Label: TAVALISSE- fostamatinib tablet


- NDC Code(s): 71332-001-01, 71332-002-01
- Packager: Rigel Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated November 5, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TAVALISSE® safely and effectively. See full prescribing information for TAVALISSE.
TAVALISSE® (fostamatinib disodium hexahydrate) tablets, for oral use
Initial U.S. Approval: 2018INDICATIONS AND USAGE
TAVALISSE is a kinase inhibitor indicated for the treatment of thrombocytopenia in adult patients with chronic immune thrombocytopenia (ITP) who have had an insufficient response to a previous treatment. (1)
DOSAGE AND ADMINISTRATION
- Initiate TAVALISSE at 100 mg orally twice daily with or without food. After 4 weeks, increase to 150 mg twice daily, if needed, to achieve platelet counts of at least 50 × 109/L as necessary to reduce the risk of bleeding. (2.1)
- Manage adverse reactions using dose reduction, interruption of treatment, or discontinuation. (2.3)
- Discontinue TAVALISSE after 12 weeks of treatment if the platelet count does not increase to a level sufficient to avoid clinically important bleeding. (2.5)
DOSAGE FORMS AND STRENGTHS
Tablets: 100 mg, 150 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hypertension: Monitor blood pressure every 2 weeks until stable, then monthly. Manage hypertension using standard antihypertensive treatment and, if needed, interrupt, reduce or discontinue TAVALISSE. (5.1)
- Hepatotoxicity: Monitor LFTs monthly. If LFT levels are elevated, interrupt, reduce or discontinue TAVALISSE. (5.2)
- Diarrhea: Manage diarrhea with supportive measures. If diarrhea becomes severe, interrupt, reduce or discontinue TAVALISSE. (5.3)
- Neutropenia: Monitor ANC monthly, and for infection. If neutrophil count decreases below 1.0 × 109/L, interrupt, reduce or discontinue TAVALISSE. (5.4)
- Embryo-Fetal Toxicity: TAVALISSE can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.5)
ADVERSE REACTIONS
The most common adverse reactions (≥5% and more than placebo) are diarrhea, hypertension, nausea, respiratory infection, dizziness, ALT/AST increased, rash, abdominal pain, fatigue, chest pain and neutropenia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Rigel Pharmaceuticals, Inc. at 1-800-983-1329 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Monitoring
2.3 Dose Modification for Adverse Reactions
2.4 Dose Modification for Drug Interactions
2.5 Discontinuation
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypertension
5.2 Hepatotoxicity
5.3 Diarrhea
5.4 Neutropenia
5.5 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TAVALISSE
7.2 Effect of TAVALISSE on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Initiate TAVALISSE at a dose of 100 mg taken orally twice daily. After a month, if platelet count has not increased to at least 50 × 109/L, increase TAVALISSE dose to 150 mg twice daily.
Use the lowest dose of TAVALISSE to achieve and maintain a platelet count at least 50 × 109/L as necessary to reduce the risk of bleeding.
TAVALISSE may be taken with or without food. In the case of a missed dose of TAVALISSE, instruct patients to take their next dose at its regularly scheduled time.
2.2 Monitoring
After obtaining baseline assessments:
- Monitor CBCs, including platelet counts, monthly until a stable platelet count (at least 50 × 109/L) is achieved. Thereafter, continue to monitor CBCs, including neutrophils, regularly.
- Monitor liver function tests (LFTs) (e.g., ALT, AST, and bilirubin) monthly.
- Monitor blood pressure every 2 weeks until establishment of a stable dose, then monthly thereafter.
2.3 Dose Modification for Adverse Reactions
TAVALISSE dose modification is recommended based on individual safety and tolerability. Management of some adverse reactions may require dose-interruption, reduction, or discontinuation.
A dose reduction schedule is provided in Table 1, based on daily dose. For example, if a patient is on the maximum dose at the time of an adverse reaction, the first dose reduction would be from 300 mg/day to 200 mg/day.
Table 1: Dose Reduction Schedule Daily Dose Administered as: AM PM 300 mg/day 150 mg 150 mg 200 mg/day 100 mg 100 mg 150 mg/day 150 mg* --- 100 mg/day† 100 mg* --- The recommended dose modifications for adverse reactions are provided in Table 2.
Table 2: Recommended Dose Modifications and Management for Specific Adverse Reactions Adverse Reaction Recommended Action ALT = alanine aminotransferase; AST = aspartate aminotransferase; BP = blood pressure; BL = bilirubin; ULN = upper limit of normal; LFT = liver function tests (AST, ALT, total BL with fractionation if elevated, alkaline phosphatase); AST/ALT = AST or ALT Hypertension Stage 1: systolic between 130-139 or diastolic between 80-89 mmHg - Initiate or increase dosage of antihypertensive medication for patients with increased cardiovascular risk, and adjust as needed until BP is controlled.
- If the BP target is not met after 8 weeks, reduce TAVALISSE to next lower daily dose (refer to Table 1).
Stage 2: systolic at least 140 or diastolic at least 90 mmHg - Initiate or increase dosage of antihypertensive medication, and adjust as needed until BP is controlled.
- If BP remains 140/90 mmHg or higher for more than 8 weeks, reduce TAVALISSE to next lower daily dose (refer to Table 1).
- If BP remains 160/100 mmHg or higher for more than 4 weeks despite aggressive antihypertensive therapy, interrupt or discontinue TAVALISSE.
