Label: INFLAMMATION REDUCTION PACK kit





-
Contains inactivated NDC Code(s)
NDC Code(s): 69176-010-03 - Packager: TMIG, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated October 27, 2015
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- Kit Description
-
Ranitidine hydrochloride
The active ingredient in ranitidine tablets USP 150 mg and 300 mg is ranitidine hydrochloride (HCl), USP, a histamine H2-receptor antagonist. Chemically it is N[2-[[[5-[(dimethylamino)methyl]-2-furanyl]methyl]thio]ethyl]-N'-methyl-2-nitro-1,1-ethenediamine, HCl. It has the following structure:
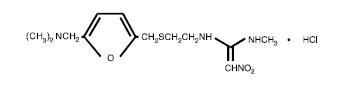
The empirical formula is C13H22N4O3S•HCl, representing a molecular weight of 350.87.
Ranitidine HCl USP is a white to pale yellow, granular substance that is soluble in water. It has a slightly bitter taste and sulfur like odor.
Each ranitidine tablet USP 150 mg for oral administration contains 168 mg of ranitidine HCl USP equivalent to 150 mg of ranitidine. Each tablet also contains the inactive ingredients microcrystalline cellulose, croscarmellose sodium, colloidal silicon dioxide, magnesium stearate, FD&C Red # 40 Aluminum Lake, hypromellose, titanium dioxide, triacetin.
-
Diclofenac sodium
Diclofenac sodium delayed-release tablets, USP are a benzeneacetic acid derivative. The chemical name is 2-[(2,6-dichlorophenyl)amino] benzeneacetic acid, monosodium salt. The molecular weight is 318.13. Its molecular formula is C14H10Cl2NNaO2, and it has the following structural formula
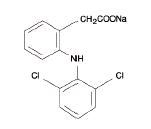
Each enteric-coated tablet for oral administration contains 50 mg or 75 mg of diclofenac sodium, USP. In addition, each tablet contains the following inactive ingredients: aluminum hydrate, colloidal silicon dioxide, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, polyvinyl acetate phthalate, propylene glycol, silica, sodium alginate, sodium starch glycolate (Type A), stearic acid, synthetic black iron oxide, talc, and titanium dioxide.
-
Lidocaine 2.5% and Prilocaine 2.5%
Lidocaine 2.5% and Prilocaine 2.5% Cream, USP is an emulsion in which the oil phase is a eutectic mixture of lidocaine and prilocaine cream in a ratio of 1:1 by weight. This eutectic mixture has a melting point below room temperature and therefore both local anesthetics exist as a liquid oil rather than as crystals. It is packaged in 5 gram and 30 gram tubes.
Lidocaine is chemically designated as acetamide, 2-(diethylamino)-N-(2,6-dimethylphenyl), has an octanol: water partition ratio of 43 at pH 7.4, and has the following structure: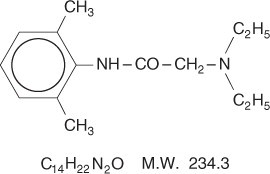
Prilocaine is chemically designated as propanamide, N-(2-methylphenyl)-2-(propylamino), has an octanol: water partition ratio of 25 at pH 7.4, and has the following structure:
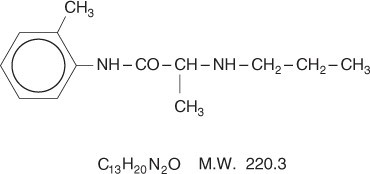
Each gram of lidocaine and prilocaine cream contains lidocaine 25 mg, prilocaine 25 mg, polyoxyethylene fatty acid esters (as emulsifiers), carboxypolymethylene (as a thickening agent), sodium hydroxide to adjust to a pH approximating 9, and purified water to 1 gram. Lidocaine and prilocaine cream contains no preservative, however it passes the USP antimicrobial effectiveness test due to the pH. The specific gravity of lidocaine and prilocaine cream is 1.00.
-
RANITIDINE HYDROCHLORIDE
Ranitidine is a competitive, reversible inhibitor of the action of histamine at the histamine H2-receptors, including receptors on the gastric cells. Ranitidine does not lower serum Ca++ in hypercalcemic states. Ranitidine is not an anticholinergic agent.
Pharmacokinetics:
Absorption:
Ranitidine is 50% absorbed after oral administration, compared to an intravenous (IV) injection with mean peak levels of 440 to 545 ng/mL occurring 2 to 3 hours after a 150-mg dose. Absorption is not significantly impaired by the administration of food or antacids. Propantheline slightly delays and increases peak blood levels of ranitidine, probably by delaying gastric emptying and transit time. In one study, simultaneous administration of high-potency antacid (150 mmol) in fasting subjects has been reported to decrease the absorption of ranitidine.
Distribution:
The volume of distribution is about 1.4 L/kg. Serum protein binding averages 15%.
Metabolism:
In humans, the N-oxide is the principal metabolite in the urine; however, this amounts to <4% of the dose. Other metabolites are the S-oxide (1%) and the desmethyl ranitidine (1%). The remainder of the administered dose is found in the stool. Studies in patients with hepatic dysfunction (compensated cirrhosis) indicate that there are minor, but clinically insignificant, alterations in ranitidine half-life, distribution, clearance, and bioavailability.
Excretion:
The principal route of excretion is the urine, with approximately 30% of the orally administered dose collected in the urine as unchanged drug in 24 hours. Renal clearance is about 410 mL/min, indicating active tubular excretion. The elimination half-life is 2.5 to 3 hours. Four patients with clinically significant renal function impairment (creatinine clearance 25 to 35 mL/min) administered 50 mg of ranitidine intravenously had an average plasma half-life of 4.8 hours, a ranitidine clearance of 29 mL/min, and a volume of distribution of 1.76 L/kg. In general, these parameters appear to be altered in proportion to creatinine clearance (see DOSAGE AND ADMINISTRATION ).
Geriatrics:
The plasma half-life is prolonged and total clearance is reduced in the elderly population due to a decrease in renal function. The elimination half-life is 3 to 4 hours. Peak levels average 526 ng/mL following a 150-mg twice-daily dose and occur in about 3 hours (see PRECAUTIONS: Geriatric Use and DOSAGE AND ADMINISTRATION: Dosage Adjustment for Patients With Impaired Renal Function ).
Pediatrics:
There are no significant differences in the pharmacokinetic parameter values for ranitidine in pediatric patients (from 1 month up to 16 years of age) and healthy adults when correction is made for body weight. The average bioavailability of ranitidine given orally to pediatric patients is 48% which is comparable to the bioavailability of ranitidine in the adult population. All other pharmacokinetic parameter values (t1/2, Vd, and CL) are similar to those observed with intravenous ranitidine use in pediatric patients. Estimates of Cmax and Tmax are displayed in Table 1.
Table 1. Ranitidine Pharmacokinetics in Pediatric Patients Following Oral Dosing Population (age) n Dosage Form (dose) Cmax (ng/mL) Tmax (hours) Gastric or duodenal ulcer (3.5 to 16 years) 12 Tablets (1 to 2 mg/kg) 54 to 492 2.0 Plasma clearance measured in 2 neonatal patients (less than 1 month of age) was considerably lower (3 mL/min/kg) than children or adults and is likely due to reduced renal function observed in this population (see PRECAUTIONS: Pediatric Use and DOSAGE AND ADMINISTRATION: Pediatric Use ).
Pharmacodynamics:
Serum concentrations necessary to inhibit 50% of stimulated gastric acid secretion are estimated to be 36 to 94 ng/mL. Following a single oral dose of 150 mg, serum concentrations of ranitidine are in this range up to 12 hours. However, blood levels bear no consistent relationship to dose or degree of acid inhibition.
Antisecretory Activity:
1. Effects on Acid Secretion:
Ranitidine inhibits both daytime and nocturnal basal gastric acid secretions as well as gastric acid secretion stimulated by food, betazole, and pentagastrin, as shown in Table 2.
Table 2. Effect of Oral Ranitidine on Gastric Acid Secretion Time After Dose, hours % Inhibition of Gastric Acid Output by Dose, mg 75 - 80 100 150 200 Basal Up to 4 99 95 Nocturnal Up to 13 95 96 92 Betazole Up to 3 97 99 Pentagastrin Up to 5 58 72 72 80 Meal Up to 3 73 79 95 It appears that basal-, nocturnal-, and betazole-stimulated secretions are most sensitive to inhibition by ranitidine, responding almost completely to doses of 100 mg or less, while pentagastrin- and food-stimulated secretions are more difficult to suppress.
2. Effects on Other Gastrointestinal Secretions:
Pepsin: Oral ranitidine does not affect pepsin secretion. Total pepsin output is reduced in proportion to the decrease in volume of gastric juice.
Intrinsic Factor: Oral ranitidine has no significant effect on pentagastrin-stimulated intrinsic factor secretion.
Serum Gastrin: Ranitidine has little or no effect on fasting or postprandial serum gastrin.
Other Pharmacologic Actions:
- Gastric bacterial flora - increase in nitrate-reducing organisms, significance not known.
- Prolactin levels - no effect in recommended oral or intravenous (IV) dosage, but small, transient, dose-related increases in serum prolactin have been reported after IV bolus injections of 100 mg or more.
- Other pituitary hormones - no effect on serum gonadotropins, TSH, or GH. Possible impairment of vasopressin release.
- No change in cortisol, aldosterone, androgen, or estrogen levels.
- No antiandrogenic action.
- No effect on count, motility, or morphology of sperm.
Pediatrics:
Oral doses of 6 to 10 mg/kg/day in 2 or 3 divided doses maintain gastric pH>4 throughout most of the dosing interval.
Active Duodenal Ulcer:
In a multicenter, double-blind, controlled, US study of endoscopically diagnosed duodenal ulcers; earlier healing was seen in the patients treated with ranitidine as shown in Table 3.
Table 3. Duodenal Ulcer Patient healing Rates * All patients were permitted antacids as needed for relief of pain. †p < 0.0001. Ranitidine * Placebo* Number Entered Healed/Evaluable Number Entered Healed/Evaluable Outpatients 195 69/182 (38%) † 188 31/164 (19%) Week 2 Week 4 137/187 (73%) † 76/168 (45%) In these studies, patients treated with ranitidine reported a reduction in both daytime and nocturnal pain, and they also consumed less antacid than the placebo-treated patients.
Table 4. Mean Daily Doses of Antacid Ulcer Healed Ulcer Not Healed Ranitidine 0.06 0.71 Placebo 0.71 1.43 Foreign studies have shown that patients heal equally well with 150 mg twice daily and 300 mg at bedtime (85% versus 84%, respectively) during a usual 4-week course of therapy. If patients require extended therapy of 8 weeks, the healing rate may be higher for 150 mg twice daily as compared to 300 mg at bedtime (92% versus 87%, respectively).
Studies have been limited to short-term treatment of acute duodenal ulcer. Patients whose ulcers healed during therapy had recurrences of ulcers at the usual rates.
Maintenance Therapy in Duodenal Ulcer:
Ranitidine has been found to be effective as maintenance therapy for patients following healing of acute duodenal ulcers. In 2 independent, double-blind, multicenter, controlled trials, the number of duodenal ulcers observed was significantly less in patients treated with ranitidine (150 mg at bedtime) than in patients treated with placebo over a 12-month period.
Table 5. Duodenal Ulcer Prevalence % = Life table estimate.
RAN = ranitidine (ranitidine tablets USP)
PLC = placebo*p < 0.05 (ranitidine tablets USP versus comparator) Double-Blind, Multicenter, Placebo-Controlled Trials Multicenter trial Drug Duodenal Ulcer Prevalence No. of Patients 0 - 4 Months 0 - 8 Months 0 - 12 Months USA RAN 20% * 24% * 35% * 138 PLC 44% 54% 59% 139 Foreign RAN 12% * 21% * 28% * 174 PLC 56% 64% 68% 165 As with other H2-antagonists, the factors responsible for the significant reduction in the prevalence of duodenal ulcers include prevention of recurrence of ulcers, more rapid healing of ulcers that may occur during maintenance therapy, or both.
