Label: PROLASTIN-C LIQUID (alpha1-proteinase inhibitor- human injection, solution


-
NDC Code(s):
13533-705-01,
13533-705-11,
13533-705-31,
13533-705-32, view more13533-705-51, 13533-705-52
- Packager: GRIFOLS USA, LLC
- Category: PLASMA DERIVATIVE
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated July 5, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PROLASTIN-C LIQUID safely and effectively. See full prescribing information for PROLASTIN-C LIQUID.
PROLASTIN-C LIQUID (Alpha1-Proteinase Inhibitor [Human])
Solution for Intravenous Injection
Initial U.S. Approval: 1987INDICATIONS AND USAGE
PROLASTIN®-C LIQUID is an Alpha1-Proteinase Inhibitor (Human) (Alpha1-PI) indicated for chronic augmentation and maintenance therapy in adults with clinical evidence of emphysema due to severe hereditary deficiency of Alpha1-PI (alpha1-antitrypsin deficiency). (1)
Limitations of Use:
- The effect of augmentation therapy with any Alpha1-PI, including PROLASTIN-C LIQUID, on pulmonary exacerbations and on the progression of emphysema in Alpha1-PI deficiency has not been conclusively demonstrated in randomized, controlled clinical trials.
- Clinical data demonstrating the long-term effects of chronic augmentation or maintenance therapy with PROLASTIN-C LIQUID are not available.
- PROLASTIN-C LIQUID is not indicated as therapy for lung disease in patients in whom severe Alpha1-PI deficiency has not been established.
DOSAGE AND ADMINISTRATION
For intravenous use only. (2)
DOSAGE FORMS AND STRENGTHS
For injection: approximately 500 mg (10 mL), 1,000 mg (20 mL) and 4,000 mg (80 mL) of a solution for injection in single-dose vials. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Severe hypersensitivity and anaphylactic reactions may occur in IgA deficient patients with antibodies against IgA. Discontinue administration of the product and initiate appropriate emergency treatment if hypersensitivity reactions occur. (5.1)
- Because PROLASTIN-C LIQUID is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent.(5.2)
ADVERSE REACTIONS
The most common adverse reactions during PROLASTIN-C LIQUID clinical trials in > 5% of subjects were diarrhea and fatigue, each of which occurred in 2 subjects (6%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Grifols Therapeutics LLC at 1-800-520-2807 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Preparation and Handling
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Transmissible Infectious Agents
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
PROLASTIN®-C LIQUID is an Alpha1-Proteinase Inhibitor (Human) (Alpha1-PI) indicated for chronic augmentation and maintenance therapy in adults with clinical evidence of emphysema due to severe hereditary deficiency of Alpha1-PI (alpha1-antitrypsin deficiency).
Limitations of Use
- The effect of augmentation therapy with any Alpha1-PI, including PROLASTIN-C LIQUID, on pulmonary exacerbations and on the progression of emphysema in Alpha1-PI deficiency has not been conclusively demonstrated in randomized, controlled clinical trials.
- Clinical data demonstrating the long-term effects of chronic augmentation or maintenance therapy with PROLASTIN-C LIQUID are not available.
- PROLASTIN-C LIQUID is not indicated as therapy for lung disease in patients in whom severe Alpha1-PI deficiency has not been established.
- The effect of augmentation therapy with any Alpha1-PI, including PROLASTIN-C LIQUID, on pulmonary exacerbations and on the progression of emphysema in Alpha1-PI deficiency has not been conclusively demonstrated in randomized, controlled clinical trials.
-
2 DOSAGE AND ADMINISTRATION
For intravenous use only.
2.1 Dose
- The recommended dose of PROLASTIN-C LIQUID is 60 mg/kg body weight administered intravenously once weekly.
- Dose ranging studies using efficacy endpoints have not been performed with any Alpha1-PI product.
- The carton and the label on each vial of PROLASTIN-C LIQUID show the actual amount of functionally active Alpha1-PI in milligrams (as determined by the capacity to neutralize porcine pancreatic elastase).
2.2 Preparation and Handling
- Allow unopened PROLASTIN-C LIQUID to warm up to room temperature before administration.
- Remove the plastic flip top from the vial.
- Swab the exposed stopper surface with alcohol and allow to dry.
- Inspect the PROLASTIN-C LIQUID visually for particulate matter and discoloration prior to pooling. The product may contain a few protein particles. The solution is clear, colorless or pale yellow or pale green. Do not use if the product is discolored or cloudy.
- Pool PROLASTIN-C LIQUID from several vials to achieve the intended mg/kg body weight dose into an empty, sterile intravenous solution container using aseptic technique.
- Keep pooled solution at room temperature for administration within three hours.
- Discard unused portion.
2.3 Administration
- Visually inspect parenteral drug products for particulate matter and discoloration prior to administration, whenever solution and container permit. The product may contain a few protein particles. Do not use if discolored or cloudy.
- Filter the solution during administration using an intravenous administration set with a suitable 5 to 15 micron infusion filter (not supplied).
- Infuse PROLASTIN-C LIQUID separately, without mixing with other agents or diluting solutions.
- Infuse PROLASTIN-C LIQUID intravenously at 0.08 mL/kg/min as determined by patient response and comfort. The recommended dosage of 60 mg/kg takes approximately 15 minutes to infuse.
