Label: RINVOQ- upadacitinib tablet, extended release
RINVOQ- upadacitinib solution




-
NDC Code(s):
0074-1043-14,
0074-1043-28,
0074-2306-30,
0074-2306-70, view more0074-2310-20, 0074-2310-30, 0074-2320-01, 0074-2320-70
- Packager: AbbVie Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 10, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RINVOQ/RINVOQ LQ safely and effectively. See full prescribing information for RINVOQ/RINVOQ LQ.
RINVOQ® (upadacitinib) extended-release tablets, for oral use RINVOQ® LQ (upadacitinib) oral solution
Initial U.S. Approval: 2019
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS (MACE), and THROMBOSIS
See full prescribing information for complete boxed warning.
-
Increased risk of serious bacterial, fungal, viral, and opportunistic infections leading to hospitalization or death, including tuberculosis (TB). Interrupt treatment with RINVOQ/RINVOQ LQ if serious infection occurs until the infection is controlled. Test for latent TB before and during therapy; treat latent TB prior to use. Monitor all patients for active TB during treatment, even patients with initial negative, latent TB test. (5.1)
-
Higher rate of all-cause mortality, including sudden cardiovascular death with another Janus kinase (JAK) inhibitor vs. tumor necrosis factor (TNF) blockers in rheumatoid arthritis (RA) patients. (5.2)
-
Malignancies have occurred in patients treated with RINVOQ. Higher rate of lymphomas and lung cancers with another JAK inhibitor vs. TNF blockers in RA patients. (5.3)
-
Higher rate of MACE (defined as cardiovascular death, myocardial infarction, and stroke) with another JAK inhibitor vs. TNF blockers in RA patients. (5.4)
- Thrombosis has occurred in patients treated with RINVOQ. Increased incidence of pulmonary embolism, venous and arterial thrombosis with another JAK inhibitor vs. TNF blockers. (5.5)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
RINVOQ/RINVOQ LQ is a Janus kinase (JAK) inhibitor.
- RINVOQ is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers. (1.1)
Limitations of Use
RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.1)
- RINVOQ/RINVOQ LQ is indicated for the treatment of adults and pediatric patients 2 years of age and older with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers. (1.2)
Limitations of Use
RINVOQ/RINVOQ LQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.2)
- RINVOQ is indicated for the treatment of adults and pediatric patients 12 years of age and older with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies are inadvisable. (1.3)
Limitations of Use
RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants. (1.3)
- RINVOQ is indicated for the treatment of adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response or intolerance to one or more TNF blockers. If TNF blockers are clinically inadvisable, patients should have received at least one approved systemic therapy prior to use of RINVOQ. (1.4)
Limitations of Use
RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for UC, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.4)
- RINVOQ is indicated for the treatment of adults with moderately to severely active Crohn’s disease (CD) who have had an inadequate response or intolerance to one or more TNF blockers. If TNF blockers are clinically inadvisable, patients should have received at least one approved systemic therapy prior to use of RINVOQ. (1.5)
Limitations of Use
RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for CD, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.5)
- RINVOQ is indicated for the treatment of adults with active ankylosing spondylitis who have had an inadequate response or intolerance to one or more TNF blockers. (1.6)
Limitations of Use
RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.6)
- RINVOQ is indicated for the treatment of adults with active non-radiographic axial spondyloarthritis with objective signs of inflammation who have had an inadequate response or intolerance to TNF blocker therapy. (1.7)
Limitations of Use
RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.7)
- RINVOQ/RINVOQ LQ is indicated for the treatment of patients 2 years of age and older with active polyarticular juvenile idiopathic arthritis who have had an inadequate response or intolerance to one or more TNF blockers. (1.8)
Limitations of Use
RINVOQ/RINVOQ LQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.8)
- RINVOQ is indicated for the treatment of adults with giant cell arteritis (1.9)
Limitations of Use
RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine. (1.9)
DOSAGE AND ADMINISTRATION
-
RINVOQ LQ oral solution is not substitutable with RINVOQ extended-release tablets (2.2, 2.10).
- Changes between RINVOQ LQ oral solution and RINVOQ extended-release tablets should be made by the healthcare provider.
- Prior to treatment update immunizations and consider evaluating for active and latent tuberculosis, viral hepatitis, hepatic function, and pregnancy status (2.1)
- Avoid initiation or interrupt RINVOQ/RINVOQ LQ if absolute lymphocyte count is less than 500 cells/mm3, absolute neutrophil count is less than 1000 cells/mm3, or hemoglobin level is less than 8 g/dL. (2.1, 2.14)
Rheumatoid Arthritis, Ankylosing Spondylitis, and Non-radiographic Axial Spondyloarthritis
Psoriatic Arthritis
-
Pediatric Patients 2 to less than 18 Years of Age Weighing at Least 10 kg: The recommended dosage is based on body weight (2.4)
- Adults: The recommended dosage of RINVOQ is 15 mg once daily. (2.4)
Atopic Dermatitis
-
Pediatric Patients 12 Years of Age and Older Weighing at Least 40 kg and Adults Less Than 65 Years of Age: Initiate treatment with RINVOQ 15 mg orally once daily. If an adequate response is not achieved, consider increasing the dosage to 30 mg orally once daily. (2.5)
-
Adults 65 Years of Age and Older: Recommended dosage of RINVOQ is 15 mg once daily. (2.5)
- Severe Renal Impairment: Recommended dosage of RINVOQ is 15 mg once daily. (2.12)
Ulcerative Colitis
-
Adults: The recommended induction dosage of RINVOQ is 45 mg once daily for 8 weeks. The recommended maintenance dosage of RINVOQ is 15 mg once daily. A maintenance dosage of 30 mg once daily may be considered for patients with refractory, severe, or extensive disease. Discontinue RINVOQ if adequate therapeutic response is not achieved with the 30 mg dosage. Use the lowest effective dosage needed to maintain response. (2.6)
- See the Full Prescribing Information for the recommended dosage in patients with renal or hepatic impairment and for dosage modification due to drug interactions. (2.12, 2.13)
Crohn’s Disease
-
Adults: The recommended induction dosage of RINVOQ is 45 mg once daily for 12 weeks. The recommended maintenance dosage of RINVOQ is 15 mg once daily. A maintenance dosage of 30 mg once daily may be considered for patients with refractory, severe, or extensive disease. Discontinue RINVOQ if an adequate therapeutic response is not achieved with the 30 mg dosage. Use the lowest effective dosage needed to maintain response. (2.7)
- See the Full Prescribing Information for the recommended dosage in patients with renal or hepatic impairment and for dosage modification due to drug interactions. (2.12, 2.13)
Polyarticular Juvenile Idiopathic Arthritis
- The recommended dosage is based on body weight (2.10)
Giant Cell Arteritis
- The recommended dosage of RINVOQ is 15 mg once daily in combination with a tapering course of corticosteroids. RINVOQ 15 mg once daily can be used as monotherapy following discontinuation of corticosteroids (2.11)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
-
Serious Infections: Avoid use in patients with active, serious infection, including localized infections. (5.1)
-
Hypersensitivity: Serious hypersensitivity reactions (e.g., anaphylaxis) have been reported. Discontinue if a serious hypersensitivity reaction occurs. (5.6)
-
Gastrointestinal (GI) Perforations: Monitor patients at risk for GI perforations and promptly evaluate patients with symptoms. (5.7)
-
Laboratory Abnormalities: Monitoring recommended due to potential changes in lymphocytes, neutrophils, hemoglobin, liver enzymes and lipids. (5.8)
-
Embryo-Fetal Toxicity: May cause fetal harm based on animal studies. Advise female patients of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.9, 8.1, 8.3)
-
Vaccinations: Avoid use with live vaccines. (5.10)
- Medication Residue in Stool: Observed in stool or ostomy output in patients with shortened GI transit times. Monitor patients clinically and consider alternative treatment if inadequate therapeutic response. (5.11)
ADVERSE REACTIONS
-
Rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and non-radiographic axial spondyloarthritis: Adverse reactions (≥ 1%) were: upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, and headache. (6.1)
-
Giant cell arteritis: Adverse reactions (≥ 5%) are upper respiratory tract infections, headache, fatigue, peripheral edema, cough, anemia, rash, herpes zoster, and nausea. (6.1)
-
Atopic dermatitis: Adverse reactions (≥ 1%) are: upper respiratory tract infections, acne, herpes simplex, headache, blood creatine phosphokinase increased, cough, hypersensitivity, folliculitis, nausea, abdominal pain, pyrexia, increased weight, herpes zoster, influenza, fatigue, neutropenia, myalgia, and influenza like illness. (6.1)
-
Ulcerative colitis: Adverse reactions (≥ 5%) reported during induction or maintenance are: upper respiratory tract infections, increased blood creatine phosphokinase, acne, neutropenia, elevated liver enzymes, pyrexia, and rash. (6.1)
- Crohn’s disease: Adverse reactions (≥ 5%) reported during induction or maintenance are: upper respiratory tract infections, anemia, pyrexia, acne, herpes zoster, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2025
-
Increased risk of serious bacterial, fungal, viral, and opportunistic infections leading to hospitalization or death, including tuberculosis (TB). Interrupt treatment with RINVOQ/RINVOQ LQ if serious infection occurs until the infection is controlled. Test for latent TB before and during therapy; treat latent TB prior to use. Monitor all patients for active TB during treatment, even patients with initial negative, latent TB test. (5.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, and THROMBOSIS
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis
1.2 Psoriatic Arthritis
1.3 Atopic Dermatitis
1.4 Ulcerative Colitis
1.5 Crohn’s Disease
1.6 Ankylosing Spondylitis
1.7 Non-radiographic Axial Spondyloarthritis
1.8 Polyarticular Juvenile Idiopathic Arthritis
1.9 Giant Cell Arteritis
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluations and Immunizations Prior to Treatment Initiation
2.2 Important Administration Instructions
2.3 Recommended Dosage in Rheumatoid Arthritis
2.4 Recommended Dosage in Psoriatic Arthritis
2.5 Recommended Dosage in Atopic Dermatitis
2.6 Recommended Dosage in Ulcerative Colitis
2.7 Recommended Dosage in Crohn’s Disease
2.8 Recommended Dosage in Ankylosing Spondylitis
2.9 Recommended Dosage in Non-radiographic Axial Spondyloarthritis
2.10 Recommended Dosage in Polyarticular Juvenile Idiopathic Arthritis
2.11 Recommended Dosage in Giant Cell Arteritis
2.12 Recommended Dosage in Patients with Renal Impairment or Hepatic Impairment
2.13 Dosage Modifications Due to Drug Interactions
2.14 Dosage Interruption
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Mortality
5.3 Malignancy and Lymphoproliferative Disorders
5.4 Major Adverse Cardiovascular Events
5.5 Thrombosis
5.6 Hypersensitivity Reactions
5.7 Gastrointestinal Perforations
5.8 Laboratory Abnormalities
5.9 Embryo-Fetal Toxicity
5.10 Vaccinations
5.11 Medication Residue in Stool
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 Inhibitors
7.2 Strong CYP3A4 Inducers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
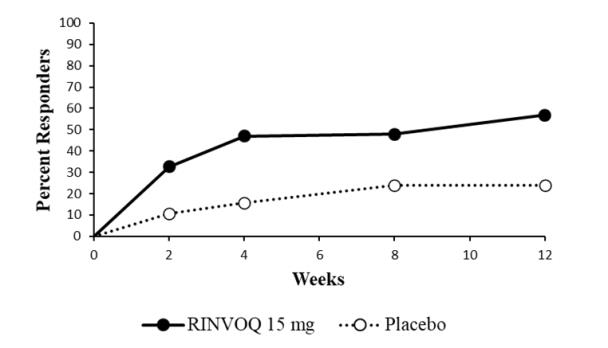
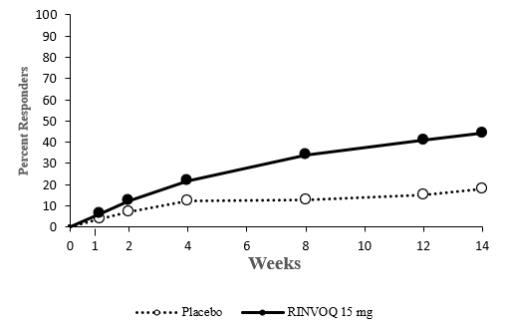
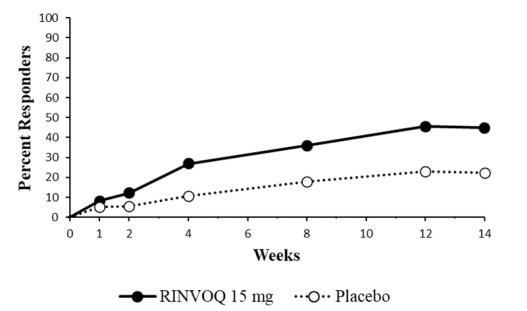
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis
14.2 Psoriatic Arthritis
14.3 Atopic Dermatitis
14.4 Ulcerative Colitis
14.5 Crohn’s Disease
14.6 Ankylosing Spondylitis
14.7 Non-radiographic Axial Spondyloarthritis
14.8 Polyarticular Juvenile Idiopathic Arthritis
14.9 Giant Cell Arteritis
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, and THROMBOSIS
SERIOUS INFECTIONS
Patients treated with RINVOQ/RINVOQ LQ are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt RINVOQ/RINVOQ LQ until the infection is controlled.
Reported infections include:
-
Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before RINVOQ/RINVOQ LQ use and during therapy. Treatment for latent infection should be considered prior to RINVOQ/RINVOQ LQ use.
-
Invasive fungal infections, including cryptococcosis and pneumocystosis.
- Bacterial, viral, including herpes zoster, and other infections due to opportunistic pathogens.
The risks and benefits of treatment with RINVOQ/RINVOQ LQ should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with RINVOQ/RINVOQ LQ, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1)].
MORTALITY
In a large, randomized, postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another Janus kinase (JAK) inhibitor to tumor necrosis factor (TNF) blockers, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed with the JAK inhibitor [see Warnings and Precautions (5.2)].
MALIGNANCIES
Lymphoma and other malignancies have been observed in patients treated with RINVOQ. In RA patients treated with another JAK inhibitor, a higher rate of malignancies (excluding non-melanoma skin cancer (NMSC)) was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk [see Warnings and Precautions (5.3)].
MAJOR ADVERSE CARDIOVASCULAR EVENTS
In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. Patients who are current or past smokers are at additional increased risk. Discontinue RINVOQ/RINVOQ LQ in patients that have experienced a myocardial infarction or stroke [see Warnings and Precautions (5.4)].
THROMBOSIS
Thromboses, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis, have occurred in patients treated for inflammatory conditions with JAK inhibitors, including RINVOQ. Many of these adverse events were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of thrombosis was observed when compared with TNF blockers. Avoid RINVOQ/RINVOQ LQ in patients at risk. Patients with symptoms of thrombosis should discontinue RINVOQ/RINVOQ LQ and be promptly evaluated [see Warnings and Precautions (5.5)].
-
Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before RINVOQ/RINVOQ LQ use and during therapy. Treatment for latent infection should be considered prior to RINVOQ/RINVOQ LQ use.
-
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis
RINVOQ® is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
- Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic disease-modifying antirheumatic drugs (DMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine.
1.2 Psoriatic Arthritis
RINVOQ/RINVOQ LQ is indicated for the treatment of adults and pediatric patients 2 years of age and older with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers.
- Limitations of Use: RINVOQ/RINVOQ LQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine.
1.3 Atopic Dermatitis
RINVOQ is indicated for the treatment of adults and pediatric patients 12 years of age and older with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies are inadvisable.
- Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
1.4 Ulcerative Colitis
RINVOQ is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis (UC) who have had an inadequate response or intolerance to one or more TNF blockers. If TNF blockers are clinically inadvisable, patients should have received at least one approved systemic therapy prior to use of RINVOQ.
- Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for UC, or with potent immunosuppressants such as azathioprine and cyclosporine.
1.5 Crohn’s Disease
RINVOQ is indicated for the treatment of adult patients with moderately to severely active Crohn’s disease (CD) who have had an inadequate response or intolerance to one or more TNF blockers. If TNF blockers are clinically inadvisable, patients should have received at least one approved systemic therapy prior to use of RINVOQ.
- Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biological therapies for CD, or with potent immunosuppressants such as azathioprine and cyclosporine.
1.6 Ankylosing Spondylitis
RINVOQ is indicated for the treatment of adults with active ankylosing spondylitis who have had an inadequate response or intolerance to one or more TNF blockers.
- Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine.
1.7 Non-radiographic Axial Spondyloarthritis
RINVOQ is indicated for the treatment of adults with active non-radiographic axial spondyloarthritis with objective signs of inflammation who have had an inadequate response or intolerance to TNF blocker therapy.
- Limitations of Use: RINVOQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine.
1.8 Polyarticular Juvenile Idiopathic Arthritis
RINVOQ/RINVOQ LQ is indicated for the treatment of patients 2 years of age and older with active polyarticular juvenile idiopathic arthritis (pJIA) who have had an inadequate response or intolerance to one or more TNF blockers.
- Limitations of Use: RINVOQ/RINVOQ LQ is not recommended for use in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluations and Immunizations Prior to Treatment Initiation
Prior to RINVOQ/RINVOQ LQ treatment initiation, consider performing the following evaluations:
- Active and latent tuberculosis (TB) infection evaluation - If positive, treat for TB prior to RINVOQ/RINVOQ LQ use [see Warnings and Precautions (5.1)].
- Viral hepatitis screening in accordance with clinical guidelines – RINVOQ/RINVOQ LQ initiation is not recommended in patients with active hepatitis B or hepatitis C [see Warnings and Precautions (5.1)].
- A complete blood count – RINVOQ/RINVOQ LQ initiation is not recommended in patients with an absolute lymphocyte count less than 500 cells/mm3, absolute neutrophil count less than 1000 cells/mm3, or hemoglobin level less than 8 g/dL [see Dosage and Administration (2.14) and Warnings and Precautions (5.8)].
- Baseline hepatic function: RINVOQ/RINVOQ LQ initiation is not recommended for patients with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
- Pregnancy Status: Verify the pregnancy status of females of reproductive potential prior to starting treatment [see Warnings and Precautions (5.9) and Use in Specific Populations (8.1, 8.3)].
Update immunizations according to current immunization guidelines [see Warnings and Precautions (5.10)].
2.2 Important Administration Instructions
- RINVOQ LQ oral solution is not substitutable with RINVOQ extended-release tablets [see Dosage and Administration (2.4, 2.10)].
- Changes between RINVOQ LQ oral solution and RINVOQ extended-release tablets should be made by the health care provider.
- RINVOQ/RINVOQ LQ should be taken orally with or without food [see Clinical Pharmacology (12.3)].
- RINVOQ tablets should be swallowed whole. RINVOQ tablets should not be split, crushed, or chewed.
- RINVOQ LQ should be administered using the provided press-in bottle adapter and oral dosing syringe [see Instructions for Use].
- RINVOQ LQ is dosed twice daily [see Dosage and Administration (2.4, 2.10)].
2.3 Recommended Dosage in Rheumatoid Arthritis
The recommended dosage of RINVOQ is 15 mg once daily.
2.4 Recommended Dosage in Psoriatic Arthritis
Pediatric Patients 2 to Less Than 18 Years of Age
The recommended dosage is based on body weight (Table 1).
Table 1: RINVOQ/RINVOQ LQ Dosage for Pediatric Patients 2 Years to Less Than 18 Years of Age with Psoriatic Arthritis Patient Weight RINVOQ LQ RINVOQ 10 kg to less than 20 kg 3 mg (3 mL oral solution) twice daily Not recommended 20 kg to less than 30 kg 4 mg (4 mL oral solution) twice daily Not recommended 30 kg and greater 6 mg (6 mL oral solution) twice daily 15 mg (one 15 mg tablet) once daily RINVOQ LQ oral solution is not substitutable with RINVOQ extended-release tablets. Changes between RINVOQ LQ oral solution and RINVOQ extended-release tablets should be made by the health care provider.
Adults 18 Years of Age and Older
The recommended dosage of RINVOQ is 15 mg once daily.
2.5 Recommended Dosage in Atopic Dermatitis
Pediatric Patients 12 Years of Age and Older Weighing at Least 40 kg and Adults Less Than 65 Years of Age
Initiate treatment with RINVOQ 15 mg once daily. If an adequate response is not achieved, consider increasing the dosage to 30 mg once daily. Discontinue RINVOQ if an adequate response is not achieved with the 30 mg dose. Use the lowest effective dose needed to maintain response.
Adults 65 Years of Age and Older
The recommended dosage of RINVOQ is 15 mg once daily.
2.6 Recommended Dosage in Ulcerative Colitis
Adult Patients: Induction
The recommended induction dosage of RINVOQ is 45 mg once daily for 8 weeks.
Adult Patients: Maintenance
The recommended dosage of RINVOQ for maintenance treatment is 15 mg once daily. A dosage of 30 mg once daily may be considered for patients with refractory, severe or extensive disease. Discontinue RINVOQ if an adequate therapeutic response is not achieved with the 30 mg dosage. Use the lowest effective dosage needed to maintain response.
2.7 Recommended Dosage in Crohn’s Disease
Adult Patients: Induction
The recommended induction dosage of RINVOQ is 45 mg once daily for 12 weeks.
Adult Patients: Maintenance
The recommended dosage of RINVOQ for maintenance treatment is 15 mg once daily. A dosage of 30 mg once daily may be considered for patients with refractory, severe or extensive disease. Discontinue RINVOQ if an adequate therapeutic response is not achieved with the 30 mg dosage. Use the lowest effective dosage needed to maintain response.
2.8 Recommended Dosage in Ankylosing Spondylitis
The recommended dosage of RINVOQ is 15 mg once daily.
2.9 Recommended Dosage in Non-radiographic Axial Spondyloarthritis
The recommended dosage of RINVOQ is 15 mg once daily.
2.10 Recommended Dosage in Polyarticular Juvenile Idiopathic Arthritis
The recommended dosage is based on body weight (Table 2).
Table 2: RINVOQ/RINVOQ LQ Dosage for Patients 2 years and older with pJIA Patient Weight RINVOQ LQ RINVOQ 10 kg to less than 20 kg 3 mg (3 mL oral solution) twice daily Not recommended 20 kg to less than 30 kg 4 mg (4 mL oral solution) twice daily Not recommended 30 kg and greater 6 mg (6 mL oral solution) twice daily 15 mg (one 15 mg tablet) once daily RINVOQ LQ oral solution is not substitutable with RINVOQ extended-release tablets. Changes between RINVOQ LQ oral solution and RINVOQ extended-release tablets should be made by the health care provider.
2.11 Recommended Dosage in Giant Cell Arteritis
The recommended dosage of RINVOQ is 15 mg once daily in combination with a tapering course of corticosteroids.
RINVOQ 15 mg once daily can be used as monotherapy following discontinuation of corticosteroids.
2.12 Recommended Dosage in Patients with Renal Impairment or Hepatic Impairment
Renal Impairment
Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis, Non-radiographic Axial Spondyloarthritis, pJIA, and Giant Cell Arteritis:
- No dosage adjustment is needed for patients with mild, moderate, or severe renal impairment.
Atopic Dermatitis:
- For patients with severe renal impairment [estimated glomerular filtration rate (eGFR) 15 to < 30 mL/min/1.73m2] the recommended dosage of RINVOQ is 15 mg once daily [see Use in Specific Populations (8.6)].
- No dosage adjustment is needed for patients with mild or moderate renal impairment (eGFR ≥ 30 mL/min/1.73m2).
- RINVOQ is not recommended for use in patients with end stage renal disease (eGFR < 15 mL/min/1.73m2) [see Use in Specific Populations (8.6)].
Ulcerative Colitis:
- For patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73m2), the recommended dosage of RINVOQ is:
• Induction: 30 mg once daily for 8 weeks
• Maintenance: 15 mg once daily
- No dosage adjustment is needed for patients with mild or moderate renal impairment (eGFR ≥ 30 mL/min/1.73m2).
- RINVOQ is not recommended for use in patients with end stage renal disease (eGFR < 15 mL/min/1.73m2) [see Use in Specific Populations (8.6)].
Crohn’s Disease:
- For patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73m2), the recommended dosage of RINVOQ is:
• Induction: 30 mg once daily for 12 weeks
• Maintenance: 15 mg once daily
- No dosage adjustment is needed for patients with mild or moderate renal impairment (eGFR ≥ 30 mL/min/1.73m2).
- RINVOQ is not recommended for use in patients with end stage renal disease (eGFR < 15 mL/min/1.73m2) [see Use in Specific Populations (8.6)].
Hepatic Impairment
RINVOQ/RINVOQ LQ is not recommended for use in patients with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.7)].
Rheumatoid Arthritis, Psoriatic Arthritis, Atopic Dermatitis, Ankylosing Spondylitis, Non-radiographic Axial Spondyloarthritis, pJIA, and Giant Cell Arteritis:
No dosage adjustment is needed for patients with mild or moderate hepatic impairment (Child-Pugh A or B).
