Label: IBSRELA- tenapanor hydrochloride tablet


- NDC Code(s): 73154-050-06, 73154-050-60
- Packager: Ardelyx, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated May 28, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IBSRELA® safely and effectively. See full prescribing information for IBSRELA.
IBSRELA (tenapanor) tablets, for oral use
Initial U.S. Approval: 2019WARNING: RISK OF SERIOUS DEHYDRATION IN PEDIATRIC PATIENTS
See full prescribing information for complete boxed warning.
- IBSRELA is contraindicated in patients less than 6 years of age; in young juvenile rats, tenapanor caused death presumed to be due to dehydration. (4, 8.4)
- Avoid use of IBSRELA in patients 6 years to less than 12 years of age. (5.1, 8.4)
- The safety and effectiveness of IBSRELA have not been established in pediatric patients less than 18 years of age. (8.4)
INDICATIONS AND USAGE
IBSRELA is a sodium/hydrogen exchanger 3 (NHE3) inhibitor indicated for treatment of irritable bowel syndrome with constipation (IBS-C) in adults. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 50 mg tenapanor. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Diarrhea: Patients may experience severe diarrhea. If severe diarrhea occurs, suspend dosing and rehydrate patient. (5.2)
ADVERSE REACTIONS
Most common adverse reactions (≥2%) are diarrhea, abdominal distension, flatulence and dizziness. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Ardelyx at 1-844-427-7352 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
OATP2B1 Substrates: Potential for reduced exposure of the concomitant drug (e.g., enalapril). Monitor for signs related to loss of efficacy and adjust the dosage of the concomitantly administered drug as needed. (7.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK OF SERIOUS DEHYDRATION IN PEDIATRIC PATIENTS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Serious Dehydration in Pediatric Patients
5.2 Diarrhea
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 OATP2B1 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK OF SERIOUS DEHYDRATION IN PEDIATRIC PATIENTS
- IBSRELA is contraindicated in patients less than 6 years of age; in nonclinical studies in young juvenile rats administration of tenapanor caused deaths presumed to be due to dehydration [see Contraindications (4), Use in Specific Populations (8.4)].
- Avoid use of IBSRELA in patients 6 years to less than 12 years of age [see Warnings and Precautions (5.1), Use in Specific Populations (8.4)].
- The safety and effectiveness of IBSRELA have not been established in patients less than 18 years of age [see Use in Specific Populations (8.4)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
The recommended dosage of IBSRELA in adults is 50 mg orally twice daily.
Administration Instructions
- Take IBSRELA immediately prior to breakfast or the first meal of the day and immediately prior to dinner [see Clinical Pharmacology (12.2)].
- If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take 2 doses at the same time.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
IBSRELA is contraindicated in:
- Patients less than 6 years of age due to the risk of serious dehydration [see Warnings and Precautions (5.1), Use in Specific Populations (8.4)].
- Patients with known or suspected mechanical gastrointestinal obstruction.
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Serious Dehydration in Pediatric Patients
IBSRELA is contraindicated in patients below 6 years of age. The safety and effectiveness of IBSRELA in patients less than 18 years of age have not been established. In young juvenile rats (less than 1 week old; approximate human age equivalent of less than 2 years of age), decreased body weight and deaths occurred, presumed to be due to dehydration, following oral administration of tenapanor. There are no data available in older juvenile rats (human age equivalent 2 years to less than 12 years).
Avoid the use of IBSRELA in patients 6 years to less than 12 years of age. Although there are no data in older juvenile rats, given the deaths in younger rats and the lack of clinical safety and efficacy data in pediatric patients, avoid the use of IBSRELA in patients 6 years to less than 12 years of age [see Contraindications (4), Warnings and Precautions (5.2), Use in Specific Populations (8.4)].
