Label: INPEFA- sotagliflozin tablet




-
NDC Code(s):
70183-220-07,
70183-220-30,
70183-220-90,
70183-221-07, view more70183-221-30, 70183-221-31, 70183-240-07, 70183-240-30, 70183-240-90, 70183-241-30
- Packager: Lexicon Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated January 15, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use INPEFA safely and effectively. See full prescribing information for INPEFA.
INPEFA® (sotagliflozin) tablets, for oral use
Initial U.S. Approval: 2023INDICATIONS AND USAGE
INPEFA is a sodium-glucose cotransporter 2 (SGLT2) inhibitor indicated to reduce the risk of cardiovascular death, hospitalization for heart failure, and urgent heart failure visit in adults with:
DOSAGE AND ADMINISTRATION
Correct volume status before starting INPEFA at 200 mg daily and titrate to 400 mg as tolerated. (2.2) In patients with decompensated heart failure, begin dosing when patients are hemodynamically stable. (2.1)
Withhold INPEFA at least 3 days, if possible, prior to major surgery or procedures associated with prolonged fasting. (2.3)
DOSAGE FORMS AND STRENGTHS
Tablets: 200 mg and 400 mg (3)
CONTRAINDICATIONS
- History of serious hypersensitivity reaction to INPEFA. (4)
WARNINGS AND PRECAUTIONS
- Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis: Consider ketone monitoring in patients with type 1 diabetes mellitus and consider ketone monitoring in others at risk for ketoacidosis, as indicated. Assess for ketoacidosis regardless of presenting blood glucose levels and discontinue INPEFA if ketoacidosis is suspected. Monitor patients for resolution of ketoacidosis before restarting. (5.1)
- Volume Depletion: Before initiating, correct volume status. Monitor for signs and symptoms of hypotension during therapy. (5.2)
- Urosepsis and Pyelonephritis: Monitor for signs and symptoms during therapy and treat promptly. (5.3)
- Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues: Lower dose of insulin or insulin secretagogue may be required. (5.4)
- Necrotizing Fasciitis of the Perineum (Fournier's Gangrene): Monitor for pain, tenderness, erythema, or swelling in the genital or perineal area, along with fever or malaise. Discontinue INPEFA and treat urgently. (5.5)
- Genital Mycotic Infections: Monitor and treat as appropriate. (5.6)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 5%) are urinary tract infection, volume depletion, diarrhea, and hypoglycemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Lexicon at 1-855-330-2573 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
Pregnancy: Advise females of the potential risk to a fetus especially during the second and third trimesters. (8.1)
Lactation: INPEFA is not recommended when breastfeeding. (8.2)
Geriatrics: Higher incidence of adverse reactions related to volume depletion. (5.2, 8.5)
Renal Impairment: Higher incidence of adverse reactions related to volume depletion. (5.2, 8.6)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Prior to Initiation of INPEFA
2.2 Recommended Dosage
2.3 Temporary Interruption for Surgery
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis
5.2 Volume Depletion
5.3 Urosepsis and Pyelonephritis
5.4 Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
5.5 Necrotizing Fasciitis of the Perineum (Fournier's Gangrene)
5.6 Genital Mycotic Infections
5.7 Positive Urine Glucose Test
5.8 Interference with 1,5-anhydroglucitol (1,5-AG) Assay
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Digoxin
7.2 Uridine 5'-diphospho-glucuronosyltransferase (UGT) Inducer
7.3 Lithium
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 SOLOIST Study
14.2 SCORED Study
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Prior to Initiation of INPEFA
Assess volume status and, if necessary, correct volume depletion prior to initiation of INPEFA [see Warnings and Precautions (5.2) and Use in Specific Populations (8.5, 8.6)].
Assess renal function prior to initiation of INPEFA and then as clinically indicated [see Warnings and Precautions (5.2)].
For patients with decompensated heart failure, dosing may begin as soon as the patient is hemodynamically stable, including during hospitalization or urgent outpatient treatment or immediately upon discharge.
2.2 Recommended Dosage
The recommended starting dose of INPEFA is 200 mg orally once daily not more than one hour before the first meal of the day.
Up-titrate after at least 2 weeks to 400 mg orally once daily as tolerated [see Clinical Studies (14)]. Down-titrate to 200 mg as necessary [see Adverse Reactions (6.1), Warnings and Precautions (5) and Use in Specific Populations (8.6)].
Swallow tablets whole. Do not cut, crush, or chew tablets.
If a dose of INPEFA is missed by more than 6 hours, take the next dose as prescribed the next day.
2.3 Temporary Interruption for Surgery
Withhold INPEFA at least 3 days, if possible, prior to major surgery or procedures associated with prolonged fasting. Resume INPEFA when the patient is clinically stable and has resumed oral intake [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
INPEFA 200 mg printed tablet is an oval, blue, film-coated tablet imprinted in black ink with “LX200” on one side.
INPEFA 200 mg debossed tablet is an oval, blue, film-coated tablet debossed with “LEX” on one side and “200” on the other side.
INPEFA 400 mg debossed tablet is an oval, yellow, film-coated tablet debossed with “LEX” on one side and “400” on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis
In patients with type 1 diabetes mellitus, INPEFA significantly increases the risk of diabetic ketoacidosis, a life-threatening event, beyond the background rate. In placebo-controlled trials of patients with type 1 diabetes mellitus, the risk of ketoacidosis was markedly increased in patients who received sodium glucose cotransporter 2 (SGLT2) inhibitors compared to patients who received placebo; this risk may be greater with higher doses of INPEFA. INPEFA is not indicated for glycemic control.
Type 2 diabetes mellitus and pancreatic disorders (e.g., history of pancreatitis or pancreatic surgery) are also risk factors for ketoacidosis. There have been postmarketing reports of fatal events of ketoacidosis in patients with type 2 diabetes using SGLT2 inhibitors.
Precipitating conditions for diabetic ketoacidosis or other ketoacidosis include acute febrile illness, reduced caloric intake, ketogenic diet, surgery, insulin dose reduction, volume depletion, and alcohol abuse.
Signs and symptoms are consistent with dehydration and severe metabolic acidosis and include nausea, vomiting, abdominal pain, generalized malaise, and shortness of breath. Blood glucose levels at presentation may be below those typically expected for diabetic ketoacidosis (e.g., less than 250 mg/dL). Ketoacidosis and glucosuria may persist longer than typically expected. Urinary glucose excretion persists for 3 days after discontinuing INPEFA [see Clinical Pharmacology (12.3)]; however, there have been postmarketing reports of ketoacidosis and glucosuria lasting greater than 6 days and some up to 2 weeks after discontinuation of SGLT2 inhibitors.
Consider ketone monitoring in patients with type 1 diabetes mellitus and consider ketone monitoring in others at risk for ketoacidosis if indicated by the clinical situation. Assess for ketoacidosis regardless of presenting blood glucose levels in patients who present with signs and symptoms consistent with severe metabolic acidosis. If ketoacidosis is suspected, discontinue INPEFA, promptly evaluate, and treat ketoacidosis, if confirmed. Monitor patients for resolution of ketoacidosis before restarting INPEFA.
Withhold INPEFA, if possible, in temporary clinical situations that could predispose patients to ketoacidosis. Resume INPEFA when the patient is clinically stable and has resumed oral intake [see Dosage and Administration (2.3)].
Educate all patients on the signs and symptoms of ketoacidosis and instruct patients to discontinue INPEFA and seek medical attention immediately if signs and symptoms occur.