Ulcerative Colitis:
For patients with mild to moderate hepatic impairment (Child-Pugh A or B) the recommended dosage of RINVOQ is:
-
Induction: 30 mg once daily for 8 weeks
- Maintenance: 15 mg once daily
Crohn’s Disease:
For patients with mild to moderate hepatic impairment (Child-Pugh A or B) the recommended dosage of RINVOQ is:
-
Induction: 30 mg once daily for 12 weeks
- Maintenance: 15 mg once daily
2.13 Dosage Modifications Due to Drug Interactions
Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis, Non-radiographic Axial Spondyloarthritis, pJIA, and Giant Cell Arteritis
No dosage adjustment is needed in patients receiving strong CYP3A4 inhibitors [see Drug Interactions (7.1)].
Atopic Dermatitis
The recommended dosage of RINVOQ in patients receiving strong CYP3A4 inhibitors is 15 mg once daily [see Drug Interactions (7.1)].
Ulcerative Colitis
The recommended dosage of RINVOQ in patients with ulcerative colitis receiving strong CYP3A4 inhibitors [see Drug Interactions (7.1)]:
-
Induction: 30 mg once daily for 8 weeks
- Maintenance: 15 mg once daily
Crohn’s Disease
The recommended dosage of RINVOQ in patients with Crohn’s disease receiving strong CYP3A4 inhibitors [see Drug Interactions (7.1)]:
-
Induction: 30 mg once daily for 12 weeks
- Maintenance: 15 mg once daily
2.14 Dosage Interruption
Infections
If a patient develops a serious infection, including serious opportunistic infection, interrupt RINVOQ/RINVOQ LQ treatment until the infection is controlled [see Warnings and Precautions (5.1)].
Laboratory Abnormalities
Interruption of dosing may be needed for management of laboratory abnormalities as described in Table 3 [see Warnings and Precautions (5.8)].
Table 3: Recommended Dosage Interruptions for Laboratory Abnormalities Laboratory Measure Action Absolute Neutrophil Count (ANC) Interrupt treatment if ANC is less than 1000 cells/mm3; treatment may be restarted once ANC returns above this value Absolute Lymphocyte Count (ALC) Interrupt treatment if ALC is less than 500 cells/mm3; treatment may be restarted once ALC returns above this value Hemoglobin (Hb) Interrupt treatment if Hb is less than 8 g/dL; treatment may be restarted once Hb returns above this value Hepatic transaminases Interrupt treatment if drug-induced liver injury is suspected, until this diagnosis is excluded. - Active and latent tuberculosis (TB) infection evaluation - If positive, treat for TB prior to RINVOQ/RINVOQ LQ use [see Warnings and Precautions (5.1)].
-
3 DOSAGE FORMS AND STRENGTHS
RINVOQ extended-release tablets:
- 15 mg upadacitinib: purple, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a15’ on one side.
- 30 mg upadacitinib: red, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a30’ on one side.
- 45 mg upadacitinib: yellow to mottled yellow, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a45’ on one side.
RINVOQ LQ oral solution:
- 1 mg/mL upadacitinib; clear, colorless to light yellow solution in bottle of 180 mL.
- 15 mg upadacitinib: purple, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a15’ on one side.
-
4 CONTRAINDICATIONS
RINVOQ/RINVOQ LQ is contraindicated in patients with known hypersensitivity to upadacitinib or any of its excipients [see Warnings and Precautions (5.6)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Serious and sometimes fatal infections have been reported in patients receiving RINVOQ. The most frequent serious infections reported with RINVOQ included pneumonia and cellulitis [see Adverse Reactions (6.1)]. Among opportunistic infections, tuberculosis, multidermatomal herpes zoster, oral/esophageal candidiasis, and cryptococcosis, were reported with RINVOQ. A higher rate of serious infections was observed with RINVOQ 30 mg compared to RINVOQ 15 mg.
Avoid use of RINVOQ/RINVOQ LQ in patients with an active, serious infection, including localized infections. Consider the risks and benefits of treatment prior to initiating RINVOQ/RINVOQ LQ in patients:
- with chronic or recurrent infection
- who have been exposed to tuberculosis
- with a history of a serious or an opportunistic infection
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
- with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with RINVOQ/RINVOQ LQ. Interrupt RINVOQ/RINVOQ LQ if a patient develops a serious or opportunistic infection.
A patient who develops a new infection during treatment with RINVOQ/RINVOQ LQ should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, the patient should be closely monitored, and RINVOQ/RINVOQ LQ should be interrupted if the patient is not responding to antimicrobial therapy. RINVOQ/RINVOQ LQ may be resumed once the infection is controlled.
Tuberculosis
Evaluate and test patients for latent and active tuberculosis (TB) infection prior to administration of RINVOQ/RINVOQ LQ. Patients with latent TB should be treated with standard antimycobacterial therapy before initiating RINVOQ/RINVOQ LQ. RINVOQ/RINVOQ LQ should not be given to patients with active TB. Consider anti-TB therapy prior to initiation of RINVOQ/RINVOQ LQ in patients with previously untreated latent TB or active TB in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent TB but who have risk factors for TB infection.
Consultation with a physician with expertise in the treatment of TB is recommended to aid in the decision about whether initiating anti-TB therapy is appropriate for an individual patient.
During RINVOQ/RINVOQ LQ use, monitor patients for the development of signs and symptoms of TB, including patients who tested negative for latent TB infection prior to initiating therapy.
Viral Reactivation
Viral reactivation, including cases of herpes virus reactivation (e.g., herpes zoster) and hepatitis B virus reactivation, were reported in clinical trials with RINVOQ [see Adverse Reactions (6.1)]. The risk of herpes zoster appears to be higher in patients treated with RINVOQ in Japan. If a patient develops herpes zoster, consider temporarily interrupting RINVOQ/RINVOQ LQ until the episode resolves.
Screening for viral hepatitis and monitoring for reactivation should be performed in accordance with clinical guidelines before starting and during therapy with RINVOQ/RINVOQ LQ. Patients who were positive for hepatitis C antibody and hepatitis C virus RNA, were excluded from clinical trials. Patients who were positive for hepatitis B surface antigen or hepatitis B virus DNA were excluded from clinical trials. However, cases of hepatitis B reactivation were still reported in patients enrolled in the Phase 3 trials of RINVOQ. If hepatitis B virus DNA is detected while receiving RINVOQ/RINVOQ LQ, a liver specialist should be consulted.
5.2 Mortality
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of all-cause mortality, including sudden cardiovascular death, was observed in patients treated with the JAK inhibitor compared with TNF blockers.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ/RINVOQ LQ.
5.3 Malignancy and Lymphoproliferative Disorders
Malignancies, including lymphomas, were observed in clinical trials of RINVOQ [see Adverse Reactions (6.1)].
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding NMSC) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. In this study, current or past smokers had an additional increased risk of overall malignancies.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ/RINVOQ LQ, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Non-Melanoma Skin Cancer
NMSCs have been reported in patients treated with RINVOQ. Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
Exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen.
5.4 Major Adverse Cardiovascular Events
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, a higher rate of major adverse cardiovascular events (MACE) defined as cardiovascular death, non-fatal myocardial infarction (MI), and non-fatal stroke was observed with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with RINVOQ/RINVOQ LQ, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur. Discontinue RINVOQ/RINVOQ LQ in patients that have experienced a myocardial infarction or stroke.
5.5 Thrombosis
Thromboses, including deep venous thrombosis (DVT), pulmonary embolism (PE), and arterial thrombosis, have occurred in patients treated for inflammatory conditions with JAK inhibitors, including RINVOQ. Many of these adverse events were serious and some resulted in death [see Adverse Reactions (6.1)].
In a large, randomized, postmarketing safety study of another JAK inhibitor in RA patients 50 years of age and older with at least one cardiovascular risk factor, higher rates of overall thrombosis, DVT, and PE were observed compared to those treated with TNF blockers.
If symptoms of thrombosis occur, patients should discontinue RINVOQ/RINVOQ LQ and be evaluated promptly and treated appropriately. Avoid RINVOQ/RINVOQ LQ in patients that may be at increased risk of thrombosis.
5.6 Hypersensitivity Reactions
Serious hypersensitivity reactions such as anaphylaxis and angioedema were reported in patients receiving RINVOQ in clinical trials. If a clinically significant hypersensitivity reaction occurs, discontinue RINVOQ/RINVOQ LQ and institute appropriate therapy [see Adverse Reactions (6.1)].
5.7 Gastrointestinal Perforations
Gastrointestinal perforations have been reported in clinical trials with RINVOQ [see Adverse Reactions (6.1)].
Monitor RINVOQ/RINVOQ LQ-treated patients who may be at risk for gastrointestinal perforation (e.g., patients with a history of diverticulitis and those taking concomitant medications including NSAIDs or corticosteroids). Evaluate promptly patients presenting with new onset abdominal pain for early identification of gastrointestinal perforation.
5.8 Laboratory Abnormalities
Neutropenia
Treatment with RINVOQ was associated with an increased incidence of neutropenia (ANC less than 1000 cells/mm3).
Evaluate neutrophil counts at baseline and thereafter according to routine patient management. Avoid RINVOQ/RINVOQ LQ initiation and interrupt RINVOQ/RINVOQ LQ treatment in patients with a low neutrophil count (i.e., ANC less than 1000 cells/mm3) [see Dosage and Administration (2.1, 2.14)].
Lymphopenia
ALC less than 500 cells/mm3 were reported in RINVOQ-treated patients in clinical trials.
Evaluate lymphocyte counts at baseline and thereafter according to routine patient management. Avoid RINVOQ/RINVOQ LQ initiation or interrupt RINVOQ/RINVOQ LQ treatment in patients with a low lymphocyte count (i.e., less than 500 cells/mm3) [see Dosage and Administration (2.1, 2.14)].
Anemia
Decreases in hemoglobin levels to less than 8 g/dL were reported in RINVOQ-treated patients in clinical trials.
Evaluate hemoglobin at baseline and thereafter according to routine patient management. Avoid RINVOQ/RINVOQ LQ initiation or interrupt RINVOQ/RINVOQ LQ treatment in patients with a low hemoglobin level (i.e., less than 8 g/dL) [see Dosage and Administration (2.1, 2.14)].
Lipids
Treatment with RINVOQ was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol [see Adverse Reactions (6.1)]. Elevations in LDL cholesterol decreased to pre-treatment levels in response to statin therapy. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Assess lipid parameters approximately 12 weeks after initiation of treatment, and thereafter according to the clinical guidelines for hyperlipidemia. Manage patients according to clinical guidelines for the management of hyperlipidemia.
Liver Enzyme Elevations
Treatment with RINVOQ was associated with increased incidence of liver enzyme elevations compared to treatment with placebo.
Evaluate liver enzymes at baseline and thereafter according to routine patient management. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury.
If increases in ALT or AST are observed during routine patient management and drug-induced liver injury is suspected, RINVOQ/RINVOQ LQ should be interrupted until this diagnosis is excluded.
5.9 Embryo-Fetal Toxicity
Based on findings in animal studies, RINVOQ/RINVOQ LQ may cause fetal harm when administered to a pregnant woman. Administration of upadacitinib to rats and rabbits during organogenesis caused increases in fetal malformations. Verify the pregnancy status of patients of reproductive potential prior to starting treatment. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception during treatment with RINVOQ/RINVOQ LQ and for 4 weeks following completion of therapy [see Use in Specific Populations (8.1, 8.3)].
5.10 Vaccinations
Avoid use of live vaccines during or immediately prior to RINVOQ/RINVOQ LQ therapy initiation. Prior to initiating RINVOQ/RINVOQ LQ treatment, it is recommended that patients be brought up to date with all immunizations, including prophylactic varicella zoster or herpes zoster vaccinations, in agreement with current immunization guidelines.
5.11 Medication Residue in Stool
Reports of medication residue in stool or ostomy output have occurred in patients taking RINVOQ. Most reports described anatomic (e.g., ileostomy, colostomy, intestinal resection) or functional gastrointestinal conditions with shortened gastrointestinal transit times. Instruct patients to contact their healthcare provider if medication residue is observed repeatedly. Monitor patients clinically and consider alternative treatment if there is an inadequate therapeutic response.
- with chronic or recurrent infection
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Mortality [see Warnings and Precautions (5.2)]
- Malignancy and Lymphoproliferative Disorders [see Warnings and Precautions (5.3)]
- Major Adverse Cardiovascular Events [see Warnings and Precautions (5.4)]
- Thrombosis [see Warnings and Precautions (5.5)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.6)]
- Gastrointestinal Perforations [see Warnings and Precautions (5.7)]
- Laboratory Abnormalities [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Patients with Rheumatoid Arthritis
A total of 3833 adult patients with rheumatoid arthritis were treated with RINVOQ 15 mg or upadacitinib 30 mg tablets once daily in the Phase 3 clinical trials of whom 2806 were exposed for at least one year.
Patients could advance or switch to RINVOQ 15 mg from placebo, or be rescued to RINVOQ from active comparator or placebo from as early as Week 12 depending on the trial design.
A total of 2630 patients received at least 1 dose of RINVOQ 15 mg, of whom 1860 were exposed for at least one year. In trials RA-I, RA-II, RA-III and RA-V, 1213 patients received at least 1 dose of RINVOQ 15 mg, of which 986 patients were exposed for at least one year, and 1203 patients received at least 1 dose of upadacitinib 30 mg, of which 946 were exposed for at least one year.
Table 4: Adverse Reactions Reported in ≥ 1% of Rheumatoid Arthritis Patients Treated with RINVOQ 15 mg in Placebo-controlled Trials Adverse Reaction Placebo RINVOQ
15 mgN = 1042
(%)N = 1035
(%)Upper respiratory tract infection (URTI)* 9.5 13.5 Nausea 2.2 3.5 Cough 1.0 2.2 Pyrexia 0 1.2 *URTI includes: acute sinusitis, laryngitis, nasopharyngitis, oropharyngeal pain,
pharyngitis, pharyngotonsillitis, rhinitis, sinusitis, tonsillitis, viral upper respiratory tract
infectionOther adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg group and at a higher rate than in the placebo group through Week 12 included pneumonia, herpes zoster, herpes simplex (includes oral herpes), and oral candidiasis.
Four integrated datasets are presented in the Specific Adverse Reaction section:
Placebo-controlled Trials: Trials RA-III, RA-IV, and RA-V were integrated to represent safety through 12/14 weeks for placebo (n=1042) and RINVOQ 15 mg (n=1035). Trials RA-III and RA-V were integrated to represent safety through 12 weeks for placebo (n=390), RINVOQ 15 mg (n=385), and upadacitinib 30 mg (n=384). Trial RA-IV did not include the 30 mg dose and, therefore, safety data for upadacitinib 30 mg can only be compared with placebo and RINVOQ 15 mg rates from pooling trials RA-III and RA-V.
MTX-controlled Trials: Trials RA-I and RA-II were integrated to represent safety through 12/14 weeks for MTX (n=530), RINVOQ 15 mg (n=534), and upadacitinib 30 mg (n=529).
12-Month Exposure Dataset: Trials RA-I, II, III, and V were integrated to represent the long-term safety of RINVOQ 15 mg (n=1213) and upadacitinib 30 mg (n=1203).
Exposure adjusted incidence rates were adjusted by trial for all the adverse events reported in this section.
Specific Adverse Reactions
Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, infections were reported in 218 patients (95.7 per 100 patient-years) treated with placebo and 284 patients (127.8 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, infections were reported in 99 patients (136.5 per 100 patient-years) treated with placebo, 118 patients (164.5 per 100 patient-years) treated with RINVOQ 15 mg, and 126 patients (180.3 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Infections were reported in 127 patients (119.5 per 100 patient-years) treated with MTX monotherapy, 104 patients (91.8 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 128 patients (115.1 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Infections were reported in 615 patients (83.8 per 100 patient-years) treated with RINVOQ 15 mg and 674 patients (99.7 per 100 patient-years) treated with upadacitinib 30 mg.
Serious Infections
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, serious infections were reported in 6 patients (2.3 per 100 patient-years) treated with placebo, and 12 patients (4.6 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, serious infections were reported in 1 patient (1.2 per 100 patient-years) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 7 patients (8.2 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Serious infections were reported in 2 patients (1.6 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 8 patients (6.4 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Serious infections were reported in 38 patients (3.5 per 100 patient-years) treated with RINVOQ 15 mg and 59 patients (5.6 per 100 patient-years) treated with upadacitinib 30 mg.
The most frequently reported serious infections were pneumonia and cellulitis.
Tuberculosis
Placebo-controlled Trials and MTX-controlled Trials: In the placebo-controlled period, there were no active cases of tuberculosis reported in the placebo, RINVOQ 15 mg, and upadacitinib 30 mg groups. In the MTX-controlled period, there were no active cases of tuberculosis reported in the MTX monotherapy, RINVOQ 15 mg monotherapy, and upadacitinib 30 mg monotherapy groups.
12-Month Exposure Dataset: Active tuberculosis was reported for 2 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg. Cases of extra-pulmonary tuberculosis were reported.
Opportunistic Infections (excluding tuberculosis)
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, opportunistic infections were reported in 3 patients (1.2 per 100 patient-years) treated with placebo, and 5 patients (1.9 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, opportunistic infections were reported in 1 patient (1.2 per 100 patient-years) treated with placebo, 2 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg, and 6 patients (7.1 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Opportunistic infections were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 0 patients treated with RINVOQ 15 mg monotherapy, and 4 patients (3.2 per 100 patient-years) treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Opportunistic infections were reported in 7 patients (0.6 per 100 patient-years) treated with RINVOQ 15 mg and 15 patients (1.4 per 100 patient-years) treated with upadacitinib 30 mg.
Malignancies
Placebo-controlled Trials: In RA-III, RA-IV, and RA-V, malignancies excluding NMSC were reported in 1 patient (0.4 per 100 patient-years) treated with placebo, and 1 patient (0.4 per 100 patient-years) treated with RINVOQ 15 mg. In RA-III and RA-V, malignancies excluding NMSC were reported in 0 patients treated with placebo, 1 patient (1.1 per 100 patient-years) treated with RINVOQ 15 mg, and 3 patients (3.5 per 100 patient-years) treated with upadacitinib 30 mg.
MTX-controlled Trials: Malignancies excluding NMSC were reported in 1 patient (0.8 per 100 patient-years) treated with MTX monotherapy, 3 patients (2.4 per 100 patient-years) treated with RINVOQ 15 mg monotherapy, and 0 patients treated with upadacitinib 30 mg monotherapy.
12-Month Exposure Dataset: Malignancies excluding NMSC were reported in 13 patients (1.2 per 100 patient-years) treated with RINVOQ 15 mg and 14 patients (1.3 per 100 patient-years) treated with upadacitinib 30 mg.
Gastrointestinal Perforations
Placebo-controlled Trials: There were no gastrointestinal perforations (based on medical review) reported in patients treated with placebo, RINVOQ 15 mg, and upadacitinib 30 mg.
MTX-controlled Trials: There were no cases of gastrointestinal perforations reported in the MTX and RINVOQ 15 mg group through 12/14 weeks. Two cases of gastrointestinal perforations were observed in the upadacitinib 30 mg group.
12-Month Exposure Dataset: Gastrointestinal perforations were reported in 1 patient treated with RINVOQ 15 mg and 4 patients treated with upadacitinib 30 mg.
Thrombosis
Placebo-controlled Trials: In RA-IV, venous thrombosis (pulmonary embolism or deep vein thrombosis) was observed in 1 patient treated with placebo and 1 patient treated with RINVOQ 15 mg. In RA-V, venous thrombosis was observed in 1 patient treated with RINVOQ 15 mg. There were no observed cases of venous thrombosis reported in RA-III. No cases of arterial thrombosis were observed through 12/14 weeks.
MTX-controlled Trials: In RA-II, venous thrombosis was observed in 0 patients treated with MTX monotherapy, 1 patient treated with RINVOQ 15 mg monotherapy and 0 patients treated with upadacitinib 30 mg monotherapy through Week 14. In RA-II, no cases of arterial thrombosis were observed through 12/14 weeks. In RA-I, venous thrombosis was observed in 1 patient treated with MTX, 0 patients treated with RINVOQ 15 mg and 1 patient treated with upadacitinib 30 mg through Week 24. In RA-I, arterial thrombosis was observed in 1 patient treated with upadacitinib 30 mg through Week 24.
12-Month Exposure Dataset: Venous thrombosis events were reported in 5 patients (0.5 per 100 patient-years) treated with RINVOQ 15 mg and 4 patients (0.4 per 100 patient-years) treated with upadacitinib 30 mg. Arterial thrombosis events were reported in 0 patients treated with RINVOQ 15 mg and 2 patients (0.2 per 100 patient-years) treated with upadacitinib 30 mg.
Laboratory Abnormalities
Hepatic Transaminase Elevations
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, alanine transaminase (ALT) and aspartate transaminase (AST) elevations ≥ 3 x upper limit of normal (ULN) in at least one measurement were observed in 2.1% and 1.5% of patients treated with RINVOQ 15 mg, and in 1.5% and 0.7% of patients treated with placebo, respectively. In RA-III and RA-V, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 1.0% of patients treated with RINVOQ 15 mg, 1.0% and 0% of patients treated with upadacitinib 30 mg and in 1.3% and 1.0% of patients treated with placebo, respectively.
In MTX-controlled trials, for up to 12/14 weeks, ALT and AST elevations ≥ 3 x ULN in at least one measurement were observed in 0.8% and 0.4% of patients treated with RINVOQ 15 mg, 1.7% and 1.3% of patients treated with upadacitinib 30 mg and in 1.9% and 0.9% of patients treated with MTX, respectively.
Lipid Elevations
Upadacitinib treatment was associated with dose-related increases in total cholesterol, triglycerides and LDL cholesterol. Upadacitinib was also associated with increases in HDL cholesterol. Elevations in LDL and HDL cholesterol peaked by Week 8 and remained stable thereafter. In controlled trials, for up to 12/14 weeks, changes from baseline in lipid parameters in patients treated with RINVOQ 15 mg and upadacitinib 30 mg, respectively, are summarized below:
- Mean LDL cholesterol increased by 14.81 mg/dL and 17.17 mg/dL.
- Mean HDL cholesterol increased by 8.16 mg/dL and 9.01 mg/dL.
- The mean LDL/HDL ratio remained stable.
- Mean triglycerides increased by 13.55 mg/dL and 14.44 mg/dL.
Creatine Phosphokinase Elevations
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related increases in creatine phosphokinase (CPK) values were observed. CPK elevations > 5 x ULN were reported in 1.0%, and 0.3% of patients over 12/14 weeks in the RINVOQ 15 mg and placebo groups, respectively. Most elevations >5 x ULN were transient and did not require treatment discontinuation. In RA-III and RA-V, CPK elevations > 5 x ULN were observed in 0.3% of patients treated with placebo, 1.6% of patients treated with RINVOQ 15 mg, and none in patients treated with upadacitinib 30 mg.
Neutropenia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in neutrophil counts, below 1000 cells/mm3 in at least one measurement occurred in 1.1% and <0.1% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in neutrophil counts below 1000 cells/mm3 in at least one measurement occurred in 0.3% of patients treated with placebo, 1.3% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg. In clinical trials, treatment was interrupted in response to ANC less than 1000 cells/mm3.
Lymphopenia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, dose-related decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.9% and 0.7% of patients in the RINVOQ 15 mg and placebo groups, respectively. In RA-III and RA-V, decreases in lymphocyte counts below 500 cells/mm3 in at least one measurement occurred in 0.5% of patients treated with placebo, 0.5% of patients treated with RINVOQ 15 mg, and 2.4% of patients treated with upadacitinib 30 mg.
Anemia
In placebo-controlled trials (RA-III, RA-IV, and RA-V) with background DMARDs, for up to 12/14 weeks, hemoglobin decreases below 8 g/dL in at least one measurement occurred in <0.1% of patients in both the RINVOQ 15 mg and placebo groups. In RA-III and RA-V, hemoglobin decreases below 8 g/dL in at least one measurement were observed in 0.3% of patients treated with placebo, and none in patients treated with RINVOQ 15 mg and upadacitinib 30 mg.
Adverse Reactions in Patients with Psoriatic Arthritis
A total of 1827 adult patients with psoriatic arthritis were treated with RINVOQ 15 mg or upadacitinib 30 mg tablets once daily in clinical trials, representing 1639.2 patient-years of exposure, of whom 722 were exposed to upadacitinib for at least one year. In the two Phase 3 trials, 907 patients received at least 1 dose of RINVOQ 15 mg, of whom 359 were exposed for at least one year.
Two placebo-controlled trials were integrated (640 patients on RINVOQ 15 mg once daily and 635 patients on placebo) to evaluate the safety of RINVOQ 15 mg in comparison to placebo for up to 24 weeks after treatment initiation.
Overall, the safety profile observed in patients with active psoriatic arthritis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis. During the 24-week placebo-controlled period, the frequencies of herpes zoster and herpes simplex were ≥1% (1.1% and 1.4%, respectively) with RINVOQ 15 mg and 0.8% and 1.3%, respectively with placebo. A higher incidence of acne and bronchitis was also observed in patients treated with RINVOQ 15 mg (1.3% and 3.9%, respectively) compared to placebo (0.3% and 2.7%, respectively).