5.2 Diarrhea
Diarrhea was the most common adverse reaction in two randomized, double-blind, placebo-controlled trials of IBS-C. Severe diarrhea was reported in 2.5% of IBSRELA-treated patients [see Adverse Reactions (6.1)]. If severe diarrhea occurs, suspend dosing and rehydrate patient.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described below reflect data from 1203 adult patients with IBS-C in two randomized, double-blind, placebo-controlled clinical trials (Trial 1 and Trial 2). Patients were randomized to receive placebo or IBSRELA 50 mg twice daily for up to 52 weeks. Demographic characteristics were comparable between treatment groups in the two trials [see Clinical Studies (14)].
Most Common Adverse Reactions
The most common adverse reactions reported in at least 2% of patients in IBSRELA-treated patients and at an incidence greater than placebo during the 26-week double-blind placebo-controlled treatment period of Trial 1 are shown in Table 1.
Table 1: Most Common Adverse Reactions* in Patients with IBS-C in Trial 1 (26 Weeks) Adverse Reactions IBSRELA
N=293
%Placebo
N=300
%- *
- Reported in at least 2% of patients in IBSRELA-treated patients and at an incidence greater than placebo
Diarrhea 16 4 Abdominal Distension 3 <1 Flatulence 3 1 Dizziness 2 <1 The adverse reaction profile was similar during the 12-week double-blind placebo-controlled treatment period of Trial 2 (610 patients: 309 IBSRELA-treated and 301 placebo-treated) with diarrhea (15% with IBSRELA vs 2% with placebo) and abdominal distension (2% with IBSRELA vs 0% with placebo) as the most common adverse reactions.
Adverse Reaction of Special Interest – Severe Diarrhea
Severe diarrhea was reported in 2.5% of IBSRELA-treated patients compared to 0.2% of placebo-treated patients during the 26 weeks of Trial 1 and the 12 weeks of Trial 2 [see Warnings and Precautions (5.2)].
Patients with Renal Impairment
In Trials 1 and 2, there were 368 patients (31%) with baseline renal impairment (defined as eGFR less than 90 mL/min/1.73m2). In patients with renal impairment, diarrhea, including severe diarrhea, was reported in 20% (39/194) of IBSRELA-treated patients and 0.6% (1/174) of placebo-treated patients. In patients with normal renal function at baseline, diarrhea, including severe diarrhea, was reported in 13% (53/407) of IBSRELA-treated patients and 3.5% (15/426) of placebo-treated patients. No other differences in the safety profile were reported in the renally impaired subgroup. The incidence of diarrhea and severe diarrhea in IBSRELA-treated patients did not correspond to the severity of renal impairment.
Adverse Reactions Leading to Discontinuation
Discontinuations due to adverse reactions occurred in 7.6% of IBSRELA-treated patients and 0.8% of placebo-treated patients during the 26 weeks of Trial 1 and the 12 weeks of Trial 2. The most common adverse reaction leading to discontinuation was diarrhea: 6.5% of IBSRELA-treated patients compared to 0.7% of placebo-treated patients.
Less Common Adverse Reactions
Adverse reactions reported in less than 2% of IBSRELA-treated patients and at an incidence greater than placebo during the 26 weeks of Trial 1 and the 12 weeks of Trial 2 were: rectal bleeding and abnormal gastrointestinal sounds.
Hyperkalemia
In a trial of another patient population with chronic kidney disease (defined by eGFR from 25 to 70 mL/min/1.73m2) and Type 2 diabetes mellitus, three serious adverse reactions of hyperkalemia resulting in hospitalization were reported in 3 patients (2 IBSRELA-treated patients and 1 placebo-treated patient).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of IBSRELA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity reactions: pruritis, rash, and urticaria
-
7 DRUG INTERACTIONS
7.1 OATP2B1 Substrates
Tenapanor is an inhibitor of intestinal uptake transporter, OATP2B1 [see Clinical Pharmacology (12.3)]. Drugs which are substrates of OATP2B1 may have reduced exposures when concomitantly taken with IBSRELA. Monitor for signs related to loss of efficacy and adjust the dosage of concomitantly administered drug as needed.