5.2 Volume Depletion
INPEFA can cause intravascular volume depletion which may sometimes manifest as symptomatic hypotension or acute transient changes in creatinine. There have been postmarketing reports of acute kidney injury, some requiring hospitalization and dialysis, in patients with type 2 diabetes mellitus receiving SGLT2 inhibitors. Patients with impaired renal function (eGFR < 60 mL/min/1.73 m2), elderly patients, or patients on loop diuretics may be at increased risk for volume depletion or hypotension [see Adverse Reactions (6.1) and Use in Specific Populations (8.5, 8.6)]. Before initiating INPEFA in patients with one or more of these characteristics, assess volume status and renal function. Monitor for signs and symptoms of hypotension, and renal function after initiating therapy.
5.3 Urosepsis and Pyelonephritis
Treatment with SGLT2 inhibitors, including INPEFA, increases the risk for urinary tract infections. Serious urinary tract infections including urosepsis and pyelonephritis requiring hospitalization have been reported. Evaluate patients for signs and symptoms of urinary tract infections and treat promptly, if indicated [see Adverse Reactions (6.1)].
5.4 Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
Insulin and insulin secretagogues are known to cause hypoglycemia. INPEFA may increase the risk of hypoglycemia when combined with insulin or an insulin secretagogue [see Adverse Reactions (6.1)]. Therefore, a lower dose of insulin or insulin secretagogue may be required to minimize the risk of hypoglycemia when these agents are used in combination with INPEFA.
5.5 Necrotizing Fasciitis of the Perineum (Fournier's Gangrene)
Reports of necrotizing fasciitis of the perineum (Fournier's Gangrene), a rare but serious and life-threatening necrotizing infection requiring urgent surgical intervention, have been identified in postmarketing surveillance in patients with diabetes mellitus receiving SGLT2 inhibitors. Cases have been reported in both females and males. Serious outcomes have included hospitalization, multiple surgeries, and death.
Patients treated with INPEFA presenting with pain, tenderness, erythema, or swelling in the genital or perineal area, along with fever or malaise, should be assessed for necrotizing fasciitis. If suspected, start treatment immediately with broad-spectrum antibiotics and, if necessary, surgical debridement. Discontinue INPEFA, closely monitor blood glucose levels, and provide appropriate alternative therapy for heart failure.
5.6 Genital Mycotic Infections
INPEFA increases the risk of genital mycotic infections. Patients with a history of genital mycotic infections were more likely to develop genital mycotic infections [see Adverse Reactions (6.1)]. Monitor and treat appropriately.
-
6 ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in the labeling:
- Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis [see Warnings and Precautions (5.1)]
- Volume Depletion [see Warnings and Precautions (5.2)]
- Urosepsis and Pyelonephritis [see Warnings and Precautions (5.3)]
- Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues [see Warnings and Precautions (5.4)]
- Necrotizing Fasciitis of the Perineum (Fournier's Gangrene) [see Warnings and Precautions (5.5)]
- Genital Mycotic Infections [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In the phase 3 (SOLOIST [see Clinical Studies (14.1)] and SCORED [see Clinical Studies (14.2)]) placebo-controlled trials, 5,896 subjects received INPEFA.
In the SOLOIST study, 336 patients (56%) reached the 400 mg dose. In the SCORED study, 3,934 patients (74%) reached the 400 mg dose.
In the SOLOIST study, 5.6% of patients in the INPEFA group and 5.4% of patients in the placebo group discontinued therapy due to adverse events (AEs). In the SCORED study, 5.0% of patients in the INPEFA group and 4.5% of patients in the placebo group discontinued therapy due to AEs.
Table 1 Adverse Reactions Reported in ≥ 2% of Patients Treated with INPEFA and Greater Than Placebo in Either SOLOIST or SCORED Adverse Reaction SOLOIST
N = 1,216SCORED
N = 10,577Placebo (%)
N = 611INPEFA (%)
N = 605Placebo (%)
N = 5,286INPEFA (%)
N = 5,291Urinary tract infection 7.2 8.6 11.0 11.5 Volume depletion 8.8 9.3 4.0 5.2 Diarrhea 4.1 6.9 6.0 8.4 Hypoglycemia 2.8 4.3 7.9 7.7 Dizziness 2.5 2.6 2.8 3.3 Genital mycotic infection 0.2 0.8 0.9 2.4 Changes in Laboratory Test Values During Treatment
Increase in Serum Creatinine and Decrease in eGFR
Initiation of SGLT2 inhibitors, including INPEFA, causes a small increase in serum creatinine and decrease in eGFR. These changes in serum creatinine and eGFR generally occur within 4 weeks of starting therapy and then stabilize regardless of baseline kidney function. Changes that do not fit this pattern should prompt further evaluation to exclude the possibility of acute kidney injury. In studies that included patients with type 2 diabetes mellitus with moderate renal impairment, the acute effect on eGFR reversed after treatment discontinuation, suggesting acute hemodynamic changes may play a role in the renal function changes observed with INPEFA [see Warnings and Precautions (5.2) and Use in Specific Populations (8.6)].
-
7 DRUG INTERACTIONS
7.1 Digoxin
There is an increase in the exposure of digoxin when coadministered with INPEFA 400 mg. Patients taking INPEFA with concomitant digoxin should be monitored appropriately [see Clinical Pharmacology (12.3)].
7.2 Uridine 5'-diphospho-glucuronosyltransferase (UGT) Inducer
Glucuronidation by UGT1A9, to form the 3-O-glucuronide, was identified as a major metabolic pathway for sotagliflozin. The coadministration of rifampicin, an inducer of UGTs, with a single dose of 400 mg sotagliflozin resulted in a decrease in the exposure to sotagliflozin. This decrease in exposure to sotagliflozin may decrease efficacy [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal data showing renal effects, INPEFA is not recommended during the second and third trimesters of pregnancy.
Available data with INPEFA in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with untreated heart failure in pregnancy [see Clinical Considerations].
In rats, renal changes were observed when sotagliflozin was administered during a period of renal development corresponding to the late second and third trimesters of human pregnancy. Exposure approximately 5 times the clinical exposure at the maximum recommended human dose (MRHD) of 400 mg once daily caused increased kidney weights and renal pelvis and tubule dilatations that were partially reversible [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies in rats and rabbits, sotagliflozin was administered for intervals coinciding with the first trimester period of organogenesis in humans.
Sotagliflozin was not teratogenic when administered at doses up to 100 mg/kg/day in pregnant rats during embryonic organogenesis (40 times the human exposure at the MRHD). Higher exposures (350 mg/kg or 161 times the human exposure at the MRHD) resulted in embryo-lethality, effects on fetal growth, and cardiovascular and skeletal fetal abnormalities commensurate with maternal toxicity.
Sotagliflozin was not teratogenic when administered at doses up to 200 mg/kg/day in pregnant rabbits (9 times the human exposure at the MRHD).
In a prenatal and postnatal development study in pregnant and lactating rats, sotagliflozin was administered at oral doses up to 100 mg/kg/day from gestation Day 6 through to lactation Day 20 (weaning). An increased incidence of dilated kidneys with discoloration and dilated ureters was observed at doses ≥ 30 mg/kg (≥ 4 times the human exposure at the MRHD). Sotagliflozin did not adversely affect developmental landmarks, sexual maturation, or reproductive performance of the offspring at doses up to 40 times the human exposure at the MRHD.
Sotagliflozin dosed directly to juvenile rats from postnatal Day (PND) 21 until PND 90 at doses of 3, 10, 30, and 75 mg/kg/day caused dose-related increased kidney weights for males given ≥ 10 mg/kg/day and females given ≥ 30 mg/kg/day and was correlated with renal tubular and pelvis dilation for animals given ≥ 30 mg/kg/day. These findings were fully or partially reversed after a 29-day recovery period. These outcomes occurred with drug exposure during periods of renal development in rats that correspond to the late second and third trimesters of human development.