Adverse Reactions in Patients with Atopic Dermatitis
Three Phase 3 (AD-1, AD-2, and AD-3) and one Phase 2b (AD-4) randomized, double-blind, placebo-controlled, multicenter trials evaluated the safety of RINVOQ in patients with moderate-to-severe atopic dermatitis. The majority of patients were White (68%) and male (57%). The mean age was 34 years (ranged from 12 to 75 years) and 13% of the patients were 12 to less than 18 years. In these 4 trials, 2612 patients were treated with RINVOQ 15 mg tablets or 30 mg tablets orally once daily, with or without concomitant topical corticosteroids (TCS).
In the Phase 3 clinical trials (AD-1, AD-2, and AD-3), a total of 1239 patients received RINVOQ 15 mg, of whom 791 were exposed for at least one year and 1246 patients received RINVOQ 30 mg, of whom 826 were exposed for at least one year.
Trials AD-1, AD-2, and AD-4 compared the safety of RINVOQ monotherapy to placebo through Week 16. Trial AD-3 compared the safety of RINVOQ + TCS to placebo + TCS through Week 16.
Weeks 0 to 16 (Trials AD-1 to AD-4)
In RINVOQ trials with and without TCS (Trials AD-1, 2, 3 and 4) through Week 16, the proportion of patients who discontinued treatment because of adverse reactions in the RINVOQ 15 mg, 30 mg and placebo groups were 2.3%, 2.9% and 3.8%, respectively. Table 5 summarizes the adverse reactions that occurred at a rate of at least 1% in the RINVOQ 15 mg or 30 mg groups during the first 16 weeks of treatment.
Table 5: Adverse Reactions Reported in ≥ 1% of Patients with Atopic Dermatitis Treated with RINVOQ 15 mg or 30 mg Adverse Reaction Placebo RINVOQ
15 mgRINVOQ
30 mgN = 902
(%)N = 899
(%)N = 906
(%)Upper respiratory tract
infection (URTI)*17 23 25 Acne** 2 10 16 Herpes simplex*** 2 4 8 Headache 4 6 6 Increased blood creatine
phosphokinase2 5 6 Cough 1 3 3 Hypersensitivity**** 2 2 3 Folliculitis 1 2 3 Nausea 1 3 3 Abdominal pain***** 1 3 2 Pyrexia 1 2 2 Increased Weight 1 2 2 Herpes zoster****** 1 2 2 Influenza <1 2 2 Fatigue 1 1 2 Neutropenia <1 1 2 Myalgia 1 1 2 Influenza like illness 1 1 2 * Includes: laryngitis, laryngitis viral, nasopharyngitis, oropharyngeal pain, pharyngeal abscess, pharyngitis, pharyngitis streptococcal, pharyngotonsillitis, respiratory tract infection, respiratory tract infection viral, rhinitis, rhinolaryngitis, sinusitis, tonsillitis, tonsillitis bacterial, upper respiratory tract infection, viral pharyngitis, viral upper respiratory tract infection
** Includes: acne and dermatitis acneiform
*** Includes: genital herpes, genital herpes simplex, herpes dermatitis, herpes ophthalmic, herpes simplex, nasal herpes, ophthalmic herpes simplex, herpes virus infection, oral herpes
**** Includes anaphylactic reaction, anaphylactic shock, angioedema, dermatitis exfoliative generalized, drug hypersensitivity, eyelid oedema, face oedema, hypersensitivity, periorbital swelling, pharyngeal swelling, swelling face, toxic skin eruption, type I hypersensitivity, urticaria
***** Includes abdominal pain and abdominal pain upper
****** Includes herpes zoster and varicellaOther adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg and/or 30 mg group and at a higher rate than in the placebo group through Week 16 included anemia, oral candidiasis, pneumonia, non-melanoma skin cancer, and the adverse event of retinal detachment.
The safety profile of RINVOQ through Week 52 was generally consistent with the safety profile observed at Week 16.
Overall, the safety profile observed in patients with AD treated with RINVOQ was similar to the safety profile in patients with RA. Other specific adverse reactions that were reported in patients with AD included eczema herpeticum/Kaposi’s varicelliform eruption.
Eczema Herpeticum/Kaposi’s Varicelliform Eruption
Placebo-controlled Period (16 weeks): Eczema herpeticum was reported in 4 patients (1.6 per 100 patient-years) treated with placebo, 6 patients (2.2 per 100 patient-years) treated with RINVOQ 15 mg and 7 patients (2.6 per 100 patient-years) treated with RINVOQ 30 mg.
12-Month Exposure (Weeks 0 to 52): Eczema herpeticum was reported in 18 patients (1.6 per 100 patient-years) treated with RINVOQ 15 mg and 17 patients (1.5 per 100 patient-years) treated with RINVOQ 30 mg.
Adverse Reactions in Patients with Ulcerative Colitis
RINVOQ was studied up to 8 weeks in patients with moderately to severely active ulcerative colitis in two randomized, double-blind, placebo-controlled induction studies (UC-1, UC-2) and a randomized, double-blind, placebo controlled, dose-finding study (UC-4; NCT02819635). Long term safety up to 52-weeks was evaluated in patients who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (UC-3) and a long-term extension study [see Clinical Studies (14.4)].
In the two induction studies (UC-1, UC-2) and a dose finding study (UC-4), 1097 patients were enrolled of whom 719 patients received RINVOQ 45 mg tablets once daily.
In the maintenance study (UC-3), 746 patients were enrolled of whom 250 patients received RINVOQ 15 mg tablets once daily and 251 patients received RINVOQ 30 mg tablets once daily.
Adverse reactions reported in ≥2% of patients in any treatment arm in the induction and maintenance studies are shown in Tables 6 and 7, respectively.
Table 6: Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 45 mg in Placebo-Controlled Induction Studies (UC-1, UC-2 and UC-4) Adverse Reaction Placebo RINVOQ
45 mg Once DailyN = 378
(%)N = 719
(%)Upper respiratory tract infection* 7 9 Acne* 1 6 Increased blood creatine phosphokinase 1 5 Neutropenia* <1 5 Rash* 1 4 Elevated liver enzymes** 2 3 Lymphopenia* 1 3 Folliculitis 1 2 Herpes simplex* <1 2 * Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury and cholestasis.Other adverse reactions reported in less than 2% of patients in the RINVOQ 45 mg group and at a higher rate than in the placebo group through Week 8 included herpes zoster and pneumonia.
Table 7: Adverse Reactions Reported in ≥2% of Patients with Ulcerative Colitis Treated with RINVOQ 15 mg or 30 mg in the Placebo-Controlled Maintenance Study (UC-3)1 Adverse Reaction Placebo RINVOQ
15 mg Once DailyRINVOQ
30 mg Once DailyN = 245
(%)N = 250
(%)N = 251
(%)Upper respiratory tract infection* 18 17 20 Increased blood creatine phosphokinase 2 6 8 Pyrexia 3 3 6 Neutropenia* 2 3 6 Elevated liver enzymes** 1 6 4 Rash* 4 5 5 Herpes zoster 0 5 6 Folliculitis 2 2 4 Hypercholesterolemia* 1 2 4 Influenza 1 3 3 Herpes simplex* 1 2 3 Lymphopenia* 2 3 2 Hyperlipidemia* 0 2 2 1 Patients who were responders to 8 weeks induction therapy with RINVOQ 45 mg once daily
* Composed of several similar terms
** Elevated liver enzymes composed of elevated ALT, AST, GGT, ALP, liver transaminases, hepatic enzymes, bilirubin, drug-induced liver injury, and cholestasis.The adverse reaction of non-melanoma skin cancer was reported in 1% of patients in the RINVOQ 30 mg group and none of the patients in the RINVOQ 15 mg or placebo group through Week 52.
The safety profile of RINVOQ in the long-term extension study was similar to the safety profile observed in the placebo-controlled induction and maintenance periods.
Overall, the safety profile observed in patients with ulcerative colitis treated with RINVOQ was generally similar to the safety profile in patients with RA and AD.
Specific Adverse Reactions
Serious Infections
Induction Studies: In UC-1, UC-2, and UC-4, serious infections were reported in 5 patients (8.4 per 100 patient-years) treated with placebo and 9 patients (8.4 per 100 patient-years) treated with RINVOQ 45 mg through 8 weeks.
Placebo-controlled Maintenance Study: In UC-3, serious infections were reported in 8 patients (5.9 events per 100 patient-years) treated with placebo, 9 patients (5.0 events per 100 patient-years) treated with RINVOQ 15 mg, and 8 patients (3.7 events per 100 patient-years) treated with RINVOQ 30 mg through 52 weeks.
Laboratory Abnormalities
Hepatic Transaminase Elevations
In studies UC-1, UC-2, and UC-4, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 1.5% of patients treated with RINVOQ 45 mg, and 0% of patients treated with placebo for 8 weeks. AST elevations to ≥ 3 x ULN occurred in 1.5% of patients treated with RINVOQ 45 mg, and 0.3% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN occurred in 0.4% of patients treated with RINVOQ 45 mg and 0% of patients treated with placebo.
In UC-3, elevations of ALT to ≥ 3 x ULN in at least one measurement were observed in 4.4% of patients treated with RINVOQ 30 mg, 2% of patients treated with RINVOQ 15 mg, and 1.2% of patients treated with placebo for 52 weeks. Elevations of AST to ≥ 3 x ULN in at least one measurement were observed in 2% of patients treated with RINVOQ 30 mg, 1.6% of patients treated with RINVOQ 15 mg and 0.4% of patients treated with placebo. Elevations of ALT to ≥ 5 x ULN were observed in 1.2% of patients treated with 30 mg, 0.4% of patients treated with 15 mg, and 0.4% of patients treated with placebo.
Overall, laboratory abnormalities observed in patients with ulcerative colitis treated with RINVOQ were similar to those described in patients with RA.
Adverse Reactions in Patients with Crohn’s Disease
RINVOQ was studied up to 12 weeks in patients with moderately to severely active CD in two randomized, double-blind, placebo-controlled induction studies (CD-1, CD-2). Long term safety up to 52 weeks was evaluated in patients who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (CD-3), with additional data provided from a long-term extension (LTE) period [see Clinical Studies (14.5)].
In the two induction studies (CD-1, CD-2), 1021 patients were enrolled, of whom 674 patients received RINVOQ 45 mg tablets once daily during the placebo-controlled period.
In the maintenance study (CD-3), 673 patients were enrolled, of whom 221 patients received RINVOQ 15 mg tablets once daily and 229 patients received RINVOQ 30 mg tablets once daily during the randomized, placebo-controlled period.
Overall, the safety profile observed in patients with Crohn’s disease treated with RINVOQ was consistent with the known safety profile for RINVOQ in other indications.
Adverse reactions reported in ≥2% of patients treated with RINVOQ and at a higher rate than placebo in the induction and maintenance studies are shown in Tables 8 and 9, respectively.
Table 8: Adverse Reactions Reported in ≥2% of Patients with Crohn’s Disease Treated with RINVOQ 45 mg in Placebo-Controlled Induction Studies (CD-1 and CD-2) Adverse Reaction Placebo RINVOQ
45 mg Once DailyN = 347
(%)N = 674
(%)Upper respiratory tract infection* 8 13 Anemia* 6 7 Acne* 2 6 Pyrexia 3 4 Increased blood creatine phosphokinase 1 3 Influenza 1 3 Herpes simplex* 1 3 Leukopenia* 1 2 Neutropenia* <1 2 Herpes zoster 0 2 * Composed of several similar terms Adverse reactions reported in less than 2% of patients in the RINVOQ 45 mg group and at a higher rate than in the placebo group through Week 12 included folliculitis, hypercholesterolemia, bronchitis, pneumonia, oral candidiasis, and hyperlipidemia.
Table 9: Adverse Reactions Reported in ≥2% of Patients with Crohn’s Disease Treated with RINVOQ 15 mg or 30 mg in the Placebo-Controlled Maintenance Study (CD-3)1 Adverse Reaction Placebo RINVOQ
15 mg Once DailyRINVOQ
30 mg Once DailyN = 223
(%)N = 221
(%)N = 229
(%)Upper respiratory tract infection* 11 14 12 Pyrexia 2 3 7 Herpes zoster* 2 3 5 Headache* 1 3 5 Acne* 3 2 5 Gastroenteritis* 2 3 3 Fatigue 2 3 3 Increased blood creatine phosphokinase 1 2 3 Elevated liver enzymes2 <1 2 3 Leukopenia* <1 1 2 Neutropenia* <1 1 2 Bronchitis* 0 1 2 Pneumonia* 1 4 1 Cough 2 3 1 1 Patients who were responders to 12 weeks induction therapy with RINVOQ 45 mg once daily.
2 Elevated liver enzymes includes alanine aminotransferase increased, aspartate aminotransferase increased, blood alkaline phosphatase increased, transaminases increased, blood bilirubin increased.
* Composed of several similar termsAdverse reactions reported in less than 2% of patients in the RINVOQ 15 mg or 30 mg group and at a higher rate than in the placebo group through Week 52 included hyperlipidemia, oral candidiasis, and hypercholesterolemia.
The safety profile of RINVOQ in the long-term extension study was similar to the safety profile observed in the placebo-controlled induction and maintenance periods.
Specific Adverse Reactions
Serious Infections
Induction Studies: In CD-1 and CD-2, serious infections were reported in 6 patients (8 per 100 patient-years) treated with placebo and 13 patients (9 per 100 patient-years) treated with RINVOQ 45 mg through 12 weeks of the placebo-controlled period.
Maintenance Study/LTE: In the long-term placebo-controlled period, serious infections were reported in 10 patients (7 per 100 patient-years) treated with placebo, 7 patients (4 per 100 patient-years) treated with RINVOQ 15 mg, and 13 patients (6 per 100 patient-years) treated with RINVOQ 30 mg.
Gastrointestinal Perforations
Induction Studies: During the induction studies in all patients treated with RINVOQ 45 mg (N=938), gastrointestinal perforation was reported in 4 patients (2 per 100 patient-years). In the placebo-controlled induction period, in CD-1 and CD-2, gastrointestinal perforation was reported in no patients treated with placebo (N=347) and 1 patient (1 per 100 patient-years) treated with RINVOQ 45 mg (N=674) through 12 weeks.
Maintenance Study/LTE: In the long-term placebo-controlled period, gastrointestinal perforation was reported in 1 patient (1 per 100 patient-years) treated with placebo, 1 patient (<1 per 100 patient-years) treated with RINVOQ 15 mg, and 1 patient (<1 per 100 patient-years) treated with RINVOQ 30 mg.
Patients who received placebo or RINVOQ 15 mg for maintenance therapy and lost response were treated with rescue RINVOQ 30 mg (N=336). Among these patients, gastrointestinal perforation was reported in 3 patients (1 per 100 patient-years) through long-term treatment.
Adverse Reactions in Patients with Ankylosing Spondylitis
A total of 596 patients with ankylosing spondylitis were treated with RINVOQ 15 mg tablets in the two clinical trials representing 577.3 patient-years of exposure, of whom 220 were exposed to RINVOQ 15 mg for at least one year.
Overall, the safety profile observed in patients with active ankylosing spondylitis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis and psoriatic arthritis. During the 14-week placebo-controlled period in Trial AS-I, the frequency of headache was 5.4% with RINVOQ 15 mg and 2.1% with placebo. During the 14-week placebo-controlled period in Trial AS-II, the frequency of headache was 3.3% with RINVOQ 15 mg and 1.4% with placebo.
Adverse Reactions in Patients with Non-radiographic Axial Spondyloarthritis
A total of 187 patients with non-radiographic axial spondyloarthritis were treated with RINVOQ 15 mg tablets in the clinical trial representing 116.6 patient-years of exposure, of whom 31 were exposed to RINVOQ 15 mg for at least one year.
Overall, the safety profile observed in patients with active non-radiographic axial spondyloarthritis treated with RINVOQ 15 mg was consistent with the safety profile observed in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis.
Adverse Reactions in Patients with Polyarticular Juvenile Idiopathic Arthritis
A total of 83 pediatric patients with juvenile idiopathic arthritis (JIA) with active polyarthritis were treated with RINVOQ/RINVOQ LQ in the clinical trial, representing 123.7 patient-years of exposure, of whom 48 were exposed to RINVOQ/RINVOQ LQ for at least one year.
Overall, the safety profile observed in pediatric patients with JIA with active polyarthritis treated with RINVOQ/RINVOQ LQ was consistent with the known safety profile of RINVOQ.
Adverse Reactions in Patients with Giant Cell Arteritis
In the Phase 3 study, 209 patients with giant cell arteritis received at least 1 dose of RINVOQ 15 mg, of whom 122 were exposed for at least one year during the 52-week placebo-controlled period. The safety profile observed in patients with giant cell arteritis was generally consistent with the known safety profile for RINVOQ.
Table 10: Adverse Reactions Reported in ≥ 5% of Patients with Giant Cell Arteritis Treated with RINVOQ 15 mg in the Placebo-controlled Study Adverse Reaction Placebo
RINVOQ 15 mg
N = 112
(%)N = 209
(%)Upper respiratory tract infection (URTI)a 20.5 21.5 Headache 11.6 16.3 Fatigue 5.4 9.1 Peripheral edemab 2.7 8.6 Coughc 3.6 7.2 Anemiad 2.7 6.7 Rashe 2.7 5.7 Herpes zosterf 2.7 5.3 Nausea 3.6 5.3 a Includes acute sinusitis, laryngitis, nasopharyngitis, oropharyngeal pain, pharyngitis, viral pharyngitis, pharyngotonsillitis, rhinitis, sinusitis, tonsillitis, viral upper respiratory tract infection, upper respiratory tract infection
b Includes edema and edema peripheral
c Includes cough and productive cough
d Includes anemia, iron deficiency anemia, blood iron decreased, hemoglobin decreased, mean cell volume increased
e Includes rash, rash erythematous, rash macular, rash maculo-papular, rash vesicular
f Includes herpes zoster, herpes zoster oticus, ophthalmic herpes zosterSpecific Adverse Reactions
Opportunistic Infections (excluding tuberculosis and herpes zoster)
In the 52-week placebo-controlled period, opportunistic infections were reported in 1 patient (1.1 per 100 patient-years) treated with placebo and 4 patients (2.3 per 100 patient-years) treated with RINVOQ 15 mg.
Thrombosis
In the 52-week placebo-controlled period, venous thromboembolic events (pulmonary embolism or deep vein thrombosis) were observed in 4 patients (4.3 per 100 patient-years) treated with placebo and 7 patients (3.9 per 100 patient-years) treated with RINVOQ 15 mg. Events of arterial thrombosis were observed in 2 patients (1.1 per 100 patient-years) treated with RINVOQ 15 mg and 0 patients treated with placebo.
- Serious Infections [see Warnings and Precautions (5.1)]
-
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 Inhibitors
Upadacitinib exposure is increased when it is co-administered with a strong CYP3A4 inhibitor (such as ketoconazole, clarithromycin, and grapefruit), which may increase the risk of adverse reactions [see Clinical Pharmacology (12.3)]. Monitor patients with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondylarthritis, pJIA, or giant cell arteritis closely for adverse reactions when co-administering RINVOQ/RINVOQ LQ with strong CYP3A4 inhibitors. Food or drink containing grapefruit should be avoided during treatment with RINVOQ/RINVOQ LQ.
For patients with atopic dermatitis, coadministration of RINVOQ 30 mg once daily with strong CYP3A4 inhibitors is not recommended.
For patients with ulcerative colitis or Crohn’s disease taking strong CYP3A4 inhibitors, reduce the RINVOQ induction dosage to 30 mg once daily. The recommended maintenance dosage is 15 mg once daily [see Dosage and Administration (2.13)].
7.2 Strong CYP3A4 Inducers
Upadacitinib exposure is decreased when it is co-administered with strong CYP3A4 inducers (such as rifampin), which may lead to reduced therapeutic effect [see Clinical Pharmacology (12.3)]. Coadministration of RINVOQ/RINVOQ LQ with strong CYP3A4 inducers is not recommended.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Surveillance Program
There is a pregnancy surveillance program for RINVOQ/RINVOQ LQ that monitors pregnancy outcomes in women exposed to RINVOQ/RINVOQ LQ. If RINVOQ/RINVOQ LQ exposure occurs during pregnancy, healthcare providers or patients should report the pregnancy by calling 1-800-633-9110.
Risk Summary
Available data from the pharmacovigilance safety database and postmarketing case reports on use of RINVOQ in pregnant women are not sufficient to evaluate a drug-associated risk for major birth defects or miscarriage. Based on animal studies, RINVOQ/RINVOQ LQ has the potential to adversely affect a developing fetus. Advise patients of reproductive potential and pregnant patients of the potential risk to the fetus.
In animal embryo-fetal development studies, oral upadacitinib administration to pregnant rats and rabbits at exposures equal to or greater than approximately 1.6 and 15 times the 15 mg tablet dose, 0.8 and 7.6 times the 30 mg tablet dose, and 0.6 and 5.6 times the maximum recommended human dose (MRHD) of 45 mg (on an AUC basis) resulted in dose-related increases in skeletal malformations (rats only), an increased incidence of cardiovascular malformations (rabbits only), increased post-implantation loss (rabbits only), and decreased fetal body weights in both rats and rabbits. No developmental toxicity was observed in pregnant rats and rabbits treated with oral upadacitinib during organogenesis at exposures approximately 0.29 and 2.2 times the 15 mg dose, 0.15 times and 1.1 times the 30 mg dose, and at 0.11 and 0.82 times the MRHD (on an AUC basis). In a pre- and post-natal development study in pregnant female rats, oral upadacitinib administration at exposures approximately 3 times the 15 mg dose, 1.4 times the 30 mg dose, and the same as the MRHD (on an AUC basis) resulted in no maternal or developmental toxicity (see Data).
The background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages are 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that increased disease activity is associated with the risk of developing adverse pregnancy outcomes in women with rheumatoid arthritis or inflammatory bowel disease. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Data
Animal Data
In an oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 5, 25, and 75 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that consisted of misshapen humerus and bent scapula) at exposures equal to or greater than approximately 1.7 times the 15 mg tablet dose, 0.9 times the 30 mg tablet dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 5 mg/kg/day and higher). Additional skeletal malformations (bent forelimbs/hindlimbs and rib/vertebral defects) and decreased fetal body weights were observed in the absence of maternal toxicity at an exposure approximately 84 times the 15 mg dose, 43 times the 30 mg dose, and 31 times the MRHD (on an AUC basis at a maternal oral dose of 75 mg/kg/day).
In a second oral embryo-fetal development study, pregnant rats received upadacitinib at doses of 1.5 and 4 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Upadacitinib was teratogenic (skeletal malformations that included bent humerus and scapula) at exposures approximately 1.6 times the 15 mg dose, 0.8 times the 30 mg dose, and 0.6 times the MRHD (on an AUC basis at maternal oral doses of 4 mg/kg/day). No developmental toxicity was observed in rats at an exposure approximately 0.29 times the 15 mg tablet dose, 0.15 times the 30 mg tablet dose, and 0.11 times the MRHD (on an AUC basis at a maternal oral dose of 1.5 mg/kg/day).
In an oral embryo-fetal developmental study, pregnant rabbits received upadacitinib at doses of 2.5, 10, and 25 mg/kg/day during the period of organogenesis from gestation day 7 to 19. Embryolethality, decreased fetal body weights, and cardiovascular malformations were observed in the presence of maternal toxicity at an exposure approximately 15 times the 15 mg tablet dose, 7.6 times the 30 mg tablet dose, and 5.6 times the MRHD (on an AUC basis at a maternal oral dose of 25 mg/kg/day). Embryolethality consisted of increased post-implantation loss that was due to elevated incidences of both total and early resorptions. No developmental toxicity was observed in rabbits at an exposure approximately 2.2 times the 15 mg tablet dose, 1.1 times the 30 mg tablet dose, and 0.82 times the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
In an oral pre- and post-natal development study, pregnant female rats received upadacitinib at doses of 2.5, 5, and 10 mg/kg/day from gestation day 6 through lactation day 20. No maternal or developmental toxicity was observed in either mothers or offspring, respectively, at an exposure approximately 3 times the 15 mg tablet dose, 1.4 times the 30 mg tablet dose, and at approximately the same exposure as the MRHD (on an AUC basis at a maternal oral dose of 10 mg/kg/day).
8.2 Lactation
Risk Summary
There are no data on the presence of upadacitinib in human milk, the effects on the breastfed infant, or the effects on milk production. Available pharmacodynamic/toxicological data in animals have shown excretion of upadacitinib in milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions in the breastfed infant, advise patients that breastfeeding is not recommended during treatment with RINVOQ/RINVOQ LQ, and for 6 days (approximately 10 half-lives) after the last dose.
Data
A single oral dose of 10 mg/kg radiolabeled upadacitinib was administered to lactating female Sprague-Dawley rats on post-partum days 7-8. Drug exposure was approximately 30-fold greater in milk than in maternal plasma based on AUC0-t values. Approximately 97% of drug-related material in milk was parent drug.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to starting treatment with RINVOQ/RINVOQ LQ [see Use in Specific Populations (8.1)].