Enalapril is a substrate of OATP2B1. When enalapril was coadministered with tenapanor (30 mg twice daily for five days, a dosage 0.6 times the recommended dosage), the peak exposure (Cmax) of enalapril and its active metabolite, enalaprilat, decreased by approximately 70% and total systemic exposures (AUC) decreased by approximately 50% to 65% compared to when enalapril was administered alone [see Clinical Pharmacology (12.3)].
Monitor blood pressure and increase the dosage of enalapril, if needed, when IBSRELA is coadministered with enalapril.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Tenapanor is minimally absorbed systemically, with plasma concentrations below the limit of quantification (less than 0.5 ng/mL) following oral administration [see Clinical Pharmacology (12.3)]. Therefore, maternal use is not expected to result in fetal exposure to the drug. The available data on IBSRELA exposure from a small number of pregnant women have not identified any drug associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. In reproduction studies with tenapanor in pregnant rats and rabbits, no adverse fetal effects were observed in rats at 0.1 times the maximum recommended human dose and in rabbits at doses up to 8.8 times the maximum recommended human dose (based on body surface area).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the United States general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryofetal development study in rats, tenapanor was administered orally to pregnant rats during the period of organogenesis at dose levels of 1, 10 and 30 mg/kg/day. Tenapanor doses of 10 and 30 mg/kg/day were not tolerated by the pregnant rats and was associated with mortality and moribundity with body weight loss. The 10 and 30 mg/kg dose group animals were sacrificed early, and the fetuses were not examined for intrauterine parameters and fetal morphology. No adverse fetal effects were observed in rats at 1 mg/kg/day (approximately 0.1 times the maximum recommended human dose) and in rabbits at doses up to 45 mg/kg/day (approximately 8.8 times the maximum recommended human dose, based on body surface area).
In a pre- and post-natal developmental study in mice, tenapanor at doses up to 200 mg/kg/day (approximately 9.7 times the maximum recommended human dose, based on body surface area) had no effect on pre- and post-natal development.
8.2 Lactation
Risk Summary
Tenapanor and its major metabolite, M1, were not detected in the breast milk of lactating women (see Data). In adults, concentrations of tenapanor were below the limit of quantitation in plasma following multiple doses of IBSRELA [see Clinical Pharmacology (12.3)]. Maternal use of IBSRELA is not expected to result in exposure to tenapanor or its major metabolite in breastfed infants. There is no information on the effects of tenapanor or its major metabolite on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for IBSRELA and any potential adverse effects on the breastfed infant from IBSRELA or from the underlying maternal condition.
Data
A clinical lactation study was conducted in seven healthy adult women who were 22 to 37 years of age. Following oral administration of IBSRELA 50 mg twice daily for 3 days, the concentrations of tenapanor and its major metabolite were below the limit of quantitation (<1 ng/mL and <1 ng/mL) in all breast milk samples collected over 24 hours post-dosing.
8.4 Pediatric Use
IBSRELA is contraindicated in patients less than 6 years of age. Avoid IBSRELA in patients 6 years to less than 12 years of age [see Contraindications (4), Warnings and Precautions (5.1)].
The safety and effectiveness of IBSRELA in patients less than 18 years of age have not been established.
In nonclinical studies, deaths occurred in young juvenile rats (less than 1 week-old-rats approximate human age equivalent of less than 2 years of age) following oral administration of tenapanor, as described below in Juvenile Animal Toxicity Data.
Juvenile Animal Toxicity Data
In a 21-day oral dose range finding toxicity study in juvenile rats, tenapanor was administered to neonatal rats (post-natal day (PND) 5) at doses of 5 and 10 mg/kg/day. Tenapanor was not tolerated in male and female pups and the study was terminated on PND 16 due to mortalities and decreased body weight (24% to 29% reduction in females at the respective dose groups and 33% reduction in males in the 10 mg/kg/day group, compared to control).