- The recommended dose of PROLASTIN-C LIQUID is 60 mg/kg body weight administered intravenously once weekly.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis, may occur. Monitor vital signs and observe the patient carefully throughout the infusion. Early signs and symptoms of hypersensitivity reactions may include pruritus; generalized urticaria; flushing; swollen lips, tongue, or uvula; wheezing; tightness of the chest; dyspnea; hypotension; and syncope. If hypersensitivity symptoms occur, promptly stop PROLASTIN-C LIQUID infusion and begin appropriate therapy. Have epinephrine and other appropriate therapy available for the treatment of any acute anaphylactic or anaphylactoid reaction. [see Patient Counseling Information (17)]
PROLASTIN-C LIQUID may contain trace amounts of IgA. Patients with known antibodies to IgA, which can be present in patients with selective or severe IgA deficiency, have a greater risk of developing potentially severe hypersensitivity and anaphylactic reactions.
5.2 Transmissible Infectious Agents
Because PROLASTIN-C LIQUID is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. This also applies to unknown or emerging viruses and other pathogens. The risk of transmission of infectious agents has been reduced by screening plasma donors for prior exposure to certain infectious agents, by testing for the presence of certain virus infections, and by including steps in the manufacturing process with the demonstrated capacity to inactivate and /or remove certain infectious agents. Despite these measures, this product may still potentially transmit disease.
Report all infections thought by a physician possibly to have been transmitted by this product to Grifols Therapeutics LLC (1-800-520-2807).
-
6 ADVERSE REACTIONS
The most serious adverse reaction observed during clinical trials with PROLASTIN-C was an abdominal and extremity rash in one subject. [see Warnings and Precautions (5.1)]
The most common adverse reactions observed at a rate of > 5% in subjects receiving PROLASTIN-C LIQUID were diarrhea and fatigue, each of which occurred at a rate of 6% (two subjects each).
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in practice.
One clinical trial was conducted with PROLASTIN-C LIQUID: a 16 week, multicenter, randomized, double-blind crossover study to assess the safety, immunogenicity, and pharmacokinetic comparability of PROLASTIN-C LIQUID to PROLASTIN-C in 32 subjects.
Adverse reactions (as defined in the footnote to Table 1) occurring in >5% of subjects during the 16 week double-blind crossover treatment period are shown in Table 1.
Table 1: Adverse Reactions Occurring in >5% of Subjects during the Double-Blinded Crossover Treatment - *
- An adverse reaction is defined as any adverse event that occurred where either a) the event was not considered “unrelated” to administration of the product, or b) the occurrence was during or within 72 hours of the end of the previous infusion of the product, or c) the investigator's causality assessment of the event was missing or indeterminate, or d) the incidence during treatment with 1 investigational product was 130% or more of the incidence during treatment with the other investigational product.
- †
- Source: the randomized double-blinded comparator trial of PROLASTIN-C LIQUID vs PROLASTIN-C.
PROLASTIN®-C LIQUID
(N=32)PROLASTIN®-C
(N=31)Adverse Reaction*,† No. of Subjects with Adverse Reaction
(percentage of all subjects)No. of Subjects with Adverse Reaction
(percentage of all subjects)Diarrhea 2 (6) 0 Fatigue 2 (6) 0 Table 2 below displays the adverse reaction (defined as per Table 1) rate as a percentage of infusions received during the 16 week double-blinded treatment period.
Table 2: Adverse Reaction Frequency as a Percent of All Infusions and Occurring More than Once in the PROLASTIN®-C LIQUID Group during the 16 Week Double Blinded Treatment Period - *
- Source: the randomized double-blinded comparator trial of PROLASTIN-C LIQUID vs PROLASTIN-C.
PROLASTIN®-C LIQUID
No. of infusions: 252PROLASTIN®-C
No. of infusions: 245Adverse Reaction* No. of Adverse Reactions
(percentage of all infusions)No. of Adverse Reactions
(percentage of all infusions)Diarrhea 3 (1.2) 0 Fatigue 2 (0.8) 0 A total of 23 COPD exacerbations were reported for a total of 18 individual subjects. Twelve subjects (12/32, 38%) during PROLASTIN-C LIQUID treatment experienced 13 COPD exacerbations, and 9 subjects (9/31, 29%) during PROLASTIN-C treatment had 10 COPD exacerbations. Three COPD exacerbations occurred during the Follow-Up Period after PROLASTIN-C LIQUID treatment and 1 COPD exacerbation occurred in the Follow-up period after PROLASTIN-C treatment. The overall rate of pulmonary exacerbations during treatment with either product was 1.9 exacerbations per subject-year. No exacerbation was considered to be serious, except for one event after PROLASTIN-C treatment during the Follow-Up period (due to hospitalization).
Two separate prior clinical trials were conducted with PROLASTIN-C: 1.) a 20 week, open-label, single arm safety study in 38 subjects (single-arm open-label trial), and 2.) a 16 week, randomized, double-blind, crossover pharmacokinetic comparability study vs. PROLASTIN in 24 subjects, followed by an 8 week open-label treatment with PROLASTIN-C (randomized double-blinded comparator trial). Thus, a total of 93 subjects were exposed to PROLASTIN-C in clinical trials.