Contraception
Females
Based on animal studies, upadacitinib may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use effective contraception during treatment with RINVOQ/RINVOQ LQ and for 4 weeks after the final dose.
8.4 Pediatric Use
Ankylosing Spondylitis, Non-radiographic Axial Spondyloarthritis, Ulcerative Colitis, and Crohn’s Disease
The safety and effectiveness of RINVOQ/RINVOQ LQ in pediatric patients with ankylosing spondylitis, non-radiographic axial spondyloarthritis, ulcerative colitis, or Crohn’s disease have not been established.
Polyarticular Juvenile Idiopathic Arthritis and Psoriatic Arthritis
The safety and effectiveness of RINVOQ/RINVOQ LQ in pediatric patients 2 to less than 18 years of age with pJIA and psoriatic arthritis have been established.
The use of RINVOQ/RINVOQ LQ in these age groups is supported by evidence from well-controlled studies of RINVOQ in adults with rheumatoid arthritis and psoriatic arthritis, pharmacokinetic data from adult patients with rheumatoid arthritis and psoriatic arthritis and 51 pediatric patients with JIA with active polyarthritis, and safety data from 83 pediatric patients 2 to < 18 years of age with JIA with active polyarthritis. Upadacitinib plasma exposures in pediatric patients with pJIA and psoriatic arthritis at the recommended dosage are predicted to be comparable to those observed in adults with rheumatoid arthritis and psoriatic arthritis based on population pharmacokinetic modeling and simulation [see Dosage and Administration (2.4, 2.10), Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14.8)].
The safety and effectiveness of RINVOQ/RINVOQ LQ in pediatric patients less than 2 years of age with pJIA or psoriatic arthritis have not been established.
Atopic Dermatitis
The safety and effectiveness of RINVOQ in pediatric patients 12 years of age and older weighing at least 40 kg with atopic dermatitis have been established. A total of 344 pediatric patients aged 12 to 17 years with moderate to severe atopic dermatitis were randomized across three trials (AD-1, AD-2 and AD-3) to receive either RINVOQ 15 mg (N=114) or 30 mg (N=114) or matching placebo (N=116) in monotherapy or combination with topical corticosteroids. Efficacy was consistent between the pediatric patients and adults [see Clinical Studies (14.3)]. The adverse reaction profile in the pediatric patients was similar to the adults [see Adverse Reactions (6.1)].
The safety and effectiveness of RINVOQ in pediatric patients less than 12 years of age with atopic dermatitis have not been established.
The safety and effectiveness of RINVOQ LQ in pediatric patients with atopic dermatitis have not been established.
8.5 Geriatric Use
Rheumatoid Arthritis and Psoriatic Arthritis
Of the 4381 patients treated in the five clinical trials, a total of 906 rheumatoid arthritis patients were 65 years of age or older, including 146 patients 75 years and older. Of the 1827 patients treated in the two psoriatic arthritis Phase 3 clinical trials, a total of 274 patients were 65 years of age or older, including 34 patients 75 years and older. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of overall adverse events, including serious infections, in patients 65 years of age and older.
Atopic Dermatitis
Of the 2583 patients treated in the three Phase 3 clinical trials, a total of 120 patients with atopic dermatitis were 65 years of age or older, including 6 patients 75 years of age. No differences in effectiveness were observed between these patients and younger patients; however, there was a higher rate of serious infections and malignancies in those patients 65 years of age or older in the 30 mg dosing group in the long-term trials.
Ulcerative Colitis
Of the 1097 patients treated in the controlled clinical trials, a total of 95 patients with ulcerative colitis were 65 years and older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with ulcerative colitis to determine whether they respond differently from younger adult patients.
Crohn’s Disease
Of the 1021 patients who were treated in the controlled induction clinical trials, a total of 39 patients with Crohn’s disease were 65 years of age or older, and no patients were 75 years of age or older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with Crohn’s disease to determine whether they respond differently from younger adult patients.
Ankylosing Spondylitis
Of the 607 patients treated in the controlled clinical trials, a total of 32 patients with ankylosing spondylitis were 65 years and older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with ankylosing spondylitis to determine whether they respond differently from younger adult patients.
Non-radiographic Axial Spondyloarthritis
Of the 313 patients treated in a phase 3 clinical trial, a total of 9 patients with non-radiographic axial spondyloarthritis were 65 years and older. Clinical studies of RINVOQ did not include sufficient numbers of patients 65 years of age and older with non-radiographic axial spondyloarthritis to determine whether they respond differently from younger adult patients.
8.6 Renal Impairment
For patients with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, pJIA, or giant cell arteritis no dosage adjustment is needed in patients with mild (eGFR 60 to < 90 mL/min/1.73 m2), moderate (eGFR 30 to < 60 mL/min/1.73 m2), or severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2).
For patients with atopic dermatitis, the maximum recommended dosage of RINVOQ is 15 mg once daily for patients with severe renal impairment. No dosage adjustment is needed in patients with mild or moderate renal impairment.
For patients with ulcerative colitis or Crohn’s disease, the recommended dosage of RINVOQ for severe renal impairment is 30 mg once daily for induction and 15 mg once daily for maintenance. No dosage adjustment is needed in patients with mild or moderate renal impairment [see Dosage and Administration (2.12)].
RINVOQ/RINVOQ LQ has not been studied in patients with end stage renal disease (eGFR <15 mL/min/1.73m2). Use in patients with atopic dermatitis, ulcerative colitis, or Crohn’s disease with end stage renal disease is not recommended [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The use of RINVOQ/RINVOQ LQ has not been studied in patients with severe hepatic impairment (Child Pugh C), and is therefore not recommended [see Dosage and Administration (2.12) and Clinical Pharmacology (12.3)].
For patients with rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, ankylosing spondylitis, non-radiographic axial spondyloarthritis, pJIA, or giant cell arteritis, no dosage adjustment is needed in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment.
For patients with ulcerative colitis or Crohn’s disease, the recommended dosage of RINVOQ for mild to moderate hepatic impairment is 30 mg once daily for induction and 15 mg once daily for maintenance [see Dosage and Administration (2.12)].
-
11 DESCRIPTION
RINVOQ and RINVOQ LQ are formulated with upadacitinib, a JAK inhibitor.
Upadacitinib has the following chemical name: (3S,4R)-3-Ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide hydrate (2:1).
The strength of upadacitinib is based on anhydrous upadacitinib. The solubility of upadacitinib in water is 38 to less than 0.2 mg/mL across a pH range of 2 to 9 at 37 oC.
Upadacitinib has a molecular weight of 389.38 g/mol and a molecular formula of C17H19F3N6O • ½ H2O. The chemical structure of upadacitinib is:
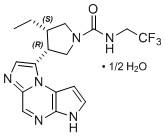
RINVOQ 15 mg extended-release tablets for oral administration are purple, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a15’ on one side. Each tablet contains the following inactive ingredients: colloidal silicon dioxide, ferrosoferric oxide, hypromellose, iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyvinyl alcohol, polyethylene glycol, talc, tartaric acid and titanium dioxide.
RINVOQ 30 mg extended-release tablets for oral administration are red, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a30’ on one side. Each tablet contains the following inactive ingredients: colloidal silicon dioxide, hypromellose, iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyvinyl alcohol, polyethylene glycol, talc, tartaric acid and titanium dioxide.
RINVOQ 45 mg extended-release tablets for oral administration are yellow to mottled yellow, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a45’ on one side. Each tablet contains the following inactive ingredients: colloidal silicon dioxide, hypromellose, iron oxide yellow, iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyvinyl alcohol, polyethylene glycol, talc, tartaric acid and titanium dioxide.
RINVOQ LQ oral solution for oral administration is a 1 mg/mL clear, colorless to light yellow solution. Each 1 mL RINVOQ LQ contains 1 mg of upadacitinib as free base (equivalent to 1.02 mg upadacitinib hemihydrate) and the following inactive ingredients: citric acid anhydrous, purified water, sodium benzoate, sodium citrate dihydrate, and sucralose.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Upadacitinib is a Janus kinase (JAK) inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor-receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Within the signaling pathway, JAKs phosphorylate and activate signal transducers and activators of transcription (STATs) which modulate intracellular activity including gene expression. Upadacitinib modulates the signaling pathway at the point of JAKs, preventing the phosphorylation and activation of STATs.
JAK enzymes transmit cytokine signaling through their pairing (e.g., JAK1/JAK2, JAK1/JAK3, JAK1/TYK2, JAK2/JAK2, JAK2/TYK2). In a cell-free isolated enzyme assay, upadacitinib had greater inhibitory potency at JAK1 and JAK2 relative to JAK3 and TYK2. In human leukocyte cellular assays, upadacitinib inhibited cytokine-induced STAT phosphorylation mediated by JAK1 and JAK1/JAK3 more potently than JAK2/JAK2 mediated STAT phosphorylation. The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness is not currently known.
12.2 Pharmacodynamics
Inhibition of IL-6 Induced STAT3 and IL-7 Induced STAT5 Phosphorylation
In healthy volunteers, the administration of upadacitinib (immediate release formulation) resulted in a dose- and concentration-dependent inhibition of IL-6 (JAK1/JAK2)-induced STAT3 and IL-7 (JAK1/JAK3)-induced STAT5 phosphorylation in whole blood. The maximal inhibition was observed 1 hour after dosing which returned to near baseline by the end of dosing interval.
Lymphocytes
In patients with rheumatoid arthritis, treatment with upadacitinib was associated with a small, transient increase in mean ALC from baseline up to Week 36 which gradually returned to, at or near baseline levels with continued treatment.
Immunoglobulins
In patients with rheumatoid arthritis, small decreases from baseline in mean IgG and IgM levels were observed with upadacitinib treatment in the controlled period; however, the mean values at baseline and at all visits were within the normal reference range.
Cardiac Electrophysiology
At 2.5 times the mean exposure of the maximum therapeutic dose, 45 mg once daily dose, there was no clinically relevant effect on the QTc interval.
12.3 Pharmacokinetics
Upadacitinib plasma exposures are proportional to dose over the therapeutic dose range. After a single dose administration of RINVOQ 15 mg, 30 mg, and 45 mg tablets under fasting condition in healthy subjects, mean Cmax was 31.6 ng/mL, 71.8 ng/mL, and 90.7 ng/mL, respectively, and mean AUCinf was 265 ng·h/mL, 543 ng·h/mL, and 752 ng·h/mL, respectively. Steady-state plasma concentrations are achieved within 4 days with minimal accumulation after once daily administration. Following the administration of the recommended pediatric dosage (Table 1, Table 2) in pJIA and psoriatic arthritis patients, the mean steady-state Cmax is predicted to be 47.6 ng/mL and the mean steady-state AUC0-24 is predicted to be 342 ng·h/mL. Upadacitinib pharmacokinetics are comparable between rheumatoid arthritis, psoriatic arthritis, atopic dermatitis, ulcerative colitis, Crohn’s disease, ankylosing spondylitis, non-radiographic axial spondyloarthritis, and giant cell arteritis patients.
RINVOQ tablets and RINVOQ LQ are not bioequivalent; therefore, the 2 dosage forms are not interchangeable on a milligram-per-milligram basis [see Dosage and Administration (2.2)].
Absorption
Following oral administration of RINVOQ extended-release tablets, upadacitinib is absorbed with a median Tmax of 2 to 4 hours. Following oral administration of 6 mg RINVOQ LQ, upadacitinib is absorbed with a median Tmax of 1 hour.
Coadministration of RINVOQ tablets with a high-fat/high-calorie meal had no clinically relevant effect on upadacitinib exposures (increased AUCinf by 29% and Cmax by 39% to 60%). Coadministration of RINVOQ LQ with food is not expected to have a clinically relevant effect on upadacitinib exposure [see Dosage and Administration (2.2)].
Distribution
Upadacitinib is 52% bound to plasma proteins. Upadacitinib partitions similarly between plasma and blood cellular components with a blood to plasma ratio of 1.0.
Elimination
Metabolism
Upadacitinib metabolism is mediated by mainly CYP3A4 with a potential minor contribution from CYP2D6. The pharmacologic activity of upadacitinib is attributed to the parent molecule. In a human radiolabeled study, unchanged upadacitinib accounted for 79% of the total radioactivity in plasma while the main metabolite detected (product of monooxidation followed by glucuronidation) accounted for 13% of the total plasma radioactivity. No active metabolites have been identified for upadacitinib.
Excretion
Following single dose administration of [14C]-upadacitinib immediate-release solution, upadacitinib was eliminated predominantly as the unchanged parent drug in urine (24%) and feces (38%). Approximately 34% of upadacitinib dose was excreted as metabolites. Upadacitinib mean terminal elimination half-life ranged from 8 to 14 hours.
Specific Populations
Body Weight, Gender, and Race
Body weight, gender, race, and ethnicity did not have a clinically meaningful effect on upadacitinib exposure in adult patient populations.
Pediatric Patients
In pediatric patients with JIA with active polyarthritis, upadacitinib clearance increased with increasing body weight. Age (over the range of 2 to < 18 years old) had no additional effect on upadacitinib pharmacokinetics after accounting for the effect of body weight. Upadacitinib plasma exposures in pediatric patients with pJIA and psoriatic arthritis following the recommended pediatric dosage are predicted to be comparable to those observed in adult patients with rheumatoid arthritis and psoriatic arthritis, respectively.
No meaningful difference in the systemic exposure of upadacitinib was observed in pediatric patients with atopic dermatitis 12 years of age and older weighing at least 40 kg compared to adults.
Geriatric Patients
No clinically meaningful differences in the pharmacokinetics of upadacitinib were observed in geriatric patients (≥ 65 years of age) compared to younger adult patients.
Patients with Renal Impairment
Upadacitinib mean AUCinf after a single dose administration of 15 mg upadacitinib tablets was 18%, 33%, and 44% higher in patients with mild (eGFR 60 to < 90 mL/min/1.73 m2), moderate (eGFR 30 to < 60 mL/min/1.73 m2), and severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2), respectively, compared to subjects with normal renal function (eGFR ≥ 90 mL/min/1.73 m2). Upadacitinib mean Cmax was similar among subjects with normal and impaired renal function. In patients receiving RINVOQ/RINVOQ LQ, mild and moderate renal impairment is not expected to have a clinically relevant effect on upadacitinib exposure [see Dosage and Administration (2.12) and Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
Upadacitinib mean AUCinf after a single dose administration of 15 mg upadacitinib tablets was 28% and 24% higher in patients with mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment, respectively, compared to subjects with normal liver function. Upadacitinib mean Cmax was unchanged in patients with mild hepatic impairment and 43% higher in patients with moderate hepatic impairment compared to subjects with normal liver function. Upadacitinib was not studied in patients with severe hepatic impairment (Child-Pugh C) [see Dosage and Administration (2.12) and Use in Specific Populations (8.7)].
Drug Interaction Studies
Potential for Other Drugs to Influence the Pharmacokinetics of Upadacitinib
Upadacitinib is metabolized in vitro by CYP3A4 with a minor contribution from CYP2D6. The effect of co-administered drugs on upadacitinib plasma exposures is provided in Table 11 [see Drug Interactions (7)].
Table 11: Change in Pharmacokinetics of Upadacitinib in the Presence of Co-administered Drugs Co-
administered
DrugRegimen
of Co-
administered
DrugRatio
(90% CI)aCmax AUC Methotrexate 10 to 25 mg/week 0.97
(0.86-1.09)0.99
(0.93- 1.06)Strong CYP3A4 inhibitor:
Ketoconazole400 mg once
daily x 6 days1.70
(1.55-1.89)1.75
(1.62-1.88)Strong CYP3A4 inducer:
Rifampin600 mg once
daily x 9 days0.49
(0.44-0.55)0.39
(0.37-0.42)OATP1B inhibitor:
Rifampin600 mg single dose 1.14
(1.02-1.28)1.07
(1.01-1.14)CI: Confidence interval
a Ratios for Cmax and AUC compare co-administration of the medication with upadacitinib vs.
administration of upadacitinib alone.pH modifying medications (e.g., antacids or proton pump inhibitors) are not expected to affect upadacitinib plasma exposures based on in vitro assessments and population pharmacokinetic analyses. CYP2D6 metabolic phenotype had no effect on upadacitinib pharmacokinetics (based on population pharmacokinetic analyses), indicating that inhibitors of CYP2D6 have no clinically relevant effect on upadacitinib exposures.
Potential for Upadacitinib to Influence the Pharmacokinetics of Other Drugs
In vitro studies indicate that upadacitinib does not inhibit the activity of cytochrome P450 (CYP) enzymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) at clinically relevant concentrations. In vitro studies indicate that upadacitinib induces CYP3A4 but does not induce CYP2B6 or CYP1A2 at clinically relevant concentrations. In vitro studies indicate that upadacitinib does not inhibit the transporters P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, and MATE2K at clinically relevant concentrations.
Clinical studies indicate that upadacitinib has no clinically relevant effects on the pharmacokinetics of co-administered drugs. Following upadacitinib 30 mg and 45 mg tablets once daily, the effects on each CYP enzymes (CYP1A2, CYP3A, CYP2C9, and CYP2C19) were similar between two doses except for the effect on CYP2D6. Following upadacitinib 30 mg and 45 mg tablets once daily, a weak induction of CYP3A4 was observed. A weak inhibition of CYP2D6 was observed at upadacitinib 45 mg but not at 30 mg. Summary of results from clinical studies which evaluated the effect of upadacitinib on other drugs is provided in Table 12.
Table 12: Change in Pharmacokinetics of Co-administered Drugs or In Vivo Markers of CYP Activity in the Presence of Upadacitinib
Co-administered
Drug or CYP
Activity MarkerMultiple-Dose
Regimen of
UpadacitinibRatio
(90% CI)aCmax AUC Methotrexate 6 mg to 24 mg BIDb 1.03
(0.86-1.23)1.14
(0.91-1.43)Sensitive CYP1A2 Substrate:
Caffeine45 mg QDc 1.05
(0.97-1.14)1.04
(0.95-1.13)Sensitive CYP3A Substrate:
Midazolam30 mg QDc 0.74
(0.68-0.80)0.74
(0.68-0.80)Sensitive CYP3A Substrate:
Midazolam45 mg QDc 0.75
(0.69 -0.83)0.76
(0.69 -0.83)Sensitive CYP2D6 Substrate:
Dextromethorphan30 mg QDc 1.09
(0.98-1.21)1.07
(0.95-1.22)Sensitive CYP2D6 Substrate:
Dextromethorphan45 mg QDc 1.30
(1.13-1.50)1.35
(1.18-1.54)Sensitive CYP2C9 Substrate:
S-Warfarin45 mg QDc 1.18
(1.05-1.33)1.12
(1.05-1.20)Sensitive CYP2C19 Marker:
5-OH Omeprazole to
Omeprazole metabolic ratio45 mg QDc -- 0.96
(0.90-1.02)CYP2B6 Substrate:
Bupropion30 mg QDc 0.87
(0.79-0.96)0.92
(0.87-0.98)Rosuvastatin 30 mg QDc 0.77
(0.63-0.94)0.67
(0.56-0.82)Atorvastatin 30 mg QDc 0.88
(0.79-0.97)0.77
(0.70-0.85)Ethinylestradiol 30 mg QDc 0.96
(0.89-1.02)1.11
(1.04-1.19)Levonorgestrel 30 mg QDc 0.96
(0.87-1.06)0.96
(0.85-1.07)CYP: cytochrome P450; CI: Confidence interval; BID: twice daily; QD: once daily
a Ratios for Cmax and AUC compare co-administration of the medication with upadacitinib vs. administration of medication alone.
b Immediate-release formulation
c Extended-release tablet formulation -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of upadacitinib was evaluated in Sprague-Dawley rats and Tg.rasH2 mice. No evidence of tumorigenicity was observed in male or female rats that received upadacitinib for up to 101 weeks at oral doses up to 15 or 20 mg/kg/day, respectively (approximately 4 and 10 times the 15 mg tablet dose, 2 and 5 times the 30 mg tablet dose, and 1.6 and 4 times the maximum recommended human dose (MRHD) of 45 mg on an AUC basis, respectively). No evidence of tumorigenicity was observed in male or female Tg.rasH2 mice that received upadacitinib for 26 weeks at oral doses up to 20 mg/kg/day.
Mutagenesis
Upadacitinib tested negative in the following genotoxicity assays: the in vitro bacterial mutagenicity assay (Ames assay), in vitro chromosome aberration assay in human peripheral blood lymphocytes, and in vivo rat bone marrow micronucleus assay.
Impairment of Fertility
Upadacitinib had no effect on fertility in male or female rats at oral doses up to 50 mg/kg/day in males and 75 mg/kg/day in females (approximately 42 and 84 times the 15 mg dose, 22 and 43 times the 30 mg dose, and 16 and 31 times the MRHD, respectively, on an AUC basis). However, maintenance of pregnancy was adversely affected at oral doses of 25 mg/kg/day and 75 mg/kg/day based upon dose-related findings of increased post-implantation losses (increased resorptions) and decreased numbers of mean viable embryos per litter (approximately 22 and 84 times the 15 mg tablet dose, 11 and 43 times the 30 mg tablet dose, and 8 and 31 times the MRHD on an AUC basis, respectively). The number of viable embryos was unaffected in female rats that received upadacitinib at an oral dose of 5 mg/kg/day and were mated to males that received the same dose (approximately 2 times the 15 mg dose, 0.9 times the 30 mg dose, and at 0.6 times the MRHD on an AUC basis).
-
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis
The efficacy and safety of RINVOQ 15 mg once daily were assessed in five Phase 3 randomized, double-blind, multicenter trials in patients with moderately to severely active rheumatoid arthritis and fulfilling the ACR/EULAR 2010 classification criteria. Patients 18 years of age and older were eligible to participate. The presence of at least 6 tender and 6 swollen joints and evidence of systemic inflammation based on elevation of hsCRP was required at baseline. Although other doses have been studied, the recommended dosage of RINVOQ is 15 mg once daily.
Trial RA-I (NCT02706873) was a 24-week monotherapy trial in 947 patients with moderately to severely active rheumatoid arthritis who were naïve to methotrexate (MTX). Patients received RINVOQ 15 mg or upadacitinib 30 mg orally once daily or MTX as monotherapy. At Week 26, non-responding patients on upadacitinib could be rescued with the addition of MTX, while patients on MTX could be rescued with the addition of blinded RINVOQ 15 mg or upadacitinib 30 mg once daily. The primary endpoint was the proportion of patients who achieved an ACR50 response at Week 12. Key secondary endpoints included disease activity score (DAS28-CRP) ≤3.2 at Week 12, DAS28-CRP <2.6 at Week 24, change from baseline in HAQ-DI at Week 12, and change from baseline in van der Heijde-modified total Sharp Score (mTSS) at Week 24.
Trial RA-II (NCT02706951) was a 14-week monotherapy trial in 648 patients with moderately to severely active rheumatoid arthritis who had an inadequate response to MTX. Patients received RINVOQ 15 mg or upadacitinib 30 mg once daily monotherapy or continued their stable dose of MTX monotherapy. At Week 14, patients who were randomized to MTX were advanced to RINVOQ 15 mg or upadacitinib 30 mg once daily monotherapy in a blinded manner based on pre-determined assignment at baseline. The primary endpoint was the proportion of patients who achieved an ACR20 response at Week 14. Key secondary endpoints included DAS28-CRP ≤3.2, DAS28-CRP <2.6, and change from baseline in HAQ-DI at Week 14.
Trial RA-III (NCT02675426) was a 12-week trial in 661 patients with moderately to severely active rheumatoid arthritis who had an inadequate response to conventional disease modifying anti-rheumatic drugs (cDMARDs). Patients received RINVOQ 15 mg or upadacitinib 30 mg once daily or placebo added to background cDMARD therapy. At Week 12, patients who were randomized to placebo were advanced to RINVOQ 15 mg or upadacitinib 30 mg once daily in a blinded manner based on pre-determined assignment at baseline. The primary endpoint was the proportion of patients who achieved an ACR20 response at Week 12. Key secondary endpoints included DAS28-CRP ≤3.2, DAS28-CRP<2.6, and change from baseline in HAQ-DI at Week 12.
Trial RA-IV (NCT02629159) was a 48-week trial in 1629 patients with moderately to severely active rheumatoid arthritis who had an inadequate response to MTX. Patients received RINVOQ 15 mg once daily, active comparator, or placebo added to background MTX. From Week 14, non-responding patients on RINVOQ 15 mg could be rescued to active comparator in a blinded manner, and non-responding patients on active comparator or placebo could be rescued to RINVOQ 15 mg in a blinded manner. At Week 26, all patients randomized to placebo were switched to RINVOQ 15 mg once daily in a blinded manner. The primary endpoint was the proportion of patients who achieved an ACR20 response at Week 12 versus placebo. Key secondary endpoints versus placebo included DAS28-CRP ≤3.2, DAS28-CRP <2.6, change from baseline in HAQ-DI at Week 12, and change from baseline in mTSS at Week 26.