In a second dose range finding study, tenapanor doses of 0.1, 0.5, 2.5, or 5 mg/kg/day were administered to neonatal rats from PND 5 through PND 24. Treatment-related mortalities were observed at 0.5, 2.5, and 5 mg/kg/day doses. These premature deaths were observed as early as PND 8, with majority of deaths occurring between PND 15 and 25. In the 5 mg/kg/day group, mean body weights were 47% lower for males on PND 23 and 35% lower for females on PND 22 when compared to the controls. Slightly lower mean tibial lengths (5% to 11%) were noted in males and females in the 0.5, 2.5, and 5 mg/kg/day dose groups on PND 25 and correlated with the decrements in body weight noted in these groups. Lower spleen, thymus, and/or ovarian weights were noted at the 0.5, 2.5 and 5 mg/kg/day doses. Tenapanor-related gastrointestinal distension and microscopic bone findings of increased osteoclasts, eroded bone, and/or decreased bone in sternum and/or femorotibial joint were noted in males and females in the 0.5, 2.5 and 5 mg/kg/day dose groups [see Contraindications (4), Warnings and Precautions (5.1)].
8.5 Geriatric Use
Of the 1203 patients in placebo-controlled clinical trials of IBSRELA, 100 (8%) were 65 years of age and older. No overall differences in safety or effectiveness were observed between elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
10 OVERDOSAGE
Based on nonclinical data, overdose of IBSRELA may result in gastrointestinal adverse effects such as diarrhea as a result of exaggerated pharmacology with a risk for dehydration if diarrhea is severe or prolonged [see Warnings and Precautions (5.1)].
-
11 DESCRIPTION
IBSRELA (tenapanor) tablets contain tenapanor hydrochloride as an active ingredient. Tenapanor hydrochloride is a sodium/hydrogen exchanger 3 (NHE3) inhibitor for oral use. The chemical name for tenapanor hydrochloride is 12,15-Dioxa-2,7,9-triazaheptadecanamide, 17-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-, hydrochloride (1:2). Tenapanor hydrochloride has the molecular formula of C50H68Cl6N8O10S2, the molecular weight of 1218 Daltons, and the chemical structure below:
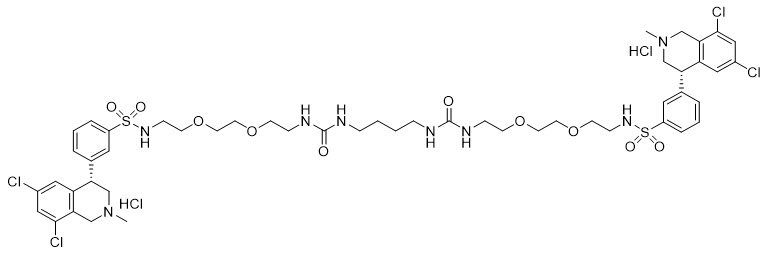
Tenapanor hydrochloride is a white to off-white to light brown hygroscopic amorphous solid. It is practically insoluble in water.
IBSRELA tablets contain 50 mg of tenapanor (equivalent to 53.2 mg of tenapanor hydrochloride). Inactive ingredients in the tablet are colloidal silicon dioxide, low-substituted hydroxypropyl cellulose, microcrystalline cellulose, propyl gallate, stearic acid, tartaric acid, and the coating agent OPADRY®, which consists of hypromellose, titanium dioxide and triacetin.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tenapanor is a locally acting inhibitor of the sodium/hydrogen exchanger 3 (NHE3), an antiporter expressed on the apical surface of the small intestine and colon primarily responsible for the absorption of dietary sodium. In vitro and animal studies indicate its major metabolite, M1, is not active against NHE3. By inhibiting NHE3 on the apical surface of the enterocytes, tenapanor reduces absorption of sodium from the small intestine and colon, resulting in an increase in water secretion into the intestinal lumen, which accelerates intestinal transit time and results in a softer stool consistency.
Tenapanor has also been shown to reduce abdominal pain by decreasing visceral hypersensitivity and by decreasing intestinal permeability in animal models. In rat model of colonic hypersensitivity, tenapanor reduced visceral hyperalgesia and normalized colonic sensory neuronal excitability.
