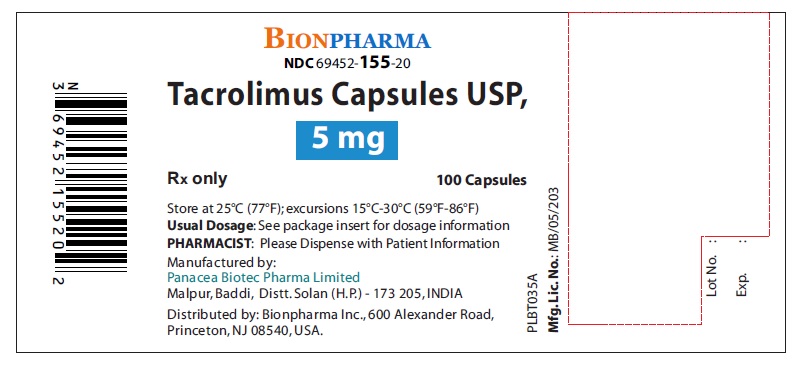
Label: TACROLIMUS capsule, gelatin coated






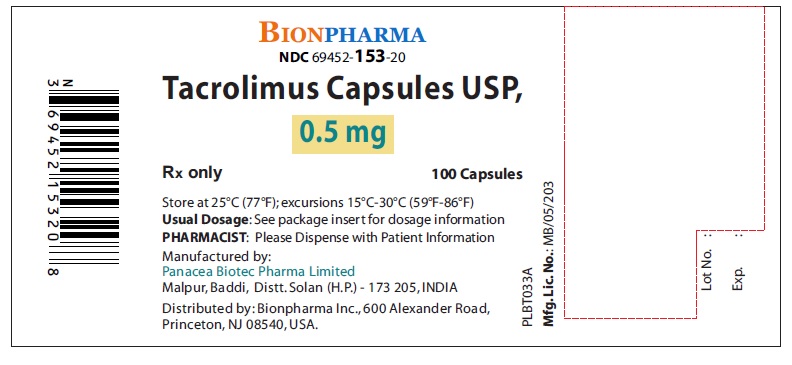
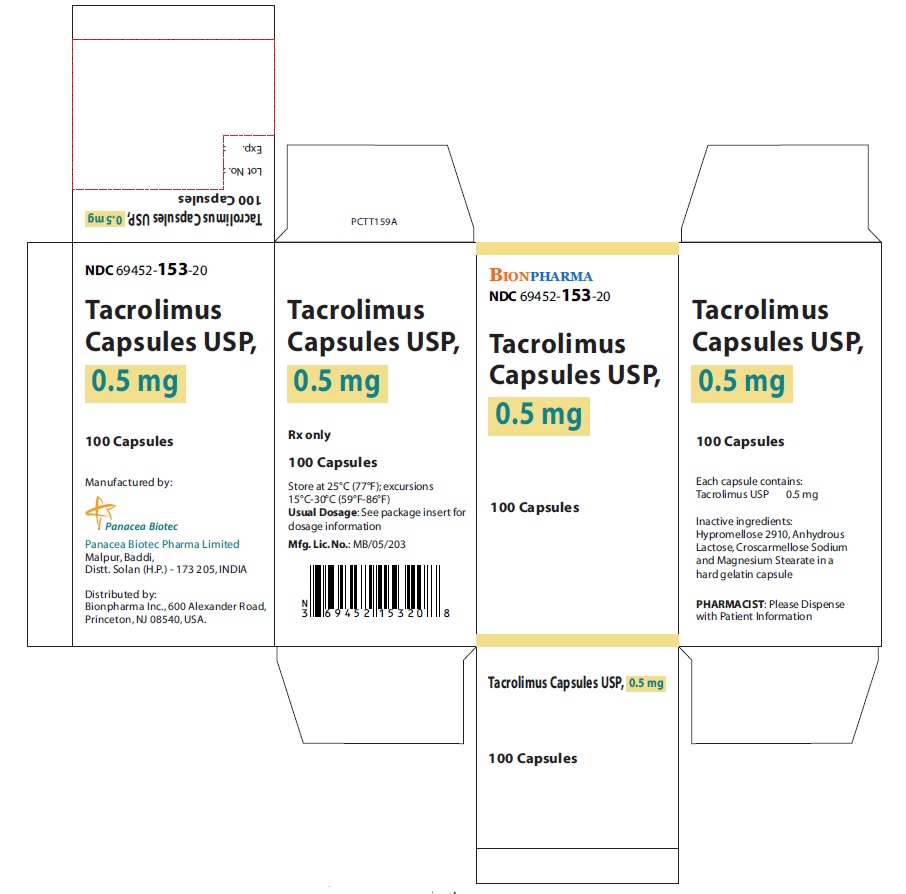
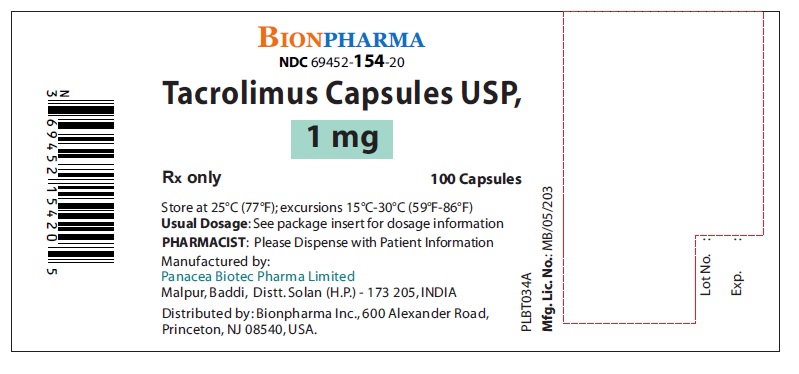
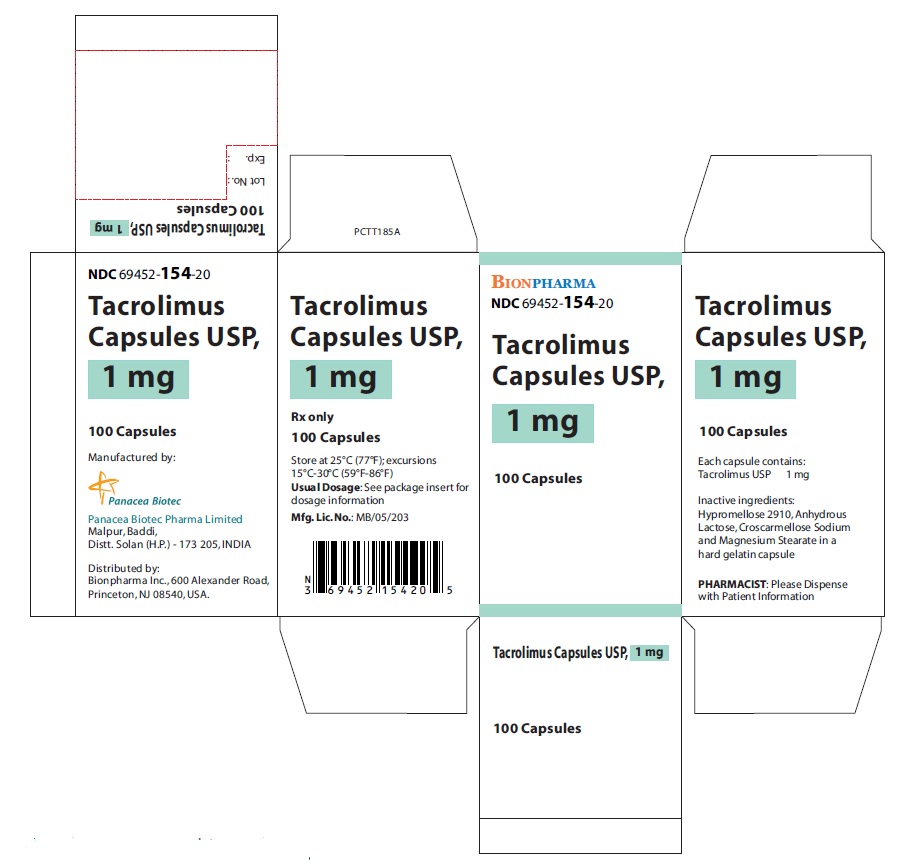
- NDC Code(s): 69452-153-20, 69452-154-20, 69452-155-20
- Packager: BionPharma Inc.,
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated December 25, 2020
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TACROLIMUS CAPSULES safely and effectively. See full prescribing information for TACROLIMUS CAPSULES.