Gastric Ulcer:
In a multicenter, double-blind, controlled, US study of endoscopically diagnosed gastric ulcers, earlier healing was seen in the patients treated with ranitidine as shown in Table 6.
Table 6. Gastric Ulcer Patient Healing Rates * All patients were permitted antacids as needed for relief of pain. †p = 0.009. Ranitidine * Placebo* Number Entered Healed/Evaluable Number Entered Healed/Evaluable Outpatients 92 16/83 (19%) 94 10/83 (12%) Week 2 Week 6 50/73 (68%) † 35/69 (51%) In this multicenter trial, significantly more patients treated with ranitidine became pain free during therapy.
Maintenance of Healing of Gastric Ulcers:
In 2 multicenter, double-blind, randomized, placebo-controlled, 12-month trials conducted in patients whose gastric ulcers had been previously healed, ranitidine 150 mg at bedtime was significantly more effective than placebo in maintaining healing of gastric ulcers.
Pathological Hypersecretory Conditions (such as Zollinger-Ellison syndrome):
Ranitidine inhibits gastric acid secretion and reduces occurrence of diarrhea, anorexia, and pain in patients with pathological hypersecretion associated with Zollinger-Ellison syndrome, systemic mastocytosis, and other pathological hypersecretory conditions (e.g., postoperative, "short-gut" syndrome, idiopathic). Use of ranitidine was followed by healing of ulcers in 8 of 19 (42%) patients who were intractable to previous therapy.
Gastroesophageal Reflux Disease (GERD):
In 2 multicenter, double-blind, placebo-controlled, 6-week trials performed in the United States and Europe, ranitidine 150 mg twice daily was more effective than placebo for the relief of heartburn and other symptoms associated with GERD. Ranitidine-treated patients consumed significantly less antacid than did placebo-treated patients.
The US trial indicated that ranitidine 150 mg twice daily significantly reduced the frequency of heartburn attacks and severity of heartburn pain within 1 to 2 weeks after starting therapy. The improvement was maintained throughout the 6-week trial period. Moreover, patient response rates demonstrated that the effect on heartburn extends through both the day and night time periods.
In 2 additional US multicenter, double-blind, placebo-controlled, 2-week trials, ranitidine 150 mg twice daily was shown to provide relief of heartburn pain within 24 hours of initiating therapy and a reduction in the frequency of severity of heartburn.
Erosive Esophagitis:
In 2 multicenter, double-blind, randomized, placebo-controlled, 12-week trials performed in the United States, ranitidine 150 mg 4 times daily was significantly more effective than placebo in healing endoscopically diagnosed erosive esophagitis and in relieving associated heartburn. The erosive esophagitis healing rates were as follows:
Table 7. Erosive Esophagitis Patient Healing Rates * All patients were permitted antacids as needed for relief of pain. †p < 0.001 versus placebo. Healed/Evaluable Placebo * n=229 Ranitidine 150 mg 4 times daily * n=215 Week 4 43/198 (22%) 96/206 (47%) † Week 8 63/176 (36%) 142/200 (71%) † Week 12 92/159 (58%) 162/192 (84%) † No additional benefit in healing of esophagitis or in relief of heartburn was seen with a ranitidine dose of 300 mg 4 times daily.
Maintenance of Healing of Erosive Esophagitis:
In 2 multicenter, double-blind, randomized, placebo-controlled, 48-week trials conducted in patients whose erosive esophagitis had been previously healed, ranitidine 150 mg twice daily was significantly more effective than placebo in maintaining healing of erosive esophagitis.
INDICATIONS AND USAGE
Ranitidine tablets USP are indicated in:
- Short-term treatment of active duodenal ulcer. Most patients heal within 4 weeks. Studies available to date have not assessed the safety of ranitidine in uncomplicated duodenal ulcer for periods of more than 8 weeks.
- Maintenance therapy for duodenal ulcer patients at reduced dosage after healing of acute ulcers. No placebo-controlled comparative studies have been carried out for periods of longer than 1 year.
- The treatment of pathological hypersecretory conditions (e.g., Zollinger-Ellison syndrome and systemic mastocytosis).
- Short-term treatment of active, benign gastric ulcer. Most patients heal within 6 weeks and the usefulness of further treatment has not been demonstrated. Studies available to date have not assessed the safety of ranitidine in uncomplicated, benign gastric ulcer for periods of more than 6 weeks.
- Maintenance therapy for gastric ulcer patients at reduced dosage after healing of acute ulcers. Placebo-controlled studies have been carried out for 1 year.
- Treatment of GERD. Symptomatic relief commonly occurs within 24 hours after starting therapy with ranitidine 150 mg twice daily.
- Treatment of endoscopically diagnosed erosive esophagitis. Symptomatic relief of heartburn commonly occurs within 24 hours of therapy initiation with ranitidine 150 mg 4 times daily.
- Maintenance of healing of erosive esophagitis. Placebo-controlled trials have been carried out for 48 weeks.
Concomitant antacids should be given as needed for pain relief to patients with active duodenal ulcer; active, benign gastric ulcer; hypersecretory states; GERD; and erosive esophagitis.
CONTRAINDICATIONS
Ranitidine tablets USP are contraindicated for patients known to have hypersensitivity to the drug or any of the ingredients (see PRECAUTIONS ).
General:
- Symptomatic response to therapy with ranitidine does not preclude the presence of gastric malignancy.
- Since ranitidine is excreted primarily by the kidney, dosage should be adjusted in patients with impaired renal function (see DOSAGE AND ADMINISTRATION ). Caution should be observed in patients with hepatic dysfunction since ranitidine is metabolized in the liver.
- Rare reports suggest that ranitidine may precipitate acute porphyric attacks in patients with acute porphyria. Ranitidine should therefore be avoided in patients with a history of acute porphyria.
Laboratory Tests:
False-positive tests for urine protein with MULTISTIX® may occur during therapy with ranitidine, and therefore testing with sulfosalicylic acid is recommended.
Drug Interactions:
Ranitidine has been reported to affect the bioavailability of other drugs through several different mechanisms such as competition for renal tubular secretion, alteration of gastric pH, and inhibition of cytochrome P450 enzymes.
Procainamide: Ranitidine, a substrate of the renal organic cation transport system, may affect the clearance of other drugs eliminated by this route. High doses of ranitidine (e.g., such as those used in the treatment of Zollinger-Ellison syndrome) have been shown to reduce the renal excretion of procainamide and N-acetylprocainamide resulting in increased plasma levels of these drugs. Although this interaction is unlikely to be clinically relevant at usual ranitidine doses, it may be prudent to monitor for procainamide toxicity when administered with oral ranitidine at a dose exceeding 300 mg per day.
Warfarin: There have been reports of altered prothrombin time among patients on concomitant warfarin and ranitidine therapy. Due to the narrow therapeutic index, close monitoring of increased or decreased prothrombin time is recommended during concurrent treatment with ranitidine.
Ranitidine may alter the absorption of drugs in which gastric pH is an important determinant of bioavailability. This can result in either an increase in absorption (e.g., triazolam, midazolam, glipizide) or a decrease in absorption (e.g., ketoconazole, atazanavir, delavirdine, gefitinib). Appropriate clinical monitoring is recommended.
Atazanavir: Atazanavir absorption may be impaired based on known interactions with other agents that increase gastric pH. Use with caution. See atazanavir label for specific recommendations.
Delavirdine: Delavirdine absorption may be impaired based on known interactions with other agents that increase gastric pH. Chronic use of H2-receptor antagonists with delavirdine is not recommended.
Gefitinib : Gefitinib exposure was reduced by 44% with the coadministration of ranitidine and sodium bicarbonate (dosed to maintain gastric pH above 5.0). Use with caution.
Glipizide: In diabetic patients, glipizide exposure was increased by 34% following a single 150-mg dose of oral ranitidine. Use appropriate clinical monitoring when initiating or discontinuing ranitidine.
Ketoconazole: Oral ketoconazole exposure was reduced by up to 95% when oral ranitidine was coadministered in a regimen to maintain a gastric pH of 6 or above. The degree of interaction with usual dose of ranitidine (150 mg twice daily) is unknown.
Midazolam: Oral midazolam exposure in 5 healthy volunteers was increased by up to 65% when administered with oral ranitidine at a dose of 150 mg twice daily. However, in another interaction study in 8 volunteers receiving IV midazolam, a 300 mg oral dose of ranitidine increased midazolam exposure by about 9%. Monitor patients for excessive or prolonged sedation when ranitidine is coadministered with oral midazolam.
Triazolam: Triazolam exposure in healthy volunteers was increased by approximately 30% when administered with oral ranitidine at a dose of 150 mg twice daily. Monitor patients for excessive or prolonged sedation.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
There was no indication of tumorigenic or carcinogenic effects in life-span studies in mice and rats at dosages up to 2,000 mg/kg per day.
Ranitidine was not mutagenic in standard bacterial tests ( Salmonella, Escherichia coli ) for mutagenicity at concentrations up to the maximum recommended for these assays.
In a dominant lethal assay, a single oral dose of 1,000 mg/kg to male rats was without effect on the outcome of 2 matings per week for the next 9 weeks.
Pregnancy:
Teratogenic Effects:
Pregnancy Category B. Reproduction studies have been performed in rats and rabbits at doses up to 160 times the human dose and have revealed no evidence of impaired fertility or harm to the fetus due to ranitidine. There are, however no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers:
Ranitidine is secreted in human milk. Caution should be exercised when ranitidine is administered to a nursing mother.
Pediatric Use:
The safety and effectiveness of ranitidine have been established in the age-group of 1 month to 16 years for the treatment of duodenal and gastric ulcers, gastroesophageal reflux disease and erosive esophagitis, and the maintenance of healed duodenal and gastric ulcer. Use of ranitidine in this age-group is supported by adequate and well-controlled studies in adults, as well as additional pharmacokinetic data in pediatric patients and an analysis of the published literature (see CLINICAL PHARMACOLOGY: Pediatrics and DOSAGE AND ADMINISTRATION: Pediatric Use ).
Safety and effectiveness in pediatric patients for the treatment of pathological hypersecretory conditions or the maintenance of healing of erosive esophagitis have not been established.
Safety and effectiveness in neonates (less than 1 month of age) have not been established (see CLINICAL PHARMACOLOGY: Pediatrics ).
Geriatric Use:
Of the total number of subjects enrolled in US and foreign controlled clinical trials of oral formulations of ranitidine, for which there were subgroup analyses, 4,197 were 65 and over, while 899 were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, caution should be exercised in dose selection, and it may be useful to monitor renal function. (see CLINICAL PHARMACOLOGY: Pharmacokinetics: Geriatrics and DOSAGE AND ADMINISTRATION: Dosage Adjustment for Patients with Impaired Renal Function ).
The following have been reported as events in clinical trials or in the routine management of patients treated with ranitidine. The relationship to therapy with ranitidine has been unclear in many cases. Headache, sometimes severe, seems to be related to administration of ranitidine.
Central Nervous System:
Rarely, malaise, dizziness, somnolence, insomnia, and vertigo. Rare cases of reversible mental confusion, agitation, depression, and hallucinations have been reported, predominantly in severely ill elderly patients. Rare cases of reversible blurred vision suggestive of a change in accommodation have been reported. Rare reports of reversible involuntary motor disturbances have been received.
Cardiovascular:
As with other H2-blockers, rare reports of arrhythmias such as tachycardia, bradycardia, atrioventricular block, and premature ventricular beats.
Gastrointestinal:
Constipation, diarrhea, nausea/vomiting, abdominal discomfort/pain, and rare reports of pancreatitis.
Hepatic:
There have been occasional reports of hepatocellular, cholestatic, or mixed hepatitis, with or without jaundice. In such circumstances, ranitidine should be immediately discontinued. These events are usually reversible, but in rare circumstances death has occurred. Rare cases of hepatic failure have also been reported. In normal volunteers, SGPT values were increased to at least twice the pretreatment levels in 6 of 12 subjects receiving 100 mg intavenously 4 times daily for 7 days, and in 4 of 24 subjects receiving 50 mg intravenously 4 times daily for 5 days.