The most serious adverse reaction observed during clinical trials with PROLASTIN-C was an abdominal and extremity rash in one subject. The rash resolved subsequent to outpatient treatment with antihistamines and steroids. Two instances of a less severe, pruritic abdominal rash were observed upon rechallenge despite continued antihistamine and steroid treatment, which led to withdrawal of the subject from the trial.
Grifols assessed the randomized double-blinded comparator trial of PROLASTIN and PROLASTIN-C for adverse reactions (as defined in the footnote to Table 3) occurring during each 8 week double-blind crossover treatment period, as shown in Table 3.
Table 3: Adverse Reactions Occurring during the First 8 Weeks of Each Double-Blinded Treatment - *
- An adverse reaction is defined as any adverse event where either a) the incidence with PROLASTIN-C was greater than with PROLASTIN, or b) the occurrence was within 72 hours of treatment, or c) the event was otherwise considered related or possibly related to the drug.
- †
- Source: the randomized double-blinded comparator trial.
PROLASTIN®-C
No. of subjects: 24PROLASTIN®
No. of subjects: 24Adverse Reaction*,† No. of Subjects with Adverse Reaction
(percentage of all subjects)No. of Subjects with Adverse Reaction
(percentage of all subjects)Upper respiratory tract infection 3 (12.5%) 1 (4.2%) Headache 1 (4.2%) 2 (8.3%) Pruritus 1 (4.2%) 0 Urticaria 1 (4.2%) 0 Nausea 1 (4.2%) 0 Peripheral edema 1 (4.2%) 0 Pyrexia 1 (4.2%) 0 Table 4 below displays the adverse reaction (defined as per Table 3) rate as a percentage of infusions received during the 8 weeks of each double-blinded treatment.
Table 4: Adverse Reaction Frequency as a Percent of All Infusions during the First 8 Weeks of Each Double-Blinded Infusion Treatment - *
- Source: the randomized double-blinded comparator trial.
PROLASTIN®-C
No. of infusions: 188PROLASTIN®
No. of infusions: 192Adverse Reaction* No. of Adverse Reactions
(percentage of all infusions)No. of Adverse Reactions
(percentage of all infusions)Upper respiratory tract infection 3 (1.6%) 1 (0.5%) Headache 1 (0.5%) 3 (1.6%) Pruritus 1 (0.5%) 0 Urticaria 1 (0.5%) 0 Nausea 1 (0.5%) 0 Peripheral edema 1 (0.5%) 0 Pyrexia 1 (0.5%) 0 Table 5 below displays the adverse reactions occurring in two or more subjects during the single-arm open-label trial.
Table 5: Adverse Reactions Occurring in Two or More Subjects (>5%) during the 20 Week Single-Arm Open-Label Trial PROLASTIN®-C
No. of subjects: 38Adverse Reaction*,† No. of Subjects with Adverse Reaction
(percentage of all subjects)Upper respiratory tract infection 6 (15.8%) Urinary tract infection 5 (13.2%) Nausea 4 (10.5%) Chest pain 3 (7.9%) Back pain 2 (5.3%) Chills 2 (5.3%) Cough 2 (5.3%) Dizziness 2 (5.3%) Dyspnea 2 (5.3%) Headache 2 (5.3%) Hot flush 2 (5.3%) Oral candidiasis 2 (5.3%) Ten exacerbations of chronic obstructive pulmonary disease were reported by 8 subjects in the 24 week crossover pharmacokinetic study. During the 16 week double-blind crossover phase, 4 subjects (17%) had a total of 4 exacerbations during PROLASTIN-C treatment and 4 subjects (17%) had a total of 4 exacerbations during PROLASTIN treatment. Two additional exacerbations in 2 subjects (8%) occurred during the 8 week open-label treatment period with PROLASTIN-C. The overall rate of pulmonary exacerbations during treatment with either product was 0.9 exacerbations per subject-year.
Immunogenicity
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to PROLASTIN-C LIQUID with the incidence of antibodies to other products may be misleading.
In the randomized, crossover pharmacokinetic clinical trial, no immunogenicity response was observed in subjects dosed with PROLASTIN-C LIQUID or PROLASTIN-C.
In the single-arm, open-label safety clinical trial, three treatment naïve subjects out of 36 subjects evaluated developed antibody to Alpha1-PI at week 24 after receiving PROLASTIN-C. A fourth subject (non-naïve) was positive prior to and after receiving PROLASTIN-C, but levels were unchanged during the study. None of the four antibody specimens was able to neutralize the protease inhibitor capacity of PROLASTIN-C. In the randomized, crossover pharmacokinetic clinical trial comparing PROLASTIN-C and PROLASTIN, none of 24 subjects developed antibodies to PROLASTIN-C.
6.2 Postmarketing Experience
Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
Expected postmarketing experience for PROLASTIN-C LIQUID is based on the reactions reported for PROLASTIN-C. The reactions which have been chosen for inclusion due to their seriousness, frequency of reporting, possible causal connection to PROLASTIN®-C, or a combination of these factors, are:
- General/Body as a Whole: Fatigue, malaise, influenza-like illness, pain, asthenia
- Immune system: Hypersensitivity including anaphylactoid/anaphylactic reactions
- Cardiovascular: Tachycardia
- Musculoskeletal: Arthralgia, myalgia
- Gastrointestinal: Vomiting, diarrhea
- Investigation: Blood pressure increased
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data with PROLASTIN-C LIQUID use in pregnant women to inform a drug-associated risk. Animal reproduction studies have not been conducted with PROLASTIN-C LIQUID. It is not known whether PROLASTIN-C LIQUID can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. PROLASTIN-C LIQUID should be given to a pregnant woman only if clearly needed.
