less than 2 mg/dL by high doses of allopurinol. These values are far below the saturation levels, at which point their precipitation would be expected to occur (above 7 mg/dL). The increased xanthine and hypoxanthine in the urine in patients who were treated with oral allopurinol have not been accompanied by problems of nephrolithiasis; however, there are isolated case reports of xanthine crystalluria.
Drug Interaction Studies
Fluorouracil: Based on non-clinical data, allopurinol may decrease anti-tumor activity due to suppression of phosphorylation of 5-fluorouracil.
Pegloticase: Concomitant use of allopurinol and pegloticase may potentially blunt the rise of serum uric acid levels required for monitoring the safe use of pegloticase.
Cytotoxic Agents: Enhanced bone marrow suppression by cyclophosphamide and other cytotoxic agents has been reported among patients with neoplastic disease, except leukemia, in the presence of allopurinol.
Thiazide Diuretics: Reports that the concomitant administration of allopurinol and thiazide diuretics contributed to increased allopurinol toxicity were reviewed; however, a causal mechanism or cause-and-effect relationship was not found.
12.3 Pharmacokinetics
Following single 100 mg and 300 mg intravenous and oral administration of allopurinol, the relative intravenous Cmax was approximately 3-fold and 3.8-fold and AUC0-inf was approximately 1.9-fold higher for allopurinol at both dosages, respectively. The relative intravenous oxypurinol Cmax and AUC0-inf was approximately 1 compared to oral administration at both dosages.
The Cmax and AUC0-inf for both allopurinol and oxypurinol following intravenous administration of allopurinol were dose proportional in the dose range of 100 to 300 mg.
Distribution
The steady-state allopurinol volume of distribution (mean ± S.D.) is approximately 0.87 ± 0.13 L/Kg following intravenous administration.
Elimination
The half-life (mean ± S.D.) of allopurinol and oxypurinol are approximately 1.21 ± 0.33 and 23.5 ± 4.5 hours following intravenous administration, respectively. The net renal clearance of oxypurinol about 30 mL/min.
Metabolism
Allopurinol is a weak CYP1A2 inhibitor. Allopurinol is rapidly eliminated from the systemic circulation primarily via oxidative metabolism to oxypurinol.
The oxypurinol (alloxanthine) metabolite is also a xanthine oxidase inhibitor and is present in systemic circulation in much higher concentrations and for a much longer period than allopurinol. In general, the ratio of the area under the plasma concentration vs time curve (AUC0-inf) between oxypurinol and allopurinol was in the magnitude of 30 to 40.
Drug Interaction Studies
Capecitabine: Concomitant use with allopurinol may decrease concentration of capecitabine's active metabolites, which may decrease capecitabine efficacy.
Cyclosporine: Concomitant use of allopurinol increases cyclosporine concentrations which may increase the risk of adverse reactions.
Mercaptopurine or Azathioprine: Allopurinol inhibits xanthine oxidase mediated metabolism of mercaptopurine and azathioprine. Concomitant use of allopurinol increases the exposure of either mercaptopurine or azathioprine which may increase the risk of their adverse reactions including myelosuppression.
Theophylline: Concomitant use of allopurinol doses greater than or equal to 600 mg/day may decrease the clearance of theophylline.
Uricosuric Agents: Uricosuric agents increase the excretion of the active allopurinol metabolite oxypurinol. Concomitant use with uricosuric agents decreases oxypurinol exposure which may reduce the inhibition of xanthine oxidase by oxypurinol and increases the urinary excretion of uric acid.
Warfarin: Allopurinol may inhibit the metabolism of warfarin, possibly enhancing its anticoagulant effect.
12.5 Pharmacogenomics
The HLA-B*58:01 allele is a genetic marker for severe skin reactions indicative of hypersensitivity to allopurinol [see Warnings and Precautions (5.1)]. The frequency of the HLA-B*58:01 allele ranges from 8 to 10% in Han Chinese populations, about 8% in Thai populations, and about 6% in Korean populations based upon published literature and available databases. The frequency of the HLA-B*58:01 allele is about 4% in Blacks, about 1 to 2 % in indigenous peoples of the Americas and Hispanic populations, and <1% in people from European descent and Japanese.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Allopurinol was administered at doses up to 20 mg/kg/day to mice and rats for the majority of their life span. No evidence of carcinogenicity was seen in either mice or rats (at doses about 1/6 or 1/3 the recommended human dose on a mg/m2 basis, respectively).