8.2 Lactation
Risk Summary
There are no data on the presence of INPEFA in human milk, the effects on the breastfed infant, or the effects on milk production. Sotagliflozin is present in rat milk (see Data). When a drug is present in animal milk, it is likely to be present in human milk. Since human kidney maturation occurs in utero and during the first 2 years of life when lactational exposure may occur, there may be risk to the developing human kidney.
Because of the potential for serious adverse reactions in a breastfed infant, advise women that breastfeeding is not recommended while taking INPEFA.
Data
In rats, after a single oral dose of radiolabeled sotagliflozin to dams on gestation Day 13 or 18, low to moderate levels of radioactivity were present in fetal tissues. In rat milk, the mean milk/plasma concentration ratios ranged from 0.5 to 2. Exposure to radioactivity was approximately 30% greater in milk than in plasma based on AUC0-inf values.
8.4 Pediatric Use
The safety and effectiveness of INPEFA in pediatric patients under 18 years of age have not been established.
8.5 Geriatric Use
No INPEFA dosage change is recommended based on age.
In the SOLOIST study, a total of 241 (40%) patients treated with INPEFA were between 65 and < 75 years of age, and 174 (29%) were ≥ 75 years of age. In the SCORED study, a total of 2,470 (47%) patients treated with INPEFA were between 65 and < 75 years of age, and 1,240 (23%) were ≥ 75 years of age.
No overall differences in efficacy were detected between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Elderly patients may be at increased risk for volume depletion adverse reactions, including hypotension. In patients ≥ 65 years of age, a higher proportion of patients treated with INPEFA had adverse reactions of volume depletion [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
8.6 Renal Impairment
INPEFA was evaluated in 5,292 patients with chronic kidney disease (eGFR 25 to 60 mL/min/1.73 m2) in the SCORED study and in 426 patients with heart failure with eGFR < 60 mL/min/1.73 m2 in the SOLOIST study. The safety profile of INPEFA across eGFR subgroups in these studies was consistent with the known safety profile. There was an increase in volume-related adverse events (e.g., hypotension, dizziness) in patients with eGFR < 30 mL/min/1.73 m2 relative to the overall safety population [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
Efficacy and safety studies with INPEFA did not enroll patients with an eGFR less than 25 mL/min/1.73 m2 or on dialysis. After starting therapy in these studies, patients were discontinued if eGFR fell below 15 mL/min/1.73 m2 or were initiated on chronic dialysis.
8.7 Hepatic Impairment
In a clinical pharmacology study in patients with hepatic impairment, the exposure in mild hepatic impairment was not increased, but was approximately 3-fold as high in moderate and approximately 6-fold as high in severely hepatic-impaired subjects compared to subjects with normal hepatic function [see Clinical Pharmacology (12.3)].
No dosage adjustment is necessary in patients with mild hepatic impairment.
The safety and efficacy of INPEFA have not been established in patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3)]. INPEFA is not recommended in patients with moderate or severe hepatic impairment.
-
10 OVERDOSAGE
There were no confirmed reports of symptomatic overdose with sotagliflozin during the clinical development program of INPEFA.
In the event of an overdose with INPEFA, contact the Poison Control Center. Employ the usual supportive measures as dictated by the patient's clinical status.
The removal of sotagliflozin by hemodialysis has not been studied.
-
11 DESCRIPTION
INPEFA tablets for oral administration contain sotagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor.
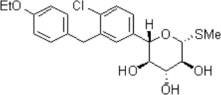
The chemical name of sotagliflozin is (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol.
Its molecular formula is C21H25ClO5S and the molecular weight is 424.94. The structural formula is:
Sotagliflozin is a white to off-white solid. It is practically insoluble in water.
Each film-coated tablet of INPEFA contains 200 mg or 400 mg of sotagliflozin and the following inactive ingredients. The core of the tablet contains colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and talc. The film coating for the 200 mg tablet contains: indigo carmine aluminum lake, polyethylene glycol, polyvinyl alcohol (partly hydrolyzed), talc, and titanium dioxide. The film coating for the 400 mg tablet contains: hypromellose, lactose monohydrate, titanium dioxide, triacetin, and yellow iron oxide. The 200 mg printed tablet also includes black ink which contains: ammonium hydroxide, black iron oxide, isopropyl alcohol, N-butyl alcohol, propylene glycol, and shellac.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sotagliflozin is an inhibitor of SGLT2 and SGLT1. Inhibiting SGLT2 reduces renal reabsorption of glucose and sodium which may influence several physiological functions such as lowering both pre-and afterload of the heart and downregulating sympathetic activity. Inhibiting SGLT1 reduces intestinal absorption of glucose and sodium which likely contributes to diarrhea. The mechanism for sotagliflozin's cardiovascular benefits has not been established.
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a randomized, placebo-controlled, active-comparator, crossover study, 58 healthy subjects were administered a single oral dose of sotagliflozin 800 mg or sotagliflozin 2000 mg (5 times the maximum recommended dose), moxifloxacin, and placebo. No increase in QT corrected for heart rate (QTc) was observed with either 800 mg or 2000 mg sotagliflozin.
12.3 Pharmacokinetics
Plasma Cmax and AUC of sotagliflozin increased in a dose-proportional manner in the therapeutic dose range of 200 mg to 400 mg once daily. Accumulation of sotagliflozin was observed with an approximate 50-100% increase in both Cmax and area under the concentration-time curve from 0 to 24 hours (AUC0-24h) at steady state verses the first day of dosing.
Absorption
The absolute bioavailability of oral sotagliflozin 400 mg dose was approximately 25% (90% confidence interval [CI] 16% to 39%) for area under the concentration-time curve from time 0 to last measurable concentration (AUC0-last). The contribution of enterohepatic circulation to the overall exposure is estimated to be approximately 50%. The median time to maximum plasma concentration (Tmax) ranged from 1.25 to 3 hours, over a single-dose range of 200 mg to 2000 mg. Following administration of multiple doses (400 mg and 800 mg), the median Tmax values ranged from 2.5 to 4 hours.
Food Effect
When sotagliflozin tablets were administered with a high-caloric breakfast compared to fasting conditions, plasma exposure to sotagliflozin as measured by Cmax and AUC0-inf increased by 149% and 50%, respectively. Multiple doses of sotagliflozin 400 mg given immediately before breakfast, 30 minutes prior to breakfast, and 1-hour before breakfast in healthy subjects showed a consistent effect of sotagliflozin on urine glucose excretion (UGE), insulin, and postprandial glucose (PPG) across all dose schedules. It is recommended that INPEFA be taken not more than one hour before the first meal of the day.
Distribution
Both sotagliflozin and its major human metabolite sotagliflozin 3-O-glucuronide exhibited high binding to human plasma proteins in vitro (> 93% bound), which was not dependent on the concentration of sotagliflozin and sotagliflozin 3-O-glucuronide and was not influenced by the degree of renal or hepatic function.
Following administration of a single 400 mg dose of [14C]-sotagliflozin, the mean blood/plasma ratios for total radioactivity were 0.5 and 0.4 for Cmax and AUC0-last, respectively, and the mean whole blood to plasma concentration ratio of sotagliflozin ranged from 0.5 to 0.6.
The mean apparent volume of distribution of sotagliflozin following administration of a single 400 mg oral dose of [14C]-sotagliflozin was 9000 L.
Elimination
Metabolism
Following the administration of a single dose of 400 mg [14C]-sotagliflozin in healthy subjects, sotagliflozin was extensively metabolized predominantly to sotagliflozin 3-O-glucuronide and it represented 94.3% of the radioactivity in plasma.
The key enzymes responsible for the metabolism of sotagliflozin were UGT1A9 and, to a lesser extent, CYP3A4. Following incubation of sotagliflozin with UGT1A9, the 3-O-glucuronide metabolite of sotagliflozin was the main conjugate observed. No acyl glucuronides of sotagliflozin were identified.