12.2 Pharmacodynamics
Cardiac Electrophysiology
At 3 times the mean maximum exposure of M1 at the recommended dosage, there were no clinically relevant effects on the QTc interval.
Food Effect
Administration of IBSRELA 5 to 10 minutes before a meal increased the 24-hour stool sodium excretion compared to taking IBSRELA in the fed or fasting condition [see Dosage and Administration (2)]. In clinical trials, IBSRELA was administered immediately prior to the first meal of the day and immediately prior to dinner.
12.3 Pharmacokinetics
Absorption
Tenapanor is minimally absorbed following repeated twice daily oral administration. Plasma concentrations of tenapanor were below the limit of quantitation (less than 0.5 ng/mL) in the majority of samples from healthy subjects following single and repeated oral administration of IBSRELA 50 mg twice daily. Therefore, standard pharmacokinetic parameters such as area under the curve (AUC), maximum concentration (Cmax), and half-life (t1/2) could not be determined.
Distribution
Plasma protein binding of tenapanor and its major metabolite, M1, is approximately 99% and 97%, respectively, in vitro.
Elimination
Metabolism
Tenapanor is metabolized primarily by CYP3A4/5 and low levels of its major metabolite, M1, are detected in plasma. The Cmax of M1 is approximately 13 ng/mL after single dose of IBSRELA 50 mg and 15 ng/mL at steady state following repeated dosing of IBSRELA 50 mg twice daily in healthy subjects.
Excretion
Following administration of a single 15 mg radiolabeled 14C-tenapanor dose to healthy subjects, approximately 70% of the radioactivity was excreted in feces within 120 hours post-dose and 79% within 240 hours post-dose, mostly as the parent drug accounting for 65% of dose within 144 hours post-dose. Approximately 9% of the administered dose was recovered in urine, primarily as metabolites. M1 is excreted in urine unchanged accounting for 1.5% of dose within 144 hours post-dose.
Specific Populations
Patients with Hepatic Impairment
Following a single dose of tenapanor 100 mg in patients with moderate hepatic impairment (Child-Pugh B), plasma concentrations of tenapanor were mostly below the limit of quantitation (< 0.5 ng/mL) and the pharmacokinetic parameters for tenapanor could not be determined. The geometric mean AUC and Cmax of the major metabolite, M1, were approximately 33% and 27% lower, respectively, in patients with moderate hepatic impairment compared to those of healthy subjects. The decrease in M1 systemic exposure is not clinically relevant.
Drug Interaction Studies
CYP Metabolism Mediated Drug Interactions
Tenapanor and M1 did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6 in vitro.
Tenapanor and M1 did not induce CYP1A2 and CYP2B6 in vitro.
No significant inhibition or induction of CYP3A4 enzyme using midazolam as a substrate was observed when IBSRELA 50 mg was administered twice a day for 13 days in healthy subjects.
Following co-administration of a single dose of IBSRELA 50 mg with repeated doses of itraconazole 200 mg, a CYP3A4 inhibitor, the mean AUC and Cmax of M1 was decreased 50% in healthy subjects. The decrease in M1 systemic exposure is not clinically relevant. Plasma concentrations of tenapanor were mostly below the limit of quantitation (less than 0.5 ng/mL) after co-administration of itraconazole.
No significant effect on CYP2C9 activity using warfarin as a substrate was observed when tenapanor 30 mg was administered twice a day (a dosage 0.6 times the recommended dosage) for 12 days in healthy subjects.
Membrane Transporter Mediated Drug Interactions
Tenapanor inhibited OATP2B1, but is not an inhibitor of P-gp, BCRP, OATP1B1, and OATP1B3. M1 did not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2-K.
M1 is a substrate of P-gp. Tenapanor is not a substrate of P-gp, BCRP, OATP1B1, and OATP1B3. M1 is not a substrate of BCRP, OAT1, OAT3, OCT2, MATE1 and MATE2-K.
No significant effect on PepT1 activity using cefadroxil as a substrate was observed when IBSRELA 50 mg was administered twice a day for 12 days in healthy subjects.