TACROLIMUS Capsules, USP, for oral use
Initial U.S. Approval: 1994
Rx olnyINDICATIONS AND USAGE
Tacrolimus Capsules are a calcineurin-inhibitor immuno-suppressant indicated for the prophylaxis of organ rejection in adult and pediatric patients receiving allogeneic liver, kidney or heart transplants, in combination with other immunosuppressants. ( 1.1)
DOSAGE AND ADMINISTRATION
Patient Population Initial Oral Dosage
(formulation)Whole Blood
Trough Concentration rangeADULT Kidney Transplant with azathioprine 0.2 mg/kg/day capsules divided in two doses, every 12 hours Month 1-3: 7-20 ng/mL Month 4-12: 5-15 ng/mL With MMF/IL-2 receptor antagonist 0.1 mg/kg/day capsules, divided in two doses, every 12 hours Month 1-12: 4-11 ng/mL Liver Transplant With corticosteroids only 0.1-0.15 mg/kg/day capsules, divided in two doses, every 12 hours Month 1-12: 5-20 ng/mL Heart Transplant With azathioprine or MMF 0.075 mg/kg/day capsules, divided in two doses, every 12 hours Month 1-3: 10-20 ng/mL Month ≥ 4: 5-15 ng/mL PEDIATRIC Kidney Transplant 0.3 mg/kg/day capsules, divided in two doses, every 12 hours Month 1-12: 5-20 ng/mL Liver Transplant 0.15-0.2 mg/kg/day capsules 0.2 mg/kg/day, divided in two doses, every 12 hours Month 1-12: 5-20 ng/mL Heart Transplant 0.3 mg/kg/day* capsules divided in two doses, every 12 hours Month 1-12: 5-20 ng/mL MMF= Mycophenolate mofetil
*0.1 mg/kg/day if cell depleting induction treatment is administered
- Administer capsules consistently with or without food. ( 2.1)
- Therapeutic drug monitoring is recommended. ( 2.1, 2.6)
- Avoid eating grapefruit or drinking grapefruit juice. ( 2.1)
- See dosing adjustments for African-American patients ( 2.2), hepatic and renal impaired. ( 2.4, 2.5)
- For complete dosing information see the full prescribing information
DOSAGE FORMS AND STRENGTHS
Capsules: 0.5 mg, 1 mg and 5 mg ( 3)
CONTRAINDICATIONS
Hypersensitivity to tacrolimus or HCO-60 (polyoxyl 60 hydrogenated castor oil) ( 4)
WARNINGS AND PRECAUTIONS
- Not Interchangeable with Extended Release Tacrolimus Products- Medication Errors: Instruct patients or caregivers to recognize the appearance of tacrolimus capsules ( 5.3)
- New Onset Diabetes After Transplant: Monitor blood glucose. ( 5.4)
- Nephrotoxicity: (acute and/or chronic); Reduce the dose; use caution with other nephrotoxic drugs. ( 5.5)
- Neurotoxicity: Including risk of Posterior Reversible Encephalopathy Syndrome (PRES); monitor for neurologic abnormalities; reduce or discontinue tacrolimus capsules and other immunosuppressants. ( 5.6)
- Hyperkalemia: Monitor serum potassium levels. Consider carefully before using with other agents also associated with hyperkalemia. ( 5.7)
- Hypertension: May require antihypertensive therapy. Monitor relevant drug-drug interactions. ( 5.8)
- Anaphylactic Reactions with IV formulation: Observe patients receiving tacrolimus injection for signs and symptoms of anaphylaxis. ( 5.9)
- Not recommended for use with Sirolimus: Not recommended in liver and heart transplant due to increased risk of serious adverse reactions. ( 5.10)
- Myocardial Hypertrophy: Consider dose reduction/ discontinuation. ( 5.13)
- Immunizations: Avoid live vaccines. ( 5.14)
- Pure Red Cell Aplasia: Consider discontinuation of tacrolimus capsules. ( 5.15)
ADVERSE REACTIONS
The most common adverse reactions (≥15%) were abnormal renal function, hypertension, diabetes mellitus, fever, CMV infection, tremor, hyperglycemia, leukopenia, infection, anemia, bronchitis, pericardial effusion, urinary tract infection, constipation, diarrhea, headache, abdominal pain, insomnia, paresthesia, peripheral edema, nausea, hyperkalemia, hypomagnesemia, and hyperlipemia. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bionpharma, Inc. at 1-888-235-BION or 1-888-235-2466 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Mycophenolic Acid Products: Can increase MPA exposure after crossover from cyclosporine to tacrolimus; monitor for MPA-related adverse reactions and adjust MMF or MPA-dose as needed. ( 7.1)
- Nelfinavir and Grapefruit Juice: Increased tacrolimus concentrations via CYP3A inhibition; avoid concomitant use. ( 7.2)
- CYP3A Inhibitors: Increased tacrolimus concentrations; monitor concentrations and adjust tacrolimus dose as needed. ( 5.11, 7.2)
- CYP3A4 Inducers: Decreased tacrolimus concentrations; monitor concentrations and adjust tacrolimus dose as needed. ( 5.11, 7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 10/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
BOXED WARNING - MALIGNANCIES AND SERIOUS INFECTIONS
1 INDICATIONS AND USAGE
1.1 Prophylaxis of Organ Rejection in Kidney, Liver, and Heart Transplant
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Dosing for Adult Kidney, Liver, or Heart Transplant Patients - Capsules
2.3 Dosing for Pediatric Kidney, Liver, and Heart Transplant Patients
2.4 Dosage Adjustment in Patients with Renal Impairment
2.5 Dosage Adjustments in Patients with Hepatic Impairment
2.6 Therapeutic Drug Monitoring
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Lymphoma and Other Malignancies
5.2 Serious Infections
5.3 Not Interchangeable With Extended-Release Tacrolimus Products - Medication Errors
5.4 New Onset Diabetes After Transplant
5.5 Nephrotoxicity
5.6 Neurotoxicity
5.7 Hyperkalemia
5.8 Hypertension
5.9 Anaphylactic Reactions with Tacrolimus Injection
5.10 Not Recommended for Use with Sirolimus
5.11 Interactions with CYP3A4 Inhibitors and Inducers
5.12 QT Prolongation
5.13 Myocardial Hypertrophy
5.14 Immunizations
5.15 Pure Red Cell Aplasia
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Postmarketing Adverse Reactions
7 DRUG INTERACTIONS
7.1 Mycophenolic Acid
7.2 Effects of Other Drugs on Tacrolimus
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Renal Impairment
8.7 Hepatic Impairment
8.8 Race or Ethnicity
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Kidney Transplantation
14.2 Liver Transplantation
14.3 Heart Transplantation
REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Tacrolimus Capsules, USP
16.4 Handling and Disposal
17 PATIENT COUNSELING INFORMATION
17.2 Development of Lymphoma and Other Malignancies
17.3 Increased Risk of Infection
17.4 New Onset Diabetes After Transplant
17.5 Nephrotoxicity
17.6 Neurotoxicity
17.7 Hyperkalemia
17.8 Hypertension
17.9 Drug Interactions
17.10 Pregnancy, Lactation and Infertility
17.11 Myocardial Hypertrophy
17.12 Immunizations
Patient Information
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- BOXED WARNING (What is this?)