Excretion
Following the administration of a single dose of 400 mg [14C]-sotagliflozin, 57% and 37% of the radioactivity was recovered in the urine and feces, respectively. These results indicate that the main route of elimination was renal. The predominant metabolite detected in urine was sotagliflozin 3-O-glucuronide, representing a mean of 33% of the administered radioactive dose. Unchanged [14C]-sotagliflozin was the predominant radioactive peak detected in fecal extracts representing a mean of 23% of the total administered radioactive dose. Sotagliflozin undergoes enterohepatic circulation. Following administration of 200 mg and 400 mg, mean terminal half-life (T1/2) ranged from 21 to 35 hours for sotagliflozin and from 19 to 26 hours for sotagliflozin 3-O-glucuronide. Effective half-life of sotagliflozin ranged from 5 to 10 hours. Steady state was generally achieved after 5 days of once daily dosing with sotagliflozin 200 mg and 400 mg.
In healthy volunteers, mean oral clearance (CL/F) of sotagliflozin ranged from 260 to 370 L/hr following administration of 200 mg and 400 mg sotagliflozin. The median population PK model predicted CL/F in type 2 diabetes mellitus patients with normal renal function was about 300 L/hr.
Specific Populations
Renal Impairment
Exposure of sotagliflozin was evaluated in a dedicated PK study in subjects with mild (eGFR 60 to < 90 mL/min/1.73 m²) and moderate (eGFR 30 to < 60 mL/min/1.73 m²) renal impairment and with normal renal function (eGFR ≥ 90 mL/min/1.73 m²). Exposure to sotagliflozin following a single dose of 400 mg was approximately 70% higher in subjects with mild and up to 170% higher in subjects with moderate renal impairment compared to subjects with normal renal function [see Use in Specific Populations (8.6)].
Hepatic Impairment
In a study with subjects with reduced hepatic function, AUC of sotagliflozin was not increased in mild (Child Pugh A) hepatic impaired subjects but was increased by approximately 3-fold in moderate (Child Pugh B) and approximately 6-fold in severe (Child Pugh C) hepatic impaired subjects compared to subjects with normal hepatic function [see Use in Specific Populations (8.7)].
Drug Interactions
In Vitro Studies
The major human metabolite of sotagliflozin, sotagliflozin 3-O-glucuronide, was shown to have an in vitro potential to inhibit CYP3A4 and CYP2D6 and to induce CYP3A4.
Sotagliflozin was shown to have inhibitory effects on P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Sotagliflozin is a substrate of the human uptake transporters organic anion transporter (OAT)3, organic-anion-transporting polypeptides (OATP)1B1, and OATP1B3, but not OAT1 and organic cation transporter 2 (OCT2). Sotagliflozin does not inhibit any of these human uptake transporters at clinically relevant plasma concentrations.
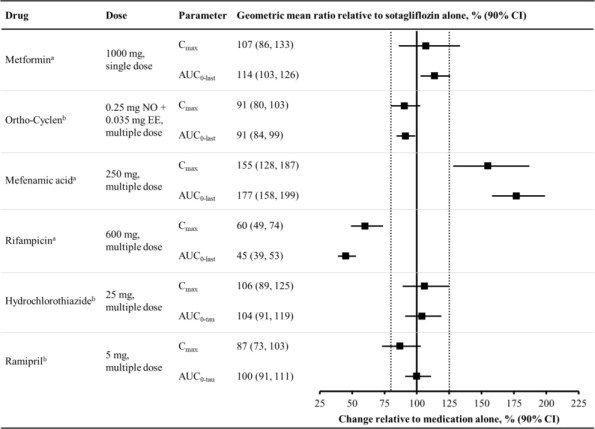
In Vivo Studies
Figure 1 summarizes the effect on sotagliflozin by concomitantly administered drugs. The observed changes in overall exposure (AUC) of sotagliflozin following coadministration with hydrochlorothiazide, ramipril, metformin, mefenamic acid, and oral contraceptives are not considered to be clinically relevant. Rifampicin (UGT inducer) decreases exposure to sotagliflozin [see Drug Interactions (7.2)].
Figure 1 Effect of Various Medications on the Pharmacokinetics of Sotagliflozin, Displayed as Geometric Mean AUCs and Cmax Ratios
Vertical reference lines indicate 100% (solid) and the 80%-125% range (dashed)
a400 mg sotagliflozin, administered as a single dose
b400 mg sotagliflozin, at steady state
AUC0-last = area under the concentration-time curve from time 0 to last measurable concentration; AUC0-tau = area under the concentration-time curve from time 0 to end of the dosing period; Cmax = maximum drug concentration; EE = ethinyl estradiol; NO = norgestimate
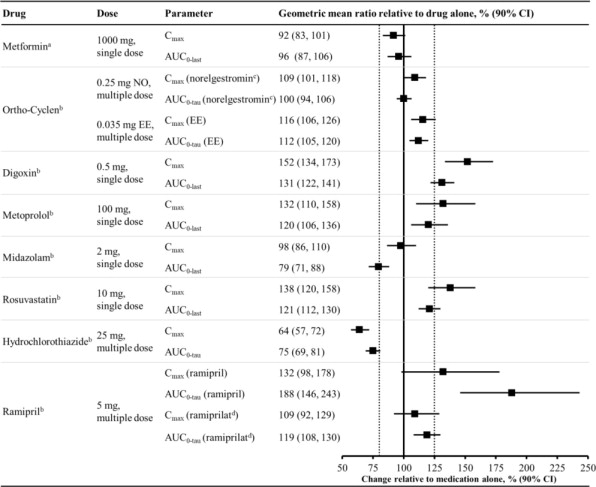
Figure 2 summarizes the effect on concomitantly administered drugs by sotagliflozin. The increases in exposure (AUC) of metoprolol (CYP2D6 substrate) and rosuvastatin (BCRP substrate), as well as the decrease in exposure to midazolam (CYP3A4 substrate) are not considered to be clinically relevant. The increased exposure (Cmax and AUC) in ramipril is not considered clinically significant because the exposure of ramiprilat, the primary active metabolite, is minimally increased.
The increase in exposure (Cmax and AUC) of digoxin, a P-gp substrate, when coadministered with sotagliflozin, requires monitoring of patients [see Drug Interactions (7.1)].
Figure 2 Effect of Sotagliflozin on the Pharmacokinetics of Various Medications, Displayed as Geometric Mean AUCs and Cmax Ratios
Vertical reference lines indicate 100% (solid) and the 80%-125% range (dashed)
a400 mg sotagliflozin, administered as a single dose
b400 mg sotagliflozin, at steady state
cNorelgestromin is the primary active metabolite of NO
dRamiprilat is the primary active metabolite of ramipril
AUC0-last = area under the concentration-time curve from time 0 to last measurable concentration; AUC0-tau = area under the concentration-time curve from time 0 to end of the dosing period; Cmax maximum drug concentration; EE = ethinyl estradiol; NO = norgestimate
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenesis was evaluated in a 6-month study in RasH2 transgenic mice and in a 2-year study in Sprague Dawley rats. Sotagliflozin did not increase the incidence of tumors in RasH2 transgenic mice at doses up to 100 mg/kg/day or in rats at doses up to 15-fold (males) and 45-fold (females) clinical exposure at the MRHD.