Hypertensive crisis: systolic over 180 and/or diastolic over 120 mmHg - Interrupt or discontinue TAVALISSE.
- Initiate or increase dosage of antihypertensive medication, and adjust as needed until BP is controlled. If BP returns to less than the target BP, resume TAVALISSE at same daily dose.
- If repeat BP is 160/100 mmHg or higher for more than 4 weeks despite aggressive antihypertensive treatment, discontinue TAVALISSE.
Hepatotoxicity AST/ALT is 3 × ULN or higher and less than 5 × ULN If patient is symptomatic (e.g., nausea, vomiting, abdominal pain): - Interrupt TAVALISSE.
- Recheck LFTs every 72 hours until ALT/AST values are no longer elevated (below 1.5 × ULN) and total BL remains less than 2 × ULN.
- Resume TAVALISSE at next lower daily dose (refer to Table 1).
If patient is asymptomatic: - Recheck LFTs every 72 hours until ALT/AST are below 1.5 × ULN) and total BL remains less than 2 × ULN.
- Consider interruption or dose reduction of TAVALISSE if ALT/AST and TBL remain in this category (AST/ALT is 3 to 5 × ULN; and total BL remains less than 2 × ULN)
- If interrupted, resume TAVALISSE at next lower daily dose (refer to Table 1) when ALT/AST are no longer elevated (below 1.5 × ULN) and total BL remains less than 2 × ULN.
AST/ALT is 5 × ULN or higher and total BL is less than 2 × ULN - Interrupt TAVALISSE.
- Recheck LFTs every 72 hours:
- If AST and ALT decrease, recheck until ALT and AST are no longer elevated (below 1.5 × ULN) and total BL remains less than 2 × ULN; resume TAVALISSE at next lower daily dose (refer to Table 1).
- If AST/ALT persist at 5 × ULN or higher for 2 weeks or more, discontinue TAVALISSE.
AST/ALT is 3 × ULN or higher and total BL is greater than 2 × ULN - Discontinue TAVALISSE.
Elevated unconjugated (indirect) BL in absence of other LFT abnormalities - Continue TAVALISSE with frequent monitoring since isolated increase in unconjugated (indirect) BL may be due to UGT1A1 inhibition
Diarrhea Diarrhea - Manage diarrhea using supportive measures (e.g., dietary changes, hydration and/or antidiarrheal medication) early after the onset until symptom(s) have resolved.
- If symptom(s) become severe (Grade 3 or above), temporarily interrupt TAVALISSE.
- If diarrhea improves to mild (Grade 1), resume TAVALISSE at the next lower daily dose (refer to Table 1).
Neutropenia Neutropenia - If absolute neutrophil count decreases (ANC less than 1.0 × 109/L) and remains low after 72 hours, temporarily interrupt TAVALISSE until resolved (ANC greater than 1.5 × 109/L).
- Resume TAVALISSE at the next lower daily dose (refer to Table 1).
2.4 Dose Modification for Drug Interactions
Concomitant use with a strong CYP3A4 inhibitor increases exposure to R406 (the major active metabolite). Monitor for toxicities of TAVALISSE that may require TAVALISSE dose modifications (see Table 1) when given concurrently with a strong CYP3A4 inhibitor [see Drug Interactions (7.1)].
2.5 Discontinuation
Discontinue TAVALISSE after 12 weeks of treatment if the platelet count does not increase to a level sufficient to avoid clinically important bleeding [see Clinical Studies (14)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypertension
Hypertension can occur with TAVALISSE treatment; hypertensive crisis occurred in 1% of patients. Patients with pre-existing hypertension may be more susceptible to the hypertensive effects of TAVALISSE.
Monitor blood pressure every 2 weeks until stable, then monthly and adjust or initiate antihypertensive therapy to ensure maintenance of blood pressure control during TAVALISSE therapy. If increased blood pressure persists despite appropriate therapy, TAVALISSE interruption, reduction or discontinuation may be necessary [see Dosage and Administration (2.3)].
5.2 Hepatotoxicity
Elevated liver function tests (LFTs), mainly ALT and AST, can occur with TAVALISSE.
In the placebo-controlled studies, laboratory testing showed maximum ALT/AST levels more than 3 × the upper limit of normal (ULN) in 9% of patients receiving TAVALISSE [see Adverse Reactions (6.1)]. For most patients, transaminases recovered to baseline levels within 2 to 6 weeks of dose-modification.
Monitor liver function tests monthly during treatment. If ALT or AST increase more than 3 × ULN, manage hepatotoxicity using TAVALISSE interruption, reduction, or discontinuation [see Dosage and Administration (2.3)].
5.3 Diarrhea
Diarrhea occurred in 31% of patients treated with TAVALISSE. Severe diarrhea occurred in 1% of patients treated with TAVALISSE. Monitor patients for the development of diarrhea. Manage diarrhea using supportive care measures, including dietary changes, hydration and/or antidiarrheal medication, early after the onset of symptoms. Interrupt, dose reduce, or discontinue TAVALISSE if diarrhea becomes severe (Grade 3 or above) [see Dosage and Administration (2.3)].
5.4 Neutropenia
Neutropenia occurred in 6% of patients treated with TAVALISSE; febrile neutropenia occurred in 1% of patients.