Musculoskeletal:
Rare reports of arthralgias and myalgias.
Hematologic:
Blood count changes (leukopenia, granulocytopenia, and thrombocytopenia) have occurred in a few patients. These were usually reversible. Rare cases of agranulocytosis, pancytopenia, sometimes with marrow hypoplasia, and aplastic anemia and exceedingly rare cases of acquired immune hemolytic anemia have been reported.
Endocrine:
Controlled studies in animals and man have shown no stimulation of any pituitary hormone by ranitidine and no antiandrogenic activity, and cimetidine-induced gynecomastia and impotence in hypersecretory patients have resolved when ranitidine has been substituted. However, occasional cases of impotence, and loss of libido have been reported in male patients receiving ranitidine, but the incidence did not differ from that in the general population. Rare cases of breast symptoms and conditions, including galactorrhea and gynecomastia, have been reported in both males and females.
Integumentary:
Rash, including rare cases of erythema multiforme. Rare cases of alopecia and vasculitis.
Respiratory:
A large epidemiological study suggested an increased risk of developing pneumonia in current users of histamine-2-receptor antagonists (H2RAs) compared to patients who had stopped H2RA treatment, with an observed adjusted relative risk of 1.63 (95% CI, 1.07 - 2.48). However, a causal relationship between use of H2RAs and pneumonia has not been established.
Other:
Rare cases of hypersensitivity reactions (e.g., bronchospasm, fever, rash, eosinophilia), anaphylaxis, angioneurotic edema, acute interstitial nephritis, and small increases in serum creatinine.
OVERDOSAGE
There has been limited experience with overdosage. Reported acute ingestions of up to 18 g orally have been associated with transient adverse effects similar to those encountered in normal clinical experience (see ADVERSE REACTIONS ). In addition, abnormalities of gait and hypotension have been reported.
When overdosage occurs, the usual measures to remove unabsorbed material from the gastrointestinal tract, clinical monitoring, and supportive therapy should be employed.
Studies in dogs receiving dosages of ranitidine in excess of 225 mg/kg/day have shown muscular tremors, vomiting, and rapid respiration. Single oral doses of 1,000 mg/kg in mice and rats were not lethal. Intravenous LD50 values in mice and rats were 77 and 83 mg/kg, respectively.
Active Duodenal Ulcer:
The current recommended adult oral dosage of ranitidine tablets USP for duodenal ulcer is 150 mg twice daily. An alternative dosage of 300 mg once daily after the evening meal or at bedtime can be used for patients in whom dosing convenience is important. The advantages of one treatment regimen compared to the other in a particular patient population have yet to be demonstrated (see Clinical Trials: Active Duodenal Ulcer ). Smaller doses have been shown to be equally effective in inhibiting gastric acid secretion in US studies, and several foreign trials have shown that 100 mg twice daily is as effective as the 150-mg dose.
Antacid should be given as needed for relief of pain (see CLINICAL PHARMACOLOGY: Pharmacokinetics ).
Maintenance of Healing of Duodenal Ulcers:
The current recommended adult oral dosage is 150 mg at bedtime.
Pathological Hypersecretory Conditions (such as Zollinger-Ellison syndrome):
The current recommended adult oral dosage is 150 mg twice daily. In some patients it may be necessary to administer ranitidine 150-mg doses more frequently. Dosages should be adjusted to individual patient needs, and should continue as long as clinically indicated. Dosages up to 6 g/day have been employed in patients with severe disease.
Benign Gastric Ulcer:
The current recommended adult oral dosage is 150 mg twice daily.
Maintenance of Healing of Gastric Ulcers:
The current recommended adult oral dosage is 150 mg at bedtime.
GERD:
The current recommended adult oral dosage is 150 mg twice daily.
Erosive Esophagitis:
The current recommended adult oral dosage is 150 mg 4 times daily.
Maintenance of Healing of Erosive Esophagitis:
The current recommended adult oral dosage is 150 mg twice daily.
Pediatric Use:
The safety and effectiveness of ranitidine have been established in the age-group of 1 month to 16 years. There is insufficient information about the pharmacokinetics of ranitidine in neonatal patients (less than 1 month of age) to make dosing recommendations.
The following 3 subsections provide dosing information for each of the pediatric indications.
Treatment of Duodenal and Gastric Ulcers:
The recommended oral dose for the treatment of active duodenal and gastric ulcers is 2 to 4 mg/kg twice daily to a maximum of 300 mg/day. This recommendation is derived from adult clinical studies and pharmacokinetic data in pediatric patients.
Maintenance of Healing of Duodenal and Gastric Ulcers:
The recommended oral dose for the maintenance of healing of duodenal and gastric ulcers is 2 to 4 mg/kg once daily to a maximum of 150 mg/day. This recommendation is derived from adult clinical studies and pharmacokinetic data in pediatric patients.
Treatment of GERD and Erosive Esophagitis:
Although limited data exist for these conditions in pediatric patients, published literature supports a dosage of 5 to 10 mg/kg/day, usually given as 2 divided doses.
Dosage Adjustment for Patients With Impaired Renal Function:
On the basis of experience with a group of subjects with severely impaired renal function treated with ranitidine, the recommended dosage in patients with a creatinine clearance <50 mL/min is 150 mg every 24 hours. Should the patient's condition require, the frequency of dosing may be increased to every 12 hours or even further with caution. Hemodialysis reduces the level of circulating ranitidine. Ideally, the dosing schedule should be adjusted so that the timing of a scheduled dose coincides with the end of hemodialysis.
Elderly patients are more likely to have decreased renal function, therefore caution should be exercised in dose selection, and it may be useful to monitor renal function (see CLINICAL PHARMACOLGOY: Pharmacokinetics: Geriatrics and PRECAUTIONS: Geriatric Use ).
HOW SUPPLIED
Ranitidine tablets USP 150 mg (ranitidine HCl USP equivalent to 150 mg of ranitidine) are pink colored, circular, biconvex, beveled edge film coated tablets with "G51" engraved on one side and "150" on the other side. They are available in bottles of 60 (NDC 68462-248-60), 100 (NDC 68462-248-01), and 500 (NDC 68462-248-05) tablets.
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15 o to 30 o C (59 o to 86 o F) [see USP Controlled Room Temperature]. Protect from light. Replace cap securely after each opening.
Manufactured by:
Shasun Pharmaceuticals Limited,
Unit-II,
R.S. No. 32, 33 & 34, Shasun Road, Periyakalapet,
Puducherry - 605 014. India.Manufactured for:
Glenmark Generics Inc., USA
Mahwah, NJ 07430
Questions? 1 (888)721-7115
www.glenmarkgenerics.comApril 2011
MULTISTIX is a registered trademark of Bayer Healthcare LLC.
-
DICLOFENAC SODIUM
Pharmacodynamics
Diclofenac sodium delayed-release tablets, USP are a non-steroidal anti-inflammatory drug (NSAID) that exhibits anti-inflammatory, analgesic, and antipyretic activities in animal models. The mechanism of action of diclofenac sodium delayed-release, like that of other NSAIDs, is not completely understood but may be related to prostaglandin synthetase inhibition.
Pharmacokinetics
Absorption
Diclofenac is 100% absorbed after oral administration compared to IV administration as measured by urine recovery. However, due to first-pass metabolism, only about 50% of the absorbed dose is systemically available (see Table 1). Food has no significant effect on the extent of diclofenac absorption. However, there is usually a delay in the onset of absorption of 1 to 4.5 hours and a reduction in peak plasma levels of <20%.
Table 1. Pharmacokinetic Parameters for Diclofenac Normal Healthy Adults PK Parameter (20 to 48 yrs.) Coefficient of Mean Variation (%) Absolute Bioavailability (%) [N = 7] 55 40 Tmax (hr) [N = 56] 2.3 69 Oral Clearance (CL/F; mL/min) [N = 56] 582 23 Renal Clearance (% unchanged drug in urine) <1 -- [N = 7] Apparent Volume of Distribution (V/F; L/kg) [N = 56] 1.4 58 Terminal Half-life (hr) [N = 56] 2.3 48 Distribution
The apparent volume of distribution (V/F) of diclofenac sodium is 1.4 L/kg.
Diclofenac is more than 99% bound to human serum proteins, primarily to albumin. Serum protein binding is constant over the concentration range (0.15 to 105 mcg/mL) achieved with recommended doses.
Diclofenac diffuses into and out of the synovial fluid. Diffusion into the joint occurs when plasma levels are higher than those in the synovial fluid, after which the process reverses and synovial fluid levels are higher than plasma levels. It is not known whether diffusion into the joint plays a role in the effectiveness of diclofenac.
Metabolism
Five diclofenac metabolites have been identified in human plasma and urine. The metabolites include 4'-hydroxy-, 5-hydroxy-, 3'-hydroxy-, 4',5-dihydroxy- and 3'-hydroxy-4'-methoxy diclofenac. The major diclofenac metabolite, 4'-hydroxy-diclofenac, has very weak pharmacologic activity. The formation of 4'-hydroxy- diclofenac is primarily mediated by CPY2C9. Both diclofenac and its oxidative metabolites undergo glucuronidation or sulfation followed by biliary excretion. Acylglucuronidation mediated by UGT2B7 and oxidation mediated by CPY2C8 may also play a role in diclofenac metabolism. CYP3A4 is responsible for the formation of minor metabolites, 5-hydroxy- and 3'-hydroxy-diclofenac. In patients with renal dysfunction, peak concentrations of metabolites 4'-hydroxy- and 5-hydroxy-diclofenac were approximately 50% and 4% of the parent compound after single oral dosing compared to 27% and 1% in normal healthy subjects.
Excretion
Diclofenac is eliminated through metabolism and subsequent urinary and biliary excretion of the glucuronide and the sulfate conjugates of the metabolites. Little or no free unchanged diclofenac is excreted in the urine. Approximately 65% of the dose is excreted in the urine and approximately 35% in the bile as conjugates of unchanged diclofenac plus metabolites. Because renal elimination is not a significant pathway of elimination for unchanged diclofenac, dosing adjustment in patients with mild to moderate renal dysfunction is not necessary. The terminal half-life of unchanged diclofenac is approximately 2 hours.
Drug Interactions
When co-administered with voriconazole (inhibitor of CYP2C9, 2C19 and 3A4 enzyme), the Cmax and AUC of diclofenac increased by 114% and 78%, respectively (see PRECAUTIONS, Drug Interactions ).
Special Populations
Pediatric : The pharmacokinetics of diclofenac sodium delayed-release has not been investigated in pediatric patients.
Race : Pharmacokinetic differences due to race have not been identified.
Hepatic Insufficiency : Hepatic metabolism accounts for almost 100% of diclofenac sodium delayed-release elimination, so patients with hepatic disease may require reduced doses of diclofenac sodium delayed-release compared to patients with normal hepatic function.
Renal Insufficiency : Diclofenac pharmacokinetics has been investigated in subjects with renal insufficiency. No differences in the pharmacokinetics of diclofenac have been detected in studies of patients with renal impairment. In patients with renal impairment (inulin clearance 60 to 90, 30 to 60, and <30 mL/min; N=6 in each group), AUC values and elimination rate were comparable to those in healthy subjects.
INDICATIONS AND USAGE
Carefully consider the potential benefits and risks of diclofenac sodium delayed-release tablets, USP and other treatment options before deciding to use diclofenac sodium delayed-release tablets, USP. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS ).
Diclofenac sodium delayed-release tablets are indicated:
- For relief of the signs and symptoms of osteoarthritis
- For relief of the signs and symptoms of rheumatoid arthritis
- For acute or long-term use in the relief of signs and symptoms of ankylosing spondylitis
Diclofenac sodium delayed-release tablets, USP are contraindicated in patients with known hypersensitivity to diclofenac.
Diclofenac sodium delayed-release should not be given to patients who have experienced asthma, urticaria, or other allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal, anaphylactic-like reactions to NSAIDs have been reported in such patients (see WARNINGS, Anaphylactoid Reactions , and PRECAUTIONS, Preexisting Asthma ).
Diclofenac sodium delayed-release is contraindicated in the setting of coronary artery bypass graft (CABG) surgery (see WARNINGS ).