-
14 CLINICAL STUDIES
14.1 SOLOIST Study
The SOLOIST (Effects of Sotagliflozin on Clinical Outcomes in Hemodynamically Stable Patients with Type 2 Diabetes Post Worsening Heart Failure) study (NCT03521934) was a randomized, double-blind, placebo-controlled, parallel-group, multicenter study in patients with type 2 diabetes mellitus who had been admitted to the hospital, a heart failure unit, infusion center, or emergency department for worsening heart failure to determine if INPEFA reduces the risk of total occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visit. Patients were randomized to treatment if they met the following criteria for clinical stability: no need for oxygen therapy, a systolic blood pressure of at least 100 mmHg, no need for intravenous inotropic or vasodilator therapy (excluding nitrates) and transitioned from intravenous to oral diuretic therapy.
Of the 1,222 randomized patients, 608 were randomized to INPEFA and 614 to placebo. Assigned treatment was initiated in the hospital or within a median of 2 days following hospital discharge.
The dose of study drug was to be up-titrated from 200 mg to 400 mg sotagliflozin or matching placebo as soon as 2 weeks or up until 8 months after initiation of treatment. The dose was increased to 400 mg once daily for 336 patients (56%) in the sotagliflozin group and 325 patients (53%) in the placebo group. The median time to up-titration was 16 days. Up-titration was to be performed based on the judgment of the investigator, who considered whether the patient's clinical condition was satisfactory, whether the drug was well tolerated, and whether AEs typical of SGLT2 inhibitors had occurred, such as those associated with volume depletion.
At baseline, median age was 70 years, 34% were female, 93% were White and 4% were Black or African American. Median A1C was 7.1%, median body mass index (BMI) was 31 kg/m2, and median eGFR was 50 mL/min/1.73 m2. Median left ventricular ejection fraction (LVEF) was 35% (79% with LVEF < 50%), median N-terminal pro B-type natriuretic peptide (NT-proBNP) was 1806 pg/mL, and the median Kansas City Cardiomyopathy Questionnaire-12 (KCCQ-12) score was 41.
At baseline, 86% were treated with at least one antihyperglycemic medication, including 52% with a biguanide, 36% with insulin, 19% with a sulfonylurea, 16% with a dipeptidyl peptidase-4 (DPP4) inhibitor, and 3% with a glucagon-like peptide 1 (GLP-1) receptor agonist. At baseline, 91% were treated with inhibitors of the renin-angiotensin-aldosterone system, 92% with a beta blocker, 95% with a loop diuretic, and 10% with another diuretic.
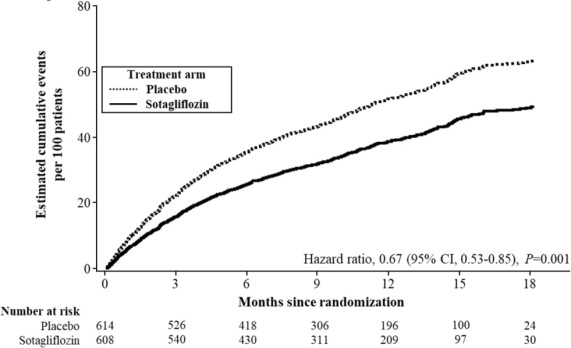
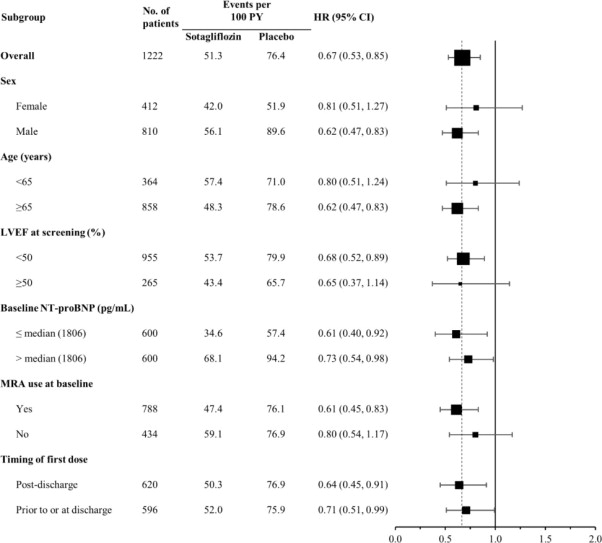
INPEFA was superior to placebo in reducing the risk of the primary composite endpoint (Hazard Ratio [HR] 0.67 [95% confidence interval (CI) 0.53, 0.85]; p = 0.001). (Table 2).
Table 2 Treatment Effect for the Primary Composite, Components and Secondary Endpoint in the SOLOIST Study Primary Endpointa Event Rates per 100 Patient-years Hazard Ratio
(95% CI)INPEFA
N = 608Placebo
N = 614a Based on investigator-reported events in all randomized patients, analyzed according to the treatment group allocated by randomization.
b Predefined primary endpoint.
c Predefined secondary endpoint and tested with multiplicity control.
d Time-to-event analysis was performed; event rates are percentages of patients with events.
Total occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visitb 51.3 76.4 0.67 (0.53, 0.85)
p = 0.001Primary Endpoint Components Cardiovascular deathc,d 8.4 9.4 0.84 (0.58, 1.23) Hospitalization for heart failure 33.7 51.9 0.65 (0.49, 0.87) Urgent heart failure visit 6.9 12.1 0.60 (0.34, 1.06) Secondary Endpointc Hospitalization for heart failure and urgent heart failure visitc 40.6 63.9 0.64 (0.50, 0.84)
p = 0.0009Figure 3 displays a cumulative events plot of the primary composite endpoint. The INPEFA and placebo event curves diverged early and remained separated over the study period.
Figure 3 Primary Composite Endpoint (Total Occurrences of Cardiovascular Death, Hospitalization for Heart Failure, and Urgent Heart Failure Visit) Over Time in the SOLOIST Study
Primary composite endpoint was based on investigator-reported events in all randomized patients, analyzed according to the treatment group allocated by randomization.
The results of the primary composite endpoint were generally consistent across prespecified subgroups (Figure 4), including screening LVEF < 50 or ≥ 50% and timing of first dose (post-discharge versus prior to discharge).
Figure 4 Treatment Effect for Primary Composite Endpoint (Total Occurrences of Cardiovascular Death, Hospitalization for Heart Failure, and Urgent Heart Failure Visit) Subgroup Analysis (SOLOIST Study)
HF = heart failure; LVEF = left ventricular ejection fraction; MRA-mineralocorticoid receptor antagonist; NT-proBNP = N-terminal pro B-type natriuretic peptide; PY = patient-years
Primary composite endpoint was based on investigator-reported events in all randomized patients, analyzed according to the treatment group allocated by randomization.
Discharge may have been from hospital or urgent treatment facility where urgent heart failure visit occurred.
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and all of which were pre-specified. The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
14.2 SCORED Study
The SCORED (Effects of Sotagliflozin on Cardiovascular and Renal Events in Patients with Type 2 Diabetes Mellitus, Cardiovascular Risk Factors and Moderately Impaired Renal Function) study (NCT03315143) was a randomized, double-blind, placebo-controlled, parallel-group, multicenter study in patients with type 2 diabetes mellitus (A1C > 7%), chronic kidney disease (eGFR 25 to 60 mL/min/1.73 m2), and additional cardiovascular risk factors, such as a history of heart failure, obesity, dyslipidemia, hypertension, or elevated cardiac and inflammatory biomarkers, to determine if INPEFA reduces the risk of total occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visit. Of the 10,584 randomized patients, 5,292 were randomized to INPEFA and 5,292 to placebo.
The dose of study drug was to be up-titrated from 200 mg to 400 mg sotagliflozin or matching placebo as soon as 4 weeks or up until 6 months after initiation of treatment. The dose was increased to 400 mg once daily for 3,934 patients (74%) in the INPEFA group and for 3,987 patients (75%) in the placebo group. The median time to up-titration was 29 days. Up-titration was to be performed based on the judgment of the investigator, who considered whether the patient's clinical condition was satisfactory, whether the drug was well tolerated, and whether AEs typical of SGLT2 inhibitors had occurred, such as those associated with volume depletion.