In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of PROLASTIN-C LIQUID in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for PROLASTIN-C LIQUID and any potential adverse effects on the breastfed infant from PROLASTIN-C LIQUID or from the underlying maternal condition.
-
11 DESCRIPTION
PROLASTIN-C LIQUID is a sterile, concentrate of Alpha1-PI for intravenous infusion. The solution is clear, colorless or pale yellow or pale green. The actual amount of functionally active Alpha1-PI in milligrams, as determined by capacity to neutralize porcine pancreatic elastase, is printed on the carton and vial label of PROLASTIN-C LIQUID. The specific activity of PROLASTIN-C LIQUID is ≥ 0.7 mg functional Alpha1-PI per mg of total protein. PROLASTIN-C LIQUID has a purity of ≥ 90% Alpha1-PI (Alpha1-PI protein/total protein). PROLASTIN-C LIQUID has a pH of 6.6–7.4, a sodium phosphate content of 0.013–0.025 M, and is stabilized with 0.20-0.30 M of alanine. The total sodium concentration is ≤ 100 mEq/L. PROLASTIN-C LIQUID contains no preservative.
PROLASTIN-C LIQUID is produced from pooled human plasma through modifications of the PROLASTIN process using purification by polyethylene glycol (PEG) precipitation, anion exchange chromatography, and cation exchange chromatography. All Source Plasma used in the manufacture of PROLASTIN-C LIQUID is non-reactive (negative) by FDA-licensed serological test methods for hepatitis B surface antigen (HBsAg) and antibodies to hepatitis C virus (HCV) and human immunodeficiency virus types 1 and 2 and negative by FDA-licensed Nucleic Acid Technologies (NAT) for HCV and human immunodeficiency virus type 1 (HIV-1). In addition, all Source Plasma is negative for hepatitis B virus (HBV) by either an FDA-licensed or investigational NAT assay. The goal of the investigational HBV NAT test is to detect low levels of viral nucleic acid; however, the significance of a negative result for the investigational HBV NAT test has not been established. By in-process NAT, all Source Plasma is negative for hepatitis A virus (HAV). As a final plasma safety step, all plasma manufacturing pools are tested by serological test methods and NAT.
To evaluate further the virus safety profile of PROLASTIN-C LIQUID, in vitro studies have been conducted to validate the capacity of the manufacturing process to reduce the infectious titer of a wide range of viruses with diverse physicochemical properties. These studies evaluated the inactivation/removal of clinically relevant viruses, including human immunodeficiency virus type 1 (HIV-1) and hepatitis A virus (HAV), as well as the following model viruses: bovine viral diarrhea virus (BVDV), a surrogate for hepatitis C virus; pseudorabies virus (PRV), a surrogate for large enveloped DNA viruses (e.g., herpes viruses); vesicular stomatitis virus (VSV), a model for enveloped viruses; reovirus type 3 (Reo3), a non-specific model for non-enveloped viruses; and porcine parvovirus (PPV), a model for human parvovirus B19.
The PROLASTIN-C LIQUID manufacturing process has several steps (Cold Ethanol Fractionation, PEG Precipitation, and Depth Filtration) that are important for purifying Alpha1-PI as well as removing potential virus contaminants. Two additional steps, Solvent/Detergent Treatment and 15 nm Virus Removal Nanofiltration, are included in the process as dedicated pathogen reduction steps. The Solvent/Detergent Treatment step effectively inactivates enveloped viruses (such as HIV-1, VSV, HBV, and HCV). The 15 nm Virus Removal Nanofiltration step has been implemented to reduce the risk of transmission of enveloped and non-enveloped viruses as small as 18 nm. Table 6 presents the virus reduction capacity of each process step and the accumulated virus reduction for the process as determined in viral validation studies in which virus was deliberately added to a process model in order to study virus reduction. In addition, the Solvent/Detergent Treatment step inactivates ≥ 5.4 log10 of West Nile virus, a clinically relevant enveloped virus.
Table 6: Virus Reduction (Log10) for the PROLASTIN®-C LIQUID Manufacturing Process Process Step Enveloped Viruses Non-enveloped Viruses HIV-1 BVDV PRV VSV Reo3 HAV PPV Cold Ethanol Fractionation 1.5 1.7 2.5 ND* ≥ 2.1 1.4 1.0 PEG Precipitation 4.3 2.8 3.3 ND 3.3 3.0 3.2 Depth Filtration ≥ 4.7 4.0 ≥ 4.8 ND ≥ 4.0 ≥ 2.8 ≥ 4.4 Solvent/Detergent Treatment ≥ 6.2 ≥ 4.6 ≥ 4.3 5.1 NA† NA NA 15 nm Virus Removal Nanofiltration ≥ 6.9 ≥ 4.7 ≥ 5.2 ≥ 5.1 ≥ 4.3 ≥ 5.5 4.2 Accumulated Virus Reduction ≥ 23.6 ≥ 17.8 ≥ 20.1 ≥ 10.2 ≥ 13.7 ≥ 12.7 ≥ 12.8 Additionally, the manufacturing process was investigated for its capacity to decrease the infectivity of an experimental agent of transmissible spongiform encephalopathy (TSE), considered as a model for the variant Creutzfeldt-Jakob disease (vCJD) and Creutzfeldt-Jakob disease (CJD) agents. Studies of the PROLASTIN-C LIQUID manufacturing process demonstrate that a minimum of 6 log10 reduction of TSE infectivity is achieved. These studies provide reasonable assurance that low levels of vCJD/CJD agent infectivity, if present in the starting material, would be removed.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Alpha1-PI deficiency is an autosomal, co-dominant, hereditary disorder characterized by low serum and lung levels of Alpha1-PI. Smoking is an important risk factor for the development of emphysema in patients with Alpha1-PI deficiency.(1,2) Because emphysema affects many, but not all individuals with the more severe genetic variants of Alpha1-PI deficiency, augmentation therapy with Alpha1-PI is indicated only in patients with severe Alpha1-PI deficiency who have clinically evident emphysema.