Monitor the ANC monthly, and for infection during treatment. Manage toxicity with TAVALISSE interruption, reduction or discontinuation [see Dosage and Administration (2.3)].
5.5 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, TAVALISSE can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of fostamatinib to pregnant rats and rabbits during organogenesis caused adverse developmental outcomes including embryo-fetal mortality (post-implantation loss), alterations to growth (lower fetal weights), and structural abnormalities (variations and malformations) at maternal exposures (AUCs) approximately 0.3 and 10 times the human exposure at the maximum recommended human dose (MRHD), respectively. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for at least 1 month after the last dose. [see Use in Specific Populations (8.1) and Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following clinically important adverse reactions, that can become serious are described elsewhere in the labeling:
- Hypertension [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Diarrhea [see Warnings and Precautions (5.3)]
- Neutropenia [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
TAVALISSE was studied in two randomized, double-blind, placebo-controlled trials that were identical in design. The data described below reflect exposure to TAVALISSE in 102 patients with chronic ITP who had received one or more prior ITP treatment(s). Groups were stratified with respect to splenectomy and severity of thrombocytopenia. Patients randomized to the TAVALISSE arm received 100 mg orally twice daily. Based upon platelet count and tolerability, if a patient's platelet count did not increase to at least 50 × 109/L, the TAVALISSE dose could be increased to 150 mg twice daily after one month. In the placebo controlled studies, the median duration of TAVALISSE exposure in these studies was 86 days (range 8 to 183) [see Clinical Studies (14) for additional details for patients on TAVALISSE].
In the ITP double-blind studies, serious adverse drug reactions were febrile neutropenia, diarrhea, pneumonia, and hypertensive crisis, which each occurred in 1% of patients receiving TAVALISSE. In addition, severe adverse reactions observed in patients receiving TAVALISSE included dyspnea and hypertension (both 2%); and neutropenia, arthralgia, chest pain, diarrhea, dizziness, nephrolithiasis, pain in extremity, toothache, syncope and hypoxia (all 1%) [see Warnings and Precautions (5.1)]. Table 3 presents the common adverse reactions from these studies.
Table 3: Incidence of Common (≥ 5%) Adverse Reactions from Double-Blind Clinical Studies (FIT 1 and FIT 2) Adverse Reaction TAVALISSE
(N=102)Placebo
(N=48)Mild
%Moderate
%Severe
%TOTAL
%Mild
%Moderate
%Severe
%TOTAL
%ALT = Alanine aminotransferase
AST = Aspartate aminotransferase
Note: Common adverse reactions defined as all adverse reactions occurring at a rate of ≥ 5% of patients in the TAVALISSE group and greater than placebo rate.- *
- Includes diarrhea and frequent bowel movement.
- †
- Includes hypertension, blood pressure (BP) increased, BP diastolic abnormal, and BP diastolic increased.
- ‡
- Includes upper respiratory tract infection, respiratory tract infection, lower respiratory tract infection, and viral upper respiratory tract infection.
- §
- Includes rash, rash erythematous and rash macular.
- ¶
- Includes abdominal pain, and abdominal pain upper.
- #
- Includes neutropenia and neutrophil count decreased.
Diarrhea* 21 10 1 31 13 2 0 15 Hypertension† 17 9 2 28 10 0 2 13 Nausea 16 3 0 19 8 0 0 8 Dizziness 8 2 1 11 6 2 0 8 ALT increased 5 6 0 11 0 0 0 0 AST increased 5 4 0 9 0 0 0 0 Respiratory infection‡ 7 4 0 11 6 0 0 6 Rash§ 8 1 0 9 2 0 0 2 Abdominal pain¶ 5 1 0 6 2 0 0 2 Fatigue 4 2 0 6 0 2 0 2 Chest pain 2 3 1 6 2 0 0 2 Neutropenia# 3 2 1 6 0 0 0 0 Table 4: Elevations in Hepatic Transaminases During Placebo-Controlled Clinical Studies Enzyme Maximum Level of Elevation Number of Patients (%) TAVALISSE
(N=102)Placebo
(N=48)Alanine aminotransferase (ALT) and/or Aspartate aminotransferase (AST) >3 and ≤5 × ULN 3 (3) 0 >5 and ≤10 × ULN 5 (5) 0 ≥10 × ULN 1 (1) 0 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TAVALISSE
Strong CYP3A4 Inhibitors
Concomitant use with strong CYP3A4 inhibitors increases exposure to R406 (the major active metabolite), which may increase the risk of adverse reactions. Monitor for toxicities of TAVALISSE that may require dose reduction (see Table 1) when given concurrently with a strong CYP3A4 inhibitor [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
Strong CYP3A4 Inducers
Concomitant use with a strong CYP3A4 inducer reduces exposure to R406. Concomitant use of TAVALISSE with strong CYP3A4 inducers is not recommended [see Clinical Pharmacology (12.3)].
7.2 Effect of TAVALISSE on Other Drugs
CYP3A4 Substrates
Concomitant use of TAVALISSE may increase concentrations of some CYP3A4 substrate drugs. Monitor for toxicities of CYP3A4 substrate drug that may require dosage reduction when given concurrently with TAVALISSE [see Clinical Pharmacology (12.3)].
BCRP Substrates
Concomitant use of TAVALISSE may increase concentrations of BCRP substrate drugs (e.g., rosuvastatin). Monitor for toxicities of BCRP substrate drug that may require dosage reduction when given concurrently with TAVALISSE [see Clinical Pharmacology (12.3)].