At baseline, median age was 68 years, 45% were female, 82% were White, 3% Black or African American, 6% Asian, and 4% American Indian or Native Alaskan. Median A1C was 8.3%, median BMI was 32 kg/m2, median eGFR was 45 mL/min/1.73 m2 (8% with eGFR < 30, 44% with eGFR 30 to < 45, and 48% with eGFR ≥ 45 mL/min/1.73 m2), and median urinary albumin-to-creatinine ratio (UACR) was 82 mg/g (32% with UACR ≥ 300 mg/g). A history of heart failure was present in 31%, prior myocardial infarction had occurred in 20%, a prior cerebrovascular event had occurred in 9%, and coronary revascularization had been performed in 22% of patients prior to study entry.
At baseline, 97% were treated with at least one antihyperglycemic medication, including 56% with a biguanide, 64% with insulin, 27% with a sulfonylurea, 20% with a DPP4 inhibitor, and 6% with a GLP-1 receptor agonist. At baseline, 88% were treated with inhibitors of the renin-angiotensin-aldosterone system, 14% with a beta blocker, 42% with a calcium channel blocker, 35% with a loop diuretic, and 30% with another diuretic.
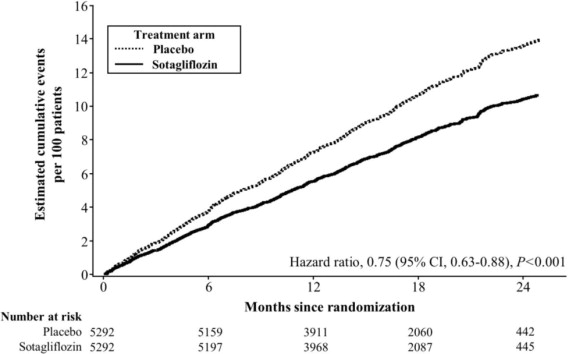
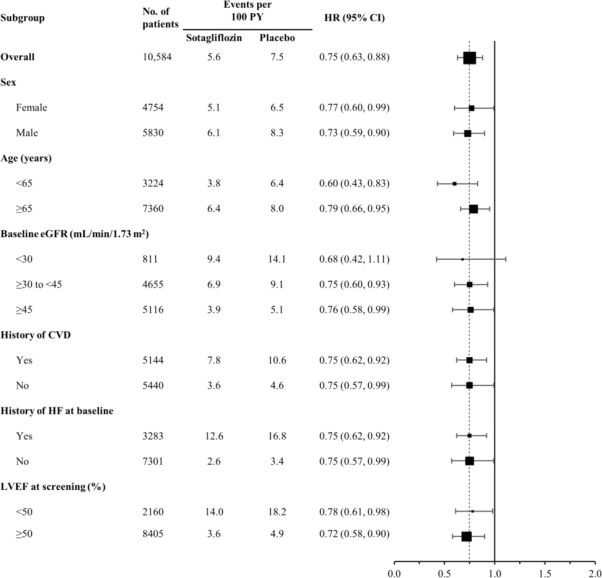
INPEFA was superior to placebo in reducing the risk of the primary composite endpoint (HR 0.75 [95% CI 0.63, 0.88]; p < 0.001) (Table 3).
Table 3 Treatment Effect for the Primary Composite, Components and Secondary Endpoint in the SCORED Study Primary Endpointa Event Rates (per 100 Patient-years) Hazard Ratio
(95% CI)INPEFA
N = 5,292Placebo
N = 5,292a Based on investigator-reported events in all randomized patients, analyzed according to the treatment group allocated by randomization.
b Predefined primary endpoint.
c Predefined secondary endpoint and tested with multiplicity control.
d Time-to-event analysis was performed; event rates are percentages of patients with events.
Total occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visitb 5.6 7.5 0.75 (0.63, 0.88)
p < 0.001Primary Endpoint Components Cardiovascular deathc,d 2.9 3.2 0.90 (0.73, 1.12) Hospitalization for heart failure 2.8 4.2 0.66 (0.53, 0.82) Urgent heart failure visit 0.7 0.9 0.73 (0.48, 1.11) Secondary Endpointc Hospitalization for heart failure or urgent heart failure visitc 3.5 5.1 0.67 (0.55, 0.82)
p = 0.0001Figure 5 displays a cumulative events plot of the primary composite endpoint. The INPEFA and placebo event curves separated early and continued to diverge over the study period following randomization.
Figure 5 Primary Composite Endpoint (Total Occurrences of Cardiovascular Death, Hospitalization for Heart Failure, and Urgent Heart Failure Visit) Over Time in the SCORED Study
Primary composite endpoint was based on investigator-reported events in all randomized patients, analyzed according to the treatment group allocated by randomization.
The results of the primary composite endpoint were generally consistent across prespecified subgroups, including history of cardiovascular disease, history of heart failure, and screening LVEF < 50 or ≥ 50% (Figure 6).
Figure 6 Treatment Effect for Primary Composite Endpoint (Total Occurrences of Cardiovascular Death, Hospitalization for Heart Failure, and Urgent Heart Failure Visit) Subgroup Analysis (SCORED Study)
CVD = cardiovascular disease; eGFR = estimated glomerular filtration rate; LVEF = left ventricular ejection fraction; HF = heart failure; PY = patient-years
Primary composite endpoint was based on investigator-reported events in all randomized patients, analyzed according to the treatment group allocated by randomization.
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and all of which were pre-specified. The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
INPEFA tablets are oval and film-coated.
Printed Tablets Debossed Tablets Strength (mg) Color Printing Bottle/30 Deboss Bottle/30 Blister/30
(3 x 10)200 Blue LX200
on one sideNDC 70183-220-30 LEX on one side, 200 on other side NDC 70183-221-30 NDC 70183-221-31
400 Yellow -- -- LEX on one side, 400 on other side NDC 70183-241-30 -- -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Risk of Diabetic Ketoacidosis in Patients with Type 1 Diabetes Mellitus and Other Ketoacidosis
In patients with type 1 diabetes mellitus, inform them that using INPEFA can increase their risk of life-threatening diabetic ketoacidosis. For all other patients, inform them that INPEFA can cause potentially fatal ketoacidosis and that type 2 diabetes mellitus is a risk factor.
Educate all patients on precipitating factors (such as infection, reduced caloric intake, ketogenic diet, surgery, insulin dose reduction, dehydration, and alcohol abuse) and symptoms of ketoacidosis (including nausea, vomiting, abdominal pain, tiredness, and labored breathing). Inform patients that blood glucose may be normal even in the presence of ketoacidosis.
Advise patients that they may be asked to monitor ketones. If symptoms of ketoacidosis occur, instruct patients to discontinue INPEFA and seek medical attention immediately [see Warnings and Precautions (5.1)].
Volume Depletion
Inform patients that symptomatic hypotension may occur with INPEFA and advise them to contact their healthcare provider if they experience such symptoms [see Warnings and Precautions (5.2)]. Inform patients that dehydration may increase the risk for hypotension, and to have adequate fluid intake.
Serious Urinary Tract Infections
Inform patients of the potential for urinary tract infections, which may be serious. Provide them with information on the symptoms of urinary tract infections. Advise them to seek medical advice promptly if such symptoms occur [see Warnings and Precautions (5.3)].
Hypoglycemia with Concomitant Use with Insulin and Insulin Secretagogues
Inform patients that the incidence of hypoglycemia is increased when INPEFA is used in combination with insulin and that a lower dose of insulin may be required to reduce the risk of hypoglycemia [see Warnings and Precautions (5.4)].