Only some Alpha1-PI alleles are associated with clinically apparent Alpha1-PI deficiency.(3,4) Approximately 95% of all severely deficient patients are homozygous for the PiZ allele.(4) Individuals with the PiZZ variant typically have serum Alpha1-PI levels less than 35% of the average normal level. Individuals with the Pi(null)(null) variant have undetectable Alpha1-PI protein in their serum. Individuals with these low serum Alpha1-PI levels, i.e., less than 11 µM, have a markedly increased risk for developing emphysema over their lifetimes. In addition, PiSZ individuals, whose serum Alpha1-PI levels range from approximately 9 to 23 µM,(5) are considered to have moderately increased risk for developing emphysema, regardless of whether their serum Alpha1-PI levels are above or below 11 µM.
Augmenting the levels of functional protease inhibitor by intravenous infusion is an approach to therapy for patients with Alpha1-PI deficiency. The intended theoretical goal is to provide protection to the lower respiratory tract by correcting the imbalance between neutrophil elastase and protease inhibitors. Whether augmentation therapy with any Alpha1-PI product actually protects the lower respiratory tract from progressive emphysematous changes has not been demonstrated in adequately powered, randomized controlled, clinical trials. Although the maintenance of blood serum levels of Alpha1-PI (antigenically measured) above 11 µM has been historically postulated to provide therapeutically relevant anti-neutrophil elastase protection,(6) this has not been proven. Individuals with severe Alpha1-PI deficiency have been shown to have increased neutrophil and neutrophil elastase concentrations in lung epithelial lining fluid compared to normal PiMM individuals, and some PiSZ individuals with Alpha1-PI above 11 µM have emphysema attributed to Alpha1-PI deficiency. These observations underscore the uncertainty regarding the appropriate therapeutic target serum level of Alpha1-PI during augmentation therapy.
The pathogenesis of emphysema is understood as described in the “protease-antiprotease imbalance” model. Alpha1-PI is understood to be the primary antiprotease in the lower respiratory tract, where it inhibits neutrophil elastase (NE). Normal healthy individuals produce sufficient Alpha1-PI to control the NE produced by activated neutrophils and are thus able to prevent inappropriate proteolysis of the lung tissue by NE. Conditions that increase neutrophil accumulation and activation in the lung, such as respiratory infection and smoking, will in turn increase levels of NE. However, individuals who are severely deficient in endogenous Alpha1-PI are unable to maintain an appropriate antiprotease defense, and, in addition, they have been shown to have increased lung epithelial lining fluid neutrophil and NE concentrations. Because of these factors, many (but not all) individuals who are severely deficient in endogenous Alpha1-PI are subject to more rapid proteolysis of the alveolar walls leading to chronic lung disease. PROLASTIN-C LIQUID serves as Alpha1-PI augmentation therapy in the patient population with severe Alpha1-PI deficiency and emphysema, acting to increase and maintain serum and lung epithelial lining fluid levels of Alpha1-PI.
12.2 Pharmacodynamics
Chronic augmentation therapy with the predecessor product, PROLASTIN®, administered weekly at a dose of 60 mg/kg body weight, results in increased levels of Alpha1-PI and functional anti-neutrophil elastase capacity in the epithelial lining fluid of the lower respiratory tract of the lung, as compared to levels prior to commencing therapy with PROLASTIN.(7) However, the clinical benefit of the increased levels at the recommended dose has not been demonstrated in adequately powered, randomized, controlled clinical trials for any Alpha1-PI product.
PROLASTIN-C LIQUID increases antigenic and functional (anti-neutrophil elastase capacity, ANEC) serum levels.
12.3 Pharmacokinetics
The pharmacokinetic (PK) study was a randomized, double-blind, crossover trial comparing PROLASTIN-C LIQUID to PROLASTIN-C (Alpha1-Proteinase Inhibitor [Human]) conducted in 32 adult subjects age 44 to 71 years with severe Alpha1-PI deficiency. Eighteen subjects were male and 14 subjects were female. Sixteen subjects were randomized to each treatment sequence. All but one subject had the PiZZ genotype and the remaining subject was PiSZ. Twenty-eight subjects had received prior Alpha1-PI augmentation therapy and 4 subjects were naïve to Alpha1-PI augmentation therapy. Study subjects were randomly assigned to receive either 60 mg/kg body weight of functional PROLASTIN-C LIQUID or PROLASTIN-C weekly by intravenous infusion during the first 8-week treatment period. Following the last dose in the first 8-week treatment period, subjects underwent serial blood sampling for PK analysis and then crossed over to the alternate treatment for the second 8-week treatment period. Following the last treatment in the second 8-week treatment period, subjects underwent serial blood sampling for PK analysis. In addition, blood samples were drawn for trough levels before infusion at Weeks 6, 7, 8, and 9, as well as before infusion at Weeks 14, 15, 16, and 17. A final PK sample was drawn at Week 20 (4 weeks after the last dose) to correct for endogenous Alpha1-PI levels.