Trial RA-V (NCT02706847) was a 12-week trial in 499 patients with moderately to severely active rheumatoid arthritis who had an inadequate response or intolerance to biologic DMARDs. Patients received RINVOQ 15 mg or upadacitinib 30 mg once daily or placebo added to background cDMARD therapy. At Week 12, patients who were randomized to placebo were advanced to RINVOQ 15 mg or upadacitinib 30 mg once daily in a blinded manner based on pre-determined assignment at baseline. The primary endpoint was the proportion of patients who achieved an ACR20 response at Week 12. Key secondary endpoints included DAS28-CRP ≤3.2 and change from baseline in HAQ-DI at Week 12.
Clinical Response
The percentages of RINVOQ-treated patients achieving ACR20, ACR50, and ACR70 responses, and DAS28(CRP) < 2.6 in all trials are shown in Table 13.
Patients treated with RINVOQ 15 mg, alone or in combination with cDMARDs, achieved higher ACR response rates compared to MTX monotherapy or placebo, respectively, at the primary efficacy timepoint (Table 13).
In Trial IV, the percent of patients achieving ACR20 response by visit is shown in Figure 1.
In Trials RA-III and RA-V, higher ACR20 response rates were observed at 1 week with RINVOQ 15 mg versus placebo.
Treatment with RINVOQ 15 mg, alone or in combination with cDMARDs, resulted in greater improvements in the ACR components compared to MTX or placebo at the primary efficacy timepoint (Table 14).
Table 13: Clinical Response in RA Patients in Trials RA-I, RA-II, RA-III, RA-IV and RA V Trial RA-I
MTX-NaïveTrial RA-II
MTX-IRTrial RA-III
cDMARD-IRTrial RA-IV
MTX-IRTrial RA-V
bDMARD-IRMonotherapy Monotherapy Background
cDMARDsBackground
MTXBackground
cDMARDsMTX RINVOQ
15 mg
%
Δ (95% CI)MTX RINVOQ
15 mg
%
Δ (95% CI)PBO RINVOQ
15 mg
%
Δ (95% CI)PBO RINVOQ
15 mg
%
Δ (95% CI)PBO RINVOQ
15 mg
%
Δ (95% CI)N 314 317 216 217 221 221 651 651 169 164 Week ACR20 12a/14b 54 76
22 (14, 29)41 68
26 (17, 36)36 64
28 (19, 37)36 71
34 (29, 39)28 65
36 (26, 46)24c/26d 59 79
20 (13, 27)36 67
32 (27, 37)ACR50 12a/14b 28 52
24 (16, 31)15 42
27 (18, 35)15 38
23 (15, 31)15 45
30 (26, 35)12 34
22 (14, 31)24c/26d 33 60
27 (19, 34)21 54
33 (28, 38)ACR70 12a/14b 14 32
18 (12, 25)3 23
20 (14, 26)6 21
15 (9, 21)5 25
20 (16, 24)7 12
5 (-1, 11)24c/26d 18 44
26 (19, 33)10 35
25 (21, 29)DAS28-CRP <2.6 12a/14b 14 36
22 (15, 28)8 28
20 (13, 27)10 31
21 (14, 28)6 29
23 (19, 27)9 29
19 (11, 27)24c/26d 18 48
30 (23, 37)9 41
32 (27, 36)Abbreviations: ACR20 (or 50 or 70) = American College of Rheumatology ≥20% (or ≥50% or ≥70%) improvement; bDMARD = biologic disease-modifying anti-rheumatic drug; CRP = c-reactive protein; DAS28 = Disease Activity Score 28 joints; cDMARDs = conventional disease-modifying anti-rheumatic drugs; MTX = methotrexate; PBO = placebo; IR = inadequate responder
Patients who discontinued randomized treatment, or had cross-over between randomized treatments, or were missing data at week of evaluation were imputed as non-responders in the analyses.
a Trial RA-I, Trial RA-III, Trial RA-IV, Trial RA-V
b Trial RA-II
c Trial RA-I
d Trial RA-IVTable 14: Components of ACR Response at Primary Efficacy Timepointa Trial RA-I
MTX-NaïveTrial RA-IIb
MTX-IRTrial RA-III
cDMARD-IRTrial RA-IV
MTX-IRTrial RA-V
bDMARD-IRMonotherapy Monotherapy Background
cDMARDsBackground
MTXBackground
cDMARDsMTX RINVOQ
15 mgMTX RINVOQ
15 mgPBO RINVOQ
15 mgPBO RINVOQ
15 mgPBO RINVOQ
15 mgN 314 317 216 217 221 221 651 651 169 164 Number of tender joints (0-68) Baseline 26
(16)25
(14)25
(16)24
(15)25
(15)25
(14)26
(14)26
(15)28
(15)28
(16)Week
12/1413
(15)9
(12)15
(16)10
(13)16
(17)12
(14)16
(15)10
(13)18
(17)11
(14)Number of swollen joints (0-66) Baseline 17
(11)17
(10)17
(12)16
(11)15
(9)16
(10)16
(9)17
(10)16
(10)17
(11)Week
12/146
(8)5
(7)9
(11)6
(9)9
(10)7
(10)9
(9)5
(7)9
(10)6
(8)Painc Baseline 66
(21)68
(21)63
(21)62
(23)62
(21)64
(19)65
(21)66
(21)69
(21)68
(20)Week
12/1441
(25)31
(25)49
(25)36
(27)51
(26)33
(24)49
(25)33
(24)55
(28)41
(28)Patient global assessmentc Baseline 66
(21)67
(22)60
(22)62
(22)60
(20)63
(22)64
(21)64
(22)66
(23)67
(20)Week
12/1442
(25)31
(24)48
(26)37
(27)50
(26)32
(24)48
(24)33
(24)54
(28)40
(26)Disability Index (HAQ-DI)d Baseline 1.60
(0.67)1.60
(0.67)1.47
(0.66)1.47
(0.66)1.42
(0.63)1.48
(0.61)1.61
(0.61)1.63
(0.64)1.56
(0.60)1.67
(0.64)Week
12/141.08
(0.72)0.76
(0.69)1.19
(0.69)0.86
(0.67)1.13
(0.70)0.85
(0.66)1.28
(0.67)0.98
(0.68)1.33
(0.66)1.24
(0.77)Physician global assessmentc Baseline 69
(16)67
(17)62
(17)66
(18)64
(18)64
(16)66
(18)66
(17)67
(17)69
(17)Week
12/1432
(22)22
(19)37
(24)26
(21)41
(24)26
(21)41
(25)27
(21)39
(25)29
(22)CRP (mg/L) Baseline 21.2
(22.1)23.0
(27.4)14.5
(17.3)14.0
(16.5)12.6
(14.0)16.6
(19.2)18.0
(21.5)17.9
(22.5)16.3
(21.1)16.3
(18.6)Week
12/1410.9
(14.9)4.2
(8.8)12.8
(21.4)3.7
(7.8)13.1
(15.5)4.6
(9.6)16.2
(19.8)5.5
(10.9)13.9
(17.3)5.0
(14.0)Abbreviations: ACR = American College of Rheumatology; bDMARD = biologic disease-modifying anti-rheumatic drug; CRP = c-reactive protein; cDMARDs = conventional disease-modifying anti-rheumatic drugs; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate responder; MTX = methotrexate; PBO = placebo
a Data shown are mean (standard deviation).
b Primary efficacy timepoint is at Week 14.
c Visual analog scale: 0 = best, 100 = worst.
d Health Assessment Questionnaire-Disability Index: 0=best, 3=worst; 20 questions; 8 categories: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities.Figure 1. Percent of Patients Achieving ACR20 in Trial RA-IV
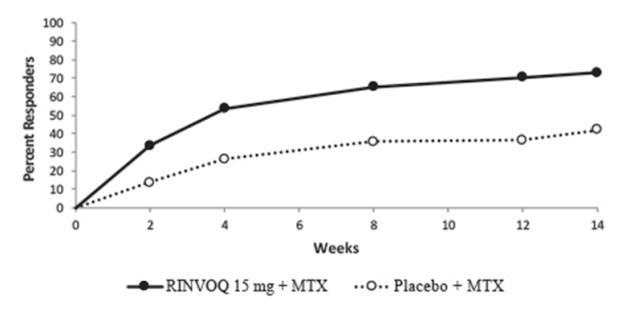
Abbreviations: ACR20 = American College of Rheumatology ≥20% improvement; MTX = methotrexate
Patients who discontinued randomized treatment, or were missing ACR20 results, or were lost-to-follow-up or withdrawn from the trial were imputed as non-responders.In RA-I and RA-IV, a higher proportion of patients treated with RINVOQ 15 mg alone or in combination with MTX, achieved DAS28-CRP < 2.6 compared to MTX or placebo at the primary efficacy timepoint (Table 15).
Table 15: Proportion of Patients with DAS28-CRP Less Than 2.6 with Number of Residual Active Joints at Primary Efficacy Timepoint Trial RA-I
MTX-naïveMonotherapy DAS28-CRP Less Than 2.6 MTX
N = 314RINVOQ 15 mg
N = 317Proportion of responders at Week 12 (n) 14% (43) 36% (113) Of responders, proportion with 0 active joints (n) 51% (22) 45% (51) Of responders, proportion with 1 active joint (n) 35% (15) 23% (26) Of responders, proportion with 2 active joints (n) 9% (4) 17% (19) Of responders, proportion with 3 or more active joints (n) 5% (2) 15% (17) Trial RA-IV
MTX-IRBackground MTX DAS28-CRP Less Than 2.6 PBO
N = 651RINVOQ 15 mg
N = 651Proportion of responders at Week 12 (n) 6% (40) 29% (187) Of responders, proportion with 0 active joints (n) 60% (24) 48% (89) Of responders, proportion with 1 active joint (n) 20% (8) 23% (43) Of responders, proportion with 2 active joints (n) 15% (6) 13% (25) Of responders, proportion with 3 or more active joints (n) 5% (2) 16% (30) Abbreviations: CRP = c-reactive protein; DAS28 = Disease Activity Score 28 joints; MTX = methotrexate; PBO = placebo; IR = inadequate responder Radiographic Response
Inhibition of progression of structural joint damage was assessed using the modified Total Sharp Score (mTSS) and its components, the erosion score and joint space narrowing score, at Week 26 in Trial RA-IV and Week 24 in Trial RA-I. The proportion of patients with no radiographic progression (mTSS change from baseline ≤ 0) was also assessed.
In Trial RA-IV, treatment with RINVOQ 15 mg inhibited the progression of structural joint damage compared to placebo in combination with cDMARDs at Week 26 (Table 16). Analyses of erosion and joint space narrowing scores were consistent with overall results.
In the placebo plus MTX group, 76% of the patients experienced no radiographic progression at Week 26 compared to 83% of the patients treated with RINVOQ 15 mg.
In Trial RA-I, treatment with RINVOQ 15 mg monotherapy inhibited the progression of structural joint damage compared to MTX monotherapy at Week 24 (Table 16). Analyses of erosion and joint space narrowing scores were consistent with overall results.
In the MTX monotherapy group, 78% of the patients experienced no radiographic progression at Week 24 compared to 87% of the patients treated with RINVOQ 15 mg monotherapy.
Table 16: Radiographic Changes Trial RA-IV
MTX-IRBackground MTX mTSS PBO
(N=651)
Mean (SD)RINVOQ 15 mg
(N=651)
Mean (SD)Estimated Difference vs PBO
at Week 26
(95% CI)aBaseline 35.9 (52) 34.0 (50) Week 26b 0.78 (0.1) 0.15 (0.1) -0.63 (-0.92, -0.34) Trial RA-I
MTX-naïveMonotherapy MTX
(N=309)
Mean (SD)RINVOQ 15 mg
(N=309)
Mean (SD)Estimated Difference vs MTX
at Week 24
(95% CI)cBaseline 13.3 (31) 18.1 (38) Week 24d 0.67 (2.8) 0.14 (1.4) -0.53 (-0.85, -0.20) Abbreviations: mTSS = modified Total Sharp Score, MTX = methotrexate; PBO = placebo; SD = standard deviation; IR = inadequate responders; bDMARDs = biologic disease modifying anti-rheumatic drugs; LS = least squares; CI = confidence intervals
a LS means and 95% CI based on a random coefficient model fit to the mTSS value adjusting for time, treatment group, prior bDMARDs use, treatment group-by-time interaction, with random slopes and random intercept.
b Estimated linear rate of structural progression by Week 26 and standard errors are presented.
c LS means and 95% CI based on a linear regression model fit to change from baseline in mTSS adjusting for treatment group, baseline mTSS, and geographic region.
d Mean change from baseline and standard deviation are presented.Physical Function Response
Treatment with RINVOQ 15 mg, alone or in combination with cDMARDs, resulted in a greater improvement in physical function at Week 12/14 compared to all comparators as measured by HAQ-DI.
Other Health-Related Outcomes
In all trials except for Trial RA-V, patients receiving RINVOQ 15 mg had greater improvement from baseline in physical component summary (PCS) score, mental component summary (MCS) scores, and in all 8 domains of the Short Form Health Survey (SF-36) compared to placebo in combination with cDMARDs or MTX monotherapy at Week 12/14.
Fatigue was assessed by the Functional Assessment of Chronic Illness Therapy-Fatigue score (FACIT-F) in Trials RA-I, RA-III, and RA-IV. Improvement in fatigue at Week 12 was observed in patients treated with RINVOQ 15 mg compared to patients on placebo in combination with cDMARDs or MTX monotherapy.
14.2 Psoriatic Arthritis
The efficacy and safety of RINVOQ 15 mg once daily were assessed in two Phase 3 randomized, double-blind, multicenter, placebo-controlled trials in patients 18 years of age or older with moderately to severely active psoriatic arthritis. All patients had active psoriatic arthritis for at least 6 months based upon the Classification Criteria for Psoriatic Arthritis (CASPAR), at least 3 tender joints and at least 3 swollen joints, and active plaque psoriasis or history of plaque psoriasis. Although another dose has been studied, the recommended dose of RINVOQ is 15 mg once daily for psoriatic arthritis.
Trial PsA-I (NCT03104400) was a 24-week trial in 1705 patients with moderately to severely active psoriatic arthritis who had an inadequate response or intolerance to at least one non-biologic DMARD. Patients received RINVOQ 15 mg or upadacitinib 30 mg once daily, adalimumab, or placebo, alone or in combination with background non-biologic DMARDs. At Week 24, all patients randomized to placebo were switched to RINVOQ 15 mg or upadacitinib 30 mg once daily in a blinded manner. The primary endpoint was the proportion of patients who achieved an ACR20 response at Week 12.
Trial PsA-II (NCT03104374) was a 24-week trial in 642 patients with moderately to severely active psoriatic arthritis who had an inadequate response or intolerance to at least one biologic DMARD. Patients received RINVOQ 15 mg or upadacitinib 30 mg once daily or placebo, alone or in combination with background non-biologic DMARDs. At Week 24, all patients randomized to placebo were switched to RINVOQ 15 mg or upadacitinib 30 mg once daily in a blinded manner. The primary endpoint was the proportion of patients who achieved an ACR20 response at Week 12.
Clinical Response
In both trials, patients treated with RINVOQ 15 mg achieved significantly higher ACR20 responses compared to placebo at Week 12 (Table 17, Figure 2). A higher proportion of patients treated with RINVOQ 15 mg achieved ACR50 and ACR70 responses at Week 12 compared to placebo.
Treatment with RINVOQ 15 mg resulted in improvements in the ACR components compared to placebo at the primary efficacy timepoint (Table 18).
Table 17: Clinical Response Trial Trial PsA-I
non-biologic DMARD-IRTrial PsA-II
bDMARD-IRTreatment Group PBO
%RINVOQ
15 mg
%
Δ (95% CI)PBO
%RINVOQ
15 mg
%
Δ (95% CI)N 423 429 212 211 ACR20 Week 12 36 71
35 (28, 41)24 57
33 (24, 42)ACR50 Week 12 13 38
24 (19, 30)5 32
27 (20, 34)ACR70 Week 12 2 16
13 (10, 17)1 9
8 (4, 12)Abbreviations: ACR20 (or 50 or 70) = American College of Rheumatology ≥20% (or ≥50% or ≥70%) improvement, bDMARD = biologic disease-modifying anti-rheumatic drug; IR = inadequate responder; PBO = placebo
Patients who discontinued randomized treatment or were missing data at week of evaluation were imputed as non-responders in the analyses.Table 18: Components of ACR Responsea Trial Trial PsA-I
non-biologic DMARD-IRTrial PsA-II
bDMARD-IRTreatment Group PBO
(N=423)RINVOQ
15 mg
(N=429)PBO
(N=212)RINVOQ
15 mg
(N=211)Number of tender/painful joints (0-68) Baseline 20.0 (14.3) 20.4 (14.7) 25.3 (17.6) 24.9 (17.3) Week 12 12.5 (13.3) 8.8 (12.5) 19.3 (18.5) 12.6 (15.6) Number of swollen joints (0-66) Baseline 11.0 (8.2) 11.6 (9.3) 12.0 (8.9) 11.3 (8.2) Week 12 5.6 (7.2) 3.5 (6.0) 7.3 (9.4) 4.4 (5.7) Patient assessment of painb Baseline 6.1 (2.1) 6.2 (2.1) 6.6 (2.1) 6.4 (2.1) Week 12 5.1 (2.3) 3.8 (2.4) 5.9 (2.3) 4.4 (2.5) Patient global assessmentb Baseline 6.3 (2.0) 6.6 (2.0) 6.8 (2.0) 6.8 (1.9) Week 12 5.2 (2.2) 3.8 (2.3) 6.1 (2.3) 4.5 (2.5) Disability index (HAQ-DI)c Baseline 1.1 (0.6) 1.2 (0.7) 1.2 (0.7) 1.1 (0.6) Week 12 1.0 (0.7) 0.7 (0.6) 1.1 (0.6) 0.8 (0.7) Physician global assessmentb Baseline 6.5 (1.6) 6.7 (1.6) 6.5 (1.8) 6.5 (1.8) Week 12 4.3 (2.2) 3.1 (2.0) 5.0 (2.2) 3.4 (2.1) hsCRP (mg/L) Baseline 11.5 (15.8) 11.0 (14.9) 10.4 (18.5) 11.2 (18.6) Week 12 10.1 (15.2) 4.2 (9.9) 9.4 (13.4) 4.3 (7.9) Abbreviations: ACR = American College of Rheumatology; hsCRP = high sensitivity c-reactive protein; HAQ-DI = Health Assessment Questionnaire-Disability Index; IR = inadequate responder; PBO = placebo
a Data shown are mean (standard deviation).
b Numeric rating scale (NRS): 0 = best, 10 = worst
c Health Assessment Questionnaire-Disability Index: 0=best, 3=worst; 20 questions; 8 categories: dressing and grooming, arising, eating, walking, hygiene, reach, grip, and activities.The percentage of patients achieving ACR20 response by visit is shown in Figure 2.
Figure 2. Percent of Patients Achieving ACR20 in Trial PsA-II
Abbreviations: ACR20 = American College of Rheumatology ≥20% improvement Patients who discontinued randomized treatment, or were missing ACR20 results, or were lost-to-follow-up or withdrawn from the trial were imputed as non-responders.
Treatment with RINVOQ 15 mg resulted in improvement in dactylitis and enthesitis in patients with pre-existing dactylitis or enthesitis.
Treatment with RINVOQ 15 mg resulted in improvement in skin manifestations in patients with PsA. However, RINVOQ has not been studied in and is not indicated for the treatment of plaque psoriasis.
Physical Function Response
In both trials, patients treated with RINVOQ 15 mg showed significant improvement in physical function from baseline compared to placebo as assessed by HAQ-DI at Week 12 (Table 17). The mean difference (95% CI) from placebo in HAQ-DI change from baseline at Week 12 was -0.28 (-0.35, -0.22) in Trial PsA-I and -0.21 (-0.30, -0.12) in Trial PsA-II.
The proportion of HAQ-DI responders (≥ 0.35 improvement from baseline in HAQ-DI score) at Week 12 in Trial PsA-I and Trial PsA-II was 58% and 45%, respectively, in patients receiving RINVOQ 15 mg and 33% and 27%, respectively, in patients receiving placebo.
Radiographic Response
In Trial PsA-I, inhibition of progression of structural damage was assessed radiographically and expressed as the change from baseline in modified Total Sharp Score (mTSS) and its components, the erosion score and the joint space narrowing score, at Week 24.
Treatment with RINVOQ 15 mg inhibited progression of structural joint damage compared to placebo at Week 24 (Table 19). Analyses of erosion and joint space narrowing scores were consistent with overall results. The proportion of patients with no radiographic progression (mTSS change ≤ 0) at Week 24 was 93% in patients receiving RINVOQ 15 mg and 89% in patients receiving placebo.
Table 19: Radiographic Changes in Trial PsA-I PBO
(N=392)
Mean (SD)
RINVOQ 15 mg
(N=407)
Mean (SD)
Estimated Difference vs PBO
at Week 24
(95% CI)amTSS Baseline 13.32 (31.2) 13.14 (42.4) Week 24b 0.23 (0.07) -0.02 (0.04) -0.25 (-0.41, -0.09) Abbreviations: CI = confidence intervals; LS = least squares; mTSS = modified Total Sharp Score; PBO = placebo; SD = standard deviation
a LS means and 95% CI based on a random coefficient model fit to the mTSS value adjusting for time, treatment group, current DMARD use (yes/no), treatment group-by-time interaction, with random slopes and random intercept.
b Estimated linear rate of structural progression by Week 24 and standard errors are presented.Other Health-Related Outcomes
Health-related quality of life was assessed by SF-36. In both trials, patients receiving RINVOQ 15 mg experienced significantly greater improvement from baseline in the Physical Component Summary score compared to placebo at Week 12. Greater improvement was also observed in the Mental Component Summary score and all 8 domains of SF-36 compared to placebo.
Patients receiving RINVOQ 15 mg showed greater improvement from baseline in fatigue, as measured by FACIT-F score, at Week 12 compared to placebo in both trials.
14.3 Atopic Dermatitis
The efficacy of RINVOQ 15 mg and 30 mg once daily, was assessed in three Phase 3 randomized, double-blind, multicenter trials (AD-1, AD-2, AD-3; NCT03569293, NCT03607422, and NCT03568318, respectively) in a total of 2584 patients (12 years of age and older). RINVOQ was evaluated in 344 pediatric patients and 2240 adult patients with moderate to severe atopic dermatitis (AD) not adequately controlled by topical medication(s).
Disease severity at baseline was defined by a validated Investigator's Global Assessment (vIGA-AD) score ≥3 in the overall assessment of AD on a severity scale of 0 to 4, an Eczema Area and Severity Index (EASI) score ≥16, a minimum body surface area (BSA) involvement of ≥10%, and weekly average Worst Pruritus Numerical Rating Scale (NRS) score ≥4. Overall, 57% of the patients were male and 69% were white. The mean age at baseline was 34 years (ranged from 12 to 75 years) and 13% of the patients were 12 to less than 18 years. At baseline, 49% of patients had a vIGA-AD score of 3 (moderate AD), and 51% of patients had a vIGA-AD score of 4 (severe AD). The baseline mean EASI score was 29 and the baseline weekly average Worst Pruritus NRS score was 7. Approximately 52% of the patients had prior exposure to systemic AD treatment.
In all three trials, patients received RINVOQ once daily oral doses of 15 mg, 30 mg, or matching placebo for 16 weeks. In Trial AD-3, patients also received RINVOQ or placebo with concomitant topical corticosteroids (TCS) for 16 weeks.
All three trials assessed the co-primary endpoints of the proportion of patients with a vIGA-AD score of 0 (clear) or 1 (almost clear) with at least a 2-point improvement and the proportion of patients with EASI-75 (improvement of at least 75% in EASI score from baseline) at Week 16. Secondary endpoints included EASI-90 and EASI-100 at Week 16, and the proportion of patients with reduction in itch (≥4-point improvement from baseline in the Worst Pruritus NRS) at Weeks 1, 4, and 16. In Trials AD-1 and AD-2, the proportion of patients with reduction in pain (≥4-point improvement in the Atopic Dermatitis Symptom Scale [ADerm-SS] Skin Pain NRS) from baseline to Week 16 was a secondary endpoint.
Clinical Response
Monotherapy Trials (AD-1 and AD-2)
The results of RINVOQ monotherapy trials (AD-1 and AD-2) are presented in Table 20. Figure 3 presents the proportion of patients with ≥ 4-point improvement in Worst Pruritus NRS at Weeks 1, 4, and 16 for Trials AD-1 and AD-2.