Necrotizing Fasciitis of the Perineum (Fournier's Gangrene)
Inform patients that necrotizing infections of the perineum (Fournier's Gangrene) have occurred with INPEFA in patients with diabetes mellitus. Counsel patients to promptly seek medical attention if they develop pain, tenderness, redness, or swelling of the genitals or the area from the genitals back to the rectum, along with a fever above 100.4°F or malaise [see Warnings and Precautions (5.5)].
Genital Mycotic Infections in Females (e.g., Vulvovaginitis)
Inform female patients that vaginal yeast infections may occur and provide them with information on the signs and symptoms of vaginal yeast infections. Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.6)].
Genital Mycotic Infections in Males (e.g., Balanitis)
Inform male patients that yeast infections of the penis (e.g., balanitis or balanoposthitis) may occur, especially in patients with prior history. Provide them with information on the signs and symptoms of balanitis and balanoposthitis (rash or redness of the glans or foreskin of the penis). Advise them of treatment options and when to seek medical advice [see Warnings and Precautions (5.6)].
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions (e.g., urticaria, anaphylactic reactions, and angioedema) have been reported with other SGLT2 inhibitors. Advise patients to immediately report any signs or symptoms suggesting allergic reaction or angioedema and to discontinue the drug until they have consulted their prescribing physician.
Pregnancy
Advise pregnant patients of the potential risk to a fetus with treatment with INPEFA.
Instruct patients to immediately inform their healthcare provider if pregnant or planning to become pregnant [see Use in Specific Populations (8.1)].
Lactation
Advise patients that use of INPEFA is not recommended while breastfeeding [see Use in Specific Populations (8.2)].
Laboratory Tests
Due to its mechanism of action, patients taking INPEFA will test positive for glucose in their urine.
Missed Dose
If a dose of INPEFA is missed by more than 6 hours, take the next dose as prescribed the next day. Advise patients not to take two doses of INPEFA at the same time.
Manufactured for:
Lexicon Pharmaceuticals, Inc. (The Woodlands, TX 77381)INPEFA is a registered trademark of Lexicon Pharmaceuticals, Inc.
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Issued 01/2024
MEDICATION GUIDE
INPEFA® (in peh' fah)
(sotagliflozin)
tablets, for oral useWhat is the most important information I should know about INPEFA?
INPEFA can cause serious side effects, including:
-
Ketoacidosis (acidic blood with increased ketones in your blood or urine). Ketoacidosis has happened in people who have type 1 or type 2 diabetes during treatment with INPEFA. Ketoacidosis can happen with INPEFA even if your blood sugar is not high. Your healthcare provider may ask you to periodically check ketones in your urine or blood. Ketoacidosis can also happen in people who are sick or who have surgery during treatment with INPEFA. Ketoacidosis is a serious condition which needs to be treated in a hospital. Ketoacidosis may lead to death.
-
Stop taking INPEFA and call your healthcare provider or get medical help right away if you get any of the following symptoms. If possible, check for ketones in your urine or blood, even if your blood sugar is less than 250 mg/dL:
- nausea
- vomiting
- trouble breathing
- stomach-area (abdominal) pain
- tiredness
- ketones in your urine or blood
-
Stop taking INPEFA and call your healthcare provider or get medical help right away if you get any of the following symptoms. If possible, check for ketones in your urine or blood, even if your blood sugar is less than 250 mg/dL:
-
Dehydration. INPEFA can cause some people to become dehydrated (the loss of body water and salt). Dehydration may cause you to feel dizzy, faint, lightheaded, or weak, especially when you stand up (orthostatic hypotension). There have been reports of sudden kidney injury due to dehydration in people with type 2 diabetes who are taking a medicine that works like INPEFA.
You may be at higher risk of dehydration if you:
- take medicines to lower your blood pressure, including water pills (diuretics)
- are 65 years of age or older
- are on a low salt diet
- have kidney problems
Talk to your healthcare provider about what you can do to prevent dehydration including how much fluid you should drink on a daily basis. Call your healthcare provider right away if you reduce the amount of food or liquid you drink, for example if you cannot eat or you start to lose liquids from your body, for example from vomiting, diarrhea, or being in the sun too long.
See “What are the possible side effects of INPEFA?” for more information about side effects. What is INPEFA?
INPEFA is a prescription medicine used to reduce the risk of death due to heart problems (cardiovascular death), hospitalization for heart failure, and urgent visits to the doctor for heart failure in adults with:
- heart failure (when the heart is weak and cannot pump enough blood to the rest of your body), or
- type 2 diabetes, chronic kidney disease, and other cardiovascular risk factors.
It is not known if INPEFA is safe and effective in children under 18 years of age. Do not take INPEFA if you:
- are allergic to sotagliflozin or any of the ingredients in INPEFA. See the end of this Medication Guide for a list of ingredients in INPEFA. Symptoms of a serious allergic reaction to INPEFA may include:
- skin rash
- raised red patches on your skin (hives)
- swelling of the face, lips, tongue, and throat that may cause difficulty in breathing or swallowing
If you have any of these symptoms, stop taking INPEFA and call your healthcare provider or go to the nearest hospital emergency room right away.
Before taking INPEFA, tell your healthcare provider about all of your medical conditions, including if you:
- have type 1 diabetes or have had diabetic ketoacidosis.
- are going to have surgery or a procedure that requires fasting. Your healthcare provider may stop your INPEFA for at least 3 days before you have surgery. Talk to your healthcare provider if you are having surgery about when to stop taking INPEFA and when to start it again.
- are eating less or there is a change in your diet.
- have or have had problems with your pancreas, including pancreatitis or surgery on your pancreas.
- drink alcohol very often or drink a lot of alcohol over a short period (“binge” drinking).
- have a history of urinary tract infections or problems urinating.
- have a history of infection of the vagina or penis.
- have kidney or liver problems.
- are pregnant or plan to become pregnant. INPEFA may harm your unborn baby. Tell your healthcare provider right away if you become pregnant during treatment with INPEFA. Your healthcare provider may switch you to a different medicine.
- are breastfeeding or plan to breastfeed. It is not known if INPEFA passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with INPEFA. You should not breastfeed during treatment with INPEFA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. INPEFA may affect the way other medicines work, and other medicines may affect how INPEFA works. How should I take INPEFA?
- Take INPEFA exactly as your healthcare provider tells you to take it.
- Do not change your dose of INPEFA without talking to your healthcare provider.
- Take INPEFA by mouth 1 time each day no more than 1 hour before your first meal of the day.
- Swallow INPEFA tablets whole. Do not cut, crush, or chew.
- If you miss a dose of INPEFA by more than 6 hours after taking INPEFA, take your next dose at your next scheduled time the next day.
- INPEFA will cause your urine to test positive for glucose.
- Your healthcare provider may do certain blood tests before you start INPEFA and during your treatment as needed.
- If you take too much INPEFA, call your healthcare provider or local poison control center, or go to the nearest emergency room right away.
What are the possible side effects of INPEFA?
INPEFA can cause serious side effects, including:
- See “What is the most important information I should know about INPEFA?”
- Serious urinary tract infections. Serious urinary tract infections that may lead to hospitalization have happened in people who are taking INPEFA. Tell your healthcare provider if you have any signs or symptoms of a urinary tract infection such as a burning feeling when passing urine, a need to urinate often, the need to urinate right away, pain in the lower part of your stomach (pelvis), or blood in the urine. Sometimes people also may have a fever, back pain, nausea, or vomiting.