Cardiovascular Effects
Cardiovascular Thrombotic Events
Clinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, including myocardial infarction (MI) and stroke, which can be fatal. Based on available data, it is unclear that the risk for CV thrombotic events is similar for all NSAIDs. The relative increase in serious CV thrombotic events over baseline conferred by NSAID use appears to be similar in those with and without known CV disease or risk factors for CV disease. However, patients with known CV disease or risk factors had a higher absolute incidence of excess serious CV thrombotic events, due to their increased baseline rate. Some observational studies found that this increased risk of serious CV thrombotic events began as early as the first weeks of treatment. The increase in CV thrombotic risk has been observed most consistently at higher doses.
To minimize the potential risk for an adverse CV event in NSAID-treated patients, use the lowest effective dose for the shortest duration possible. Physicians and patients should remain alert for the development of such events, throughout the entire treatment course, even in the absence of previous CV symptoms. Patients should be informed about the symptoms of serious CV events and the steps to take if they occur.
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID, such as diclofenac, increases the risk of serious gastrointestinal (GI) events (see WARNINGS ).
Status Post Coronary Artery Bypass Graft (CABG) Surgery
Two large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10 to 14 days following CABG surgery found an increased incidence of myocardial infarction and stroke. NSAIDs are contraindicated in the setting of CABG (see CONTRAINDICATIONS ).
Post-MI Patients
Observational studies conducted in the Danish National Registry have demonstrated that patients treated with NSAIDs in the post-MI period were at increased risk of reinfarction, CV-related death, and all-cause mortality beginning in the first week of treatment. In this same cohort, the incidence of death in the first year post MI was 20 per 100 person years in NSAID-treated patients compared to 12 per 100 person years in non-NSAID exposed patients. Although the absolute rate of death declined somewhat after the first year post-MI, the increased relative risk of death in NSAID users persisted over at least the next four years of follow-up.
Avoid the use of diclofenac sodium delayed-release in patients with a recent MI unless the benefits are expected to outweigh the risk of recurrent CV thrombotic events. If diclofenac sodium delayed-release is used in patients with a recent MI, monitor patients for signs of cardiac ischemia.
Hypertension
NSAIDs can lead to onset of new hypertension or worsening of preexisting hypertension, either of which may contribute to the increased incidence of CV events. Patients taking thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs. NSAIDs, including diclofenac sodium delayed-release should be used with caution in patients with hypertension. Blood pressure (BP) should be monitored closely during the initiation of NSAID treatment and throughout the course of therapy.
Heart Failure and Edema
The Coxib and traditional NSAID Trialists' Collaboration meta-analysis of randomized controlled trials demonstrated an approximately two-fold increase in hospitalizations for heart failure in COX-2 selective-treated patients and nonselective NSAID-treated patients compared to placebo-treated patients. In a Danish National Registry study of patients with heart failure, NSAID use increased the risk of MI, hospitalization for heart failure, and death.
Additionally, fluid retention and edema have been observed in some patients treated with NSAIDs. Use of diclofenac may blunt the CV effects of several therapeutic agents used to treat these medical conditions [e.g., diuretics, ACE inhibitors, or angiotensin receptor blockers (ARBs)] (see Drug Interactions ).
Avoid the use of diclofenac sodium delayed-release in patients with severe heart failure unless the benefits are expected to outweigh the risk of worsening heart failure. If diclofenac sodium delayed-release is used in patients with severe heart failure, monitor patients for signs of worsening heart failure.
Gastrointestinal (GI) Effects - Risk of GI Ulceration, Bleeding, and Perforation
NSAIDs, including diclofenac sodium delayed-release, can cause serious gastrointestinal (GI) adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs. Only one in five patients, who develop a serious upper GI adverse event on NSAID therapy, is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occur in approximately 1% of patients treated for 3 to 6 months, and in about 2% to 4% of patients treated for one year. These trends continue with longer duration of use, increasing the likelihood of developing a serious GI event at some time during the course of therapy. However, even short-term therapy is not without risk.
NSAIDs should be prescribed with extreme caution in those with a prior history of ulcer disease or gastrointestinal bleeding. Patients with a prior history of peptic ulcer disease and/or gastrointestinal bleeding who use NSAIDs have a greater than 10-fold increased risk for developing a GI bleed compared to patients with neither of these risk factors. Other factors that increase the risk for GI bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most spontaneous reports of fatal GI events are in elderly or debilitated patients and therefore, special care should be taken in treating this population.
To minimize the potential risk for an adverse GI event in patients treated with an NSAID, the lowest effective dose should be used for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of GI ulceration and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious GI adverse event is suspected. This should include discontinuation of the NSAID until a serious GI adverse event is ruled out. For high risk patients, alternate therapies that do not involve NSAIDs should be considered.
Renal Effects
Caution should be used when initiating treatment with diclofenac sodium delayed-release in patients with considerable dehydration.
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of a nonsteroidal anti-inflammatory drug may cause a dose-dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
Advanced Renal Disease
No information is available from controlled clinical studies regarding the use of diclofenac sodium delayed-release in patients with advanced renal disease. Therefore, treatment with diclofenac sodium delayed-release is not recommended in these patients with advanced renal disease. If diclofenac sodium delayed-release therapy must be initiated, close monitoring of the patient's renal function is advisable.
Hepatic Effects
Elevations of one or more liver tests may occur during therapy with diclofenac sodium delayed-release. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continued therapy. Borderline elevations (i.e., less than 3 times the ULN [ULN = the upper limit of the normal range]) or greater elevations of transaminases occurred in about 15% of diclofenac-treated patients. Of the markers of hepatic function, ALT (SGPT) is recommended for the monitoring of liver injury.
In clinical trials, meaningful elevations (i.e., more than 3 times the ULN) of AST (GOT) (ALT was not measured in all studies) occurred in about 2% of approximately 5,700 patients at some time during diclofenac treatment. In a large, open-label, controlled trial of 3,700 patients treated for 2 to 6 months, patients were monitored first at 8 weeks and 1,200 patients were monitored again at 24 weeks. Meaningful elevations of ALT and/or AST occurred in about 4% of patients and included marked elevations (i.e., more than 8 times the ULN) in about 1% of the 3,700 patients. In that open-label study, a higher incidence of borderline (less than 3 times the ULN), moderate (3 to 8 times the ULN), and marked (>8 times the ULN) elevations of ALT or AST was observed in patients receiving diclofenac when compared to other NSAIDs. Elevations in transaminases were seen more frequently in patients with osteoarthritis than in those with rheumatoid arthritis.
Almost all meaningful elevations in transaminases were detected before patients became symptomatic. Abnormal tests occurred during the first 2 months of therapy with diclofenac in 42 of the 51 patients in all trials who developed marked transaminase elevations.
In postmarketing reports, cases of drug-induced hepatotoxicity have been reported in the first month, and in some cases, the first 2 months of therapy, but can occur at any time during treatment with diclofenac. Postmarketing surveillance has reported cases of severe hepatic reactions, including liver necrosis, jaundice, fulminant hepatitis with and without jaundice, and liver failure. Some of these reported cases resulted in fatalities or liver transplantation.
Physicians should measure transaminases periodically in patients receiving long-term therapy with diclofenac, because severe hepatotoxicity may develop without a prodrome of distinguishing symptoms. The optimum times for making the first and subsequent transaminase measurements are not known. Based on clinical trial data and postmarketing experiences, transaminases should be monitored within 4 to 8 weeks after initiating treatment with diclofenac. However, severe hepatic reactions can occur at any time during treatment with diclofenac.
If abnormal liver tests persist or worsen, if clinical signs and/or symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, abdominal pain, diarrhea, dark urine, etc.), diclofenac sodium delayed-release should be discontinued immediately.
To minimize the possibility that hepatic injury will become severe between transaminase measurements, physicians should inform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, diarrhea, pruritus, jaundice, right upper quadrant tenderness, and "flu-like" symptoms), and the appropriate action patients should take if these signs and symptoms appear.
To minimize the potential risk for an adverse liver related event in patients treated with diclofenac sodium delayed-release, the lowest effective dose should be used for the shortest duration possible. Caution should be exercised in prescribing diclofenac sodium delayed-release with concomitant drugs that are known to be potentially hepatotoxic (e.g., antibiotics, anti-epileptics).
Anaphylactic Reactions
As with other NSAIDs, anaphylactic reactions may occur both in patients with the aspirin triad and in patients without known sensitivity to NSAIDs or known prior exposure to diclofenac sodium delayed-release. Diclofenac sodium delayed-release should not be given to patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs. (See CONTRAINDICATIONS and PRECAUTIONS, Preexisting Asthma .) Anaphylaxis-type reactions have been reported with NSAID products, including with diclofenac products, such as diclofenac sodium delayed-release. Emergency help should be sought in cases where an anaphylactic reaction occurs.
Skin Reactions
NSAIDs, including diclofenac sodium delayed-release, can cause serious skin adverse events such as exfoliative dermatitis, Stevens-Johnson Syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Patients should be informed about the signs and symptoms of serious skin manifestations and use of the drug should be discontinued at the first appearance of skin rash or any other sign of hypersensitivity.
Pregnancy
In late pregnancy, as with other NSAIDs, diclofenac sodium delayed-release should be avoided because it may cause premature closure of the ductus arteriosus.
General
Diclofenac sodium delayed-release tablets, USP cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to disease exacerbation. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
The pharmacological activity of diclofenac sodium delayed-release in reducing fever and inflammation may diminish the utility of these diagnostic signs in detecting complications of presumed noninfectious, painful conditions.
Hematological Effects
Anemia is sometimes seen in patients receiving NSAIDs, including diclofenac sodium delayed-release. This may be due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. Patients on long-term treatment with NSAIDs, including diclofenac sodium delayed-release, should have their hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia.
NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible. Patients receiving diclofenac sodium delayed-release who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants, should be carefully monitored.
Preexisting Asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm which can be fatal. Since cross-reactivity, including bronchospasm, between aspirin and other nonsteroidal anti-inflammatory drugs has been reported in such aspirin-sensitive patients, diclofenac sodium delayed-release should not be administered to patients with this form of aspirin sensitivity and should be used with caution in patients with preexisting asthma.
Information for Patients
Patients should be informed of the following information before initiating therapy with an NSAID and periodically during the course of ongoing therapy. Patients should also be encouraged to read the NSAID Medication Guide that accompanies each prescription dispensed.
- Cardiovascular Thrombotic Events Advise patients to be alert for the symptoms of cardiovascular thrombotic events, including chest pain, shortness of breath, weakness, or slurring of speech, and to report any of these symptoms to their health care provider immediately (see WARNINGS ).
- Diclofenac sodium delayed-release, like other NSAIDs, can cause GI discomfort and, rarely, more serious GI side effects, such as ulcers and bleeding, which may result in hospitalization and even death. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, patients should be alert for the signs and symptoms of ulcerations and bleeding, and should ask for medical advice when observing any indicative sign or symptoms including epigastric pain, dyspepsia, melena, and hematemesis. Patients should be apprised of the importance of this follow-up (see WARNINGS, Gastrointestinal (GI) Effects: Risk of GI Ulceration, Bleeding, and Perforation ).
- Diclofenac sodium delayed-release, like other NSAIDs, can cause serious skin side effects such as exfoliative dermatitis, SJS, and TEN, which may result in hospitalizations and even death. Although serious skin reactions may occur without warning, patients should be alert for the signs and symptoms of skin rash and blisters, fever, or other signs of hypersensitivity such as itching, and should ask for medical advice when observing any indicative signs or symptoms. Patients should be advised to stop the drug immediately if they develop any type of rash and contact their physicians as soon as possible.
- Heart Failure and Edema Advise patients to be alert for the symptoms of congestive heart failure including shortness of breath, unexplained weight gain, or edema and to contact their healthcare provider if such symptoms occur (see WARNINGS ).
- Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness, and "flu-like" symptoms). If these occur, patients should be instructed to stop therapy and seek immediate medical therapy. (see WARNINGS, Hepatic Effects ).