No significant effect on P-gp activity using digoxin as a substrate was observed when tenapanor 30 mg was administered twice a day (a dosage 0.6 times the recommended dosage) for 12 days in healthy subjects.
Following administration of a single 20 mg dose of enalapril (OATP2B1 substrate) with tenapanor 30 mg administered twice a day (a dosage 0.6 times the recommended dosage) at steady state in healthy subjects, the mean AUC and Cmax of enalapril was decreased by 64% and 69%, respectively. The mean AUC and Cmax of enalaprilat was decreased by 52% and 68%, respectively [see Drug Interactions (7.1)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of tenapanor was assessed in a 6-month carcinogenicity study in Tg rasH2 mice and in a 2-year carcinogenicity study in rats.
Tenapanor was not tumorigenic at oral doses up to 100 mg/kg/day (approximately 4.5 times the recommended human dose, based on the body surface area) in male mice and 800 mg/kg/day (approximately 39 times the maximum recommended human dose, based on the body surface area) for female mice. Tenapanor was not tumorigenic in male and female rats at oral doses up to 5 mg/kg/day (approximately 0.5 times the recommended human dose, based on the body surface area). The major metabolite of tenapanor, M1, was not tumorigenic in Tg rasH2 mice at oral doses up to 165 mg/kg/day (approximately 8 times the maximum recommended human dose, based on the body surface area)
Mutagenesis
Tenapanor was not genotoxic in the in vitro bacterial reverse mutation (Ames) assays, an in vitro chromosomal aberration assay in cultured human peripheral blood lymphocytes or the in vivo micronucleus assays in mice and rats.
Impairment of Fertility
Tenapanor had no effect on fertility or reproductive function in male rats at oral doses up to 10 mg/kg/day (approximately 0.97 times the recommended human dose, based on the body surface area) and in female mice at oral doses up to 50 mg/kg/day (approximately 2.4 times the recommended human dose, based on the body surface area).
-
14 CLINICAL STUDIES
The efficacy of IBSRELA for the treatment of IBS-C was established in two double-blind, placebo-controlled, randomized, multicenter trials in adult patients: Trial 1 (TEN-01-302; NCT02686138) and Trial 2 (TEN-01-301; NCT02621892). The intent-to-treat (ITT) analysis population included 620 patients in Trial 1 and 606 patients in Trial 2 with mean age of 46 years (range 18 to 75 years), 80% females, 64% White and 31% Black/African American. In these clinical trials, IBSRELA was administered immediately prior to breakfast or the first meal of the day and immediately prior to dinner.
To enter the trials, all patients met Rome III criteria for IBS-C and were required to meet the following clinical criteria during the 2-week baseline run-in period:
- a mean abdominal pain score of at least 3 on a 0-to-10-point numeric rating scale where a score of 0 indicates no pain and 10 indicates very severe pain
- less than 3 complete spontaneous bowel movements (CSBMs) per week, where a CSBM is defined as a spontaneous bowel movement (SBM) that is associated with a sense of complete evacuation (an SBM is a bowel movement occurring in the absence of laxative use)
- less than or equal to 5 SBMs per week
The trial designs were identical through the first 12 weeks of treatment, and thereafter differed in that Trial 1 continued for an additional 14 weeks of treatment (26 weeks double-blind treatment), whereas Trial 2 included a 4-week randomized withdrawal (RW) period.
Efficacy of IBSRELA was assessed using responder analyses based on daily diary entries.
In both trials, the primary endpoint was the proportion of responders, where a responder was defined as a patient achieving both the stool frequency and abdominal pain intensity responder criteria in the same week for at least 6 of the first 12 weeks of treatment. The stool frequency (CSBM) and abdominal pain responder criteria assessed each week were defined as:
- CSBM responder: a patient who experienced an increase of at least 1 CSBM in weekly average from baseline.
- Abdominal pain responder: a patient who experienced at least a 30% reduction in the weekly average of abdominal pain score compared with baseline.