P-Glycoprotein (P-gp) Substrates
Concomitant use of TAVALISSE may increase concentrations of P-gp substrates (e.g., digoxin). Monitor for toxicities of the P-gp substrate drug that may require dosage reduction when given concurrently with TAVALISSE [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and the mechanism of action, TAVALISSE can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)].
There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of fostamatinib to pregnant rats and rabbits during organogenesis caused adverse developmental outcomes that were directly attributed to exposure in utero to the major fostamatinib metabolite (R406) at maternal exposures (AUC) as low as 0.3 and 10 times the exposure in patients at the maximum recommended human dose (MRHD), respectively (see Data). Advise pregnant women of the potential risk to a fetus.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. An estimated background risk of major birth defects and miscarriage for the chronic ITP population is 8% and 4-11%, respectively.
Animal Data
In a fertility and early embryonic development study in female rats, fostamatinib was administered orally for 15 days before mating to Day 7 of pregnancy, which caused a slight decrease in pregnancy rates and an increase in post-implantation loss were seen at maternal doses approximately 4.2 times the dose in patients at the MRHD.
In embryo-fetal development studies, pregnant animals were orally administered fostamatinib during the period of organogenesis at doses up to 25 and 50 mg/kg/day in rats and rabbits, respectively. The adverse developmental outcomes included an increase in embryo-fetal mortality (post-implantation loss), alterations to growth (lower fetal weights), and structural abnormalities (variations and malformations). These effects occurred at maternal exposures (AUCs) of 3,763 ng.h/mL in rats and 111,105 ng.h/mL in rabbits that were approximately 0.3 and 10 times the human exposure at the MRHD in rats and rabbits, respectively.
In a peri and postnatal development study in rats, fostamatinib was orally administered at doses of 2.5, 12.5, and 25 mg/kg/day from gestation day 7 until lactation day 20. The dose of 25 mg/kg/day was associated with maternal toxicity, including decreased body weights, body weight gains, and food consumption. At doses as low as 12.5 mg/kg/day fostamatinib caused increases in newborn mortality (neonatal mortality), alterations in growth and/or development (lower neonatal weights into post-weaning and structural abnormalities [malformations]). Functional impairment (delayed sexual maturation) was observed at 25 mg/kg/day. There was no evidence of neurobehavioral defects (maze learning and shuttle box avoidance) or immunological compromise (influenza host resistance challenge) in the F1 generation or latent untoward effects in the F2 generation. The maternal doses were approximately 2.1 and 4.2 times the MHRD in patients.
8.2 Lactation
Risk Summary
There are no data on the presence of fostamatinib and/or its metabolites in human milk, the effects on the breastfed child, or on milk production. In rodents, R406 (the major active metabolite) was detected in maternal milk in concentrations 5- to 10-fold higher than in maternal plasma. Because of the potential for serious adverse reactions in a breastfed child from TAVALISSE, advise a lactating woman not to breastfeed during treatment with TAVALISSE and for at least 1 month after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Based on animal studies, TAVALISSE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. For females of reproductive potential, verify pregnancy status prior to initiating TAVALISSE.
Contraception
Females
Based on animal studies, TAVALISSE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with TAVALISSE and for at least 1 month after the last dose.
Infertility
There are no data on the effect of TAVALISSE on human fertility. Based on the finding of reduced pregnancy rates in animal studies, TAVALISSE may affect female fertility [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established. TAVALISSE is not recommended for use in patients less than 18 years of age because adverse effects on actively growing bones were observed in nonclinical studies. In subchronic, chronic, and carcinogenicity studies of TAVALISSE, chondrodystrophy of the femoral head was seen in rodents. In a study in juvenile rabbits, growth plate dysplasia was observed in the proximal femur and femoro-tibial joint, and bone marrow cellularity was reduced in the femur and sternum.
8.5 Geriatric Use
Of the 102 patients with ITP who received TAVALISSE, 28 (27%) were 65 years of age and older, while 11 (11%) were 75 years of age and older. In patients 65 years of age and older, 6 (21%) patients experienced serious adverse events and 5 (18%) experienced adverse events leading to treatment withdrawal while in patients under 65 years of age, 7 (9%) and 5 (7%) experienced serious adverse events and adverse events leading to treatment withdrawal, respectively. In patients 65 years of age and older who received TAVALISSE, 11 (39%) patients experienced hypertension versus 2 (18%) placebo compared to 17 (23%) in patients under 65 of age versus 4 (11%) placebo. No overall differences in effectiveness were observed in these patients compared to younger patients.
-
10 OVERDOSAGE
There is no specific antidote for overdose with TAVALISSE, and the amount of R406 (the pharmacologically active metabolite of fostamatinib) cleared by dialysis is negligible. In the event of an overdose, monitor patient closely for signs and symptoms of adverse reactions, and treat the reactions with supportive care [see Warnings and Precautions (5)].
-
11 DESCRIPTION
Fostamatinib is a tyrosine kinase inhibitor. TAVALISSE is formulated with the disodium hexahydrate salt of fostamatinib, a phosphate prodrug that converts to its pharmacologically active metabolite, R406, in vivo.