- Patients should be informed of the signs of an anaphylactic reaction (e.g., difficulty breathing, swelling of the face or throat). If these occur, patients should be instructed to seek immediate emergency help (see WARNINGS, Anaphylactic Reactions ).
- In late pregnancy, as with other NSAIDs, diclofenac sodium delayed-release should be avoided because it will cause premature closure of the ductus arteriosus.
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, physicians should monitor for signs or symptoms of GI bleeding. In patients on long-term treatment with NSAIDs, including diclofenac sodium delayed-release, the CBC and a chemistry profile (including transaminase levels) should be checked periodically. If clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash, etc.) or if abnormal liver tests persist or worsen, diclofenac sodium delayed-release should be discontinued.
Aspirin : When diclofenac sodium delayed-release is administered with aspirin, its protein binding is reduced. The clinical significance of this interaction is not known; however, as with other NSAIDs, concomitant administration of diclofenac and aspirin is not generally recommended because of the potential of increased adverse effects.
Methotrexate: NSAIDs have been reported to competitively inhibit methotrexate accumulation in rabbit kidney slices. This may indicate that they could enhance the toxicity of methotrexate. Caution should be used when NSAIDs are administered concomitantly with methotrexate.
Cyclosporine: Diclofenac sodium delayed-release, like other NSAIDs, may affect renal prostaglandins and increase the toxicity of certain drugs. Therefore, concomitant therapy with diclofenac sodium delayed-release may increase cyclosporine's nephrotoxicity. Caution should be used when diclofenac is administered concomitantly with cyclosporine.
ACE-Inhibitors : Reports suggest that NSAIDs may diminish the antihypertensive effect of ACE inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE inhibitors.
Furosemide : Clinical studies, as well as postmarketing observations, have shown that diclofenac sodium delayed-release can reduce the natriuretic effect of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis. During concomitant therapy with NSAIDs, the patient should be observed closely for signs of renal failure (see WARNINGS, Renal Effects ), as well as to assure diuretic efficacy.
Lithium : NSAIDs have produced an elevation of plasma lithium levels and a reduction in renal lithium clearance. The mean minimum lithium concentration increased 15% and the renal clearance was decreased by approximately 20%. These effects have been attributed to inhibition of renal prostaglandin synthesis by the NSAID. Thus, when NSAIDs and lithium are administered concurrently, subjects should be observed carefully for signs of lithium toxicity.
Warfarin : The effects of warfarin and NSAIDs on GI bleeding are synergistic, such that users of both drugs together have a risk of serious GI bleeding higher than users of either drug alone.
CYP2C9 Inhibitors or Inducers : Diclofenac is metabolized by cytochrome P450 enzymes, predominantly by CYP2C9. Co-administration of diclofenac with CYP2C9 inhibitors (e.g. voriconazole) may enhance the exposure and toxicity of diclofenac whereas co-administration with CYP2C9 inducers (e.g. rifampin) may lead to compromised efficacy of diclofenac. Use caution when dosing diclofenac with CYP2C9 inhibitors or inducers; a dosage adjustment may be warranted (see CLINICAL PHARMACOLOGY, Pharmacokinetics, Drug Interactions ).
Pregnancy
Teratogenic Effects: Pregnancy Category C
Reproductive studies conducted in rats and rabbits have not demonstrated evidence of developmental abnormalities. However, animal reproduction studies are not always predictive of human response. There are no adequate and well-controlled studies in pregnant women.
Nonteratogenic Effects : Because of the known effects of nonsteroidal anti-inflammatory drugs on the fetal cardiovascular system (closure of ductus arteriosus), use during pregnancy (particularly late pregnancy) should be avoided.
Labor and Delivery
In rat studies with NSAIDs, as with other drugs known to inhibit prostaglandin synthesis, an increased incidence of dystocia, delayed parturition, and decreased pup survival occurred. The effects of diclofenac sodium delayed-release on labor and delivery in pregnant women are unknown.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from diclofenac sodium delayed-release, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
As with any NSAIDs, caution should be exercised in treating the elderly (65 years and older).
ADVERSE REACTIONS
In patients taking diclofenac sodium delayed-release tablets, USP or other NSAIDs, the most frequently reported adverse experiences occurring in approximately 1% to 10% of patients are:
Gastrointestinal experiences including: abdominal pain, constipation, diarrhea, dyspepsia, flatulence, gross bleeding/perforation, heartburn, nausea, GI ulcers (gastric/duodenal) and vomiting.
Abnormal renal function, anemia, dizziness, edema, elevated liver enzymes, headaches, increased bleeding time, pruritus, rashes and tinnitus.
Additional adverse experiences reported occasionally include:
Body as a Whole: fever, infection, sepsis
Cardiovascular System : congestive heart failure, hypertension, tachycardia, syncope
Digestive System: dry mouth, esophagitis, gastric/peptic ulcers, gastritis, gastrointestinal bleeding, glossitis, hematemesis, hepatitis, jaundice
Hemic and Lymphatic System : ecchymosis, eosinophilia, leukopenia, melena, purpura, rectal bleeding, stomatitis, thrombocytopenia
Metabolic and Nutritional: weight changes
Nervous System: anxiety, asthenia, confusion, depression, dream abnormalities, drowsiness, insomnia, malaise, nervousness, paresthesia, somnolence, tremors, vertigo
Respiratory System: asthma, dyspnea
Skin and Appendages : alopecia, photosensitivity, sweating increased
Special Senses: blurred vision
Urogenital System: cystitis, dysuria, hematuria, interstitial nephritis, oliguria/polyuria, proteinuria, renal failure
Other adverse reactions, which occur rarely are:
Body as a Whole: anaphylactic reactions, appetite changes, death
Cardiovascular System: arrhythmia, hypotension, myocardial infarction, palpitations, vasculitis
Digestive System: colitis, eructation, fulminant hepatitis with and without jaundice, liver failure, liver necrosis, pancreatitis
Hemic and Lymphatic System: agranulocytosis, hemolytic anemia, aplastic anemia, lymphadenopathy, pancytopenia
Metabolic and Nutritional: hyperglycemia
Nervous System: convulsions, coma, hallucinations, meningitis
Respiratory System: respiratory depression, pneumonia
Skin and Appendages: angioedema, toxic epidermal necrolysis, erythema multiforme, exfoliative dermatitis, Stevens-Johnson syndrome, urticaria
Special Senses: conjunctivitis, hearing impairment
To report SUSPECTED ADVERSE EVENTS, contact Actavis at 1-800-432-8534 or FDA at 1-800-FDA-1088 or http://www.fda.gov/ for voluntary reporting of adverse reactions.
OVERDOSAGE
Symptoms following acute NSAID overdoses are usually limited to lethargy, drowsiness, nausea, vomiting, and epigastric pain, which are generally reversible with supportive care. Gastrointestinal bleeding can occur. Hypertension, acute renal failure, respiratory depression and coma may occur, but are rare. Anaphylactoid reactions have been reported with therapeutic ingestion of NSAIDs, and may occur following an overdose.
Patients should be managed by symptomatic and supportive care following a NSAID overdose. There are no specific antidotes. Emesis and/or activated charcoal (60 to 100 g in adults, 1 to 2 g/kg in children) and/or osmotic cathartic may be indicated in patients seen within 4 hours of ingestion with symptoms or following a large overdose (5 to 10 times the usual dose). Forced diuresis, alkalinization of urine, hemodialysis, or hemoperfusion may not be useful due to high protein binding.
DOSAGE AND ADMINISTRATION
Carefully consider the potential benefits and risks of diclofenac sodium delayed-release tablets, USP and other treatment options before deciding to use diclofenac sodium delayed-release. Use the lowest effective dose for the shortest duration consistent with individual patient treatment goals (see WARNINGS ).
After observing the response to initial therapy with diclofenac sodium delayed-release, the dose and frequency should be adjusted to suit an individual patient's needs.
For the relief of osteoarthritis, the recommended dosage is 100 to 150 mg/day in divided doses (50 mg b.i.d. or t.i.d., or 75 mg b.i.d.).
For the relief of rheumatoid arthritis, the recommended dosage is 150 to 200 mg/day in divided doses (50 mg t.i.d. or q.i.d., or 75 mg b.i.d.).
For the relief of ankylosing spondylitis, the recommended dosage is 100 to 125 mg/day, administered as 25 mg q.i.d., with an extra 25 mg dose at bedtime if necessary.
Different formulations of diclofenac (diclofenac sodium delayed-release tablets; diclofenac sodium extended-release tablets, USP; diclofenac potassium immediate-release tablets) are not necessarily bioequivalent even if the milligram strength is the same.
HOW SUPPLIED
Diclofenac Sodium Delayed-Release Tablets, USP are available as follows:
75 mg — Each white, round, enteric-coated tablet printed with
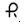 on one side and 551 on the other side with black ink contains 75 mg of Diclofenac Sodium, USP. Tablets are supplied in bottles of 60 (NDC 0228-2551-06), 100 (NDC 0228-2551-11), and 1000 (NDC 0228-2551-96).
on one side and 551 on the other side with black ink contains 75 mg of Diclofenac Sodium, USP. Tablets are supplied in bottles of 60 (NDC 0228-2551-06), 100 (NDC 0228-2551-11), and 1000 (NDC 0228-2551-96).
Store at 25ºC (77ºF); excursions permitted to 15º to 30ºC (59ºto 86ºF).
Protect from moisture.
Dispense in a tight, light-resistant container as defined in the USP.
Manufactured by:
Actavis Elizabeth LLC
Elizabeth, NJ 07207 USADistributed by:
Actavis Pharma, Inc.
Parsippany, NJ 07054 USA40-9184
Revised — August 2015
-
LIDOCAINE 2.5% AND PRILOCAINE 2.5%
Mechanism of Action: Lidocaine and prilocaine cream applied to intact skin under occlusive dressing, provides dermal analgesia by the release of lidocaine and prilocaine from the cream into the epidermal and dermal layers of the skin and by the accumulation of lidocaine and prilocaine in the vicinity of dermal pain receptors and nerve endings. Lidocaine and prilocaine are amide-type local anesthetic agents. Both lidocaine and prilocaine stabilize neuronal membranes by inhibiting the ionic fluxes required for the initiation and conduction of impulses, thereby effecting local anesthetic action.
The onset, depth and duration of dermal analgesia on intact skin provided by lidocaine and prilocaine cream depend primarily on the duration of application. To provide sufficient analgesia for clinical procedures such as intravenous catheter placement and venipuncture, lidocaine and prilocaine cream should be applied under an occlusive dressing for at least 1 hour. To provide dermal analgesia for clinical procedures such as split skin graft harvesting, lidocaine and prilocaine cream should be applied under occlusive dressing for at least 2 hours. Satisfactory dermal analgesia is achieved 1 hour after application, reaches maximum at 2 to 3 hours, and persists for 1 to 2 hours after removal. Absorption from the genital mucosa is more rapid and onset time is shorter (5 to 10 minutes) than after application to intact skin. After a 5 to 10 minute application of lidocaine and prilocaine cream to female genital mucosa, the average duration of effective analgesia to an argon laser stimulus (which produced a sharp, pricking pain) was 15 to 20 minutes (individual variations in the range of 5 to 45 minutes).
Dermal application of lidocaine and prilocaine cream may cause a transient, local blanching followed by a transient, local redness or erythema.
Pharmacokinetics: Lidocaine and prilocaine cream is a eutectic mixture of lidocaine 2.5% and prilocaine 2.5% formulated as an oil in water emulsion. In this eutectic mixture, both anesthetics are liquid at room temperature (see DESCRIPTION ) and the penetration and subsequent systemic absorption of both prilocaine and lidocaine are enhanced over that which would be seen if each component in crystalline form was applied separately as a 2.5% topical cream.
Absorption: The amount of lidocaine and prilocaine systemically absorbed from lidocaine and prilocaine cream is directly related to both the duration of application and to the area over which it is applied. In two pharmacokinetic studies, 60 g of lidocaine and prilocaine cream (1.5 g lidocaine and 1.5 g prilocaine) was applied to 400 cm2 of intact skin on the lateral thigh and then covered by an occlusive dressing. The subjects were then randomized such that one-half of the subjects had the occlusive dressing and residual cream removed after 3 hours, while the remainder left the dressing in place for 24 hours. The results from these studies are summarized below.