-
1 INDICATIONS AND USAGE
1.1 Prophylaxis of Organ Rejection in Kidney, Liver, and Heart Transplant
Tacrolimus Capsules is indicated for the prophylaxis of organ rejection, in patients receiving allogeneic kidney transplant [see Clinical Studies ( 14.1)] , liver transplants [see Clinical Studies ( 14.2)] and heart transplant [see Clinical Studies ( 14.3)] , in combination with other immunosuppressants.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
Tacrolimus Capsules should not be used without supervision by a physician with experience in immunosuppressive therapy.
Tacrolimus capsules and tacrolimus granules are not interchangeable or substitutable for other tacrolimus extended-release products. This is because rate of absorption following the administration of an extended-release tacrolimus product is not equivalent to that of an immediate-release tacrolimus drug product. Under-or overexposure to tacrolimus may result in graft rejection or other serious adverse reactions. Changes between tacrolimus immediate-release and extended-release dosage forms must occur under physician supervision [see Warnings and Precautions ( 5.3)].
Intravenous Formulation -Administration Precautions due to Risk of AnaphylaxisIntravenous use is recommended for patients who cannot tolerate oral formulations, and conversion from intravenous to oral tacrolimus capsules are recommended as soon as oral therapy can be tolerated to minimize the risk of anaphylactic reactions that occurred with injectables containing castor oil derivatives [see Warnings and Precautions ( 5.9)].
Oral Formulations (Capsules)
If patients are able to initiate oral therapy, the recommended starting doses should be initiated. Tacrolimus capsules may be taken with or without food. However, since the presence of food affects the bioavailability of tacrolimus capsules, if taken with food, it should be taken consistently the same way each time [see Clinical Pharmacology ( 12.3)].
General Administration Instructions
Patients should not eat grapefruit or drink grapefruit juice in combination with tacrolimus capsules [see Drug Interactions ( 7.2)].
Tacrolimus capsules should not be used simultaneously with cyclosporine. Tacrolimus capsules or cyclosporine should be discontinued at least 24 hours before initiating the other. In the presence of elevated tacrolimus capsules or cyclosporine concentrations, dosing with the other drug usually should be further delayed.
Therapeutic drug monitoring (TDM) is recommended for all patients receiving tacrolimus capsules [see Dosage and Administration ( 2.6)].
2.2 Dosing for Adult Kidney, Liver, or Heart Transplant Patients - Capsules
Capsules
If patients are able to tolerate oral therapy, the recommended oral starting doses should be initiated. The initial dose of tacrolimus capsules should be administered no sooner than 6 hours after transplantation in the liver and heart transplant patients. In kidney transplant patients, the initial dose of tacrolimus capsules may be administered within 24 hours of transplantation, but should be delayed until renal function has recovered.
The initial oral tacrolimus dosage recommendations for adult patients with kidney, liver, or heart transplants and whole blood trough concentration range are shown in Table 1. Perform therapeutic drug monitoring (TDM) to ensure that patients are within the ranges listed in Table 1.
Table 1. Summary of Initial Oral Tacrolimus capsules Dosing Recommendations and Whole Blood Trough Concentration Range in Adults Patient Population Tacrolimus Capsules*
Initial Oral Dosage
Whole Blood
Trough Concentration Range
kidney Transplant With azathioprine 0.2 mg/kg/day, divided in two doses, administered every 12 hours Month 1-3: 7-20 ng/mL
Month 4-12: 5-15 ng/mL
With MMF/IL-2 receptor antagonist† 0.1 mg/kg/day, divided in two doses, administered every 12 hours Month 1-12: 4-11 ng/mL Liver Transplant With corticosteroids only 0.10-0.15 mg/kg/day, divided in two doses, administered every 12 hours Month 1-12: 5-20 ng/mL Heart Transplant With azathioprine or MMF 0.075 mg/kg/day, divided in two doses, administered every 12 hours Month 1-3: 10-20 ng/mL
Month ≥ 4: 5-15 ng/mL
*African-American patients may require higher doses compared to Caucasians (see Table 2)
† In a second smaller trial, the initial dose of tacrolimus was 0.15-0.2 mg/kg/day and observed tacrolimus concentrations were 6-16 ng/mL during month 1-3 and 5-12 ng/mL during month 4-12 [see Clinical Studies ( 14.1)].
Dosing should be titrated based on clinical assessments of rejection and tolerability. Lower tacrolimus capsules dosages than the recommended initial dosage may be sufficient as maintenance therapy. Adjunct therapy with adrenal corticosteroids is recommended early post-transplant.
The data in kidney transplant patients indicate that the African-American patients required a higher dose to attain comparable trough concentrations compared to Caucasian patients ( Table 2) [see Use in Specific Populations ( 8.8) and Clinical Pharmacology ( 12.3)].