The chemical name for fostamatinib disodium hexahydrate is disodium (6-[[5-fluoro-2-(3,4,5-trimethoxyanilino) pyrimidin-4-yl]amino]-2,2-dimethyl-3-oxo-pyrido[3,2-b][1,4]oxazin-4-yl)methyl phosphate hexahydrate. The molecular formula is C23H24FN6Na2O9P∙6H2O, and the molecular weight is 732.52. The structural formula is:

Fostamatinib disodium is a white to off-white powder that is practically insoluble in pH 1.2 aqueous buffer, slightly soluble in water, and soluble in methanol.
Each TAVALISSE oral tablet contains 100 mg or 150 mg fostamatinib, equivalent to 126.2 mg or 189.3 mg fostamatinib disodium hexahydrate, respectively.
The inactive ingredients in the tablet core are mannitol, sodium bicarbonate, sodium starch glycolate, povidone, and magnesium stearate. The inactive ingredients in the film coating are polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350, talc, iron oxide yellow, and iron oxide red.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Fostamatinib is a tyrosine kinase inhibitor with demonstrated activity against spleen tyrosine kinase (SYK). The major metabolite of fostamatinib, R406, inhibits signal transduction of Fc-activating receptors and B-cell receptor. The fostamatinib metabolite R406 reduces antibody-mediated destruction of platelets.
12.2 Pharmacodynamics
Mean treatment-related increases of 2.93 mmHg in systolic blood pressure and 3.53 mmHg in diastolic blood pressure over placebo were observed following TAVALISSE doses of 100 mg twice daily for 28 days. About 31% of patients in the TAVALISSE group experienced blood pressures ≥140/90 mmHg compared to 15% of patients in the placebo group. Blood pressure returned to baseline within 1 week following TAVALISSE discontinuation in 58% (11 of 19) of patients in the TAVALISSE group who had blood pressures ≥140/90 mmHg.
12.3 Pharmacokinetics
TAVALISSE is a prodrug that is converted in the gut to the major active metabolite, R406. Mean (± standard deviation [SD]) exposure estimates of R406 are 550 (± 270) ng/mL for Cmax and 7080 (± 2670) ng∙h/mL for AUC. R406 exposure is approximately dose proportional up to 200 mg twice daily (1.3 times the 150 mg dosage). R406 accumulates approximately 2- to 3-fold upon twice daily dosing at 100–160 mg (0.67 to 1.06 times the 150 mg dosage).
Absorption
After oral administration of TAVALISSE, the absolute bioavailability of R406 was 55%. The median tmax of R406 is approximately 1.5 hours (range: 1 to 4 hours). Negligible levels of fostamatinib were found in plasma.
Effect of Food
Administration of TAVALISSE with a high-calorie, high-fat meal (deriving approximately 150, 250, and 500–600 calories from protein, carbohydrate, and fat, respectively) increased R406 AUC by 23% and Cmax by 15% [see Dosage and Administration (2.1)].
Distribution
In in vitro studies, the R406 is 98.3% protein bound in human plasma. The red blood cell to plasma concentration ratio is approximately 2.6. The mean (± SD) volume of distribution at steady-state of R406 is 256 (± 92) L.
Elimination
The mean (± SD) terminal half-life of R406 is approximately 15 (± 4.3) hours.
Metabolism
TAVALISSE is metabolized in the gut by alkaline phosphatase to the major active metabolite, R406. R406 is extensively metabolized, primarily through pathways of CYP450-mediated oxidation (by CYP3A4) and glucuronidation (by UDP glucuronosyltransferase [UGT]1A9). R406 is the predominant moiety in the systemic circulation, and there was minimal exposure to any R406 metabolites.
Excretion
Following an oral dose of TAVALISSE, approximately 80% of the R406 metabolite is excreted in feces with approximately 20% excreted in the urine. The major component excreted in urine was R406 N-glucuronide. The major components excreted in feces were R406, O-desmethyl R406 and a metabolite produced by gut bacteria from the O-desmethyl metabolite of R406.
Specific Populations
Population pharmacokinetics analyses indicate TAVALISSE is not altered based on age, sex, race/ethnicity. In addition, the pharmacokinetics of TAVALISSE is not altered in patients with renal impairment (creatinine clearance [CLcr] ≥ 30 to < 50 mL/min, estimated by Cockcroft Gault equation and end stage renal disease requiring dialysis), or hepatic impairment (Child-Pugh Class A, B and C).
Drug Interaction Studies
Clinical Pharmacology Studies
No significant interactions were seen with concomitant use of TAVALISSE with the following drugs: methotrexate (OAT1/3 transporters), midazolam (CYP3A4 substrate), microgynon (ethinyl estradiol and levonorgestrel), warfarin, pioglitazone (CYP2C8 substrate) and ranitidine (H2-antagonist that increases gastric pH).
Effect of Other Drugs on TAVALISSE
Strong CYP3A4 inhibitor: Concomitant use of ketoconazole (200 mg twice daily for 3.5 days) with a single dose of 80 mg TAVALISSE (0.53 times the 150 mg dosage) increased R406 AUC by 102% and Cmax by 37%.
Effect of TAVALISSE on Other Drugs
CYP3A4 substrate: Concomitant use of simvastatin (single dose 40 mg) with 100 mg twice daily TAVALISSE increased simvastatin AUC by 64% and Cmax by 113% and simvastatin acid AUC by 64% and Cmax by 83%.
In Vitro Studies
TAVALISSE is an inhibitor of the human P-gp efflux transporter in vitro.