TABLE 1 Absorption of Lidocaine and Prilocaine from Lidocaine and Prilocaine Cream: Normal Volunteers (N=16) *Maximum recommended duration of exposure is 4 hours. Lidocaine and
Prilocaine
Cream (g)Area
(cm2)Time on
(hrs)Drug Content
(mg)Absorbed
(mg)Cmax
(µg/mL)Tmax
(hr)60 400 3 lidocaine 1500 54 0.12 4 prilocaine 1500 92 0.07 4 60 400 24* lidocaine 1500 243 0.28 10 prilocaine 1500 503 0.14 10 When 60 g of lidocaine and prilocaine cream was applied over 400 cm2 for 24 hours, peak blood levels of lidocaine are approximately 1/20 the systemic toxic level. Likewise, the maximum prilocaine level is about 1/36 the toxic level. In a pharmacokinetic study, lidocaine and prilocaine cream was applied to penile skin in 20 adult male patients in doses ranging from 0.5 g to 3.3 g for 15 minutes. Plasma concentrations of lidocaine and prilocaine following lidocaine and prilocaine cream application in this study were consistently low (2.5 to 16 ng/mL for lidocaine and 2.5 to 7 ng/mL for prilocaine). The application of lidocaine and prilocaine cream to broken or inflamed skin, or to 2,000 cm2 or more of skin where more of both anesthetics are absorbed, could result in higher plasma levels that could, in susceptible individuals, produce a systemic pharmacologic response.
The absorption of lidocaine and prilocaine cream applied to genital mucous membranes was studied in two open-label clinical trials. Twenty-nine patients received 10 g of lidocaine and prilocaine cream applied for 10 to 60 minutes in the vaginal fornices. Plasma concentrations of lidocaine and prilocaine following lidocaine and prilocaine cream application in these studies ranged from 148 to 641 ng/mL for lidocaine and 40 to 346 ng/mL for prilocaine and time to reach maximum concentration (tmax) ranged from 21 to 125 minutes for lidocaine and from 21 to 95 minutes for prilocaine. These levels are well below the concentrations anticipated to give rise to systemic toxicity (approximately 5000 ng/mL for lidocaine and prilocaine).
Distribution: When each drug is administered intravenously, the steady-state volume of distribution is 1.1 to 2.1 L/kg (mean 1.5, ±0.3 SD, n=13) for lidocaine and is 0.7 to 4.4 L/kg (mean 2.6, ±1.3 SD, n=13) for prilocaine. The larger distribution volume for prilocaine produces the lower plasma concentrations of prilocaine observed when equal amounts of prilocaine and lidocaine are administered. At concentrations produced by application of lidocaine and prilocaine cream, lidocaine is approximately 70% bound to plasma proteins, primarily alpha-1-acid glycoprotein. At much higher plasma concentrations (1 to 4 µg/mL of free base) the plasma protein binding of lidocaine is concentration dependent. Prilocaine is 55% bound to plasma proteins. Both lidocaine and prilocaine cross the placental and blood brain barrier, presumably by passive diffusion.
Metabolism: It is not known if lidocaine or prilocaine are metabolized in the skin. Lidocaine is metabolized rapidly by the liver to a number of metabolites including monoethylglycinexylidide (MEGX) and glycinexylidide (GX), both of which have pharmacologic activity similar to, but less potent than that of lidocaine. The metabolite, 2,6-xylidine, has unknown pharmacologic activity. Following intravenous administration, MEGX and GX concentrations in serum range from 11 to 36% and from 5 to 11% of lidocaine concentrations, respectively. Prilocaine is metabolized in both the liver and kidneys by amidases to various metabolites including ortho -toluidine and N-n-propylalanine. It is not metabolized by plasma esterases. The ortho -toluidine metabolite has been shown to be carcinogenic in several animal models (see PRECAUTIONS, Carcinogenesis ). In addition, ortho -toluidine can produce methemoglobinemia following systemic doses of prilocaine approximating 8 mg/kg (see ADVERSE REACTIONS ). Very young patients, patients with glucose-6-phosphate dehydro- genase deficiencies and patients taking oxidizing drugs such as antimalarials and sulfonamides are more susceptible to methemoglobinemia (see WARNINGS, Methemoglobinemia ).
Elimination: The terminal elimination half-life of lidocaine from the plasma following IV administration is approximately 65 to 150 minutes (mean 110, ±24 SD, n=13). More than 98% of an absorbed dose of lidocaine can be recovered in the urine as metabolites or parent drug. The systemic clearance is 10 to 20 mL/min/kg (mean 13, ±3 SD, n=13). The elimination half-life of prilocaine is approximately 10 to 150 minutes (mean 70, ±48 SD, n=13). The systemic clearance is 18 to 64 mL/min/kg (mean 38, ±15 SD, n=13). During intravenous studies, the elimination half-life of lidocaine was statistically significantly longer in elderly patients (2.5 hours) than in younger patients (1.5 hours). No studies are available on the intravenous pharmacokinetics of prilocaine in elderly patients.
Pediatrics: Some pharmacokinetic (PK) data are available in infants (1 month to <2 years old) and children (2 to <12 years old). One PK study was conducted in 9 full-term neonates (mean age: 7 days and mean gestational age: 38.8 weeks). The study results show that neonates had comparable plasma lidocaine and prilocaine concentrations and blood methemoglobin concentrations as those found in previous pediatric PK studies and clinical trials. There was a tendency towards an increase in methemoglobin formation. However, due to assay limitations and very little amount of blood that could be collected from neonates, large variations in the above reported concentrations were found.
Special Populations: No specific PK studies were conducted. The half-life may be increased in cardiac or hepatic dysfunction. Prilocaine's half-life also may be increased in hepatic or renal dysfunction since both of these organs are involved in prilocaine metabolism.
CLINICAL STUDIES
Lidocaine and prilocaine cream application in adults prior to IV cannulation or venipuncture was studied in 200 patients in four clinical studies in Europe. Application for at least 1 hour provided significantly more dermal analgesia than placebo cream or ethyl chloride. Lidocaine and prilocaine cream was comparable to subcutaneous lidocaine, but was less efficacious than intradermal lidocaine. Most patients found lidocaine and prilocaine cream treatment preferable to lidocaine infiltration or ethyl chloride spray.
Lidocaine and prilocaine cream was compared with 0.5% lidocaine infiltration prior to skin graft harvesting in one open label study in 80 adult patients in England. Application of lidocaine and prilocaine cream for 2 to 5 hours provided dermal analgesia comparable to lidocaine infiltration.
Lidocaine and prilocaine cream application in children was studied in seven non-US studies (320 patients) and one US study (100 patients). In controlled studies, application of lidocaine and prilocaine cream for at least 1 hour with or without presurgical medication prior to needle insertion provided significantly more pain reduction than placebo. In children under the age of seven years, lidocaine and prilocaine cream was less effective than in older children or adults.
Lidocaine and prilocaine cream was compared with placebo in the laser treatment of facial port-wine stains in 72 pediatric patients (ages 5 to 16). Lidocaine and prilocaine cream was effective in providing pain relief during laser treatment.
Lidocaine and prilocaine cream alone was compared with lidocaine and prilocaine cream followed by lidocaine infiltration and lidocaine infiltration alone prior to cryotherapy for the removal of male genital warts. The data from 121 patients demonstrated that lidocaine and prilocaine cream was not effective as a sole anesthetic agent in managing the pain from the surgical procedure. The administration of lidocaine and prilocaine cream prior to lidocaine infiltration provided significant relief of discomfort associated with local anesthetic infiltration and thus was effective in the overall reduction of pain from the procedure only when used in conjunction with local anesthetic infiltration of lidocaine.
Lidocaine and prilocaine cream was studied in 105 full term neonates (gestational age: 37 weeks) for blood drawing and circumcision procedures. When considering the use of lidocaine and prilocaine cream in neonates, the primary concerns are the systemic absorption of the active ingredients and the subsequent formation of methemoglobin. In clinical studies performed in neonates, the plasma levels of lidocaine, prilocaine, and methemoglobin were not reported in a range expected to cause clinical symptoms.
Local dermal effects associated with lidocaine and prilocaine cream application in these studies on intact skin included paleness, redness and edema and were transient in nature (see ADVERSE REACTIONS ).
The application of lidocaine and prilocaine cream on genital mucous membranes for minor, superficial surgical procedures (eg, removal of condylomata acuminata) was studied in 80 patients in a placebo-controlled clinical trial (60 patients received lidocaine and prilocaine cream and 20 patients received placebo). Lidocaine and prilocaine cream (5 to 10 g) applied between 1 and 75 minutes before surgery, with a median time of 15 minutes, provided effective local anesthesia for minor superficial surgical procedures. The greatest extent of analgesia, as measured by VAS scores, was attained after 5 to 15 minutes' application. The application of lidocaine and prilocaine cream to genital mucous membranes as pretreatment for local anesthetic infiltration was studied in a double-blind, placebo-controlled study in 44 female patients (21 patients received lidocaine and prilocaine cream and 23 patients received placebo) scheduled for infiltration prior to a surgical procedure of the external vulva or genital mucosa. Lidocaine and prilocaine cream applied to the genital mucous membranes for 5 to 10 minutes resulted in adequate topical anesthesia for local anesthetic injection.
Individualization of Dose: The dose of lidocaine and prilocaine cream that provides effective analgesia depends on the duration of the application over the treated area.
All pharmacokinetic and clinical studies employed a thick layer of lidocaine and prilocaine cream (1 to 2 g/10 cm2). The duration of application prior to venipuncture was 1 hour. The duration of application prior to taking split thickness skin grafts was 2 hours. A thinner application has not been studied and may result in less complete analgesia or a shorter duration of adequate analgesia.
The systemic absorption of lidocaine and prilocaine is a side effect of the desired local effect. The amount of drug absorbed depends on surface area and duration of application. The systemic blood levels depend on the amount absorbed and patient size (weight) and the rate of systemic drug elimination. Long duration of application, large treatment area, small patients, or impaired elimination may result in high blood levels. The systemic blood levels are typically a small fraction (1/20 to 1/36) of the blood levels that produce toxicity. Table 2 below gives maximum recommended doses, application areas and application times for infants and children.
TABLE 2 LIDOCAINE AND PRILOCAINE CREAM MAXIMUM RECOMMENDED DOSE, APPLICATION AREA, AND APPLICATION TIME BY AGE AND WEIGHT* For Infants and Children Based on Application to Intact Skin Please note: If a patient greater than 3 months old does not meet the minimum weight requirement, the maximum total dose of lidocaine and prilocaine cream should be restricted to that which corresponds to the patient's weight. * These are broad guidelines for avoiding systemic toxicity in applying lidocaine and prilocaine cream to patients with normal intact skin and with normal renal and hepatic function. ** For more individualized calculation of how much lidocaine and prilocaine may be absorbed, physicians can use the following estimates of lidocaine and prilocaine absorption for children and adults:
The estimated mean (±SD) absorption of lidocaine is 0.045 (±0.016) mg/cm2/hr.
The estimated mean (±SD) absorption of prilocaine is 0.077 (±0.036) mg/cm2/hr.Age and Body Weight
RequirementsMaximum Total
Dose of Lidocaine and
Prilocaine creamMaximum
Application Area**Maximum
Application Time0 up to 3 months or < 5 kg 1 g 10 cm2 1 hour 3 up to 12 months and > 5 kg 2 g 20 cm2 4 hours 1 to 6 years and > 10 kg 10 g 100 cm2 4 hours 7 to 12 years and > 20 kg 20 g 200 cm2 4 hours An I.V. antiarrhythmic dose of lidocaine is 1 mg/kg (70 mg/70 kg) and gives a blood level of about 1 µg/mL. Toxicity would be expected at blood levels above 5 µg/mL. Smaller areas of treatment are recommended in a debilitated patient, a small child or a patient with impaired elimination. Decreasing the duration of application is likely to decrease the analgesic effect.