Table 2. Comparative Dose and Trough Concentrations Based on Race
Time After Transplant Caucasian n=114 Black n=56 Dose
(mg/kg)
Trough
Concentrations (ng/mL)
Dose
(mg/kg)
Trough Concentrations
(ng/mL)
Day 7 0.18 12.0 0.23 10.9 Month 1 0.17 12.8 0.26 12.9 Month 6 0.14 11.8 0.24 11.5 Month 12 0.13 10.1 0.19 11.0 Intravenous Injection
Tacrolimus injection should be used only as a continuous intravenous infusion and should be discontinued as soon as the patient can tolerate oral administration. The first dose of tacrolimus capsules should be given 8-12 hours after discontinuing the intravenous infusion.
The recommended starting dose of tacrolimus injection is 0.03-0.05 mg/kg/day in kidney and liver transplant and 0.01 mg/kg/day in heart transplant given as a continuous intravenous infusion. Adult patients should receive doses at the lower end of the dosing range. Concomitant adrenal corticosteroid therapy is recommended early post-transplantation.
The whole blood trough concentration range described in Table 1 pertain to oral administration of tacrolimus capsules only; while monitoring tacrolimus concentrations in patients receiving tacrolimus injection as a continuous intravenous infusion may have some utility, the observed concentrations will not represent comparable exposures to those estimated by the trough concentrations observed in patients on oral therapy.
Anaphylactic reactions have occurred with injectables containing castor oil derivatives, such as Tacrolimus injection. Therefore, monitoring for signs and symptoms of anaphylaxis is recommended [see Warnings and Precautions ( 5.9)].
2.3 Dosing for Pediatric Kidney, Liver, and Heart Transplant Patients
Oral formulation (capsules)
Pediatric patients in general need higher tacrolimus doses compared to adults: the higher dose requirements may decrease as the child grows older. Recommendations for the initial oral dosing for pediatric transplant patients and whole blood trough concentration range are shown in Table 3. Perform TDM to ensure that patients are within the ranges listed in Table 3.
Table 3. Summary of Initial Tacrolimus capsules Dosing Recommendations and Whole Blood Trough Concentration Range in Children
Patient Population Initial Tacrolimus Capsules USP Whole Blood Trough Concentration Range Pediatric kidney transplant patients † 0.3 mg/kg/day capsules, divided in two doses, administered every 12 hours Month 1-12: 5-20 ng/mL Pediatric liver transplant patients ‡ 0.15- 0.2 mg/kg/day, divided in two doses, administered every 12 hours Month 1-12: 5-20 ng/ mL Pediatric heart transplant patients † 0.3 mg/kg/day* capsules, divided in two doses, administered every 12 hours Month 1-12: 5-20 ng/mL *0.1 mg/kg/day if cell depleting induction treatment is administered
† See Clinical Pharmacology ( 12.3) , Tacrolimus Granules Pharmacokinetics in Pediatric Patients
‡ See Clinical Studies ( 14.2) , Liver TransplantationFor conversion of pediatric patients from tacrolimus granules to tacrolimus capsules or from tacrolimus capsules to tacrolimus granules, the total daily dose should remain the same. Following conversion from one formulation to another formulation of tacrolimus, therapeutic drug monitoring is recommended [see Dosage and Administration ( 2.6)] . If a patient is unable to receive an oral formulation, the patient may be started on tacrolimus injection. For pediatric liver transplant patients, the intravenous dose is 0.03-0.05 mg/kg/day.
2.4 Dosage Adjustment in Patients with Renal Impairment
Due to its potential for nephrotoxicity, consideration should be given to dosing tacrolimus capsules at the lower end of the therapeutic dosing range in patients who have received a liver or heart transplant and have pre-existing renal impairment. Further reductions in dose below the targeted range may be required.
In kidney transplant patients with post-operative oliguria, the initial dose of tacrolimus capsules should be administered no sooner than 6 hours and within 24 hours of transplantation, but may be delayed until renal function shows evidence of recovery [see Dosage and Administration ( 2.2) , Warnings and Precautions ( 5.5) , Use in Specific Populations ( 8.6) , and Clinical Pharmacology ( 12.3)] .
2.5 Dosage Adjustments in Patients with Hepatic Impairment
Due to the reduced clearance and prolonged half-life, patients with severe hepatic impairment (Child Pugh ≥ 10) may require lower doses of tacrolimus. Close monitoring of blood concentrations is warranted.
The use of tacrolimus capsules in liver transplant recipients experiencing post-transplant hepatic impairment may be associated with increased risk of developing renal insufficiency related to high whole blood concentrations of tacrolimus. These patients should be monitored closely and dosage adjustments should be considered. Some evidence suggests that lower doses should be used in these patients [see Dosage and Administration ( 2.2), Warnings and Precautions ( 5.5), Use in Specific Populations ( 8.7), and Clinical Pharmacology ( 12.3)].
2.6 Therapeutic Drug Monitoring
Monitoring of tacrolimus blood concentrations in conjunction with other laboratory and clinical parameters is considered an essential aid to patient management for the evaluation of rejection, toxicity, dose adjustments, and compliance. Whole blood trough concentration range can be found in Table 1.
Factors influencing frequency of monitoring include but are not limited to hepatic or renal dysfunction, the addition or discontinuation of potentially interacting drugs and the post-transplant time. Blood concentration monitoring is not a replacement for renal and liver function monitoring and tissue biopsies. Data from clinical trials show that tacrolimus whole blood concentrations were most variable during the first week post-transplantation.
The relative risks of toxicity and efficacy failure are related to tacrolimus whole blood trough concentrations. Therefore, monitoring of whole blood trough concentrations is recommended to assist in the clinical evaluation of toxicity and efficacy failure.
Methods commonly used for the assay of tacrolimus include high-performance liquid chromatography with tandem mass spectrometric detection (HPLC/MS/MS) and immunoassays. Immunoassays may react with metabolites as well as parent compound. Therefore, assay results obtained with immunoassays may have a positive bias relative to results of HPLC/MS. The bias may depend upon the specific assay and laboratory. Comparison of the concentrations in published literature to patient concentrations using the current assays must be made with detailed knowledge of the assay methods and biological matrices employed. Whole blood is the matrix of choice and specimens should be collected into tubes containing ethylene diamine tetraacetic acid (EDTA) anti-coagulant. Heparin anti-coagulation is not recommended because of the tendency to form clots on storage. Samples which are not analyzed immediately should be stored at room temperature or in a refrigerator and assayed within 7 days; see assay instructions for specifics. If samples are to be kept longer they should be deep frozen at -20°C. One study showed drug recovery > 90% for samples stored at -20°C for 6 months, with reduced recovery observed after 6 months.
-
3 DOSAGE FORMS AND STRENGTHS
Tacrolimus Capsules, USP are available in the following dosage form and strengths:
Oblong shape, hard gelatin capsules for oral administration contains tacrolimus as follows:- Tacrolimus Capsules, USP, 0.5 mg: Light yellow color, oblong shape, size “5” hard gelatin capsules printed with “PBT” and “0.5” in red ink on body and cap respectively.
- Tacrolimus Capsules, USP, 1 mg: White color, oblong shape, size “5” hard gelatin capsules printed with “PBT” and “1.0” in red ink on body and cap respectively.