Table 20: Efficacy Results of Monotherapy Trials at Week 16 in Patients with Moderate to Severe AD Trial AD-1 Trial AD-2 PBO RINVOQ
15 mgRINVOQ
30 mgPBO RINVOQ
15 mgRINVOQ
30 mgNumber of patients
randomized281 281 285 278 276 282 vIGA-AD 0/1a,b 8% 48% 62% 5% 39% 52% Difference from
PBO (95% CI)40%
(33%, 46%)54%
(47%, 60%)34%
(28%, 40%)47%
(41%, 54%)EASI-75a 16% 70% 80% 13% 60% 73% Difference from
PBO (95% CI)53%
(46%, 60%)63%
(57%, 70%)47%
(40%, 54%)60%
(53%, 66%)EASI-90a 8% 53% 66% 5% 42% 58% Difference from
PBO (95% CI)45%
(39%, 52%)58%
(51%, 64%)37%
(31%, 43%)53%
(47%, 59%)EASI-100a 2% 17% 27% 1% 14% 19% Difference from
PBO (95% CI)15%
(10%, 20%)25%
(20%, 31%)13%
(9%, 18%)18%
(13%, 23%)Number of patients with baseline Worst Pruritus NRS score ≥ 4 272 274 280 274 270 280 ≥ 4-point improvement in Worst Pruritus NRSc 12% 52% 60% 9% 42% 60% Difference from
PBO (95% CI)40%
(33%, 48%)48%
(41%, 55%)33%
(26%, 39%)50%
(44%, 57%)Number of patients with baseline ADerm-SS Skin Pain NRS score ≥ 4 233 237 249 247 237 238 ≥ 4-point improvement in ADerm-SS Skin Pain NRSd 15% 54%
63% 13% 49%
65% Difference from
PBO (95% CI)39%
(31%, 47%)49%
(41%, 56%)36%
(28%, 43%)52%
(44%, 59%)Abbreviations: ADerm-SS = Atopic Dermatitis Symptom Scale; PBO = placebo
a Based on number of patients randomized
b Responder was defined as a patient with vIGA-AD 0 or 1 (“clear” or “almost clear”) with a reduction of ≥ 2 points on a 0-4 ordinal scale
c Based on number of patients whose baseline Worst Pruritus NRS is ≥ 4
d Based on number of patients whose baseline ADerm-SS Skin Pain NRS is ≥ 4Figure 3. Proportion of Patients with Moderate to Severe AD with ≥4-point Improvement in the Worst Pruritus NRS in Monotherapy Trials
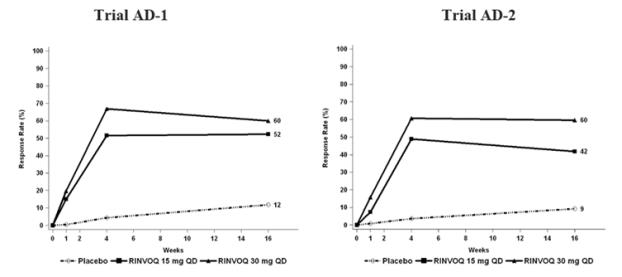
Examination of age, gender, race, weight, and prior systemic treatment with immunosuppressants did not identify differences in response to RINVOQ among these subgroups in Trials AD-1 and AD-2.
Concomitant TCS Trial (AD-3)
The results of the RINVOQ with concomitant TCS trial (AD-3) are presented in Table 21. Figure 4 presents the proportion of patients with ≥ 4-point improvement in Worst Pruritus NRS at Weeks 1, 4, and 16 for Trial AD-3.
Table 21: Efficacy Results with Concomitant TCS at Week 16 in Patients with Moderate to Severe AD Trial AD-3 PBO + TCS RINVOQ 15 mg
+ TCSRINVOQ 30 mg
+ TCSNumber of patients
randomized304 300 297 vIGA-AD 0/1a,b 11% 40% 59% Difference from
PBO (95% CI)29%
(22%, 35%)48%
(41%, 54%)EASI-75a, 26% 65% 77% Difference from
PBO (95% CI)38%
(31%, 45%)51%
(44%, 57%)EASI-90a 13% 43% 63% Difference from
PBO (95% CI)30%
(23%, 36%)50%
(43%, 56%)EASI-100a 1% 12% 23% Difference from
PBO (95% CI)11%
(7%, 14%)21%
(16%, 26%)Number of patients with baseline Worst Pruritus NRS score ≥ 4 294 288 291 ≥ 4-point improvement in Worst Pruritus NRSc 15% 52% 64% Difference from
PBO (95% CI)37%
(30%, 44%)49%
(42%, 56%)Abbreviations: PBO = placebo
a Based on number of patients randomized
b Responder was defined as a patient with vIGA-AD 0 or 1 (“clear” or “almost clear”) with a reduction of ≥ 2 points on a 0-4 ordinal scale
c Based on number of patients whose baseline Worst Pruritus NRS is ≥ 4Figure 4. Proportion of Patients with Moderate to Severe AD with ≥4-point Improvement in the Worst Pruritus NRS in Concomitant TCS Trial
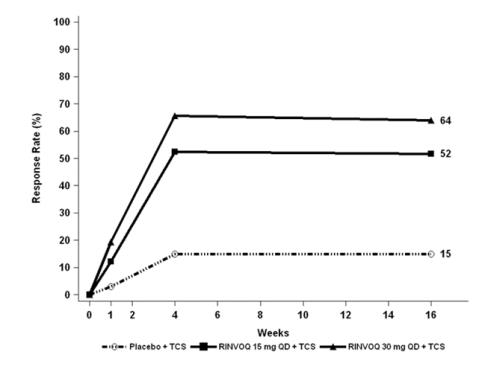
Examination of age, gender, race, weight, and prior systemic treatment with immunosuppressants did not identify differences in response to RINVOQ among these subgroups in Trial AD-3.
Pediatric Patient Population
The efficacy results of the RINVOQ monotherapy trials (AD-1 and AD-2) and the RINVOQ with concomitant TCS trial (AD-3) at Week 16 for pediatric patients 12 years of age and older are presented in Table 22 and Table 23, respectively.
Table 22: Efficacy Results of Monotherapy Trials for Pediatric Patients 12 Years of Age and Older with Moderate to Severe AD at Week 16 Trial AD-1 Trial AD-2 PBO RINVOQ
15 mgRINVOQ
30 mgPBO RINVOQ
15 mgRINVOQ
30 mgNumber of pediatric patients randomized 40 42 42 36 33 35 vIGA-AD 0/1a,b 8% 38% 69% 3% 42% 62% Difference from
PBO (95% CI)31%
(14%, 47%)62%
(45%, 78%)40%
(22%, 57%)60%
(42%, 77%)EASI-75a 8% 71% 83% 14% 67% 74% Difference from
PBO (95% CI)63%
(47%, 79%)75%
(61%, 89%)53%
(33%, 72%)61%
(42%, 79%)Number of pediatric patients with baseline Worst Pruritus NRS score ≥ 4 39 40 42 36 30 34 ≥ 4-point improvement in Worst Pruritus NRSc 15% 45% 55% 3% 33% 50% Difference from
PBO (95% CI)30%
(10%, 49%)39%
(21%, 58%)31%
(13%, 48%)47%
(30%, 65%)Abbreviations: PBO = placebo
a Based on number of pediatric patients randomized
b Responder was defined as a patient with vIGA-AD 0 or 1 (“clear” or “almost clear”) with a reduction of ≥ 2 points on a 0-4 ordinal scale
c Based on number of pediatric patients whose baseline Worst Pruritus NRS is ≥ 4Table 23: Efficacy Results with Concomitant TCS for Pediatric Patients 12 Years of Age and Older with Moderate to Severe AD at Week 16 Trial AD-3 PBO + TCS RINVOQ 15 mg
+ TCSRINVOQ 30 mg
+ TCSNumber of pediatric patients randomized 40 39 37 vIGA-AD 0/1a,b 8% 31% 65% Difference from
PBO (95% CI)23%
(7%, 40%)57%
(40%, 75%)EASI-75a 30% 56% 76% Difference from
PBO (95% CI)26%
(5%, 47%)46%
(26%, 65%)Number of pediatric patients with baseline Worst Pruritus NRS score ≥ 4 38 36 33 ≥ 4-point improvement in Worst Pruritus NRSc 13% 42% 55% Difference from
PBO (95% CI)29%
(9%, 48%)41%
(21%, 61%)Abbreviations: PBO = placebo
a Based on number of pediatric patients randomized
b Responder was defined as a patient with vIGA-AD 0 or 1 (“clear” or “almost clear”) with a reduction of ≥ 2 points on a 0-4 ordinal scale
c Based on number of pediatric patients whose baseline Worst Pruritus NRS is ≥ 414.4 Ulcerative Colitis
Induction Trials (Study UC-1 and Study UC-2)
In two identical induction trials (UC-1; NCT02819635 and UC-2; NCT03653026), patients were randomized 2:1 to receive either RINVOQ 45 mg once daily or placebo for 8 weeks. A total of 988 patients were analyzed across the two trials. These trials included adult patients with moderately to severely active ulcerative colitis who had an inadequate response, loss of response, or intolerance to oral aminosalicylates, corticosteroids, immunosuppressants, and/or biologic therapy. Enrolled patients were permitted to use stable doses of oral aminosalicylates, methotrexate, ulcerative colitis-related antibiotics, and/or oral corticosteroids (up to 30 mg/day prednisone or equivalent). At baseline, 38% of patients were receiving corticosteroids, and 68% of patients were receiving aminosalicylates. Concomitant biologic therapies, azathioprine, 6-mercaptopurine, intravenous or rectal corticosteroids were prohibited. A total of 51% of patients had previously failed treatment with or were intolerant to at least one biologic therapy. RINVOQ is indicated for patients who have an inadequate response or intolerance to one or more TNF blockers [see Indications and Usage (1.4)].
Disease activity was assessed on the modified Mayo score (mMS), a 3-component Mayo score (0-9) which consists of the following subscores (0 to 3 for each subscore): stool frequency (SFS), rectal bleeding (RBS), and findings on centrally read endoscopy score (ES). An ES of 2 was defined by marked erythema, lack of vascular pattern, any friability, and/or erosions, and a score of 3 was defined by spontaneous bleeding and ulceration. Enrolled patients had a mMS between 5 to 9 with an ES of 2 or 3; at baseline the median mMS was 7, with 61% of patients having a baseline mMS of 5 to 7 and 39% having a mMS of 8 to 9.
At baseline, 39% and 37% of patients received corticosteroids, 1% and 1% of patients received methotrexate, and 68% and 69% of patients received aminosalicylates in UC-1 and UC-2, respectively. Patient disease activity was moderate (mMS ≤7) in 61% and 60% of patients and severe (mMS >7) in 39% and 40% of patients in UC-1 and UC-2, respectively.
The primary endpoint was clinical remission defined using the mMS at Week 8. Secondary endpoints included clinical response, endoscopic improvement, and histologic endoscopic mucosal improvement (see Table 24 and Table 25).
Table 24: Proportion of Patients Meeting Primary and Key Secondary Efficacy Endpoints at Week 8 - Study UC-1 Study UC-1 Endpoint Placebo RINVOQ
45 mg Once DailyTreatment Difference vs Placebo
(95% CI)Clinical Remissiona Total Population N=154
5%N=319
26%22%b
(16, 27)Prior biologic failurec N=78
< 1%N=168
18%Without prior biologic failure N=76
9%N=151
35%Clinical Responsed Total Population N=154
27%N=319
73%46%b
(38, 54)Prior biologic failurec N=78
13%N=168
64%Without prior biologic failure N=76
42%N=151
82%Endoscopic Improvemente Total Population N=154
7%N=319
36%29%b
(23, 36)Prior biologic failurec N=78
2%N=168
27%Without prior biologic failure N=76
13%N=151
47%Histologic Endoscopic Mucosal Improvementf Total Population N=154
7%N=319
30%24%b
(17, 30)Prior biologic failurec N=78
1%N=168
23%Without prior biologic failure N=76
12%N=151
38%a Per mMS: SFS ≤1 and not greater than baseline, RBS = 0, ES of ≤ 1 without friability
b p <0.001, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors
c Prior biologic failure includes inadequate response, loss of response, or intolerance to one or more biologic treatments for ulcerative colitis.
d Per mMS: decrease ≥ 2 points and ≥ 30% from baseline and a decrease in RBS ≥ 1 from baseline or an absolute RBS ≤1
e ES ≤ 1 without friability
f ES ≤1 without friability and Geboes score ≤ 3.1 (indicating neutrophil infiltration in <5% of crypts, no crypt destruction and no erosions, ulcerations or granulation tissue)Table 25: Proportion of Patients Meeting Primary and Key Secondary Efficacy Endpoints at Week 8 - Study UC-2 Study UC-2 Endpoint Placebo RINVOQ
45 mg Once DailyTreatment Difference vs Placebo
(95% CI)Clinical Remissiona Total Population N=174
4%N=341
33%29%b
(23, 35)Prior biologic failurec N=89
2%N=173
30%Without prior biologic failure N=85
6%N=168
38%Clinical Responsed Total Population N=174
25%N=341
74%49%b
(42, 57)Prior biologic failurec N=89
19%N=173
69%Without prior biologic failure N=85
32%N=168
80%Endoscopic Improvemente Total Population N=174
8%N=341
44%35%b
(29, 42)Prior biologic failurec N=89
5%N=173
37%Without prior biologic failure N=85
12%N=168
51%Histologic Endoscopic Mucosal Improvementf Total Population N=174
6%N=341
37%30%b
(24, 36)Prior biologic failurec N=89
5%N=173
31%Without prior biologic failure N=85
7%N=168
43%a Per mMS: SFS ≤ 1 and not greater than baseline, RBS = 0, ES of ≤ 1 without friability
b p <0.001, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors
c Prior biologic failure includes inadequate response, loss of response, or intolerance to one or more biologic treatments for ulcerative colitis.
d Per mMS: decrease ≥ 2 points and ≥ 30% from baseline and a decrease in RBS ≥ 1 from baseline or an absolute RBS ≤1
e ES ≤ 1 without friability
f ES ≤1 without friability and Geboes score ≤ 3.1 (indicating neutrophil infiltration in <5% of crypts, no crypt destruction and no erosions, ulcerations or granulation tissue)Studies UC-1 and UC-2 were not designed to evaluate the relationship of histologic endoscopic mucosal improvement at Week 8 to disease progression and long-term outcomes.
Rectal Bleeding and Stool Frequency Subscores
Onset of clinical response was assessed using the SFS and RBS (partial modified Mayo Score [pmMS]). Initial response was defined as a decrease of ≥1 point and ≥30% from baseline in pmMS and a decrease in RBS ≥1 or an absolute RBS≤1. Onset of response occurred as early as Week 2 in a greater proportion of patients treated with RINVOQ 45 mg once daily compared to placebo.
Endoscopic and Histologic Assessment
Normalization of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. At Week 8, a greater proportion of patients treated with RINVOQ 45 mg once daily compared to placebo achieved endoscopic remission (UC-1: 14% vs 1%, UC-2: 18% vs 2%). Endoscopic remission with Geboes histologic score < 2.0 (indicating no neutrophils in crypts or lamina propria and no increase in eosinophil, no crypt destruction, and no erosions, ulcerations, or granulation tissue) was achieved by a greater proportion of patients treated with RINVOQ 45 mg once daily compared to placebo at Week 8 (UC-1: 11% vs 1%, UC-2: 13% vs 2%).
Abdominal Pain and Bowel Urgency
A greater proportion of patients treated with RINVOQ 45 mg once daily compared to placebo had no abdominal pain (UC-1: 47% vs 23%, UC-2: 54% vs 24%) and no bowel urgency (UC-1: 48% vs 21%, UC-2: 54% vs 26%) at Week 8.
Fatigue
In Studies UC-1 and UC-2, patients treated with RINVOQ experienced a clinically meaningful improvement in fatigue, as assessed by change from baseline in FACIT-F score, at Week 8, compared to placebo-treated patients. The effect of RINVOQ to improve fatigue after 8 weeks of induction has not been established.
Maintenance Study UC-3
In UC-3 (NCT02819635), a total of 451 patients who received RINVOQ 45 mg once daily in either UC-1, UC-2 or UC-4 and achieved clinical response were re-randomized to receive RINVOQ 15 mg, 30 mg or placebo once daily for up to 52 weeks.
The primary endpoint was clinical remission defined using mMS at Week 52. Secondary endpoints included corticosteroid-free clinical remission, endoscopic improvement, and histologic endoscopic mucosal improvement (see Table 26).
Table 26: Proportion of Patients Meeting Primary and Key Secondary Efficacy Endpoints at Week 52 in Maintenance Study UC-3 Endpoint Placebo
RINVOQ
15 mg
Once Daily
Treatment Difference
15 mg vs Placebo
(95% CI)RINVOQ
30 mg
Once Daily
Treatment Difference 30 mg vs Placebo
(95% CI)Clinical remissiona Total Population N=149
12%N=148
42%31%b
(22, 40)N=154
52%39%b
(30, 48)Prior biologic failurec N=81
7%N=71
41%N=73
49%Without prior biologic failure N=68
18%N=77
44%N=81
54%Corticosteroid-free clinical remissiond Total Population N=54
22%N=47
57%35%b
(18, 53)N=58
68%45%b
(29, 62)Prior biologic failurec N=22
14%N=17
71%N=20
73%Without prior biologic failure N=32
28%N=30
49%N=38
65%Endoscopic Improvemente Total Population N=149
14%N=148
49%34%b
(25, 44)N=154
62%46%b
(37, 56)Prior biologic failurec N=81
8%N=71
43%N=73
56%Without prior biologic failure N=68
22%N=77
54%N=81
67%Histologic Endoscopic Mucosal Improvementf Total Population N=149
12%N=148
35%24%b
(15, 33)N=154
50%37%b
(28, 47)Prior biologic failurec N=81
5%N=71
33%N=73
48%Without prior biologic failure N=68
20%N=77
37%N=81
52%a Per mMS: SFS ≤1 and not greater than baseline, RBS = 0, ES ≤1 without friability
b p <0.001, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors
c Prior biologic failure includes inadequate response, loss of response, or intolerance to one or more biologic treatments for ulcerative colitis.
d Clinical remission per mMS at Week 52 and corticosteroid free for ≥90 days immediately preceding Week 52 among patients who achieved clinical remission at the end of the induction treatment
e ES ≤ 1 without friability
f ES ≤1 without friability and Geboes score ≤ 3.1 (indicating neutrophil infiltration in <5% of crypts, no crypt destruction and no erosions, ulcerations or granulation tissue)The relationship between histologic endoscopic mucosal improvement at Week 52 and disease progression and longer-term outcomes after Week 52 was not evaluated in Study UC-3.
Endoscopic and Histologic Assessment
Normalization of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. In UC-3, a greater proportion of patients treated with RINVOQ 15 mg and 30 mg once daily compared to placebo achieved endoscopic remission at Week 52 (24% and 26% vs 6%). Endoscopic remission with Geboes histologic score < 2.0 was achieved by a greater proportion of patients treated with RINVOQ 15 mg and 30 mg once daily compared to placebo at Week 52 (18% and 19% vs 5%).
Abdominal Pain and Bowel Urgency
At Week 52, a greater proportion of patients treated with RINVOQ 15 mg and 30 mg once daily compared to placebo had no abdominal pain (46%, 55% and 21%, respectively) and no bowel urgency (56%, 64% and 17%, respectively).
14.5 Crohn’s Disease
Induction Trials (Studies CD-1 and CD-2)
In two induction trials, CD-1 (NCT03345836) and CD-2 (NCT03345849), patients were randomized 2:1 to receive RINVOQ 45 mg or placebo once daily for 12 weeks. Efficacy was assessed in a population of 857 patients (419 patients in CD-1 and 438 patients in CD-2) with moderately to severely active Crohn’s disease (CD), with baseline Crohn’s Disease Activity Index (CDAI) score of at least 220 and centrally-reviewed Simple Endoscopic Score for Crohn’s Disease (SES-CD) of ≥ 6, or ≥ 4 for isolated ileal disease, excluding the narrowing component. In CD-1, all patients had inadequate response or were intolerant to treatment with one or more biological therapies (prior biologic failure). In CD-2, 45% (197/438) of patients had inadequate response or were intolerant to treatment with one or more biological therapies (prior biologic failure). Enrolled patients in both studies were permitted to use stable doses of CD-related antibiotics, aminosalicylates, or methotrexate. Concomitant corticosteroids (up to 30 mg/day prednisone or equivalent) were permitted at enrollment; tapering was initiated at Week 4.
In CD-1, patients had a mean age of 37 years (range 18 to 74 years); 46% were female; and 72% identified as White, 21% as Asian, 6% as Black or African American, 0.5% as American Indian or Alaska Native, and 0.5% as multiple racial groups. In CD-2, patients had a mean age of 40 years (range of 18 to 74 years); 45% were female; 74% identified as White, 20% as Asian, 4% as Black or African American, and 2% as multiple racial groups.
At baseline, 36% and 37% of patients received corticosteroids, 7% and 3% of patients received methotrexate, 15% and 24% of patients received aminosalicylates, and 2% and 1% of patients received CD-related antibiotics in CD-1 and CD-2, respectively.
The co-primary endpoints were the proportion of patients achieving clinical remission (by CDAI) at Week 12, and the proportion of patients achieving endoscopic response (by SES-CD) at Week 12. Secondary efficacy endpoints included clinical response, corticosteroid-free remission, and endoscopic remission (see Table 27 and Table 28).
Table 27: Proportion of Patients Meeting Primary and Additional Efficacy Endpoints in Induction Study CD-1 CD-1 Endpoint Placebo
RINVOQ
45 mg Once Daily
Treatment Difference vs Placebo
(95% CI)Co-Primary Endpoints at Week 12 Clinical remissiona N=146
18%N=273
36%17%
(9, 25)*Endoscopic responseb N=146
3%N=273
34%30%
(24, 36)*Additional Endpoints at Week 12 Clinical response (CR-100)c N=146
31%N=273
54%22%
(13, 31)*Corticosteroid-free clinical remission in patients on corticosteroids at baselinea,d N=53
11%N=96
30%17%
(5, 29)**Endoscopic remissione N=146
3%N=273
19%15%
(10, 21)** p < 0.001, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors
** p < 0.01, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors
a CDAI < 150
b Decrease in SES-CD > 50% from baseline of the induction study (or for patients with an SES-CD of 4 at baseline of the induction study, at least a 2-point reduction from baseline of the induction study)
c Decrease of at least 100 points in CDAI from baseline
d Discontinuation of corticosteroid and achievement of clinical remission among patients on corticosteroid at baseline
e SES-CD ≤ 4 and no individual subscore >1 in any individual variableTable 28: Proportion of Patients Meeting Primary and Additional Efficacy Endpoints in Induction Study CD-2 CD-2 Endpoint Placebo
RINVOQ
45 mg Once Daily
Treatment Difference
vs Placebo
(95% CI)Co-Primary Endpoints at Week 12 Clinical remissiona Total population N=143
23%N=295
46%24%
(15, 32)*Prior biologic failure N=62
7%N=135
42%Without prior biologic failure N=81
35%N=160
50%Endoscopic responseb Total population N=143
13%N=295
46%33%
(26, 41)*Prior biologic failure N=62
10%N=135
39%Without prior biologic failure N=81
15%N=160
51%Additional Endpoints at Week 12 Clinical response (CR-100)c Total population N=143
40%N=295
64%24%
(15, 33)*Prior biologic failure N=62
25%N=135
66%Without prior biologic failure N=81
52%N=160
62%Corticosteroid-free clinical remission in patients on CS at baselinea,d Total population N=54
13%N=108
40%27%
(14, 39)*Prior biologic failure N=29
7%N=60
35%Without prior biologic failure N=25
20%N=48
46%Endoscopic remissione
Total population N=143
8%N=295
30%22%
(16, 29)*Prior biologic failure N=62
3%N=135
22%Without prior biologic failure N=81
11%N=160
37%* p < 0.001, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors
a CDAI < 150
b Decrease in SES-CD > 50% from baseline of the induction study (or for patients with an SES-CD of 4 at baseline of the induction study, at least a 2-point reduction from baseline of the induction study)
c Decrease of at least 100 points in CDAI from baseline
d Discontinuation of corticosteroid and achievement of clinical remission among patients on corticosteroid at baseline
e SES-CD ≤ 4 and no individual subscore >1 in any individual variableOnset of clinical response based on CDAI was observed as early as two weeks in Studies CD-1 and CD-2, with a greater proportion of patients achieving clinical response at Week 2 in RINVOQ-treated patients compared with placebo.
In Studies CD-1 and CD-2, reductions in stool frequency and abdominal pain were observed in a greater proportion of patients treated with RINVOQ 45 mg induction regimen compared to placebo at Week 12.
In Studies CD-1 and CD-2, patients treated with RINVOQ experienced a clinically meaningful improvement in fatigue, assessed by change from baseline in FACIT-F score, at Week 12, compared to placebo-treated patients. The effect of RINVOQ to improve fatigue after 12 weeks of induction has not been established.
Maintenance Study (CD-3)
The efficacy analysis for CD-3 (NCT03345823) evaluated 343 patients who responded to 12 weeks of RINVOQ 45 mg once daily induction treatment. Patients were re-randomized to receive a maintenance regimen of either RINVOQ 15 mg or 30 mg once daily or placebo for 52 weeks, representing a total of at least 64 weeks of therapy.