The responder rates for the primary endpoint and components of the primary endpoint (CSBM and abdominal pain), which were pre-specified key secondary endpoints, are shown in Table 2.
Table 2: Efficacy Responder Rates in Placebo-Controlled Trials (Trial 1 and Trial 2) in Adults with IBS-C: Responder for at least 6 of the First 12 Weeks of Treatment - *
- CI: Confidence Interval
- †
- A responder for these trials was defined as a patient who met both the abdominal pain and CSBM weekly responder criteria for at least 6 of the first 12 weeks.
- ‡
- A CSBM responder was defined as a patient who achieved an increase in at least 1 CSBM per week, from baseline, for a least 6 of at least 12 weeks.
- §
- An abdominal pain responder was defined as a patient who met the criteria of at least 30% reduction from baseline in weekly average of the worst daily abdominal pain, for at least 6 of the first 12 weeks.
Trial 1 IBSRELA
N=293Placebo
N=300Treatment Difference
[95% CI*]Responder† 37% 24% 13%
[6%, 20%]Components of Responder Endpoint: CSBM Responder‡ 47% 33% Abdominal Pain Responder§ 50% 38% Trial 2 Responder Rates IBSRELA
N=307Placebo
N=299Treatment Difference
[95% CI*]Responder† 27% 19% 8%
[2%, 15%]Components of Responder Endpoint: CSBM Responder‡ 34% 29% Abdominal Pain Responder§ 44% 33% In Trials 1 and 2, the proportion of responders for 9 out of the first 12 weeks, including at least 3 of the last 4 weeks, was greater in IBSRELA-treated patients compared to placebo-treated patients. In addition, in Trial 1, the proportion of responders for 13 out of 26 weeks was greater in IBSRELA-treated patients compared to placebo-treated patients.
In both trials, improvements from baseline in average weekly CSBMs and abdominal pain were observed by Week 1, with improvement maintained through the end of treatment.
In IBSRELA-treated patients re-randomized to placebo in Trial 2, CSBM frequency and abdominal pain severity worsened on average over the 4-week period but remained improved compared to baseline. Patients who continued on IBSRELA maintained their response to therapy on average over the additional 4 weeks. Patients on placebo who were re-randomized to IBSRELA had an average increase in CSBM frequency and a decrease in abdominal pain.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
IBSRELA tablets contain 50 mg tenapanor and are oval, white to off-white, debossed with "
 50" on one side and "5791" on the other side.
50" on one side and "5791" on the other side.IBSRELA is supplied in a white, opaque, high-density polyethylene bottle containing 60 tablets with a silica gel canister (as the desiccant) and screw-top polypropylene child-resistant cap lined and induction-activated aluminum foil liner (NDC 73154-050-60).
-
17 PATIENT COUNSELING INFORMATION
Advise the patients to read the FDA-approved patient labeling (Medication Guide).
Diarrhea
Instruct patients to stop IBSRELA and contact their healthcare provider if they experience severe diarrhea [see Warnings and Precautions (5.2)].
Accidental Ingestion
Accidental ingestion of IBSRELA in children, especially children less than 6 years of age, may result in severe diarrhea and dehydration. Instruct patients to store IBSRELA securely and out of reach of children [see Contraindications (4), Warnings and Precautions (5.1)].
Administration and Handling Instructions
Instruct Patients:
- To take IBSRELA immediately prior to breakfast or the first meal of the day and immediately before dinner [see Dosage and Administration (2)].
- If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take 2 doses at the same time [see Dosage and Administration (2)].
- To keep IBSRELA in a dry place. Protect from moisture. Keep in the original bottle. Do not remove desiccant from the bottle. Do not subdivide or repackage. Keep bottles tightly closed [see How Supplied/Storage and Handling (16)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: May 2025 Medication Guide
IBSRELA® (ibs rel`a)
(tenapanor)
tablets, for oral useWhat is the most important information I should know about IBSRELA? - Do not give IBSRELA to children who are less than 6 years of age. It may harm them.
- You should not give IBSRELA to children 6 years to less than 18 years of age. It may harm them.