CYP3A4 and UGT1A9 are involved in the metabolism of R406. R406 is a substrate of P-gp but not of other major transporters (OAT1/3, OCT2, OATP1B1/3, MRP2, and BCRP). R406 can inhibit CYP3A4 and BCRP, and can induce CYP2C8 activity.
R406 is an inhibitor of UGT1A1. Inhibition of UGT1A1 may result in increased unconjugated bilirubin in the absence of other LFT abnormalities.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Fostamatinib was not carcinogenic in a 2-year study in mice when administered daily by oral gavage at doses up to 500/250 mg/kg/day, and was not carcinogenic in rats when administered by oral gavage at 45 mg/kg/day.
Fostamatinib and its major active metabolite (R406) were not mutagenic in an in vitro bacterial reverse mutation (Ames) assay or clastogenic in an in vitro human lymphocyte chromosomal aberration assay or an in vivo mouse bone marrow micronucleus assay.
In a fertility study with oral fostamatinib, all mating (e.g., time to mating, breeding proficiency), sperm assessments (e.g., number and motility), and organ weight (e.g., paired testis weight) parameters in male rats were unaffected by dosages as high as 40 mg/kg/day, which is 6.7 times the MRHD. All mating and fertility parameters in female rats were unaffected by dosages as high as 11 mg/kg/day (which is 1.8 times the MRHD), but a slight decrease in pregnancy rates and an increase in post-implantation loss were seen at 25 mg/kg/day, which is 4.2 times the MRHD.
-
14 CLINICAL STUDIES
TAVALISSE was studied in two placebo-controlled efficacy and safety studies (referred to as FIT-1 [NCT02076399] and FIT-2 [NCT02076412]), and in an open-label extension study referred to as FIT-3 (NCT 02077192).
Randomized, Placebo-Controlled Studies
A total of 150 patients with persistent or chronic ITP, who had an insufficient response to previous treatment (which included corticosteroids, immunoglobulins, splenectomy, and/or a thrombopoietin receptor agonists) were enrolled in two identical, double-blind, placebo-controlled studies that were conducted in different countries. For each study, patients were randomized 2:1 to TAVALISSE or placebo for 24 weeks; randomization was stratified with respect to prior splenectomy and severity of thrombocytopenia. Stable concurrent ITP therapy (glucocorticoids [< 20 mg prednisone equivalent per day], azathioprine, or danazol) was allowed, and rescue therapy was permitted, if needed. All patients initially received study drug at 100 mg twice daily (or matching placebo). Based on platelet count and tolerability, dose escalation to 150 mg twice daily (or matching placebo) was undertaken in 88% of patients at Week 4 or later. Patients who did not respond to treatment after 12 weeks, as well as patients who completed the 24-week double blind study, were eligible to enroll in open-label extension study (FIT-3).
Patients enrolled in the placebo-controlled studies had a median age of 54 years (range: 20 to 88), and the majority were female (61%) and were White (93%). Prior ITP treatments were varied, with the most common including corticosteroids (94%), immunoglobulins (53%), and thrombopoietin receptor agonists (TPO-RA) (48%). Most patients had chronic ITP (93%), with a median time since ITP diagnosis of 8.45 years, and 35% had undergone splenectomy. At baseline, the median platelet count was 16 × 109/L (with almost half [45]%) less than 15 × 109/L) and 47% were on stable ITP therapy.
In Study FIT-1, 76 patients were randomized; 51 to the TAVALISSE group and 25 to the placebo group. In Study FIT-2, 74 patients were randomized; 50 to the TAVALISSE group and 24 to the placebo group. The efficacy of TAVALISSE was based on stable platelet response (at least 50 ×109/L on at least 4 of the 6 visits between Weeks 14 to 24). Study outcomes for FIT-1 and FIT-2 are shown in Table 5.
Table 5: Study Outcomes from Placebo-Controlled Clinical Studies Study Outcomes Study FIT-1 Study FIT-2 TAVALISSE
(N=51)Placebo
(N=25)TAVALISSE
(N=50)Placebo
(N=24)n (%) n (%) n (%) n (%) NS = Did not demonstrate a stastistically significant difference between treatment arms - *
- Includes all patients with platelet counts and excludes patients whose platelet counts were measured following rescue therapy after Week 10
- †
- Stable platelet response was prospectively defined as a platelet count of at least 50 × 109/L on at least 4 of the 6 visits between Weeks 14 and 24
- ‡
- p-value from Fisher Exact test
- §
- Patients who did not respond to treatment after 12 weeks were eligible to enroll in open-label extension study.
Stable platelet response*,† 9 (18) 0 (0) 8 (16) 1 (4) p‡ = 0.03 NS Rolled-over into FIT-3 at Week 12§ 28 (55) 22 (88) 33 (66) 19 (79) Completed study (Week 24) 12 (24) 1 (4) 13 (26) 2 (8) In the FIT-1 and FIT-2 studies a total of 47 patients in the TAVALISSE arm had received a prior TPO-RA treatment; among these patients, 8 patients (17%) achieved a stable response to TAVALISSE. All 8 patients had previously discontinued TPO-RA due to loss of effect. Rescue medication was required by 30% and 45% of patients receiving TAVALISSE or placebo, respectively.
During the placebo-controlled studies, the incidence of bleeding occurred in 29% and 37% of patients in the TAVALISSE and placebo arms, respectively. Moderate, severe and serious bleeding events are described in Table 6. All severe events led to hospitalizations.