-
Low blood sugar (hypoglycemia). If you take INPEFA with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin, your risk of getting low blood sugar is higher. The dose of your sulfonylurea medicine or insulin may need to be lowered while you take INPEFA. Signs and symptoms of low blood sugar may include:
- headache
- shaking or feeling jittery
- irritability
- fast heartbeat
- weakness
- drowsiness
- sweating
- confusion
- dizziness
- hunger
-
A rare but serious bacterial infection that causes damage to the tissue under the skin (necrotizing fasciitis) in the area between and around the anus and genitals (perineum). Necrotizing fasciitis of the perineum has happened in women and men with diabetes who take INPEFA. Necrotizing fasciitis of the perineum may lead to hospitalization, may require multiple surgeries, and may lead to death. Seek medical attention immediately if you have a fever or you are feeling very weak, tired, or uncomfortable (malaise) and you develop any of the following symptoms in the area between and around the anus and genitals:
- pain or tenderness
- swelling
- redness of skin (erythema)
-
Vaginal yeast infection. Symptoms of a vaginal yeast infection include:
- vaginal odor
- white or yellowish vaginal discharge (discharge may be lumpy or look like cottage cheese)
- vaginal itching
-
Yeast infection of the penis (balanitis or balanoposthitis). Swelling of an uncircumcised penis may develop that makes it difficult to pull back the skin around the tip of the penis. Other symptoms of yeast infection of the penis include:
- redness, itching, or swelling of the penis
- rash of the penis
- foul smelling discharge from the penis
- pain in the skin around the penis
Talk to your healthcare provider about what to do if you get symptoms of a yeast infection of the vagina or penis. Your healthcare provider may suggest you use an over-the-counter antifungal medicine. Talk to your healthcare provider right away if you use an over-the-counter antifungal medication and your symptoms do not go away.
The most common side effects of INPEFA include:-
- urinary tract infection
- dehydration
- diarrhea
- low blood sugar levels
These are not all the possible side effects of INPEFA. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store INPEFA?
- Store INPEFA at room temperature between 68°F to 77°F (20°C to 25°C).
Keep INPEFA and all medicines out of the reach of children. General information about the safe and effective use of INPEFA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use INPEFA for a condition for which it is not prescribed. Do not give INPEFA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about INPEFA that is written for health professionals.What are the ingredients in INPEFA?
Active ingredient: sotagliflozin.
Inactive ingredients: The core of the tablet contains: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and talc. The film coating for the 200 mg tablet contains: indigo carmine aluminum lake, polyethylene glycol, polyvinyl alcohol (partly hydrolyzed), talc, and titanium dioxide. The film coating for the 400 mg tablet contains: hypromellose, lactose monohydrate, titanium dioxide, triacetin, and yellow iron oxide. For tablets that are printed and not debossed, the ink contains: ammonium hydroxide, black iron oxide, isopropyl alcohol, N-butyl alcohol, propylene glycol, and shellac.
Manufactured for: Lexicon Pharmaceuticals, Inc. The Woodlands, TX, 77381.
INPEFA is a registered trademark of Lexicon Pharmaceuticals, Inc.
For more information about INPEFA, go to www.lexpharma.com or call 1-855-330-2573. -
Ketoacidosis (acidic blood with increased ketones in your blood or urine). Ketoacidosis has happened in people who have type 1 or type 2 diabetes during treatment with INPEFA. Ketoacidosis can happen with INPEFA even if your blood sugar is not high. Your healthcare provider may ask you to periodically check ketones in your urine or blood. Ketoacidosis can also happen in people who are sick or who have surgery during treatment with INPEFA. Ketoacidosis is a serious condition which needs to be treated in a hospital. Ketoacidosis may lead to death.
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
INPEFA
sotagliflozin tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70183-220 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength sotagliflozin (UNII: 6B4ZBS263Y) (sotagliflozin - UNII:6B4ZBS263Y) sotagliflozin 200 mg Inactive Ingredients Ingredient Name Strength Microcrystalline cellulose 102 (UNII: PNR0YF693Y) Microcrystalline cellulose 200 (UNII: 5XDI2TS1EZ) Croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) Talc (UNII: 7SEV7J4R1U) Magnesium stearate (UNII: 70097M6I30) polyvinyl alcohol, unspecified (UNII: 532B59J990) polyethylene glycol, unspecified (UNII: 3WJQ0SDW1A) titanium dioxide (UNII: 15FIX9V2JP) FD&C Blue No. 2 (UNII: L06K8R7DQK) Product Characteristics Color blue (BLUE) Score no score Shape OVAL (OVAL) Size 14mm Flavor Imprint Code LX200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70183-220-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 05/26/2023 2 NDC:70183-220-07 1 in 1 CARTON 05/26/2023 2 7 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC:70183-220-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 05/26/2023 05/26/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216203 05/26/2023 INPEFA
sotagliflozin tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70183-221 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength sotagliflozin (UNII: 6B4ZBS263Y) (sotagliflozin - UNII:6B4ZBS263Y) sotagliflozin 200 mg Inactive Ingredients Ingredient Name Strength Microcrystalline cellulose 102 (UNII: PNR0YF693Y) Microcrystalline cellulose 200 (UNII: 5XDI2TS1EZ) Croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) Talc (UNII: 7SEV7J4R1U) Magnesium stearate (UNII: 70097M6I30) polyvinyl alcohol, unspecified (UNII: 532B59J990) polyethylene glycol, unspecified (UNII: 3WJQ0SDW1A) titanium dioxide (UNII: 15FIX9V2JP) FD&C Blue No. 2 (UNII: L06K8R7DQK) Product Characteristics Color blue (BLUE) Score no score Shape OVAL (OVAL) Size 14mm Flavor Imprint Code LEX;200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70183-221-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 03/01/2024 2 NDC:70183-221-31 1 in 1 CARTON 12/01/2023 2 3 in 1 BLISTER PACK 2 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC:70183-221-07 1 in 1 CARTON 01/15/2024 3 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216203 05/26/2023 INPEFA
sotagliflozin tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70183-241 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength sotagliflozin (UNII: 6B4ZBS263Y) (sotagliflozin - UNII:6B4ZBS263Y) sotagliflozin 400 mg Inactive Ingredients Ingredient Name Strength Microcrystalline cellulose 102 (UNII: PNR0YF693Y) Microcrystalline cellulose 200 (UNII: 5XDI2TS1EZ) Croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) Talc (UNII: 7SEV7J4R1U) Magnesium stearate (UNII: 70097M6I30) lactose monohydrate (UNII: EWQ57Q8I5X) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) triacetin (UNII: XHX3C3X673) ferric oxide yellow (UNII: EX438O2MRT) Product Characteristics Color yellow (YELLOW) Score no score Shape OVAL (OVAL) Size 18mm Flavor Imprint Code LEX;400 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70183-241-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 12/01/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216203 05/26/2023 INPEFA
sotagliflozin tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70183-240 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength sotagliflozin (UNII: 6B4ZBS263Y) (sotagliflozin - UNII:6B4ZBS263Y) sotagliflozin 400 mg Inactive Ingredients Ingredient Name Strength Microcrystalline cellulose 102 (UNII: PNR0YF693Y) Microcrystalline cellulose 200 (UNII: 5XDI2TS1EZ) Croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) Talc (UNII: 7SEV7J4R1U) Magnesium stearate (UNII: 70097M6I30) lactose monohydrate (UNII: EWQ57Q8I5X) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) triacetin (UNII: XHX3C3X673) ferric oxide yellow (UNII: EX438O2MRT) Product Characteristics Color yellow (YELLOW) Score no score Shape OVAL (OVAL) Size 18mm Flavor Imprint Code 2457 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70183-240-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 05/26/2023 05/26/2023 2 NDC:70183-240-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 05/26/2023 05/26/2023 3 NDC:70183-240-07 1 in 1 CARTON 05/26/2023 05/26/2023 3 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216203 05/26/2023 05/26/2023 Labeler - Lexicon Pharmaceuticals, Inc. (838179638)