Allopurinol administered intravenously to rats (50 mg/kg) was not incorporated into rapidly replicating intestinal DNA. No evidence of clastogenicity was observed in an in vivo micronucleus test in rats, or in lymphocytes taken from patients treated with allopurinol (mean duration of treatment 40 months), or in an in vitro assay with human lymphocytes.
Allopurinol oral doses of 20 mg/kg/day had no effect on male or female fertility in rats or rabbits (about 1/3 or 1/2 the human dose on a mg/m2 basis, respectively).
-
14 CLINICAL STUDIES
A compassionate use trial of allopurinol conducted in the United States from 1977 through 1989 included 718 evaluable patients with malignancies requiring treatment with cytotoxic chemotherapy who were unable to ingest or retain oral medication. Of these patients, 411 had established hyperuricemia and 307 had normal serum urate levels at the time that treatment was initiated. Normal serum uric acid levels were achieved in 68% of the former (reduction of serum uric acid was documented in 93%), and were maintained throughout chemotherapy in 97% of the latter. Because of the study design, it was not possible to assess the impact of the treatment with allopurinol on the clinical outcome of the patient groups.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Allopurinol for Injection is supplied in 30 mL flint glass single-dose vials. Each vial contains 500 mg of allopurinol as a sterile, white, lyophilized powder or cake for reconstitution.
Product Code Unit of Sale Strength 380130 NDC 65219-380-30
Individually packaged500 mg/vial Store at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature].
Discard unused portion.
-
17 PATIENT COUNSELING INFORMATION
Skin Rash and Hypersensitivity
Inform patients that allopurinol may increase the risk of serious and sometimes fatal dermatologic reactions, including toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome (SJS), and drug reaction with eosinophilia and systemic symptoms (DRESS). Instruct the patient to be alert for skin rash, blisters, fever or other signs and symptoms of these hypersensitivity reactions. Advise patients to stop the allopurinol immediately if they develop any type of rash and seek medical attention [see Warnings and Precautions (5.1)].
Renal Function Impairment
Advise patients to stay well hydrated (e.g., 2 liters of liquid per day) while taking allopurinol [see Warnings and Precautions (5.2)].
Hepatotoxicity
Advise patients of the risk of hepatotoxicity and to report any signs and symptoms of liver failure, including jaundice, pruritus, bleeding, bruising, or anorexia to their healthcare provider [see Warnings and Precautions (5.3)].
Myelosuppression
Advise patients of the risk of myelosuppression and to report any signs and symptoms of infection, fever, bleeding, shortness of breath, or significant fatigue to their healthcare provider [see Warnings and Precautions (5.4)].
Drowsiness
Inform patients that drowsiness has been reported in patients taking allopurinol and to be cautious when engaging in activities where alertness is mandatory [see Warnings and Precautions (5.5)].
Pregnancy
Advise pregnant women of the potential risk to a fetus. Advise women to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with allopurinol [see Use in Specific Populations (8.1)].
Lactation
Advise women not to breastfeed during treatment with allopurinol for one week after the last dose [see Use in Specific Populations (8.2)].
Lake Zurich, IL 60047
www.fresenius-kabi.com/us
451672B
Made in India
Revised: 10/2022
- PRINCIPAL DISPLAY PANEL – SHELF CARTON
- PRINCIPAL DISPLAY PANEL – Vial Label
-
INGREDIENTS AND APPEARANCE
ALLOPURINOL
allopurinol injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:65219-380 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALLOPURINOL SODIUM (UNII: 428673RC2Z) (ALLOPURINOL - UNII:63CZ7GJN5I) ALLOPURINOL 500 mg in 25 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:65219-380-30 1 in 1 CARTON 03/14/2022 1 25 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212363 03/14/2022 Labeler - Fresenius Kabi USA, LLC (013547657) Establishment Name Address ID/FEI Business Operations GLAND PHARMA LIMITED 918601238 ANALYSIS(65219-380) , MANUFACTURE(65219-380) , PACK(65219-380)