The key pharmacokinetic parameter was the area under the serum Alpha1-PI concentration-by-antigenic-assay-time curve (AUC0-7days) following 8 weeks of treatment with PROLASTIN-C LIQUID or PROLASTIN-C. The 90% confidence interval (1.03-1.08) for the ratio of AUC0-7days for PROLASTIN-C LIQUID and PROLASTIN-C indicated that the 2 products are bioequivalent, i.e. the entire range falls within the 0.80 – 1.25 interval. AUC0-7days of the serum-equivalent Alpha1-PI concentration by functional assay and Cmax by antigenic and functional assays gave comparable results for PROLASTIN-C LIQUID and PROLASTIN-C, as shown in Table 7.
Table 7: Results of Statistical Analysis of Pharmacokinetic Parameters at Steady-State (PK Population) Treatment AUC0-7days (mg*h/mL) Antigenic Content Functional Activity Geometric
LSMGeometric
LSM Ratio90% CI of
Geometric
LSM RatioGeometric
LSMGeometric
LSM Ratio90% CI of
Geometric
LSM RatioPROLASTIN®-C
LIQUID n=30203.57 1.05 1.03, 1.08 169.86 1.04 1.01, 1.07 PROLASTIN®-C
n=28193.71 163.52 Treatment Cmax (mg/mL) PROLASTIN®-C
LIQUID n=302.517 1.04 1.00, 1.09 2.062 1.04 1.00, 1.07 PROLASTIN®-C
n=282.415 1.992 The half life (t1/2) for antigenic content was comparable, specifically 156.39 hours versus 164.10 hours for PROLASTIN-C LIQUID versus PROLASTIN-C, respectively. Similar half life was also observed when assessed by functional activity between PROLASTIN-C LIQUID versus PROLASTIN-C (126.57 hours versus 126.82 hours respectively).
Figure 1 shows the serum-equivalent concentration (functional activity) vs. time curves of Alpha1-PI after intravenous administration of PROLASTIN-C LIQUID and PROLASTIN-C.
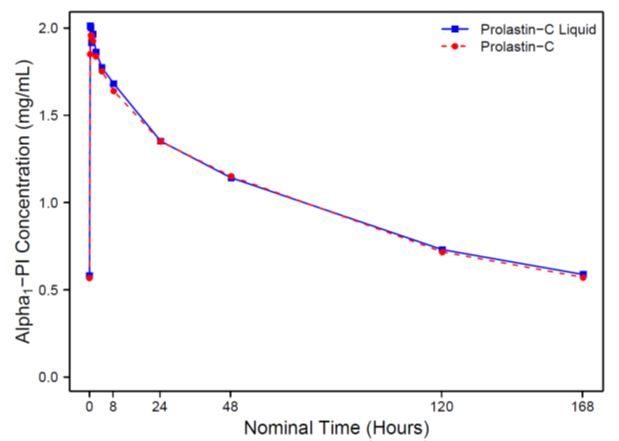
Figure 1: Mean Serum-equivalent Alpha1-PI Concentration (functional activity) vs. Time Curves Following Treatment with PROLASTIN®-C LIQUID or PROLASTIN®-C
Serum trough levels measured at steady state during the PK study using an antigenic content assay showed PROLASTIN-C LIQUID resulted in a mean trough of 17.7 µM and PROLASTIN-C resulted in a mean trough of 16.9 µM.
A randomized, double-blind, crossover pharmacokinetic (PK) study comparing PROLASTIN-C to PROLASTIN was conducted in 24 adult subjects age 40 to 72 with severe Alpha1-PI deficiency. Ten subjects were male and 14 subjects were female. All but one subject had the PiZZ genotype and the remaining subject had PiSZ. All subjects had received prior Alpha1-PI therapy with PROLASTIN for at least 1 month. The double-blind portion of the study was designed the same as the randomized, double-blind, crossover PK study comparing PROLASTIN-C LQUID to PROLASTIN-C described above.
The pharmacokinetic parameters of Alpha1-PI in plasma, based on serum-equivalent functional activity assays, showed comparability between PROLASTIN-C treatment and PROLASTIN treatment, as shown in Table 8.
Table 8: Pharmacokinetic Parameters of Alpha1-PI in Plasma Treatment AUC0-7 days
(hr*mg/mL)
Mean (%CV)Cmax
(mg/mL)
Mean (%CV)t1/2
(hr)
Mean (%CV)PROLASTIN®-C
(n=22 or 23)155.9
(17%)1.797
(10%)146.3
(16%)PROLASTIN®
(n=22 or 23)152.4
(16%)1.848
(15%)139.3
(18%)The key pharmacokinetic parameter was the area under the plasma Alpha1-PI concentration-by-antigenic-assay-time curve (AUC0-7days) following 8 weeks of treatment with PROLASTIN-C or PROLASTIN. The 90% confidence interval (0.97-1.09) for the ratio of AUC0-7days for PROLASTIN-C and PROLASTIN indicated that the 2 products are bioequivalent, i.e. the entire range falls within the 0.80 – 1.25 interval.