INDICATIONS AND USAGE
Lidocaine and prilocaine cream (a eutectic mixture of lidocaine 2.5% and prilocaine 2.5%) is indicated as a topical anesthetic for use on:
- normal intact skin for local analgesia.
- genital mucous membranes for superficial minor surgery and as pretreatment for infiltration anesthesia.
Lidocaine and prilocaine cream is not recommended in any clinical situation when penetration or migration beyond the tympanic membrane into the middle ear is possible because of the ototoxic effects observed in animal studies (see WARNINGS ).
CONTRAINDICATIONS
Lidocaine and prilocaine cream (lidocaine 2.5% and prilocaine 2.5%) is contraindicated in patients with a known history of sensitivity to local anesthetics of the amide type or to any other component of the product.
Application of lidocaine and prilocaine cream to larger areas or for longer times than those recommended could result in sufficient absorption of lidocaine and prilocaine resulting in serious adverse effects (see Individualization of Dose ).
Patients treated with class III anti-arrhythmic drugs (e.g., amiodarone, bretylium, sotalol, dofetilide) should be under close surveillance and ECG monitoring considered, because cardiac effects may be additive.
Studies in laboratory animals (guinea pigs) have shown that lidocaine and prilocaine cream has an ototoxic effect when instilled into the middle ear. In these same studies, animals exposed to lidocaine and prilocaine cream only in the external auditory canal, showed no abnormality. Lidocaine and prilocaine cream should not be used in any clinical situation when its penetration or migration beyond the tympanic membrane into the middle ear is possible.
Methemoglobinemia: Lidocaine and prilocaine cream should not be used in those rare patients with congenital or idiopathic methemoglobinemia and in infants under the age of twelve months who are receiving treatment with methemoglobin-inducing agents.
Very young patients or patients with glucose-6-phosphate dehydrogenase deficiencies are more susceptible to methemoglobinemia.
Patients taking drugs associated with drug-induced methemoglobinemia such as sulfonamides, acetaminophen, acetanilid, aniline dyes, benzocaine, chloroquine, dapsone, naphthalene, nitrates and nitrites, nitrofurantoin, nitroglycerin, nitroprusside, pamaquine, para-aminosalicylic acid, phenacetin, phenobarbital, phenytoin, primaquine, quinine, are also at greater risk for developing methemoglobinemia.
There have been reports of significant methemoglobinemia (20 to 30%) in infants and children following excessive applications of lidocaine and prilocaine cream. These cases involved the use of large doses, larger than recommended areas of application, or infants under the age of 3 months who did not have fully mature enzyme systems. In addition, a few of these cases involved the concomitant administration of methemoglobin-inducing agents. Most patients recovered spontaneously after removal of the cream. Treatment with IV methylene blue may be effective if required.
Physicians are cautioned to make sure that parents orother caregivers understand the need for careful application of lidocaine and prilocaine cream, to ensure that the doses and areas of application recommended in Table 2 are not exceeded (especially in children under the age of 3 months) and to limit the period of application to the minimum required to achieve the desired anesthesia.
Neonates and infants up to 3 months of age should be monitored for Met-Hb levels before, during, and after the application of lidocaine and prilocaine cream, provided the test results can be obtained quickly.
General: Repeated doses of lidocaine and prilocaine cream may increase blood levels of lidocaine and prilocaine. Lidocaine and prilocaine cream should be used with caution in patients who may be more sensitive to the systemic effects of lidocaine and prilocaine including acutely ill, debilitated, or elderly patients.
Lidocaine and prilocaine cream should not be applied to open wounds.
Care should be taken not to allow lidocaine and prilocaine cream to come in contact with the eye because animal studies have demonstrated severe eye irritation. Also the loss of protective reflexes can permit corneal irritation and potential abrasion. Absorption of lidocaine and prilocaine cream in conjunctival tissues has not been determined. If eye contact occurs, immediately wash out the eye with water or saline and protect the eye until sensation returns.
Patients allergic to paraaminobenzoic acid derivatives (procaine, tetracaine, benzocaine, etc.) have not shown cross sensitivity to lidocaine and/or prilocaine, however, lidocaine and prilocaine cream should be used with caution in patients with a history of drug sensitivities, especially if the etiologic agent is uncertain.
Patients with severe hepatic disease, because of their inability to metabolize local anesthetics normally, are at greater risk of developing toxic plasma concentrations of lidocaine and prilocaine.
Lidocaine and prilocaine have been shown to inhibit viral and bacterial growth. The effect of lidocaine and prilocaine cream on intradermal injections of live vaccines has not been determined.
Information for Patients: When lidocaine and prilocaine cream is used, the patient should be aware that the production of dermal analgesia may be accompanied by the block of all sensations in the treated skin. For this reason, the patient should avoid inadvertent trauma to the treated area by scratching, rubbing, or exposure to extreme hot or cold temperatures until complete sensation has returned.
Lidocaine and prilocaine cream should not be applied near the eyes or on open wounds.
Drug Interactions: Lidocaine and prilocaine cream should be used with caution in patients receiving Class I antiarrhythmic drugs (such as tocainide and mexiletine) since the toxic effects are additive and potentially synergistic.
Prilocaine may contribute to the formation of methemoglobin in patients treated with other drugs known to cause this condition (see WARNINGS, Methemoglobinemia ).
Specific interaction studies with lidocaine/prilocaine and class III anti-arrhythmic drugs (e.g., amiodarone, bretylium, sotalol, doetilide) have not been performed, but caution is advised (see WARNINGS ).
Should lidocaine and prilocaine cream be used concomitantly with other products containing lidocaine and/or prilocaine, cumulative doses from all formulations must be considered.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: Long-term studies in animals designed to evaluate the carcinogenic potential of lidocaine and prilocaine have not been conducted.
Metabolites of prilocaine have been shown to be carcinogenic in laboratory animals. In the animal studies reported below, doses or blood levels are compared with the Single Dermal Administration (SDA) of 60 g of lidocaine and prilocaine cream to 400 cm2 for 3 hours to a small person (50 kg). The typical application of lidocaine and prilocaine cream for one or two treatments for venipuncture sites (2.5 or 5 g) would be 1/24 or 1/12 of that dose in an adult or about the same mg/kg dose in an infant.
Chronic oral toxicity studies of ortho -toluidine, a metabolite of prilocaine, in mice (450 to 7200 mg/m2; 60 to 960 times SDA) and rats (900 to 4,800 mg/m2; 60 to 320 times SDA) have shown that ortho -toluidine is a carcinogen in both species. The tumors included hepatocarcinomas/adenomas in female mice, multiple occurrences of hemangiosarcomas/hemangiomas in both sexes of mice, sarcomas of multiple organs, transitional-cell carcinomas/papillomas of urinary bladder in both sexes of rats, subcutaneous fibromas/fibrosarcomas and mesotheliomas in male rats, and mammary gland fibroadenomas/adenomas in female rats. The lowest dose tested (450 mg/m2 in mice, 900 mg/m2 in rats; 60 times SDA) was carcinogenic in both species. Thus the no-effect dose must be less than 60 times SDA. The animal studies were conducted at 150 to 2,400 mg/kg in mice and at 150 to 800 mg/kg in rats. The dosages have been converted to mg/m2 for the SDA calculations above.
Mutagenesis: The mutagenic potential of lidocaine HCl has been tested in a bacterial reverse (Ames) assay in Salmonella, an in vitro chromosomal aberration assay using human lymphocytes an in vivo micronucleus test in mice. There was no indication of mutagenicity or structural damage to chromosomes in these tests.
Ortho -toluidine, a metabolite of prilocaine, at a concentration of 0.5 µg/mL, was genotoxic in Escherichia coli DNA repair and phage-induction assays. Urine concentrates from rats treated with ortho -toluidine (300 mg/kg orally; 300 times SDA) were mutagenic when examined in Salmonella typhimurium in the presence of metabolic activation. Several other tests on ortho -toluidine, including reverse mutations in five different Salmonella typhimurium strains in the presence or absence of metabolic activation and a study to detect single strand breaks in DNA of V79 Chinese hamster cells, were negative.
Impairment of Fertility: See Use in Pregnancy .
Use in Pregnancy: Teratogenic Effects: Pregnancy Category B.
Reproduction studies with lidocaine have been performed in rats and have revealed no evidence of harm to the fetus (30 mg/kg subcutaneously; 22 times SDA). Reproduction studies with prilocaine have been performed in rats and have revealed no evidence of impaired fertility or harm to the fetus (300 mg/kg intramuscularly; 188 times SDA). There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, lidocaine and prilocaine cream should be used during pregnancy only if clearly needed.
Reproduction studies have been performed in rats receiving subcutaneous administration of an aqueous mixture containing lidocaine HCl and prilocaine HCl at 1:1 (w/w). At 40 mg/kg each, a dose equivalent to 29 times SDA lidocaine and 25 times SDA prilocaine, no teratogenic, embryotoxic or fetotoxic effects were observed.
Labor and Delivery : Neither lidocaine nor prilocaine are contraindicated in labor and delivery. Should lidocaine and prilocaine cream be used concomitantly with other products containing lidocaine and/or prilocaine, cumulative doses from all formulations must be considered.
Nursing Mothers: Lidocaine, and probably prilocaine, are excreted in human milk. Therefore, caution should be exercised when lidocaine and prilocaine cream is administered to a nursing mother since the milk:plasma ratio of lidocaine is 0.4 and is not determined for prilocaine.
Pediatric Use: Controlled studies of lidocaine and prilocaine cream in children under the age of seven years have shown less overall benefit than in older children or adults. These results illustrate the importance of emotional and psychological support of younger children undergoing medical or surgical procedures.
Lidocaine and prilocaine cream should be used with care in patients with conditions or therapy associated with methemoglobinemia (see WARNINGS, Methemoglobinemia ).
When using lidocaine and prilocaine cream in young children, especially infants under the age of 3 months, care must be taken to insure that the caregiver understands the need to limit the dose and area of application, and to prevent accidental ingestion (see DOSAGE AND ADMINISTRATION and WARNINGS,Methemoglobinemia ).
In neonates (minimum gestation age: 37 weeks) and children weighing less than 20 kg, the area and duration of application should be limited (see TABLE 2 in Individualization of Dose).
Studies have not demonstrated the efficacy of lidocaine and prilocaine cream for heel lancing in neonates.
Geriatric Use: Of the total number of patients in clinical studies of lidocaine and prilocaine cream, 180 were age 65 to 74 and 138 were 75 and over. No overall differences in safety or efficacy were observed between these patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Plasma levels of lidocaine and prilocaine in geriatric and non-geriatric patients following application of a thick layer of lidocaine and prilocaine cream are very low and well below potentially toxic levels. However, there are no sufficient data to evaluate quantitative differences in systemic plasma levels of lidocaine and prilocaine between geriatric and non-geriatric patients following application of lidocaine and prilocaine cream.
Consideration should be given for those elderly patients who have enhanced sensitivity to systemic absorption (see PRECAUTIONS ).
After intravenous dosing, the elimination half-life of lidocaine is significantly longer in elderly patients (2.5 hours) than in younger patients (1.5 hours). (See CLINICAL PHARMACOLOGY ).
Localized Reactions: During or immediately after treatment with lidocaine and prilocaine cream on intact skin, the skin at the site of treatment may develop erythema or edema or may be the locus of abnormal sensation. Rare cases of discrete purpuric or petechial reactions at the application site have been reported. Rare cases of hyperpigmentation following the use of lidocaine and prilocaine cream have been reported. The relationship to lidocaine and prilocaine cream or the underlying procedure has not been established. In clinical studies on intact skin involving over 1,300 lidocaine and prilocaine cream-treated subjects, one or more such local reactions were noted in 56% of patients, and were generally mild and transient, resolving spontaneously within 1 or 2 hours. There were no serious reactions that were ascribed to lidocaine and prilocaine cream.
Two recent reports describe blistering on the foreskin in neonates about to undergo circumcision. Both neonates received 1.0 g of lidocaine and prilocaine cream.