The co-primary endpoints of clinical remission (by CDAI) and endoscopic response (by SES-CD) were assessed at Week 52. Secondary efficacy endpoints included corticosteroid-free clinical remission, maintenance of clinical remission, endoscopic remission, and achieving both clinical and endoscopic remission, at Week 52 (see Table 29).
Table 29: Proportion of Patients Meeting Primary and Additional Efficacy Endpoints at Week 52 in Maintenance Study CD-3 Endpoint
Placebo+ RINVOQ
15 mg
Once Daily
RINVOQ
30 mg Once Daily
Treatment Difference
15 mg vs
Placebo
(95% CI)Treatment
Difference
30 mg vs
Placebo
(95% CI)Co-Primary Endpoints Clinical remissiona Total population N=111
14%N=113
42%N=119
55%29%
(18, 39)*40%
(29, 51)*Prior biologic failure N=87
13%N=85
35%N=90
54%Without prior
biologic failureN=24
21%N=28
61%N=29
55%Endoscopic responseb Total population N=111
7%N=113
28%N=119
41%22%
(13, 32)*34%
(25, 44)*Prior biologic failure N=87
5%N=85
22%N=90
42%Without prior
biologic failureN=24
17%N=28
45%N=29
38%Additional Endpoints Corticosteroid-free clinical remissionc
Total population N=111
14%N=113
42%N=119
53%29%
(18, 39)*38%
(27, 49)*Prior biologic failure N=87
13%N=85
35%N=90
52%Without prior
biologic failureN=24
21%N=28
61%N=29
55%Maintenance of clinical remissiond Total population N=73
22%N=72
51%N=79
67%32%
(18, 46)*43%
(29, 57)*Prior biologic failure N=56
20%N=50
46%N=55
69%Without prior
biologic failureN=17
29%N=22
64%N=24
63%Endoscopic remissione Total population N=111
5%N=113
19%N=119
30%14%
(6, 22)*25%
(15, 34)*Prior biologic failure N=87
3%N=85
14%N=90
30%Without prior
biologic failureN=24
13%N=28
32%N=29
31%Clinical and endoscopic remission Total population N=111
4%N=113
16%N=119
26%13%
(6, 21)*22%
(14, 31)*Prior biologic failure N=87
2%N=85
13%N=90
26%Without prior
biologic failureN=24
8%N=28
25%N=29
28%+ The placebo group consisted of patients who achieved clinical response per CDAI with RINVOQ 45 mg at the end of the induction study and were randomized to receive placebo at the start of maintenance therapy.
* p < 0.001, adjusted treatment difference (95% CI) based on Cochran-Mantel-Haenszel method adjusted for randomization stratification factors
a CDAI < 150
b Decrease in SES-CD > 50% from baseline of the induction study (or for patients with an SES-CD of 4 at baseline of the induction study, at least a 2-point reduction from baseline of the induction study)
c Corticosteroid-free for 90 days prior to Week 52 and achievement of clinical remission. Among the subset of patients who were on corticosteroids at induction baseline, 48% (N=44) in RINVOQ 15 mg group, 44% (N=45) in RINVOQ 30 mg group, and 7% (N=46) in placebo were corticosteroid-free for 90 days prior to Week 52 and in clinical remission.
d Defined as achievement of clinical remission at Week 52 in patients who achieved clinical remission at the entry of the maintenance study.
e SES-CD ≤ 4 and no subscore > 1 in any individual variable
At Week 52, reductions in stool frequency and abdominal pain were observed in a greater proportion of patients treated with RINVOQ 15 mg and 30 mg compared to placebo.14.6 Ankylosing Spondylitis
The efficacy and safety of RINVOQ 15 mg once daily were assessed in two randomized, double-blind, multicenter, placebo-controlled trials in patients 18 years of age or older with active ankylosing spondylitis based upon the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 and Patient’s Assessment of Total Back Pain score ≥4.
Trial AS-I (NCT03178487) was a 14-week trial in 187 ankylosing spondylitis patients with an inadequate response to at least two nonsteroidal anti-inflammatory drugs (NSAIDs) or intolerance to or contraindication for NSAIDs and had no previous exposure to biologic DMARDs. At baseline, patients had symptoms of ankylosing spondylitis for an average of 14.4 years and approximately 16% of the patients were on a concomitant cDMARD. Patients received RINVOQ 15 mg once daily or placebo. At Week 14, all patients randomized to placebo were switched to RINVOQ 15 mg once daily. The primary endpoint was the proportion of patients achieving an Assessment of SpondyloArthritis international Society 40 (ASAS40) response at Week 14.
Trial AS-II (NCT 04169373) was a 14-week trial in 420 ankylosing spondylitis patients with an inadequate response to 1 or 2 biologic DMARDs. At baseline, patients had symptoms of ankylosing spondylitis for an average of 12.8 years and approximately 31% of the patients were on a concomitant cDMARD. Patients received RINVOQ 15 mg once daily or placebo. At Week 14, all patients randomized to placebo were switched to RINVOQ 15 mg once daily. The primary endpoint was the proportion of patients achieving an Assessment of SpondyloArthritis international Society 40 (ASAS40) response at Week 14.
Clinical Response
In both trials, a significantly greater proportion of patients treated with RINVOQ 15 mg achieved an ASAS40 response compared to placebo at Week 14 (Table 30, Figure 5).
Examination of gender, baseline body mass index (BMI), and baseline hsCRP did not identify differences in response to RINVOQ among these subgroups at Week 14.
Table 30: Clinical Response at Week 14 Trial AS-I
bDMARD-naïveTrial AS-II
bDMARD-IRPBO
(N=94)RINVOQ 15 mg
(N=93)Difference from PBO
(95% CI)PBO
(N=209)RINVOQ 15 mg
(N=211)Difference from PBO
(95% CI)ASAS40a (%) 25.5 50.5 25 (12, 38) 18.2 44.5 26 (18, 35) ASAS20a (%) 39.4 63.4 24 (10, 38) 38.3 65.4 27 (18, 36) Abbreviations: ASAS20 (or 40) = Assessment of SpondyloArthritis international Society ≥20% (or ≥40%) improvement; bDMARD = biologic disease modifying anti-rheumatic drug; IR = inadequate responders; PBO = placebo
a An ASAS20 (ASAS40) response is defined as a ≥20% (≥40%) improvement and an absolute improvement from baseline of ≥1 (≥2) unit(s) (range 0 to 10) in ≥3 of 4 domains (Patient Global, Total Back Pain, Function, and Inflammation), and no worsening in the potential remaining domain (defined as worsening ≥20% and ≥1 unit for ASAS20 or defined as worsening of > 0 units for ASAS40).
For binary endpoints, Week 14 results are based on non-responder imputation (Trial AS-I) and on non-responder imputation in conjunction with multiple imputation (Trial AS-II).Treatment with RINVOQ 15 mg resulted in improvements in the individual components of the ASAS40 response criteria compared to placebo (Table 31).
Table 31: ASAS Components and Other Measures of Disease Activitya Trial AS-I
bDMARD-naïveTrial AS-II
bDMARD-IRTreatment Group PBO RINVOQ
15 mgPBO RINVOQ
15 mgN 94 93 209 211 Patient Global Assessment of Disease Activityb Baseline 6.8 (1.66) 6.6 (1.81) 7.2 (1.40) 7.4 (1.48) Week 14 5.4 (1.97) 3.8 (2.44) 5.9 (2.13) 4.3 (2.36) Total Back Painb Baseline 6.7 (1.78) 6.8 (1.77) 7.4 (1.43) 7.5 (1.48) Week 14 5.0 (2.27) 3.7 (2.39) 5.9 (2.09) 4.4 (2.48) BASFIb Baseline 5.54 (2.17) 5.35 (2.36) 6.18 (1.87) 6.28 (2.03) Week 14 4.21 (2.26) 3.14 (2.37) 5.09 (2.21) 3.98 (2.45) Inflammationc Baseline 6.66 (1.90) 6.51 (1.99) 6.75 (1.55) 6.88 (1.84) Week 14 4.61 (2.13) 3.40 (2.16) 5.11 (2.30) 3.87 (2.50) hsCRP (mg/L) Baseline 11.02 (10.85) 8.90 (12.42) 14.71 (17.54) 15.30 (20.53) Week 14 11.72 (15.93) 2.23 (3.56) 15.31 (17.55) 3.82 (8.26) Abbreviations: ASAS = Assessment of SpondyloArthritis international Society; BASFI = Bath Ankylosing Spondylitis Functional Index; bDMARD = biologic disease modifying anti-rheumatic drug; hsCRP = high sensitivity C-reactive protein; IR = inadequate responder; PBO = placebo; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index
Results are based on as-observed data from patients who have baseline observation.
a Data shown are mean (standard deviation).
b Numeric rating scale (NRS): 0 = best, 10 = worst
c mean of BASDAI questions 5 and 6 assessing morning stiffness severity and duration: 0 = best, 10 = worstFigure 5. Percent of Patients Achieving ASAS40 in Trial AS-II*
*Results are based on non-responder imputation in conjunction with multiple imputation.
Other Health-Related Outcomes
In Trial AS-II, patients treated with RINVOQ 15 mg showed significant improvements in health-related quality of life as measured by Ankylosing Spondylitis Quality of Life (ASQoL) compared to placebo at Week 14. In Trial AS-I, improvement in ASQoL compared to placebo was also observed.
Enthesitis
In Trial AS-II, patients with pre-existing enthesitis treated with RINVOQ 15 mg showed significant improvement in enthesitis compared to placebo as measured by change from baseline in Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) at Week 14. In Trial AS-I, improvement in MASES compared to placebo was also observed.
Spinal mobility
In Trial AS-II, patients treated with RINVOQ 15 mg showed significant improvement in spinal mobility compared to placebo as measured by change from baseline in Bath Ankylosing Spondylitis Metrology Index (BASMI) at Week 14. In Trial AS-I, improvement in BASMI compared to placebo was also observed.
14.7 Non-radiographic Axial Spondyloarthritis
The efficacy and safety of RINVOQ 15 mg once daily were assessed in a randomized, double-blind, multicenter, placebo-controlled trial in patients 18 years of age or older with active non-radiographic axial spondyloarthritis. Trial nr-axSpA (NCT04169373) was a 52-week placebo-controlled trial in 314 patients (of which 313 patients received study treatment) with active non-radiographic axial spondyloarthritis with an inadequate response to at least two nonsteroidal anti-inflammatory drugs (NSAIDs) or intolerance to or contraindication for NSAIDs. Patients must have had objective signs of inflammation indicated by elevated C-reactive protein (CRP) (defined as > upper limit of normal), and/or sacroiliitis on magnetic resonance imaging (MRI), and no definitive radiographic evidence of structural damage on sacroiliac joints. Patients had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, and a Patient's Assessment of Total Back Pain score ≥ 4 based on a 0 – 10 numerical rating scale (NRS) at the Screening and baseline Visits. At baseline, approximately 29.1% of the patients were on a concomitant cDMARD. 32.9% of the patients had an inadequate response or intolerance to bDMARD therapy. Patients received RINVOQ 15 mg once daily or placebo. The primary endpoint was the proportion of patients achieving an Assessment of SpondyloArthritis international Society 40 (ASAS40) response at Week 14.
Clinical Response
In Trial nr-axSpA, a significantly greater proportion of patients treated with RINVOQ 15 mg achieved an ASAS40 response compared to placebo at Week 14 (Table 32, Figure 6).
Examination of gender, baseline BMI, symptom duration of non-radiographic axial spondyloarthritis, baseline hsCRP, MRI sacroiliitis, and prior use of bDMARDs did not identify differences in response to RINVOQ among these subgroups at Week 14.
Table 32: Clinical Response at Week 14 PBO
(N=157)
RINVOQ 15 mg
(N=156)
Difference from PBO (95% CI) ASAS40a (%) 22.3 44.9 22.5
(12.4, 32.5)ASAS20a (%) 43.3 66.7 23.2
(12.6, 33.8)Abbreviations: ASAS20 (or 40) = Assessment of SpondyloArthritis international Society ≥20% (or ≥40%) improvement; PBO = placebo
a An ASAS20 (ASAS40) response is defined as a ≥20% (≥40%) improvement and an absolute improvement from baseline of ≥1 (≥2) unit(s) (range 0 to 10) in ≥3 of 4 domains (Patient Global, Total Back Pain, Function, and Inflammation), and no worsening in the potential remaining domain (defined as worsening ≥20% and ≥1 unit for ASAS20 or defined as worsening of > 0 units for ASAS40).
For binary endpoints, results are based on non-responder imputation in conjunction with multiple imputation.Treatment with RINVOQ 15 mg resulted in improvements in the individual components of the ASAS40 response criteria compared to placebo (Table 33).
Table 33: ASAS Components and Other Measures of Disease Activitya Treatment Group PBO
(N=157)
RINVOQ 15 mg
(N=156)Patient Global Assessment of Disease Activityb Baseline 7.30 (1.38) 6.99 (1.62) Week 14 5.35 (2.31) 4.16 (2.38) Total Back Painb Baseline 7.29 (1.39) 7.23 (1.55) Week 14 5.27 (2.36) 4.29 (2.49) BASFIb Baseline 5.99 (2.14) 5.89 (2.08) Week 14 4.47 (2.42) 3.33 (2.39) Inflammationc Baseline 6.68 (1.67) 6.60 (1.83) Week 14 4.69 (2.36) 3.48 (2.51) hsCRP (mg/L) Baseline 8.75 (12.91) 9.93 (16.17) Week 14 7.25 (10.61) 2.84 (4.90) Abbreviations: ASAS = Assessment of SpondyloArthritis international Society; BASFI = Bath Ankylosing Spondylitis Functional Index; hsCRP = high sensitivity C-Reactive Protein; PBO = placebo
a Data shown are mean (standard deviation).
b Numeric rating scale (NRS): 0 = best, 10 = worst
c mean of Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) questions 5 and 6 assessing morning stiffness severity and duration: 0 = best, 10 = worstFigure 6. Percent of Patients Achieving ASAS40*
*Results are based on non-responder imputation in conjunction with multiple imputation.
Other Health-Related Outcomes
Patients treated with RINVOQ 15 mg showed significant improvements in health-related quality of life as measured by Ankylosing Spondylitis Quality of Life (ASQoL) compared to placebo at Week 14.
14.8 Polyarticular Juvenile Idiopathic Arthritis
The efficacy of RINVOQ/RINVOQ LQ in pediatric patients with JIA with active polyarthritis is based on exposure-matched extrapolation of the established efficacy of RINVOQ in rheumatoid arthritis patients. Safety and efficacy of RINVOQ/RINVOQ LQ were also assessed in a multicenter, open-label, single-arm study in 83 children (2 to < 18 years of age) with JIA with active polyarthritis (NCT03725007). The pJIA patient subtypes at study entry included rheumatoid factor negative polyarticular (68.7%), rheumatoid factor positive polyarticular (15.7%), extended oligoarticular (13.3%), and systemic JIA without systemic manifestations (2.4%). All patients received RINVOQ LQ or RINVOQ tablet dosages based on weight for up to 156 weeks. Patients treated with a stable dose of MTX were permitted to enter the study; changes in MTX dose were permitted during the study. Efficacy was assessed as supportive endpoints through Week 48. The efficacy was generally consistent with responses in patients with rheumatoid arthritis.
14.9 Giant Cell Arteritis
The efficacy and safety of RINVOQ 15 mg once daily were assessed in Trial GCA (NCT03725202), a Phase 3 randomized, double-blind, multicenter, placebo-controlled study in patients 50 years of age and older (81.8% were 65 years of age and older, 73.1% were female, 93.7% white) with new-onset or relapsing giant cell arteritis. Trial GCA was a 52-week trial in which 428 patients were randomized in a 2:1:1 ratio and dosed once daily with RINVOQ 15 mg, upadacitinib 7.5 mg, or placebo. All patients received background corticosteroid therapy. The RINVOQ and upadacitinib-treated groups followed a pre-specified corticosteroid taper regimen with the aim to reach 0 mg by 26 weeks, while the placebo-treated group followed a pre-specified corticosteroid taper regimen with the aim to reach 0 mg by 52 weeks. The primary endpoint was the proportion of patients achieving sustained remission at Week 52 as defined by the absence of giant cell arteritis signs and symptoms from Week 12 through Week 52 and adherence to the protocol-defined corticosteroid taper regimen. A key secondary endpoint was the proportion of patients achieving sustained complete remission at Week 52 as defined by the absence of giant cell arteritis signs and symptoms from Week 12 through Week 52, normalization of erythrocyte sedimentation rate and hsCRP from Week 12 through Week 52, and adherence to the protocol-defined corticosteroid taper regimen. The study included a 52-week extension for a total study duration of up to 2 years.
Clinical Response
RINVOQ 15 mg and a 26-week corticosteroid taper showed superiority in achieving corticosteroid-free sustained remission at Week 52 compared to placebo and a 52-week corticosteroid taper (Table 34).
Table 34. Clinical Response in Trial GCA Treatment Group PBO + 52-week
corticosteroid taper
N =112RINVOQ 15 mg + 26-week
corticosteroid taper
N =209Treatment Difference vs PBO (95% CI) Sustained remission at Week 52 33
29.0%97
46.4%17.1%
(6.3, 27.8)aSustained complete remission at Week 52 18
16.1%78
37.1%20.7%
(11.3, 30.2)bComponents of sustained complete remission at Week 52 Absence of GCA signs and symptomsc 33
29.8%97
46.3%Normalization of ESRd 27
23.8%98
47.0%Normalization of hsCRPe 28
25.0%110
52.6%Adherence to protocol-defined corticosteroid taper regimen
44
39.3%
122
58.4%Abbreviations: ESR = erythrocyte sedimentation rate; GCA = giant cell arteritis; hsCRP = high sensitivity C-reactive protein; PBO = placebo
a p≤0.01
b p≤0.001
c Patients who did not have any signs or symptoms of GCA recorded from Week 12 through Week 52
d ESR to ≤ 30 mm/hr (if ESR > 30 mm/hr and elevation is not attributable to GCA, this criterion can still be met) from Week 12 through Week 52
e hsCRP to < 1 mg/dL, without elevation to ≥ 1 mg/dL (on 2 consecutive visits) from Week 12 through Week 52Treatment with RINVOQ 15 mg and a 26-week corticosteroid taper resulted in improvements in the components of corticosteroid-free sustained complete remission at Week 52 compared to placebo and a 52-week corticosteroid taper (Table 34).
Cumulative Corticosteroid Dose
The total actual cumulative corticosteroid dose was lower in the RINVOQ 15 mg and a 26-week corticosteroid taper arm (mean [SD] 2283.9 [1840.0] mg and median 1615 mg) relative to the placebo and a 52-week corticosteroid taper arm (mean [SD] 3388.3 [1463.1] mg and median 2882 mg).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
RINVOQ extended-release tablets are supplied as:
- 15 mg: purple, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a15’ on one side.
30 tablets in a bottle; NDC: 0074-2306-30
- 30 mg: red, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a30’ on one side.
30 tablets in a bottle; NDC: 0074-2310-30
- 45 mg: yellow to mottled yellow, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a45’ on one side.
28 tablets in a bottle; NDC: 0074-1043-28
RINVOQ LQ oral solution is supplied as:
- A 1 mg/mL oral solution in HDPE bottles with a child-resistant cap. Each bottle contains a labeled volume of 180 mL of clear, colorless to light yellow solution. The bottle is packaged in a carton with one press-in bottle adapter and one 10 mL oral dosing syringe; NDC: 0074-2320-01
Storage and Handling
RINVOQ extended-release tablets
- Store at 2˚C to 25˚C (36˚F to 77˚F).
- Store in the original bottle in order to protect from moisture.
RINVOQ LQ oral solution
- Store between 2°C to 30°C (36°F to 86°F).
- Discard remaining oral solution 60 days after opening the bottle.
- 15 mg: purple, biconvex oblong, with dimensions of 14 x 8 mm, and debossed with ‘a15’ on one side.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Serious Infections
Inform patients that they may be more likely to develop infections when taking RINVOQ/RINVOQ LQ. Instruct patients to contact their healthcare provider immediately during treatment if they develop any signs or symptoms of an infection [see Warnings and Precautions (5.1)].
Advise patients that the risk of herpes zoster is increased in patients taking RINVOQ/RINVOQ LQ and in some cases can be serious [see Warnings and Precautions (5.1)].
Malignancies
Inform patients that RINVOQ/RINVOQ LQ may increase their risk of certain cancers and that periodic skin examinations should be performed while using RINVOQ/RINVOQ LQ.
Advise patients that exposure to sunlight and UV light should be limited by wearing protective clothing and using a broad-spectrum sunscreen [see Warnings and Precautions (5.3)].
Major Adverse Cardiovascular Events
Inform patients that RINVOQ/RINVOQ LQ may increase their risk of major adverse cardiovascular events (MACE) including myocardial infarction, stroke, and cardiovascular death. Instruct all patients, especially current or past smokers or patients with other cardiovascular risk factors, to be alert for the development of signs and symptoms of cardiovascular events [see Warnings and Precautions (5.4)].
Thrombosis
Inform patients that events of deep venous thrombosis and pulmonary embolism have been reported in clinical trials with RINVOQ. Instruct patients to seek immediate medical attention if they develop any signs or symptoms of a DVT or PE [see Warnings and Precautions (5.5)].
Hypersensitivity Reactions
Advise patients to discontinue RINVOQ/RINVOQ LQ and seek immediate medical attention if they develop any signs and symptoms of allergic reactions [see Warnings and Precautions (5.6)].
Gastrointestinal Perforations
Inform patients that gastrointestinal perforations have been reported in clinical trials with RINVOQ and that risk factors include the use of NSAIDs, corticosteroids, or history of diverticulitis. Instruct patients to seek medical care immediately if they experience new onset of abdominal pain, fever, chills, nausea, or vomiting [see Warnings and Precautions (5.7)].
Retinal Detachment
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ/RINVOQ LQ [see Adverse Reactions (6.1)].
Laboratory Abnormalities
Inform patients that RINVOQ/RINVOQ LQ may affect certain lab tests, and that blood tests are required before and during RINVOQ/RINVOQ LQ treatment [see Warnings and Precautions (5.8)].
Vaccinations
Advise patients to avoid use of live vaccines with RINVOQ/RINVOQ LQ. Instruct patients to inform their healthcare practitioner that they are taking RINVOQ/RINVOQ LQ prior to a potential vaccination [see Warnings and Precautions (5.10)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that exposure to RINVOQ/RINVOQ LQ during pregnancy may result in fetal harm. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.9) and Use in Specific Populations (8.1)].
Advise females of reproductive potential that effective contraception should be used during treatment and for 4 weeks following the final dose of RINVOQ/RINVOQ LQ [see Use in Specific Populations (8.3)].
Advise women exposed to RINVOQ/RINVOQ LQ during pregnancy that there is a pregnancy surveillance program that monitors pregnancy outcomes [see Use in Specific Populations (8.1)].
Lactation
Advise women not to breastfeed during treatment with RINVOQ/RINVOQ LQ and for 6 days after the last dose [see Use in Specific Populations (8.2)].
Administration
Advise patients that RINVOQ tablets are not substitutable with RINVOQ LQ [see Dosage and Administration (2.2)].
Advise patients not to chew, crush, or split RINVOQ tablets [see Dosage and Administration (2.2)].
For RINVOQ LQ, instruct patients and caregivers to read and follow the Instructions for Use for proper preparation, administration, storage, and disposal [See Dosage and Administration (2.2)].
Advise patients to avoid food or drink containing grapefruit during treatment with RINVOQ/RINVOQ LQ [see Drug Interactions (7.1)].
Medication Residue in Stool
Instruct patients to notify their healthcare provider if they repeatedly notice medication residue (e.g., intact RINVOQ tablet or fragments) in stool or ostomy output [see Warnings and Precautions (5.11)].
Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® is a registered trademark of AbbVie Biotechnology Ltd.
©2019-2025 AbbVie Inc.
20095150 October 2025 -
MEDICATION GUIDE
MEDICATION GUIDE RINVOQ® (RIN-VOKE)
(upadacitinib)
extended-release tablets, for oral useRINVOQ® LQ (RIN-VOKE EL-CUE)
(upadacitinib)
oral solutionWhat is the most important information I should know about RINVOQ/RINVOQ LQ?
RINVOQ/RINVOQ LQ can cause serious side effects, including:
1. Serious Infections.
RINVOQ/RINVOQ LQ is a medicine that affects your immune system. RINVOQ/RINVOQ LQ can lower the ability of your immune system to fight infections. Some people have had serious infections while taking RINVOQ, including tuberculosis (TB), and infections caused by bacteria, fungi, or viruses that can spread throughout the body. Some people have died from these infections.- Your healthcare provider should test you for TB before starting treatment with RINVOQ/RINVOQ LQ.
- Your healthcare provider should watch you closely for signs and symptoms of TB during treatment with RINVOQ/RINVOQ LQ.
- You should not start taking RINVOQ/RINVOQ LQ if you have any kind of infection unless your healthcare provider tells you it is okay. You may be at a higher risk of developing shingles (herpes zoster).
-
Before starting RINVOQ/RINVOQ LQ, tell your healthcare provider if you:
○ are being treated for an infection.
○ have had an infection that does not go away or that keeps coming back.