See "What are the possible side effects of IBSRELA?" for more information about side effects.What is IBSRELA?
IBSRELA is a prescription medicine used in adults to treat:- Irritable bowel syndrome with constipation (IBS-C).
Who should not take IBSRELA? - Do not give IBSRELA to children who are less than 6 years of age. IBSRELA can cause severe diarrhea and your child could get severe dehydration (loss of a large amount of body water and salt).
- Do not take IBSRELA if a doctor has told you that you have a bowel blockage (intestinal obstruction).
Before you take IBSRELA, tell your doctor about all your medical conditions, including if you: - are pregnant or plan to become pregnant. It is not known if IBSRELA will harm your unborn baby.
- are breastfeeding or plan to breastfeed, although IBSRELA is not expected to pass into your breast milk and to harm your baby. Talk with your doctor about the best way to feed your baby if you take IBSRELA.
How should I take IBSRELA? - Take IBSRELA exactly as your doctor tells you to take it.
- Take 1 IBSRELA tablet by mouth, 2 times each day.
- Take IBSRELA immediately before breakfast or the first meal of the day and immediately before dinner.
- If a dose is missed, skip the missed dose and take the next dose at the regular time. Do not take 2 doses at the same time.
What are the possible side effects of IBSRELA?
IBSRELA can cause serious side effects, including:- See "What is the most important information I should know about IBSRELA?"
- Diarrhea is the most common side effect of IBSRELA, and it can sometimes be severe. Stop taking IBSRELA and call your doctor if you develop severe diarrhea.
- swelling, or a feeling of fullness or pressure in your abdomen (distension).
- gas (flatulence).
- dizziness.
How should I store IBSRELA? - Store IBSRELA at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep IBSRELA in the original container and protect from moisture. Keep the container of IBSRELA tightly closed and in a dry place.
- Do not put IBSRELA in another container (repackage).
- The IBSRELA bottle contains a desiccant canister to help keep your medicine dry (protect it from moisture). Do not remove the desiccant from the bottle.
General information about the safe and effective use of IBSRELA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use IBSRELA for a condition for which it was not prescribed. Do not give IBSRELA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about IBSRELA that is written for health professionals.What are the ingredients in IBSRELA?
Active ingredient: tenapanor hydrochloride
Inactive ingredients: colloidal silicon dioxide, hypromellose, low-substituted hydroxypropyl cellulose, microcrystalline cellulose, propyl gallate, stearic acid, tartaric acid, titanium dioxide, and triacetin.
Manufactured for and distributed by Ardelyx, Inc. Waltham, MA 02451 USA
IBSRELA® is a registered trademark of Ardelyx, Inc.
Patent: www.IBSRELA-patents.com
For more information, go to www.ardelyx.com or call 1-844-427-7352 - PRINCIPAL DISPLAY PANEL - 50 mg Tablet Bottle Label - 050-60
-
PRINCIPAL DISPLAY PANEL - 50 mg Tablet Bottle Label - 050-06
PROFESSIONAL SAMPLE – NOT FOR SALE
NDC 73154-050-06IBSRELA®
(tenapanor) tablets50 mg
ATTENTION PHARMACIST: Dispense the
accompanying Medication Guide to each patient.Attention Pharmacist:
Dispense IBSRELA® in original
container to patient. Do not remove
the desiccant from inside the bottle.6 tablets
Rx Only
-
INGREDIENTS AND APPEARANCE
IBSRELA
tenapanor hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:73154-050 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TENAPANOR HYDROCHLORIDE (UNII: 50605O2ZNS) (TENAPANOR - UNII:WYD79216A6) TENAPANOR 53.2 mg Product Characteristics Color WHITE Score no score Shape OVAL Size 14mm Flavor Imprint Code A50;5791 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:73154-050-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 09/12/2019 2 NDC:73154-050-06 6 in 1 BOTTLE; Type 0: Not a Combination Product 09/12/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211801 09/12/2019 Labeler - Ardelyx, Inc. (827436556)