In patients treated with lidocaine and prilocaine cream on intact skin, local effects observed in the trials included: paleness (pallor or blanching) 37%, redness (erythema) 30%, alterations in temperature sensations 7%, edema 6%, itching 2% and rash, less than 1%.
In clinical studies on genital mucous membranes involving 378 lidocaine and prilocaine cream-treated patients, one or more application site reactions, usually mild and transient, were noted in 41% of patients. The most common application site reactions were redness (21%), burning sensation (17%) and edema (10%).
Allergic Reactions: Allergic and anaphylactoid reactions associated with lidocaine or prilocaine can occur. They are characterized by urticaria, angioedema, bronchospasm, and shock. If they occur they should be managed by conventional means. The detection of sensitivity by skin testing is of doubtful value.
Systemic (Dose Related) Reactions: Systemic adverse reactions following appropriate use of lidocaine and prilocaine cream are unlikely due to the small dose absorbed (see CLINICAL PHARMACOLOGY, Pharmacokinetics ). Systemic adverse effects of lidocaine and/or prilocaine are similar in nature to those observed with other amide local anesthetic agents including CNS excitation and/or depression (light-headedness, nervousness, apprehension, euphoria, confusion, dizziness, drowsiness, tinnitus, blurred or double vision, vomiting, sensations of heat, cold or numbness, twitching, tremors, convulsions, unconsciousness, respiratory depression and arrest). Excitatory CNS reactions may be brief or not occur at all, in which case the first manifestation may be drowsiness merging into unconsciousness. Cardiovascular manifestations may include bradycardia, hypotension and cardiovascular collapse leading to arrest.
OVERDOSAGE
Peak blood levels following a 60 g application to 400 cm2 of intact skin for 3 hours are 0.05 to 0.16 µg/mL for lidocaine and 0.02 to 0.10 µg/mL for prilocaine. Toxic levels of lidocaine (>5 µg/mL) and/or prilocaine (>6 µg/mL) cause decreases in cardiac output, total peripheral resistance and mean arterial pressure. These changes may be attributable to direct depressant effects of these local anesthetic agents on the cardiovascular system. In the absence of massive topical overdose or oral ingestion, evaluation should include evaluation of other etiologies for the clinical effects or overdosage from other sources of lidocaine, prilocaine or other local anesthetics. Consult the package inserts for parenteral Xylocaine (lidocaine HCl) or Citanest (prilocaine HCl) for further information for the management of overdose.
DOSAGE AND ADMINISTRATION
Adult Patients-Intact Skin
A thick layer of lidocaine and prilocaine cream is applied to intact skin and covered with an occlusive dressing (see INSTRUCTIONS FOR APPLICATION ).
Minor Dermal Procedures: For minor procedures such as intravenous cannulation and venipuncture, apply 2.5 grams of lidocaine and prilocaine cream (1/2 the 5 g tube) over 20 to 25 cm2 of skin surface for at least 1 hour. In controlled clinical trials using lidocaine and prilocaine cream, two sites were usually prepared in case there was a technical problem with cannulation or venipuncture at the first site.
Major Dermal Procedures: For more painful dermatological procedures involving a larger skin area such as split thickness skin graft harvesting, apply 2 grams of lidocaine and prilocaine cream per 10 cm2 of skin and allow to remain in contact with the skin for at least 2 hours.
Adult Male Genital Skin: As an adjunct prior to local anesthetic infiltration, apply a thick layer of lidocaine and prilocaine cream (1 g/10 cm2 ) to the skin surface for 15 minutes. Local anesthetic infiltration should be performed immediately after removal of lidocaine and prilocaine cream.
Dermal analgesia can be expected to increase for up to 3 hours under occlusive dressing and persist for 1 to 2 hours after removal of the cream. The amount of lidocaine and prilocaine absorbed during the period of application can be estimated from the information in Table 2, ** footnote, in Individualization of Dose .
Adult Female Patients-Genital Mucous Membranes
For minor procedures on the female external genitalia, such as removal of condylomata acuminata, as well as for use as pretreatment for anesthetic infiltration, apply a thick layer (5 to 10 grams) of lidocaine and prilocaine cream for 5 to 10 minutes.
Occlusion is not necessary for absorption, but may be helpful to keep the cream in place. Patients should be lying down during the lidocaine and prilocaine cream application, especially if no occlusion is used. The procedure or the local anesthetic infiltration should be performed immediately after the removal of lidocaine and prilocaine cream.
Pediatric Patients-Intact Skin
The following are the maximum recommended doses, application areas and application times for lidocaine and prilocaine cream based on a child's age and weight:
Age and Body Weight
RequirementsMaximum Total
Dose of Lidocaine and
Prilocaine CreamMaximum
Application AreaMaximum
Application Time0 up to 3 months or < 5 kg 1 g 10 cm2 1 hour 3 up to 12 months and > 5 kg 2 g 20 cm2 4 hours 1 to 6 years and > 10 kg 10 g 100 cm2 4 hours 7 to 12 years and > 20 kg 20 g 200 cm2 4 hours Please note: If a patient greater than 3 months old does not meet the minimum weight requirement, the maximum total dose of lidocaine and prilocaine cream should be restricted to that which corresponds to the patient's weight (see INSTRUCTIONS FOR APPLICATION ).
Practitioners should carefully instruct caregivers to avoid application of excessive amounts of lidocaine and prilocaine cream (see PRECAUTIONS ).
When applying lidocaine and prilocaine cream to the skin of young children, care must be taken to maintain careful observation of the child to prevent accidental ingestion of lidocaine and prilocaine cream or the occlusive dressing. A secondary protective covering to prevent inadvertent disruption of the application site may be useful.
Lidocaine and prilocaine cream should not be used in neonates with a gestational age less than 37 weeks nor in infants under the age of 12 months who are receiving treatment with methemoglobin-inducing agents (see WARNINGS, Methemoglobinemia).
When lidocaine and prilocaine cream (lidocaine 2.5% and prilocaine 2.5%) is used concomitantly with other products containing local anesthetic agents, the amount absorbed from all formulations must be considered (see Individualization of Dose ). The amount absorbed in the case of lidocaine and prilocaine cream is determined by the area over which it is applied and the duration of application under occlusion (see Table 2, ** footnote, in Individualization of Dose ).
Although the incidence of systemic adverse reactions with lidocaine and prilocaine cream is very low, caution should be exercised, particularly when applying it over large areas and leaving it on for longer than 2 hours. The incidence of systemic adverse reactions can be expected to be directly proportional to the area and time of exposure (see Individualization of Dose ).
To measure 1 gram of lidocaine and prilocaine cream, the Cream should be gently squeezed out of the tube as a narrow strip that is 1.5 inches (3.8 cm) long and 0.2 inches (5 mm) wide. The strip of lidocaine and prilocaine cream should be contained within the lines of the diagram shown below.

Use the number of strips that equals your dose, like the examples in the table below.
Dosing Information
1 gram = 1 strip
2 grams = 2 strips
2.5 grams = 2.5 stripsFor adult and pediatric patients, apply ONLY as prescribed by your physician.
If your child is below the age of 3 months or small for their age, please inform your doctor before applying lidocaine and prilocaine cream, which can be harmful, if applied over too much skin at one time in young children.
When applying lidocaine and prilocaine cream to the intact skin of young children, it is important that they be carefully observed by an adult in order to prevent the accidental ingestion of or eye contact with lidocaine and prilocaine cream.
Lidocaine and prilocaine cream must be applied to intact skin at least 1 hour before the start of a routine procedure and for 2 hours before the start of a painful procedure. A protective covering of the cream is not necessary for absorption but may be helpful to keep the cream in place.
If using a protective covering, your doctor will remove it, wipe off the lidocaine and prilocaine cream, and clean the entire area with an antiseptic solution before the procedure. The duration of effective skin anesthesia will be at least 1 hour after removal of the protective covering.
Precautions
- Do not apply near eyes or open wounds.
- Keep out of the reach of children.
- If your child becomes very dizzy, excessively sleepy, or develops duskiness of the face or lips after applying lidocaine and prilocaine cream, remove the cream and contact the child's physician at once.
HOW SUPPLIED
Lidocaine 2.5% and Prilocaine 2.5% Cream, USP is available as the following:
NDC No. Strength Size NDC 0591-2070-26 5 gram/tube packed individually. NDC 0591-2070-72 5 gram/tube packed in 5. NDC 0591-2070-30 30 gram/tube packed individually, in a child-resistant tube. NOT FOR OPHTHALMIC USE.
KEEP CONTAINER TIGHTLY CLOSED AT ALL TIMES WHEN NOT IN USE.
Storage: Store at 20º to 25ºC (68º to 77ºF) [see USP Controlled Room Temperature].
Rx only
Keep out of the reach of children.
For all medical inquiries contact:
ACTAVIS
Medical Communications
Parsippany, NJ 07054
1-800-272-5525Manufactured By:
IGI Laboratories Inc.
Buena, NJ 08310 USADistributed By:
Actavis Pharma, Inc.
Parsippany, NJ 07054 USAContent Updated: June 2014
76626 - PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
INFLAMMATION REDUCTION PACK
inflammation reduction pack kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69176-010 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69176-010-03 1 in 1 KIT; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 Part 2 1 Part 3 1 Part 1 of 3 RANITIDINE HYDROCHLORIDE
ranitidine hydrochloride tabletProduct Information Item Code (Source) NDC:68462-015 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RANITIDINE HYDROCHLORIDE (UNII: BK76465IHM) (RANITIDINE - UNII:884KT10YB7) RANITIDINE 150 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FD&C RED NO. 40 (UNII: WZB9127XOA) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) HYPROMELLOSES (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) Product Characteristics Color pink Score no score Shape ROUND (Circular, Biconvex, beveled edge film coated) Size 10mm Flavor Imprint Code G51;150 Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078542 10/22/1997 Part 2 of 3 EMLA CREAM
2.5% lidocaine and 2.5% prilocaine creamProduct Information Item Code (Source) NDC:05912-015 Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LIDOCAINE (UNII: 98PI200987) (LIDOCAINE - UNII:98PI200987) LIDOCAINE 25 mg in 1 g PRILOCAINE (UNII: 046O35D44R) (PRILOCAINE - UNII:046O35D44R) PRILOCAINE 25 mg in 1 g Inactive Ingredients Ingredient Name Strength PEG-55 HYDROGENATED CASTOR OIL (UNII: 0WZF1506N9) CARBOMER HOMOPOLYMER TYPE B (ALLYL SUCROSE CROSSLINKED) (UNII: Z135WT9208) WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) Product Characteristics Color white Score Shape Size Flavor Imprint Code Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA019941 03/01/2012 Part 3 of 3 DICLOFENAC SODIUM
diclofenac sodium tablet, delayed releaseProduct Information Item Code (Source) NDC:02282-015 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DICLOFENAC SODIUM (UNII: QTG126297Q) (DICLOFENAC - UNII:144O8QL0L1) DICLOFENAC SODIUM 75 mg Inactive Ingredients Ingredient Name Strength POLYSORBATE 80 (UNII: 6OZP39ZG8H) SODIUM ALGINATE (UNII: C269C4G2ZQ) STEARIC ACID (UNII: 4ELV7Z65AP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOLS (UNII: 3WJQ0SDW1A) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) TALC (UNII: 7SEV7J4R1U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYVINYL ACETATE PHTHALATE (UNII: 58QVG85GW3) ALUMINUM HYDROXIDE (UNII: 5QB0T2IUN0) Product Characteristics Color white Score no score Shape ROUND Size 9mm Flavor Imprint Code R;550 Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA074514 03/26/1996 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA074514 08/18/2015 Labeler - TMIG, Inc. (036572986) Registrant - TMIG, Inc. (036572986) Establishment Name Address ID/FEI Business Operations Actavis Elisabeth LLC 623114928 REPACK(69176-010) , MANUFACTURE(05912-015) Establishment Name Address ID/FEI Business Operations Actavis LLC 017665256 MANUFACTURE(05912-015) Establishment Name Address ID/FEI Business Operations Shasun Pharmaceuticals Limited 915786829 MANUFACTURE(68462-015) , ANALYSIS(68462-015)