- Tacrolimus Capsules, USP, 5 mg: Pink color, oblong shape, size “4” hard gelatin capsules printed with “PBT” and “5.0” in red ink on body and cap respectively.
-
4 CONTRAINDICATIONS
Tacrolimus capsules are contraindicated in patients with a hypersensitivity to tacrolimus. Tacrolimus injection is contraindicated in patients with a hypersensitivity to HCO-60 (polyoxyl 60 hydrogenated castor oil). Hypersensitivity symptoms reported include dyspnea, rash, pruritus, and acute respiratory distress syndrome [see Adverse Reactions ( 6)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Lymphoma and Other Malignancies
Patients receiving immunosuppressants, including Tacrolimus are at increased risk of developing lymphomas and other malignancies, particularly of the skin [ see Boxed Warning]. The risk appears to be related to the intensity and duration of immunosuppression rather than to the use of any specific agent.
As usual for patients with increased risk for skin cancer, examine patients for skin changese; xposure to sunlight and UV light should be limited by wearing protective clothing and using a broad spectrum sunscreen with a high protection factor.
Post-transplant lymphoproliferative disorder (PTLD) has been reported in immunosuppressed organ transplant recipients. The majority of PTLD events appear related to Epstein Barr Virus (EBV) infection. The risk of PTLD appears greatest in those individuals who are EBV seronegative, a population which includes many young children. Monitor EBV serology during treatment.
5.2 Serious Infections
Patients receiving immunosuppressants, including tacrolimus are at increased risk of developing bacterial, viral, fungal, and protozoal infections, including opportunistic infections. These infections may lead to serious, including fatal, outcomes. Serious viral infections reported include:
- Polyoma virus-associated nephropathy (PVAN), mostly due to BK virus infection
- JC virus-associated progressive multifocal leukoencephalopathy (PML)
- Cytomegalovirus infections: CMV seronegative transplant patients who receive an organ from a CMV seropositive donor disease are at higher risk of developing CMV viremia and CMV disease.
Monitor for the development of infection and adjust the immunosuppressive regimen to balance the risk of rejection with the risk of infection [see Adverse Reactions ( 6.1, 6.2)].
5.3 Not Interchangeable With Extended-Release Tacrolimus Products - Medication Errors
Medication errors, including substitution and dispensing errors, between tacrolimus immediate-release products and tacrolimus extended-release products were reported outside the U.S. This led to serious adverse reactions, including graft rejection, or other adverse reactions due to under-or over-exposure to tacrolimus. Tacrolimus is not interchangeable or substitutable with tacrolimus extended-release products. Changes between tacrolimus immediate-release and extended-release dosage forms must occur under physician supervision. Instruct patients and caregivers to recognize the appearance of tacrolimus capsules dosage forms [see Dosage Forms and Strengths ( 3)] and to confirm with the healthcare provider if a different product is dispensed.
5.4 New Onset Diabetes After Transplant
Tacrolimus was shown to cause new onset diabetes mellitus in clinical trials of kidney, liver, and heart transplantation. New onset diabetes after transplantation may be reversible in some patients. African-American and Hispanic kidney transplant patients are at an increased risk. Blood glucose concentrations should be monitored closely in patients using tacrolimus [see Adverse Reactions ( 6.1)] .
5.5 Nephrotoxicity
Tacrolimus like other calcineurin inhibitors, can cause acute or chronic nephrotoxicity. Nephrotoxicity was reported in clinical trials [see Adverse Reactions ( 6.1)]. Consider dosage reduction in patients with elevated serum creatinine and tacrolimus whole blood trough concentrations greater than the recommended range. The risk for nephrotoxicity may increase when tacrolimus is concomitantly administered with CYP3A inhibitors (by increasing tacrolimus whole blood concentrations) or drugs associated with nephrotoxicity (e.g., aminoglycosides, ganciclovir, amphotericin B, cisplatin, nucleotide reverse transcriptase inhibitors, protease inhibitors) [see Drug Interactions ( 7.2)] . Monitor renal function and consider dosage reduction if nephrotoxicity occurs.
5.6 Neurotoxicity
Tacrolimus may cause a spectrum of neurotoxicities. The most severe neurotoxicities include posterior reversible encephalopathy syndrome (PRES), delirium, seizure and coma; others include tremors, paresthesias, headache, mental status changes, and changes in motor and sensory functions [see Adverse Reactions ( 6.1, 6.2)] . As symptoms may be associated with tacrolimus whole blood trough concentrations at or above the recommended range, monitor for neurologic symptoms and consider dosage reduction or discontinuation of tacrolimus capsules if neurotoxicity occurs.
5.7 Hyperkalemia
Hyperkalemia has been reported with tacrolimus use. Serum potassium levels should be monitored. Careful consideration should be given prior to use of other agents also associated with hyperkalemia (e.g., potassium-sparing diuretics, ACE inhibitors, angiotensin receptor blockers) during tacrolimus therapy [see Adverse Reactions ( 6.1)] . Monitor serum potassium levels periodically during treatment
5.8 Hypertension
Hypertension is a common adverse effect of tacrolimus therapy and may require antihypertensive therapy [see Adverse Reactions ( 6.1)]. The control of blood pressure can be accomplished with any of the common antihypertensive agents, though careful consideration should be given prior to use of antihypertensive agents associated with hyperkalemia (e.g., potassium-sparing diuretics, ACE inhibitors, angiotensin receptor blockers) [see Warnings and Precautions ( 5.7)]. Calcium-channel blocking agents may increase tacrolimus blood concentrations and therefore require dosage reduction of tacrolimus [see Drug Interactions ( 7.2)].
5.9 Anaphylactic Reactions with Tacrolimus Injection
Anaphylactic reactions have occurred with injectables containing castor oil derivatives, including tacrolimus in a small percentage of patients (0.6%). The exact cause of these reactions is not known. Tacrolimus injection should be reserved for patients who are unable to take tacrolimus capsules orally. Monitor patients for anaphylaxis when using the intravenous route of administration [ see Dosage and Administration ( 2.1)].
5.10 Not Recommended for Use with Sirolimus
Tacrolimus capsules are not recommended for use with sirolimus:
- The use of sirolimus with tacrolimus in studies of de novo liver transplant patients was associated with an excess mortality, graft loss, and hepatic artery thrombosis (HAT) and is not recommended.
- The use of sirolimus (2 mg per day) with tacrolimus in heart transplant patients in a U.S. trial was associated with increased risk of renal function impairment, wound healing complications, and insulin-dependent post-transplant diabetes mellitus, and is not recommended [see Clinical Studies ( 14.3)].
5.11 Interactions with CYP3A4 Inhibitors and Inducers
When co-administering tacrolimus with strong CYP3A4 inhibitors (e.g., telaprevir, boceprevir, ritonavir, ketoconazole, itraconazole, voriconazole, clarithromycin) and strong inducers (e.g., rifampin, rifabutin), adjustments in the dosing regimen of tacrolimus and subsequent frequent monitoring of tacrolimus whole blood trough concentrations and tacrolimus-associated adverse reactions are recommended [see Drug Interactions ( 7)].