Serum-equivalent trough levels measured during the crossover PK study via an antigenic content assay showed PROLASTIN-C treatment resulted in a mean trough of 16.9 ± 2.3 µM and PROLASTIN resulted in a mean trough of 16.7 ± 2.7 µM. Using the functional activity assay, PROLASTIN-C resulted in a mean trough of 11.8 ± 2.2 µM and PROLASTIN resulted in a mean trough of 11.0 ± 2.2 µM.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis, mutagenesis, and impairment of fertility studies were not performed; PROLASTIN-C LIQUID is a biologic purified from human plasma.
13.2 Animal Toxicology and/or Pharmacology
Intravenous administration of five daily doses of PROLASTIN-C LIQUID to rabbits at a dose up to 600 mg/kg per day (10-fold higher dose than the recommended human dose of 60 mg/kg administered weekly), did not result in any signs of toxicity. Further, there were no differences in safety and tolerability of PROLASTIN-C and PROLASTIN-C LIQUID in nonclinical testing.
-
14 CLINICAL STUDIES
The clinical efficacy of PROLASTIN-C LIQUID in influencing the course of pulmonary emphysema or pulmonary exacerbations has not been demonstrated in adequately powered, randomized, controlled clinical trials.
A total of 23 subjects with the PiZZ variant and documented emphysema were studied in a single-arm, open label clinical trial with PROLASTIN, the predecessor product. Nineteen of the subjects received PROLASTIN, 60 mg/kg, once weekly for up to 26 weeks (average 24 weeks). Blood levels of Alpha1-PI were maintained above 11 µM. Bronchoalveolar lavage studies demonstrated statistically significant increased levels of Alpha1-PI and functional ANEC in the epithelial lining fluid of the lower respiratory tract of the lung, as compared to levels prior to dosing.
In addition to the PROLASTIN-C LIQUID/PROLASTIN-C crossover trial described above, in which 31 subjects received PROLASTIN-C, PROLASTIN-C has been studied in 62 individual subjects in 2 separate clinical trials. The first study was a crossover pharmacokinetic study. [see Clinical Pharmacology (12.3)] The second PROLASTIN-C clinical trial was a multi-center, open-label single arm safety study conducted to evaluate the safety and tolerability of PROLASTIN-C. In this study, 38 subjects were treated with weekly intravenous infusions of 60 mg/kg body weight of PROLASTIN-C for 20 weeks. Half the subjects were naïve to previous Alpha1-PI augmentation prior to study entry and the other half were receiving augmentation with PROLASTIN prior to entering the study. A diagnosis of severe Alpha1-PI deficiency was confirmed by the demonstration of the PiZZ genotype in 32 of 38 (84.2%) subjects, and 6 of 38 (15.8%) subjects presented with other alleles known to result in severe Alpha1-PI deficiency. These groups were distributed evenly between the naïve and non-naïve cohorts.
-
15 REFERENCES
-
American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:818-900.
-
Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med. 2014;7:419-27.
-
Crystal RG. α1-Antitrypsin deficiency, emphysema, and liver disease; genetic basis and strategies for therapy. J Clin Invest. 1990;85:1343-52.
-
World Health Organization. Alpha-1-antitrypsin deficiency: Memorandum from a WHO meeting. Bull World Health Organ. 1997;75:397-415.
-
Turino GM, Barker AF, Brantly ML, Cohen AB, Connelly RP, Crystal RG, et al. Clinical features of individuals with PI*SZ phenotype of α1-antitrypsin deficiency. Am J Respir Crit Care Med. 1996;154:1718-25.
-
American Thoracic Society. Guidelines for the approach to the patient with severe hereditary alpha-1-antitrypsin deficiency. Am Rev Respir Dis. 1989;140:1494-7.
-
Wewers MD, Casolaro MA, Sellers SE, Swayze SC, McPhaul KM, Wittes JT, et al. Replacement therapy for alpha1-antitrypsin deficiency associated with emphysema. N Eng J Med. 1987;316:1055-62.
-
-
16 HOW SUPPLIED/STORAGE AND HANDLING
- PROLASTIN-C LIQUID is supplied in single-dose vials with the total Alpha1-PI functional activity, in milligrams, stated on the vial label and carton.
- Components of the packaging do not contain natural rubber latex.
Carton NDC Vial NDC Approximate Alpha1–PI
Functional ActivityVolume 13533-705-31
13533-705-01
13533-705-5113533-705-32
13533-705-11
13533-705-52500 mg
1,000 mg
4,000 mg10 mL
20 mL
80 mL- Store refrigerated at 2-8°C (36-46°F) for the period indicated by the expiration date on its label.
- Product may be stored at room temperatures not exceeding 25oC (77oF) for up to one month, after which the product must be used or immediately discarded.
- Do not freeze.
- Discard unused portion.