Table 6: Incidence of Moderate, Severe and Serious Bleeding-Related Events (Placebo-Controlled Efficacy Population) Parameter TAVALISSE
Total N=101
n (%)Placebo
Total N=49
n (%)Incidence of moderate bleeding-related adverse events 9 (9) 5 (10) Incidence of severe bleeding-related adverse events 1 (1) 3 (6) Incidence of serious bleeding-related adverse events 4 (4) 5 (10) Extension Study
The FIT-3 trial is an open label extension study. Patients from FIT-1 and FIT-2 who completed 24 weeks of treatment, or who did not respond to treatment any time after 12 weeks, were eligible to enroll in this study. Patients remained blinded to their treatment assignment from the previous study (TAVALISSE or placebo), so their starting dose in this study was based on their final platelet count. Patients designated as responders (defined as achievement of platelet count of at least 50 × 109/L) at the time of roll over continued in the extension study at their current trial dose and regimen. Patients who entered the extension study as non-responders (defined as platelet count less than 50 × 109/L) received TAVALISSE 100 mg twice daily regardless of their dose and regimen in the prior study.
For the FIT-3 trial, 123 patients were enrolled, 44 patients previously randomized to placebo and 79 patients previously randomized to TAVALISSE. Stable response in this study was prospectively defined as no 2 visits, at least 4 weeks apart, with a platelet count less than 50 × 109/L, without an intervening visit with a platelet count of at least 50 × 109/L (unrelated to rescue therapy), within a period of 12 weeks following initial achievement of the target platelet count. Sixty-one of the 123 subjects (50%) have discontinued from the study early.
In a prospectively defined analysis, the 44 subjects treated with placebo in the prior study were evaluated for stable response for TAVALISSE. Ten of these subjects (23%) (including a single subject who was classified as a placebo responder in the prior study) met the criteria for stable response.
Among the subjects who achieved stable response in FIT-1, FIT-2 and FIT-3 trials, 18 subjects maintained the platelet count of at least 50 × 109/L for 12 months or longer.
-
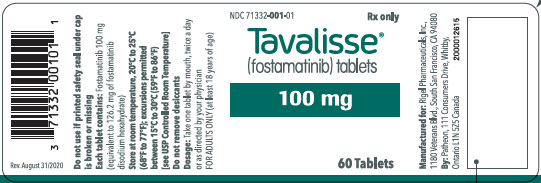
16 HOW SUPPLIED/STORAGE AND HANDLING
TAVALISSE 100 mg tablets are round, biconvex, orange, film-coated tablets debossed with "100" on one side and "R" on the reverse side.
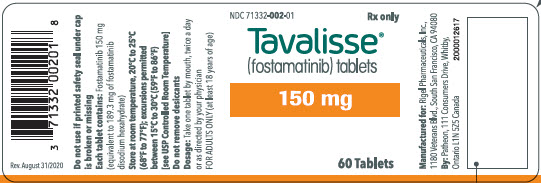
TAVALISSE 150 mg tablets are oval, biconvex, orange, film-coated tablets debossed with "150" on one side and "R" on the reverse side.
100 mg tablets: Available in bottle of 60 with 2 desiccant canisters NDC 71332-001-01 150 mg tablets: Available in bottle of 60 with 2 desiccant canisters NDC 71332-002-01 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
- Hypertension:
Inform patients that periodic monitoring of their blood pressure is required, as high blood pressure has occurred in patients taking TAVALISSE. Inform patients of the signs and symptoms of hypertension. Advise patients to undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if they experience signs or symptoms of hypertension [see Warnings and Precautions (5.1)]. - Hepatotoxicity:
Inform patients that periodic monitoring of their liver enzymes is required, and any elevations (which may indicate liver injury) will be managed appropriately, including interruption, reduction, or discontinuation of TAVALISSE [see Warnings and Precautions (5.2)]. - Diarrhea:
Advise patients to use supportive care measures, and if diarrhea becomes severe, it may necessitate interruption, reduction, or discontinuation of TAVALISSE [see Warnings and Precautions (5.3)]. - Neutropenia:
Inform patients that monitoring of their complete blood counts is required, and a decrease in neutrophils may necessitate interruption, reduction, or discontinuation of TAVALISSE [see Warnings and Precautions (5.4)]. - Advise patients to inform their healthcare providers of all their medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
- Embryo-Fetal Toxicity
Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the potential risk to a fetus [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment and for at least 1 month after receiving the last dose of TAVALISSE [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1, 8.3)]. - Lactation
Advise lactating women not to breastfeed during treatment with TAVALISSE and for at least 1 month after the last dose [see Use in Specific Populations (8.2)]. - Inform patients that TAVALISSE may be taken with or without food. In the case of a missed dose of TAVALISSE, instruct patients to take their next dose at its regularly scheduled time.
- Hypertension:
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
TAVALISSE® (TAV-a-leese)
(fostamatinib disodium hexahydrate)
tabletsThis Patient Information has been approved by the U.S. Food and Drug Administration. Issued: November 2020 What is the most important information I should know about TAVALISSE?
TAVALISSE can cause serious side effects, including:- High blood pressure (hypertension). New or worsening high blood pressure is common in people treated with TAVALISSE and can be severe. Your healthcare provider will check your blood pressure regularly during treatment with TAVALISSE. If needed, your healthcare provider may start you on blood pressure medicine or change your current medicine to treat your blood pressure. Tell your healthcare provider if you get headaches, confusion, dizziness, chest pain or shortness of breath.