5.12 QT Prolongation
Tacrolimus may prolong the QT/QTc interval and may cause Torsade de Pointes. Avoid tacrolimus in patients with congenital long QT syndrome. In patients with congestive heart failure, bradyarrhythmias, those taking certain antiarrhythmic medications or other medicinal products that lead to QT prolongation, and those with electrolyte disturbances such as hypokalemia, hypocalcemia, or hypomagnesemia, consider obtaining electrocardiograms and monitoring electrolytes (magnesium, potassium, calcium) periodically during treatment.
When co-administering tacrolimus with other substrates and/or inhibitors of CYP3A4 that also have the potential to prolong the QT interval, a reduction in tacrolimus dose, frequent monitoring of tacrolimus whole blood concentrations, and monitoring for QT prolongation is recommended.
Use of tacrolimus with amiodarone has been reported to result in increased tacrolimus whole blood concentrations with or without concurrent QT prolongation [see Drug Interactions ( 7)].
5.13 Myocardial Hypertrophy
Myocardial hypertrophy has been reported in infants, children, and adults, particularly those with high tacrolimus trough concentrations, and is generally manifested by echocardiographically demonstrated concentric increases in left ventricular posterior wall and interventricular septum thickness. This condition appears reversible in most cases following dose reduction or discontinuance of therapy. In patients who develop renal failure or clinical manifestations of ventricular dysfunction while receiving tacrolimus therapy, echocardiographic evaluation should be considered. If myocardial hypertrophy is diagnosed, dosage reduction or discontinuation of tacrolimus capsules should be considered [see Adverse Reactions ( 6.2)].
5.14 Immunizations
Whenever possible, administer the complete complement of vaccines before transplantation and treatment with Tacrolimus.
The use of live vaccines should be avoided during treatment with tacrolimus; examples include (not limited to) the following: intranasal influenza, measles, mumps, rubella, oral polio, BCG, yellow fever, varicella, and TY21a typhoid vaccines.
Inactivated vaccines noted to be safe for administration after transplantation may not be sufficiently immunogenic during treatment with tacrolimus.
5.15 Pure Red Cell Aplasia
Cases of pure red cell aplasia (PRCA) have been reported in patients treated with tacrolimus. A mechanism for tacrolimus-induced PRCA has not been elucidated. All patients reported risk factors for PRCA such as parvovirus B19 infection, underlying disease, or concomitant medications associated with PRCA. If PRCA is diagnosed, discontinuation of tacrolimus capsules should be considered [see Adverse Reactions ( 6.2)].
-
6 ADVERSE REACTIONS
The following serious and otherwise important adverse drug reactions are discussed in greater detail in other sections of labeling:
- Lymphoma and Other Malignancies [see Boxed Warning, Warnings and Precautions ( 5.1)]
- Serious Infections [see Boxed Warning, Warnings and Precautions ( 5.2)]
- New Onset Diabetes After Transplant [see Warnings and Precautions ( 5.4)]
- Nephrotoxicity [see Warnings and Precautions ( 5.5)]
- Neurotoxicity [see Warnings and Precautions ( 5.6)]
- Hyperkalemia [see Warnings and Precautions ( 5.7)]
- Hypertension [see Warnings and Precautions ( 5.8)]
- Anaphylactic Reactions with Tacrolimus Injection [see Warnings and Precautions ( 5.9)]
- Myocardial Hypertrophy [see Warnings and Precautions ( 5.13)]
- Pure Red Cell Aplasia [see Warnings and Precautions ( 5.15)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. In addition, the clinical trials were not designed to establish comparative differences across study arms with regards to the adverse reactions discussed below.
Kidney Transplantation
The incidence of adverse reactions was determined in three randomized kidney transplant trials. One of the trials used azathioprine (AZA) and corticosteroids and two of the trials used mycophenolate mofetil (MMF) and corticosteroids concomitantly for maintenance immunosuppression.
Tacrolimus based immunosuppression in conjunction with azathioprine and corticosteroids following kidney transplantation was assessed in a trial where 205 patients received tacrolimus based immunosuppression and 207 patients received cyclosporine-based immunosuppression. The trial population had a mean age of 43 years (mean ± SD was 43 ± 13 years on tacrolimus and 44 ± 12 years on cyclosporine arm), the distribution was 61% male, and the composition was White (58%), African-American (25%), Hispanic (12%), and Other (5%). The 12-month post-transplant information from this trial is presented below.
The most common adverse reactions (≥ 30%) observed in tacrolimus treated kidney transplant patients are: infection, tremor, hypertension, abnormal renal function, constipation, diarrhea, headache, abdominal pain, insomnia, nausea, hypomagnesemia, urinary tract infection, hypophosphatemia, peripheral edema, asthenia, pain, hyperlipidemia, hyperkalemia, and anemia. Based on reported adverse reactions terms related to decreased renal function, nephrotoxicity was reported in approximately 52% of kidney transplantation patients.
Adverse reactions that occurred in ≥15% of kidney transplant patients treated with tacrolimus in conjunction with azathioprine are presented below:
Table 4. Kidney Transplantation: Adverse Reactions Occurring in ≥ 15% of Patients Treated with Tacrolimus in Conjunction with Azathioprine (AZA)
Tacrolimus/AZA
(N=205)
Cyclosporine/AZA
(N=207)
Nervous System Tremor 54% 34% Headache 44% 38% Insomnia 32% 30% Paresthesia 23% 16% Dizziness 19% 16% Gastrointestinal Diarrhea 44% 41% Nausea 38% 36% Constipation 35% 43% Vomiting 29% 23% Dyspepsia 28% 20% Cardiovascular Hypertension 50% 52% Chest Pain 19% 13% Urogenital Creatinine Increased 45% 42% Urinary Tract Infection 34% 35% Metabolic and Nutritional Hypophosphatemia 49% 53% Hypomagnesemia 34% 17% Hyperlipemia 31% 38% Hyperkalemia 31% 32% Diabetes Mellitus 24% 9% Hypokalemia 22% 25% Hyperglycemia 22% 16% Edema 18% 19% Hemic and Lymphatic Anemia 30% 24% Leukopenia 15% 17% Miscellaneous Infection 45% 49% Peripheral Edema 36% 48% Asthenia 34% 30% Abdominal Pain 33% 31% Pain 32% 30% Fever 29% 29% Back Pain 24% 20% Respiratory System Dyspnea 22% 18% Cough Increased 18% 15% Musculoskeletal Arthralgia 25% 24% Skin Rash 17% 12% Pruritus 15% 7% Two trials were conducted for tacrolimus based immunosuppression in conjunction with MMF and corticosteroids. In the non-US trial (Study 1), the incidence of adverse reactions was based on 1195 kidney transplant patients that received tacrolimus (Group C, n = 403), or one of two cyclosporine (CsA) regimens (Group A, n = 384 and Group B, n = 408) in combination with MMF and corticosteroids; all patients, except those in one of the two cyclosporine groups, also received induction with daclizumab. The trial population had a mean age of 46 years (range 17 to 76), the distribution was 65% male, and the composition was 93% Caucasian. The 12-month post-transplant information from this trial is presented below.