-
17 PATIENT COUNSELING INFORMATION
- Inform patients of the signs of hypersensitivity reactions including pruritus; generalized urticaria; flushing; swollen lips, tongue, or uvula; wheezing; tightness of the chest; dyspnea; hypotension; and syncope. Advise patients to discontinue use of the product and contact their physician and/or seek immediate emergency care, depending on the severity of the reaction, if these symptoms occur. [see Warnings and Precautions (5.1)]
- Inform patients that PROLASTIN-C LIQUID is made from human plasma and may carry a risk of transmitting infectious agents that can cause disease (e.g., viruses, the vCJD agent and, theoretically, the CJD agent). Explain that the risk of PROLASTIN-C LIQUID transmitting an infectious agent has been reduced by screening plasma donors for prior exposure to certain infectious agents, by testing the donated plasma for certain current virus infections, and by inactivating and/or removing infectious agents during manufacturing. [see Warnings and Precautions (5.2)]
- Inform patients that administration of PROLASTIN-C LIQUID has been demonstrated to raise the plasma level of Alpha1-PI, but that the effect of this augmentation on pulmonary exacerbations and on the rate of progression of emphysema has not been demonstrated in adequately powered, randomized, controlled clinical trials for any Alpha1-PI product. [see Clinical Studies (14)]
- SPL UNCLASSIFIED SECTION
-
PACKAGE LABEL
NDC 13533-705-01
20 mL
Alpha1-Proteinase Inhibitor (Human)
PROLASTIN® C
LIQUID
Solution for Injection
Solvent detergent treated
Nanofiltered
Rx Only
GRIFOLS
The patient and physician
should discuss the risks
and benefits of this
product.
Dosage and administration:
Read enclosed package
insert.
Store refrigerated at 2 to 8°C
(36 to 46°F) with no more
than 1 month at room
temperatures (up to 25°C;
77°F) after which the product
must be used or immediately
discarded. Do not use after
the expiration date.
Do not freeze.
CONTENTS:
One vial of
Prolastin®-C Liquid
No preservative
For intravenous
administration only
Sterile-nonpyrogenic
U.S. License No. 1871
THIS PRODUCT IS PREPARED
FROM LARGE POOLS OF HUMAN
PLASMA WHICH MAY CONTAIN
INFECTIOUS AGENTS.
SEE PACKAGE INSERT WARNINGS.
Single dose vial
Do not use if turbid.
Discard unused portion.
Do not store after entry into vial.
Administer within 3 hours after
preparation.
1 mL contains approximately 50 mg
Alpha1-Proteinase Inhibitor (Human),
2.76 mg sodium phosphate,
22.27 mg L-alanine, and water.
If the shrink band is absent or
shows any sign of tampering, do
not use the product and notify
Grifols Therapeutics LLC
immediately.
The components of the packaging
do not contain natural rubber latex.
Grifols Therapeutics LLC
Research Triangle Park, NC
27709 USA
GRIFOLS
Carton: 3061390
LOT XXXXXXXXXX
EXP DDMMMYYYY
MG A1-P1 XXXX
SN XXXXXXXXXXXXXXXX
GTIN 00313533705015
PROLASTIN® C LIQUID

NDC 13533-705-11
Alpha1-Proteinase
Inhibitor (Human)
PROLASTIN® C LIQUID
SOLUTION FOR INJECTION
Solvent detergent treated
Nanofiltered
The patient and physician should
discuss the risks and benefits of
this product. No preservative
For intravenous administration only
Sterile-nonpyrogenic
Rx only
20 mL
Dosage and administration:
Read enclosed package insert.
Store at 2 to 8°C (36 to 46°F) with no
more than 1 month at room temperatures
(up to 25°C; 77°F), after which the product
must be used or immediately discarded.
Do not use after the expiration date.
Do not freeze.
Grifols Therapeutics LLC
Research Triangle Park, NC 27709 USA
GRIFOLS
U.S. License No. 1871
3058828
Lot / Exp. / mg α1-PI
-
INGREDIENTS AND APPEARANCE
PROLASTIN-C LIQUID
alpha1-proteinase inhibitor (human) injection, solutionProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:13533-705 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength .alpha.1-proteinase Inhibitor Human (UNII: F43I396OIS) (.alpha.1-proteinase Inhibitor Human - UNII:F43I396OIS) .alpha.1-proteinase Inhibitor Human 1000 mg in 20 mL Inactive Ingredients Ingredient Name Strength Sodium Phosphate (UNII: SE337SVY37) Alanine (UNII: OF5P57N2ZX) Water (UNII: 059QF0KO0R) Product Characteristics Color YELLOW (clear, colorless or pale yellow or pale green) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:13533-705-01 1 in 1 CARTON 1 NDC:13533-705-11 20 mL in 1 VIAL; Type 0: Not a Combination Product 2 NDC:13533-705-31 1 in 1 CARTON 2 NDC:13533-705-32 10 mL in 1 VIAL; Type 0: Not a Combination Product 3 NDC:13533-705-51 1 in 1 CARTON 3 NDC:13533-705-52 80 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103174 12/02/1987 Labeler - GRIFOLS USA, LLC (048987452) Establishment Name Address ID/FEI Business Operations Grifols Therapeutics LLC 611019113 manufacture(13533-705) Establishment Name Address ID/FEI Business Operations Instituto Grifols SA 465562213 manufacture(13533-705) Establishment Name Address ID/FEI Business Operations GRIFOLS WORLDWIDE OPERATIONS LIMITED 985528524 label(13533-705) , pack(13533-705)