○ have diabetes, chronic lung disease, HIV, or a weak immune system.
○ have TB or have been in close contact with someone with TB.
○ have had shingles (herpes zoster).
○ have or have had hepatitis B or C.
○ live or have lived, or have traveled to certain parts of the country (such as the Ohio and Mississippi River valleys and the Southwest) where there is an increased chance for getting certain kinds of fungal infections. These infections may happen or become more severe if you use RINVOQ/RINVOQ LQ. Ask your healthcare provider if you do not know if you have lived in an area where these infections are common.
○ think you have an infection or have symptoms of an infection such as:
- fever, sweating, or chills
- shortness of breath
- warm, red, or painful skin or sores on your body
- muscle aches
- feeling tired
- blood in your phlegm
- diarrhea or stomach pain
- cough
- weight loss
- burning when you urinate or urinating more often than usual
After starting RINVOQ/RINVOQ LQ, call your healthcare provider right away if you have any symptoms of an infection. RINVOQ/RINVOQ LQ can make you more likely to get infections or make worse any infections that you have. If you get a serious infection, your healthcare provider may stop your treatment with RINVOQ/RINVOQ LQ until your infection is controlled.
2. Increased risk of death in people 50 years of age and older who have at least 1 heart disease (cardiovascular) risk factor and are taking a medicine in the class of medicines called Janus kinase (JAK) inhibitors. RINVOQ/RINVOQ LQ is a JAK inhibitor medicine.
3. Cancer and immune system problems.
RINVOQ/RINVOQ LQ may increase your risk of certain cancers by changing the way your immune system works.
Lymphoma and other cancers, including skin cancers can happen in people taking RINVOQ/RINVOQ LQ. People taking a medicine in the class of medicines called Janus kinase (JAK) inhibitors have a higher risk of certain cancers including lymphoma and lung cancer, especially if you are a current or past smoker.
Tell your healthcare provider if you have ever had any type of cancer. Follow your healthcare provider’s advice about having your skin checked for skin cancer during treatment with RINVOQ/RINVOQ LQ. Limit the amount of time you spend in sunlight. Avoid using tanning beds or sunlamps. Wear protective clothing when you are in the sun and use a sunscreen with a high protection factor (SPF 30 and above). This is especially important if your skin is very fair or if you have a family history of skin cancer.
4. Increased risk of major cardiovascular events such as heart attack, stroke or death in people 50 years of age and older who have at least 1 heart disease (cardiovascular) risk factor and taking a medicine in the class of medicines called JAK inhibitors, especially if you are a current or past smoker.
Get emergency help right away if you have any symptoms of a heart attack or stroke while taking RINVOQ/RINVOQ LQ, including:
- discomfort in the center of your chest that lasts for more than a few minutes, or that goes away and comes back
- severe tightness, pain, pressure, or heaviness in your chest, throat, neck, or jaw
- pain or discomfort in your arms, back, neck, jaw, or stomach
- shortness of breath with or without chest discomfort
- breaking out in a cold sweat
- nausea or vomiting
- feeling lightheaded
- weakness in one part or on one side of your body
- slurred speech
Blood clots in the veins of your legs (deep vein thrombosis, DVT) or lungs (pulmonary embolism, PE) and arteries (arterial thrombosis) can happen in some people taking RINVOQ/RINVOQ LQ. This may be life-threatening and cause death. Blood clots in the veins of the legs (DVT) and lungs (PE) have happened more often in people who are 50 years of age and older and with at least 1 heart disease (cardiovascular) risk factor taking a medicine in the class of medicines called Janus kinase (JAK) inhibitors.- Tell your healthcare provider if you have had blood clots in the veins of your legs or lungs in the past.
- Get medical help right away if you have signs and symptoms of blood clots during treatment with RINVOQ/RINVOQ LQ, including:
○ swelling
○ pain or tenderness in one or both legs○ sudden unexplained chest or upper back pain
○ shortness of breath or difficulty breathing6. Allergic reactions. Symptoms such as rash (hives), trouble breathing, feeling faint or dizzy, or swelling of your lips, tongue, or throat, that may mean you are having an allergic reaction have been seen in people taking RINVOQ. Some of these reactions were serious. If any of these symptoms occur during treatment with RINVOQ/RINVOQ LQ, stop taking RINVOQ/RINVOQ LQ and get emergency medical help right away. 7. Tears (perforation) in the stomach or intestines. - Tell your healthcare provider if you have had diverticulitis (inflammation in parts of the large intestine) or ulcers in your stomach or intestines. Some people taking RINVOQ/RINVOQ LQ can get tears in their stomach or intestines. This happens most often in people who take nonsteroidal anti-inflammatory drugs (NSAIDs) or corticosteroids.
- Get medical help right away if you get stomach-area pain, fever, chills, nausea, or vomiting.
8. Changes in certain laboratory test results.
Your healthcare provider should do blood tests before you start taking RINVOQ/RINVOQ LQ and while you take RINVOQ/RINVOQ LQ to check for the following:
-
low neutrophil and lymphocyte counts. Neutrophils and lymphocytes are types of white blood cells that help the body fight off infections.
-
low red blood cell counts. Red blood cells carry oxygen. Low red blood cells means you may have anemia, which may make you feel weak and tired.
-
increased cholesterol levels. Your healthcare provider should do blood tests to check your cholesterol levels approximately 12 weeks after you start taking RINVOQ/RINVOQ LQ, and as needed.
- elevated liver enzymes. Liver enzymes help to tell if your liver is functioning normally. Elevated liver enzymes may indicate that your healthcare provider needs to do additional tests on your liver.
See “What are the possible side effects of RINVOQ/RINVOQ LQ?” for more information about side effects.What is RINVOQ/RINVOQ LQ?
RINVOQ/RINVOQ LQ is a prescription medicine that is a Janus kinase (JAK) inhibitor.
- RINVOQ is used to treat adults with moderate to severe rheumatoid arthritis when 1 or more medicines called tumor necrosis factor (TNF) blockers have been used, and did not work well or could not be tolerated.
- RINVOQ is used to treat adults with active psoriatic arthritis when 1 or more medicines called tumor necrosis factor (TNF) blockers have been used, and did not work well or could not be tolerated.
- RINVOQ is used to treat adults and children 12 years of age and older with moderate to severe eczema (atopic dermatitis) that did not respond to previous treatment and their eczema is not well controlled with other pills or injections, including biologic medicines, or the use of other pills or injections is not recommended.
- RINVOQ is used to treat adults with moderate to severe ulcerative colitis when 1 or more medicines called tumor necrosis factor (TNF) blockers have been used, and did not work well or could not be tolerated, or after taking a different injection or pill (systemic therapy) when your healthcare provider does not recommend TNF blockers.
- RINVOQ is used to treat adults with moderate to severe Crohn’s disease when 1 or more medicines called tumor necrosis factor (TNF) blockers have been used, and did not work well or could not be tolerated, or after taking a different injection or pill (systemic therapy) when your healthcare provider does not recommend TNF blockers.
- RINVOQ is used to treat adults with active ankylosing spondylitis when 1 or more medicines called tumor necrosis factor (TNF) blockers have been used, and did not work well or could not be tolerated.
- RINVOQ is used to treat adults with active non-radiographic axial spondyloarthritis with objective signs of inflammation when a tumor necrosis factor (TNF) blocker medicine has been used, and did not work well or could not be tolerated.
- RINVOQ/RINVOQ LQ is used to treat children 2 years of age and older with active polyarticular juvenile idiopathic arthritis (pJIA) when 1 or more medicines called tumor necrosis factor (TNF) blockers have been used, and did not work well or could not be tolerated.
- RINVOQ/RINVOQ LQ is used to treat children 2 to less than 18 years of age with active psoriatic arthritis when 1 or more medicines called tumor necrosis factor (TNF) blockers have been used, and did not work well or could not be tolerated.
- RINVOQ is used to treat adults with giant cell arteritis.
It is not known if RINVOQ is safe and effective in children under 12 years of age with atopic dermatitis.
It is not known if RINVOQ LQ is safe and effective in children with atopic dermatitis.
It is not known if RINVOQ/RINVOQ LQ is safe and effective in children under 2 years of age with pJIA or psoriatic arthritis.Do not take RINVOQ/RINVOQ LQ if you are allergic to upadacitinib or any of the ingredients in RINVOQ/RINVOQ LQ. See the end of this Medication Guide for a complete list of ingredients in RINVOQ/RINVOQ LQ.
Before taking RINVOQ/RINVOQ LQ, tell your healthcare provider about all of your medical conditions, including if you:
- See “What is the most important information I should know about RINVOQ/RINVOQ LQ?”
- have an infection.
- are a current or past smoker.
- have had a heart attack, other heart problems, or stroke.
- have liver problems.
- have kidney problems.
- have unexplained stomach (abdominal) pain, have a history of diverticulitis or ulcers in your stomach or intestines, or are taking NSAIDs.
- have low red or white blood cell counts.
- have recently received or are scheduled to receive an immunization (vaccine). People who take RINVOQ/RINVOQ LQ should not receive live vaccines.
- are pregnant or plan to become pregnant. Based on animal studies, RINVOQ/RINVOQ LQ may harm your unborn baby.
- Your healthcare provider will check whether or not you are pregnant before you start treatment with RINVOQ/RINVOQ LQ.
- You should use effective birth control (contraception) to avoid becoming pregnant during treatment with RINVOQ/RINVOQ LQ and for 4 weeks after your last dose of RINVOQ/RINVOQ LQ.
- Tell your healthcare provider if you think you are pregnant or become pregnant during treatment with RINVOQ/RINVOQ LQ.
- There is a pregnancy surveillance program for RINVOQ/RINVOQ LQ. The purpose of the program is to collect information about the health of you and your baby. If you become pregnant while taking RINVOQ/RINVOQ LQ, you are encouraged to report the pregnancy by calling 1-800-633-9110.
- are breastfeeding or plan to breastfeed. RINVOQ/RINVOQ LQ may pass into your breast milk. You and your healthcare provider should decide if you will take RINVOQ/RINVOQ LQ or breastfeed. Do not breastfeed during treatment with RINVOQ/RINVOQ LQ and for 6 days after your last dose of RINVOQ/RINVOQ LQ.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. RINVOQ/RINVOQ LQ and other medicines may affect each other causing side effects.
Especially tell your healthcare provider if you take:
- medicines for fungal infections (such as ketoconazole, itraconazole, posaconazole or voriconazole) or clarithromycin (for bacterial infections) as these medicines may increase the amount of RINVOQ/RINVOQ LQ in your blood.
- rifampicin (for bacterial infections) or phenytoin (for neurological disorders) as these medicines may decrease the effect of RINVOQ/RINVOQ LQ.
- medicines that affect your immune system (such as azathioprine and cyclosporine) as these medicines may increase your risk of infection.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take RINVOQ/RINVOQ LQ?
- Take RINVOQ/RINVOQ LQ exactly as your healthcare provider tells you to use it.
- Take RINVOQ LQ 2 times a day with or without food.
- Take RINVOQ tablets 1 time a day with or without food.
- Swallow RINVOQ tablets whole. Do not split, crush, or chew the tablets.
- RINVOQ LQ is not the same as RINVOQ tablets. Do not switch between RINVOQ LQ and RINVOQ tablets unless the change has been made by your healthcare provider.
- If you take too much RINVOQ/RINVOQ LQ, call your healthcare provider or Poison Help line at 1-800-222-1222, or go to the nearest hospital emergency room right away.
Avoid food or drink containing grapefruit during treatment with RINVOQ/RINVOQ LQ. Eating grapefruit or drinking grapefruit juice may increase the risk of side effects.
What are the possible side effects of RINVOQ/RINVOQ LQ?
RINVOQ/RINVOQ LQ may cause serious side effects, including:
- See “What is the most important information I should know about RINVOQ/RINVOQ LQ?”
• upper respiratory tract infections (common cold, sinus infections)
• shingles (herpes zoster)
• herpes simplex virus infections, including cold sores
• bronchitis• nausea
• cough
• fever
• acne
• headacheThe most common side effects of RINVOQ in people treated for giant cell arteritis include: • upper respiratory tract infections (common cold, sinus infections
• headache
• tiredness
• swelling of the feet and hands (peripheral edema)• cough
• low red blood cell count (anemia)
• rash
• shingles (herpes zoster)
• nauseaThe most common side effects of RINVOQ in people treated for atopic dermatitis include: • upper respiratory tract infections (common cold, sinus infections)
• acne
• herpes simplex virus infections, including cold sores
• headache
• increased blood levels of creatine phosphokinase
• cough
• allergic reactions
• inflammation of hair follicles
• nausea• stomach-area (abdominal) pain
• fever
• increased weight
• shingles (herpes zoster)
• flu
• tiredness
• low white blood cell count (neutropenia)
• muscle pain
• flu-like illness
The most common side effects of RINVOQ in people treated for ulcerative colitis include: • upper respiratory tract infections (common cold, sinus infections)
• acne
• herpes simplex virus infections, including cold sores
• inflammation of the hair follicles
• rash
• flu• shingles (herpes zoster)
• increased blood cholesterol levels
• increased blood levels of creatine phosphokinase
• increased liver enzyme levels
• low number of certain types of white blood cells (neutropenia, lymphopenia)
• feverThe most common side effects of RINVOQ in people treated for Crohn’s disease include: • upper respiratory tract infections (common cold, sinus infections)
• bronchitis
• pneumonia
• flu
• acne
• herpes simplex virus infections, including cold sores
• tiredness
• cough• fever
• shingles (herpes zoster)
• headache
• increased blood levels of creatine phosphokinase
• increased liver enzyme levels
• low number of red blood cells (anemia)
• low number of white blood cells (neutropenia, leukopenia)
• infection of the stomach and intestine (gastroenteritis)Separation or tear to the lining of the back part of the eye (retinal detachment) has happened in people with atopic dermatitis treated with RINVOQ. Call your healthcare provider right away if you have any sudden changes in your vision during treatment with RINVOQ/RINVOQ LQ.
Some people taking RINVOQ may see medicine residue (a whole tablet or tablet pieces) in their stool. If this happens, call your healthcare provider.
These are not all the possible side effects of RINVOQ/RINVOQ LQ.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store RINVOQ/RINVOQ LQ?- Store RINVOQ tablets between 36°F to 77°F (2°C to 25°C).
- Store RINVOQ tablets in the original bottle to protect from moisture.
- Store RINVOQ LQ between 36°F to 86°F (2°C to 30°C).
- Throw away (discard) remaining RINVOQ LQ 60 days after opening the bottle.
- Keep RINVOQ/RINVOQ LQ and all medicines out of the reach of children.
General information about the safe and effective use of RINVOQ/RINVOQ LQ.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use RINVOQ/RINVOQ LQ for a condition for which it was not prescribed.
Do not give RINVOQ/RINVOQ LQ to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about RINVOQ/RINVOQ LQ that is written for health professionals.
What are the ingredients in RINVOQ 15 mg tablets?
Active ingredient: upadacitinib
Inactive ingredients: colloidal silicon dioxide, ferrosoferric oxide, hypromellose, iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyvinyl alcohol, polyethylene glycol, talc, tartaric acid and titanium dioxide.
What are the ingredients in RINVOQ 30 mg tablets?
Active ingredient: upadacitinib
Inactive ingredients: colloidal silicon dioxide, hypromellose, iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyvinyl alcohol, polyethylene glycol, talc, tartaric acid and titanium dioxide.
What are the ingredients in RINVOQ 45 mg tablets?
Active ingredient: upadacitinib
Inactive ingredients: colloidal silicon dioxide, hypromellose, iron oxide yellow and iron oxide red, magnesium stearate, mannitol, microcrystalline cellulose, polyvinyl alcohol, polyethylene glycol, talc, tartaric acid and titanium dioxide.
What are the ingredients in RINVOQ LQ?
Active ingredient: upadacitinib
Inactive ingredients: citric acid anhydrous, purified water, sodium benzoate, sodium citrate dihydrate and sucralose.
Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® is a registered trademark of AbbVie Biotechnology Ltd.
©2019-2025 AbbVie Inc.
For more information, call 1-800 2-RINVOQ (1-800-274-6867) or go to www.RINVOQ.com.This Medication Guide has been approved by the U.S. Food and Drug Administration
20095151Revised: 10/2025 - Your healthcare provider should test you for TB before starting treatment with RINVOQ/RINVOQ LQ.
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
RINVOQ® LQ [RIN-VOKE EL-CUE]
(upadacitinib)
oral solution
This Instructions for Use contains information on how to prepare and give a dose of RINVOQ LQ oral solution.
Read this Instructions for Use before you give RINVOQ LQ to your child, and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your child’s medical condition or treatment.
Important Information You Need to Know Before Giving RINVOQ LQ
 Keep the bottle and supplies out of sight and reach of children.
Only use the syringe provided. Do not share the syringe with other people or use it with other medicines.
Call your healthcare provider or 1-800-2-RINVOQ or 1-800-274-6867 if you need help or have any questions about how to give RINVOQ LQ the right way.
Contact your healthcare provider if your child takes too much oral solution or does not receive the full dose.
Keep the bottle and supplies out of sight and reach of children.
Only use the syringe provided. Do not share the syringe with other people or use it with other medicines.
Call your healthcare provider or 1-800-2-RINVOQ or 1-800-274-6867 if you need help or have any questions about how to give RINVOQ LQ the right way.
Contact your healthcare provider if your child takes too much oral solution or does not receive the full dose.- Use RINVOQ LQ within 60 days of opening the bottle. To help you remember, write the date you opened the bottle on the carton.
-
Do not open a new bottle of RINVOQ LQ until you have finished the previous bottle. Use a new syringe when you open a new bottle.
- RINVOQ LQ oral solution is clear and colorless to light yellow in color.
- Keep these instructions and the carton your RINVOQ LQ oral solution and supplies came in for future use.
Supplies in Each Carton
Figure A
Disposal of RINVOQ LQ
Dispose of (throw away) the syringe and the bottle when you have finished the bottle or 60 days after opening. - Ask your pharmacist how to properly dispose of (throw away) any unused medicine.
- Rinse syringe then place in household trash.
Preparing RINVOQ LQ
1. Check the prescribed dose.
a. Check your child’s prescribed dose in milliliters (mL) and find this mL marking on the syringe.
b. Only use the syringe provided to give the prescribed dose.
2. Check expiration date.
a. Check the bottle and make sure the expiration date has not passed (see Figure B).
Do not use RINVOQ LQ after the expiration date printed on the carton and the bottle after “EXP.” 
Figure B
3. Check supplies.
a. Check the supplies and make sure they are not damaged (see Figure C).
b. Only use the syringe if it is clean and dry.
c. Make sure that the plunger is all the way in the syringe.
Do not use the supplies if they are wet, damaged, or appear to be tampered with.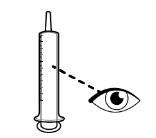
Figure C
4. Open the bottle.
a. Press down and twist the cap to remove it from the bottle (see Figure D). Do not throw away the cap.
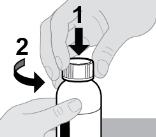
Figure D
5. Insert the adapter (first time only).
a. While holding the bottle firmly, use your thumb to push the adapter all the way down to the rim of the bottle (see Figure E).
Note: You may need to apply pressure to the adapter.
Do not remove adapter after it is inserted.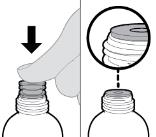
Figure E
Measuring the Dose
6. Insert the syringe into the bottle then turn it upside down.
a. Insert the tip of the syringe into the adapter.
b. With the syringe attached to the bottle, turn the bottle upside down (see Figure F).
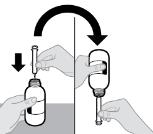
Figure F
7. Draw the oral solution into the syringe.
a. Slowly pull the plunger down (see Figure G).
b. Check the syringe for air bubbles.
Note: You may feel pressure when pulling the plunger.
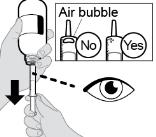
Figure G
8. Remove large air bubbles (see Figure H).
a. While holding the bottle, flick the sides of the syringe to send large air bubbles to the tip.
b. With the syringe attached to the bottle, move the plunger up and down to return air bubbles back into the bottle.
c. Repeat Step 8 until any large air bubbles are gone.
Note: Small air bubbles are normal.
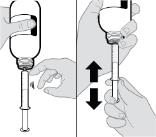
Figure H
9. Measure the dose.
a. After any large air bubbles are removed, move the plunger until it is even with the marking of the dose (see Figure I).
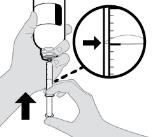
Figure I
10. Turn the bottle upright, and then remove the syringe.
a. With the syringe attached to the bottle, turn the bottle upright.
b. Hold the middle of the syringe and carefully remove it from the bottle (see Figure J).
Do not touch the plunger to avoid oral solution accidentally coming out of the syringe before you are ready to give the medicine.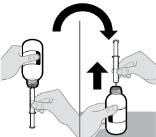
Figure J
Giving RINVOQ LQ
11. Check the dose.
a. Check that the syringe has the correct dose of oral solution (see Figure K).
b. Check the syringe for large air bubbles.
c. If the dose is not correct or you see large air bubbles, return to Step 6.
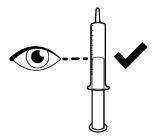
Figure K
12. Give the oral solution.
a. Place the syringe against the inside of the child’s cheek.
b. Push the plunger to give the entire dose into the child’s mouth (see Figure L).
c. Give the child a drink of water.
Note: Oral solution must be given within 1 hour of filling the syringe.
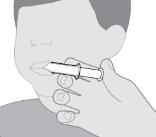
Figure L
Storing RINVOQ LQ
13. Close and store the bottle.
a. With the adapter still inserted in the bottle, screw the cap back on to the bottle to seal it (see Figure M).
b. Store the bottle upright in the carton between uses.
c. Store the bottle in the carton between 36°F to 86°F (2°C to 30°C) in a cool, dark place.
d. Keep the bottle, supplies, and all medicines out of the sight and reach of children.
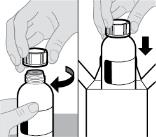
Figure M
14. Rinse and store the syringe.
a. Remove the plunger from the syringe then rinse both parts with water (see Figure N).
Do not use soap or put the syringe in the dishwasher to clean it.b. Allow separated parts to air dry on a clean surface.
c. Store the syringe in a clean, dry place.
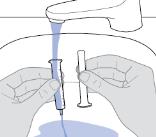
Figure N
15. When you have finished the bottle, see “Disposal of RINVOQ LQ.”

Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
RINVOQ® is a registered trademark of AbbVie Biotechnology Ltd.
©2019-2024 AbbVie Inc.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Issued: April 2024
20081174 R1
- Use RINVOQ LQ within 60 days of opening the bottle. To help you remember, write the date you opened the bottle on the carton.
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
RINVOQ
upadacitinib tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-2306 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength upadacitinib (UNII: 4RA0KN46E0) (upadacitinib - UNII:4RA0KN46E0) upadacitinib 15 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FERROSOFERRIC OXIDE (UNII: XM0M87F357) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) FERRIC OXIDE RED (UNII: 1K09F3G675) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TARTARIC ACID (UNII: W4888I119H) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color purple Score no score Shape OVAL (biconvex oblong) Size 14mm Flavor Imprint Code a15 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-2306-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 08/16/2019 2 NDC:0074-2306-70 14 in 1 BOTTLE; Type 0: Not a Combination Product 08/16/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211675 08/16/2019 RINVOQ
upadacitinib tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-2310 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength upadacitinib (UNII: 4RA0KN46E0) (upadacitinib - UNII:4RA0KN46E0) upadacitinib 30 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) FERRIC OXIDE RED (UNII: 1K09F3G675) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TARTARIC ACID (UNII: W4888I119H) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color red Score no score Shape OVAL (oblong) Size 14mm Flavor Imprint Code a30 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-2310-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 08/16/2019 2 NDC:0074-2310-20 14 in 1 BOTTLE; Type 0: Not a Combination Product 08/16/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211675 08/16/2019 RINVOQ
upadacitinib tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-1043 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength upadacitinib (UNII: 4RA0KN46E0) (upadacitinib - UNII:4RA0KN46E0) upadacitinib 45 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TARTARIC ACID (UNII: W4888I119H) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color yellow Score no score Shape OVAL Size 14mm Flavor Imprint Code a45 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-1043-28 28 in 1 BOTTLE; Type 0: Not a Combination Product 03/16/2022 2 NDC:0074-1043-14 14 in 1 BOTTLE; Type 0: Not a Combination Product 03/16/2022 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211675 08/16/2019 RINVOQ
upadacitinib solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0074-2320 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength UPADACITINIB (UNII: 4RA0KN46E0) (UPADACITINIB - UNII:4RA0KN46E0) UPADACITINIB 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) WATER (UNII: 059QF0KO0R) SODIUM BENZOATE (UNII: OJ245FE5EU) TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color yellow (Light Yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0074-2320-01 1 in 1 CARTON 04/26/2024 1 180 mL in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC:0074-2320-70 1 in 1 CARTON 04/26/2024 2 180 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218347 04/26/2024 Labeler - AbbVie Inc. (078458370)