Adverse reactions that occurred in ≥10% of kidney transplant patients treated with tacrolimus in conjunction with MMF in Study 1 [Note: This trial was conducted entirely outside of the United States. Such trials often report a lower incidence of adverse reactions in comparison to U.S. trials] are presented below:
Table 5. Kidney Transplantation: Adverse Reactions Occurring in ≥ 10% of Patients Treated with Tacrolimus in Conjunction with MMF (Study 1)
Tacrolimus Capsules USP (Group C) (N=403) Cyclosporine (Group A) (N=384) Cyclosporine (Group B) (N=408) Diarrhea 25% 16% 13% Urinary tract infection 24% 28% 24% Anemia 17% 19% 17% Hypertension 13% 14% 12% Leucopenia 13% 10% 10% Edema peripheral 11% 12% 13% Hyperlipidemia 10% 15% 13% Key: Group A = CsA/MMF/CS, B = CsA/MMF/CS/Daclizumab, C = Tac/MMF/CS/Daclizumab
CsA = Cyclosporine, CS = Corticosteroids, Tac = Tacrolimus, MMF = mycophenolate mofetilIn the U.S. trial (Study 2) with tacrolimus based immunosuppression in conjunction with MMF and corticosteroids, 424 kidney transplant patients received tacrolimus (n = 212) or cyclosporine (n = 212) in combination with MMF1 gram twice daily, basiliximab induction, and corticosteroids. The trial population had a mean age of 48 years (range 17 to 77), the distribution was 63% male, and the composition was White (74%), African-American (20%), Asian (3%), and Other (3%). The 12-month post-transplant information from this trial is presented below.
Adverse reactions that occurred in ≥ 15% of kidney transplant patients treated with tacrolimus in conjunction with MMF in Study 2 are presented below:
Table 6. Kidney Transplantation: Adverse Reactions Occurring in ≥ 15% of Patients Treated with Tacrolimus in Conjunction with MMF (Study 2)
Tacrolimus Capsules USP / MMF
(N=212)Cyclosporine / MMF
(N=212)Gastrointestinal Disorder Diarrhea 44% 26% Nausea 39% 47% Constipation 36% 41% Vomiting 26% 25% Dyspepsia 18% 15% Injury, Poisoning, and Procedural Complications Post Procedural Pain 29% 27% Incision Site Complication 28% 23% Graft Dysfunction 24% 18% Metabolism and Nutrition Disorder Hypomagnesemia 28% 22% Hypophosphatemia 28% 21% Hyperkalemia 26% 19% Hyperglycemia 21% 15% Hyperlipidemia 18% 25% Hypokalemia 16% 18% Nervous System Disorder Tremor 34% 20% Headache 24% 25% Blood and Lymphatic System Disorders Anemia 30% 28% Leukopenia 16% 12% Miscellaneous Edema Peripheral 35% 46% Hypertension 32% 35% Insomnia 30% 21% Urinary Tract Infection 26% 22% Blood Creatinine Increased 23% 23% Less frequently observed adverse reactions in kidney transplantation patients are described under the subsection “Less Frequently Reported Adverse Reactions (> 3% and < 15%) in Liver, Kidney, and Heart Transplant Studies”.
Liver Transplantation
There were two randomized comparative liver transplant trials. In the U.S. trial, 263 adult and pediatric patients received tacrolimus and steroids and 266 patients received cyclosporine-based immunosuppressive regimen (CsA/AZA). The trial population had a mean age of 44 years (range 0.4 to 70), the distribution was 52% male, and the composition was White (78%), African-American (5%), Asian (2%), Hispanic (13%), and Other (2%). In the European trial, 270 patients received tacrolimus and steroids and 275 patients received CsA/AZA. The trial population had a mean age of 46 years (range 15 to 68), the distribution was 59% male, and the composition was White (95.4%), Black (1%), Asian (2%), and Other (2%).
The proportion of patients reporting more than one adverse event was > 99% in both the tacrolimus group and the CsA/AZA group. Precautions must be taken when comparing the incidence of adverse reactions in the U.S. trial to that in the European trial. The 12-month post-transplant information from the U.S. trial and from the European trial is presented below. The two trials also included different patient populations and patients were treated with immunosuppressive regimens of differing intensities. Adverse reactions reported in ≥ 15% in tacrolimus patients (combined trial results) are presented below for the two controlled trials in liver transplantation.
The most common adverse reactions (≥ 40%) observed in tacrolimus treated liver transplant patients are: tremor, headache, diarrhea, hypertension, nausea, abnormal renal function, abdominal pain, insomnia, paresthesia, anemia, pain, fever, asthenia, hyperkalemia, hypomagnesemia, and hyperglycemia. These all occur with oral and IV administration of Tacrolimus and some may respond to a reduction in dosing (e.g., tremor, headache, paresthesia, hypertension). Diarrhea was sometimes associated with other gastrointestinal complaints such as nausea and vomiting. Based on reported adverse reactions terms related to decreased renal function, nephrotoxicity was reported in approximately 40% and 36% of liver transplantation patients receiving tacrolimus in the U.S. and European randomized trials.