- Liver problems. Changes in liver function blood tests are common with TAVALISSE. Liver problems may occur and can be severe. Your healthcare provider will regularly do blood tests to check how well your liver is working during treatment with TAVALISSE.
- Diarrhea. Diarrhea is common in people treated with TAVALISSE and can be severe. Tell your healthcare provider if you get diarrhea during treatment with TAVALISSE. Your healthcare provider may recommend changes in your diet, drinking more water, or medicine to limit these symptoms.
- Decrease in white blood cell counts (neutropenia). Decreases in your white blood cell count are common with TAVALISSE and can be severe. This may increase your risk for infection, including serious infections. Your healthcare provider will regularly do blood tests to check your white blood cell counts.
Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with TAVALISSE if you have side effects.
See "What are the possible side effects of TAVALISSE?" for more information about side effects.What is TAVALISSE?
TAVALISSE is a prescription medicine that is used to treat adults with low platelet counts due to chronic immune thrombocytopenia (ITP) when a prior treatment for ITP has not worked well enough.
It is not known if TAVALISSE is safe and effective in children.Before you take TAVALISSE, tell your healthcare provider about all of your medical conditions, including if you:
- have high blood pressure
- have liver problems
- are pregnant or plan to become pregnant. TAVALISSE can harm your unborn baby.
- Your healthcare provider will check if you are pregnant before starting treatment with TAVALISSE.
- Females who can become pregnant should use effective birth control during treatment with TAVALISSE and for at least 1 month after the last dose.
- are breastfeeding or plan to breastfeed. You should not breastfeed during treatment with TAVALISSE and for at least 1 month after the last dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking TAVALISSE with certain other medicines may affect how the other medicines work and other medicines may affect how TAVALISSE works.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine.How should I take TAVALISSE? - Take TAVALISSE exactly as your healthcare provider tells you to take it.
- Take TAVALISSE with or without food.
- If you miss a dose of TAVALISSE, take your next dose at its regularly scheduled time.
- If you take too much TAVALISSE, call your healthcare provider right away or go to the nearest hospital emergency room right away.
- Your healthcare provider will check your platelet count during your treatment with TAVALISSE and may change your dose of TAVALISSE as needed.
What are the possible side effects of TAVALISSE?
See "What is the most important information I should know about TAVALISSE?"
The most common side effects of TAVALISSE include:- nausea
- dizziness
- respiratory infection
- rash
- tiredness
- chest pain
- stomach (abdomen) pain
These are not all the side effects of TAVALISSE. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store TAVALISSE? - Store TAVALISSE at room temperature between 68°F and 77°F (20°C to 25°C).
- The bottle of TAVALISSE contains 2 desiccant canisters that help keep your medicine dry. Do not remove the desiccant canisters from the bottle.
General information about the safe and effective use of TAVALISSE
Medicines are sometimes prescribed for purposes other than those listed in the Patient Information. Do not use TAVALISSE for a condition for which it was not prescribed. Do not give TAVALISSE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about TAVALISSE that is written for health professionals.What are the ingredients in TAVALISSE?
Active ingredient: fostamatinib disodium hexahydrate
Inactive ingredients: The tablet core contains mannitol, sodium bicarbonate, sodium starch glycolate, povidone, and magnesium stearate. The film coating contains polyvinyl alcohol, titanium dioxide, polyethylene glycol 3350, talc, iron oxide yellow, and iron oxide red.
Manufactured for: Rigel Pharmaceuticals, Inc., South San Francisco, CA 94080 USA
Manufactured by: Patheon, Inc., 111 Consumers Drive, Whitby, Ontario L1N 5Z5 Canada
© Rigel Pharmaceuticals, Inc. All rights reserved. TAVALISSE is a registered trademark of Rigel Pharmaceuticals, Inc.
For more information go to www.TAVALISSE.com or call 1-800-983-1329. - PRINCIPAL DISPLAY PANEL - 100 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 150 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
TAVALISSE
fostamatinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71332-001 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FOSTAMATINIB (UNII: SQ8A3S5101) (FOSTAMATINIB - UNII:SQ8A3S5101) FOSTAMATINIB 100 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SODIUM BICARBONATE (UNII: 8MDF5V39QO) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color ORANGE Score no score Shape ROUND Size 9mm Flavor Imprint Code 100;R Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71332-001-01 60 in 1 BOTTLE; Type 0: Not a Combination Product 05/09/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209299 05/09/2018 TAVALISSE
fostamatinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71332-002 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FOSTAMATINIB (UNII: SQ8A3S5101) (FOSTAMATINIB - UNII:SQ8A3S5101) FOSTAMATINIB 150 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SODIUM BICARBONATE (UNII: 8MDF5V39QO) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color ORANGE Score no score Shape OVAL Size 15mm Flavor Imprint Code 150;R Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71332-002-01 60 in 1 BOTTLE; Type 0: Not a Combination Product 05/09/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209299 05/09/2018 Labeler - Rigel Pharmaceuticals, Inc. (967965468) Registrant - Rigel Pharmaceuticals, Inc. (967965468) Establishment Name Address ID/FEI Business Operations Patheon Whitby 205475333 MANUFACTURE(71332-001, 71332-002)