Table 7. Liver Transplantation: Adverse Reactions Occurring in ≥ 15% of Patients Treated with Tacrolimus
U.S.TRIAL EUROPEAN TRIAL Tacrolimus (N=250) Cyclosporine/AZA (N=250) Tacrolimus (N=264) Cyclosporine /AZA (N=265) Nervous System Headache 64% 60% 37% 26% Insomnia 64% 68% 32% 23% Tremor 56% 46% 48% 32% Paresthesia 40% 30% 17% 17% Gastrointestinal Diarrhea 72% 47% 37% 27% Nausea 46% 37% 32% 27% LFT Abnormal 36% 30% 6% 5% Anorexia 34% 24% 7% 5% Vomiting 27% 15% 14% 11% Constipation 24% 27% 23% 21% Cardiovascular Hypertension 47% 56% 38% 43% Urogenital Kidney Function Abnormal 40% 27% 36% 23% Creatinine Increased 39% 25% 24% 19% BUN Increased 30% 22% 12% 9% Oliguria 18% 15% 19% 12% Urinary Tract Infection 16% 18% 21% 19% Metabolic and Nutritional Hypomagnesemia 48% 45% 16% 9% Hyperglycemia 47% 38% 33% 22% Hyperkalemia 45% 26% 13% 9% Hypokalemia 29% 34% 13% 16% Hemic and Lymphatic Anemia 47% 38% 5% 1% Leukocytosis 32% 26% 8% 8% Thrombocytopenia 24% 20% 14% 19% Miscellaneous Pain 63% 57% 24% 22% Abdominal Pain 59% 54% 29% 22% Asthenia 52% 48% 11% 7% Fever 48% 56% 19% 22% Back Pain 30% 29% 17% 17% Ascites 27% 22% 7% 8% Peripheral Edema 26% 26% 12% 14% Respiratory System Pleural Effusion 30% 32% 36% 35% Dyspnea 29% 23% 5% 4% Atelectasis 28% 30% 5% 4% Skin and Appendages Pruritus 36% 20% 15% 7% Rash 24% 19% 10% 4% Table 8. Pediatric Liver Transplantation: Adverse Reactions Occurring in > 10% of Patients Treated with Tacrolimus Granules (STUDY 01-13)
Tacrolimus Granules
(N=91)Cyclosporine
(N=90)Body as a Whole Fever 46% 51% Infection 25% 29% Sepsis 22% 20% CMV Infection 15% 24% EBV Infection 26% 11% Ascites 17% 20% Peritonitis 12% 7% Cardiovascular System Hypertension 39% 47% Digestive System Liver Function Tests Abnormal 37% 28% Diarrhea 26% 26% Vomiting 15% 13% Gastrointestinal Hemorrhage 11% 12% Bile Duct Disorder 12% 8% Gastroenteritis 12% 4% Hemic and Lymphatic System Anemia 29% 19% Metabolic and Nutritional Disorders Hypomagnesemia 40% 29% Acidosis 26% 17% Hyperkalemia 12% 10% Respiratory System Pleural Effusion 22% 19% Bronchitis 11% 8% Urogenital System Kidney Function Abnormal 13% 14% Less frequently observed adverse reactions in liver transplantation patients are described under the subsection “Less Frequently Reported Adverse Reactions (> 3% and < 15%) in Liver, Kidney, and Heart Transplant Studies”
Heart Transplantation
The incidence of adverse reactions was determined based on two trials in primary orthotopic heart transplantation. In a trial conducted in Europe, 314 patients received a regimen of antibody induction, corticosteroids, and azathioprine (AZA) in combination with Tacrolimus (n = 157) or cyclosporine (n = 157) for 18 months. The trial population had a mean age of 51 years (range 18 to 65), the distribution was 82% male, and the composition was White (96%), Black (3%), and other (1%).
The most common adverse reactions (≥ 15%) observed in Tacrolimus treated heart transplant patients are: abnormal renal function, hypertension, diabetes mellitus, CMV infection, tremor, hyperglycemia, leukopenia, infection, anemia, bronchitis, pericardial effusion, urinary tract infection, and hyperlipemia. Based on reported adverse reactions terms related to decreased renal function, nephrotoxicity was reported in approximately 59% of heart transplantation patients in the European trial.
Adverse reactions in heart transplant patients in the European trial are presented below:
Table 9. Heart Transplantation: Adverse Reactions Occurring in ≥ 15% of Patients Treated with Tacrolimus Capsules USP in Conjunction with Azathioprine (AZA)
Tacrolimus Capsules USP /AZA (n=157) Cyclosporine/AZA (n=157) Cardiovascular System Hypertension 62% 69% Pericardial Effusion 15% 14% Body as a Whole CMV Infection 32% 30% Infection 24% 21% Metabolic and Nutritional Disorders Diabetes Mellitus 26% 16% Hyperglycemia 23% 17% Hyperlipemia 18% 27% Hemic and Lymphatic System Anemia 50% 36% Leukopenia 48% 39% Urogenital System Kidney Function Abnormal 56% 57% Urinary Tract Infection 16% 12% Respiratory System Bronchitis 17% 18% Nervous System Tremor 15% 6% In the European trial, the cyclosporine trough concentrations were above the pre-defined target range (i.e., 100 to 200 ng/mL) at Day 122 and beyond in 32% to 68% of the patients in the cyclosporine treatment arm, whereas the tacrolimus trough concentrations were within the pre-defined target range (i.e., 5 to 15 ng/mL) in 74% to 86% of the patients in the tacrolimus treatment arm.
In a U.S. trial, the incidence of adverse reactions was based on 331 heart transplant patients that received corticosteroids and tacrolimus in combination with sirolimus (n=109), tacrolimus in combination with MMF (n=107) or cyclosporine modified in combination with MMF (n=115) for 1 year. The trial population had a mean age of 53 years (range 18 to 75), the distribution was 78% male, and the composition was White (83%), African-American (13%) and other (4%).Only selected targeted treatment-emergent adverse reactions were collected in the U.S. heart transplantation trial. Those reactions that were reported at a rate of 15% or greater in patients treated with tacrolimus and MMF include the following: any target adverse reactions (99%), hypertension (89%), hyperglycemia requiring antihyperglycemic therapy (70%), hypertriglyceridemia (65%), anemia (hemoglobin < 10.0 g/dL) (65%), fasting blood glucose > 140 mg/dL (on two separate occasions) (61%), hypercholesterolemia (57%), hyperlipidemia (34%), WBCs < 3000 cells/mcL (34%), serious bacterial infections (30%), magnesium < 1.2 mEq/L (24%), platelet count < 75,000 cells/mcL (19%), and other opportunistic infections (15%).
Other targeted treatment-emergent adverse reactions in tacrolimus treated patients occurred at a rate of less than 15%, and include the following: Cushingoid features, impaired wound healing, hyperkalemia, Candida infection, and CMV infection/syndrome. Other less frequently observed adverse reactions in heart transplantation patients are described under the subsection “Less Frequently Reported Adverse Reactions (> 3% and < 15%) in Liver, Kidney and Heart Transplant Studies.”
New Onset Diabetes After Transplant
Kidney Transplantation
New Onset Diabetes After Transplant (NODAT) is defined as a composite of fasting plasma glucose ≥ 126 mg/dL, HbA1C ≥ 6%, insulin use ≥ 30 days, or oral hypoglycemic use. In a trial in kidney transplant patients (Study 2), NODAT was observed in 75% in the tacrolimus treated and 61% in the NEORAL-treated patients without pre-transplant history of diabetes mellitus ( Table 10) [see Clinical Studies ( 14.1).
Table 10. Incidence of New Onset Diabetes After Transplant at 1 year in Kidney Transplant Recipients in a Phase 3 Trial (Study 2)
Parameter Treatment Group Tacrolimus MMF
(n = 212)Neoral / MMF (n = 212) NODAT 112/150 (75%) 93/152 (61%) Fasting Plasma Glucose ≥ 126 mg/dL 96/150 (64%) 80/152 (53%) HbA1C ≥ 6% 59/150 (39%) 28/152 (18%) Insulin Use ≥ 30 days 9/150 (6%) 4/152 (3%) Oral Hypoglycemic Use 15/150 (10%) 5/152 (3%) In early trials of tacrolimus, Post-Transplant Diabetes Mellitus (PTDM) was evaluated with a more limited criterion of “use of insulin for 30 or more consecutive days with < 5-day gap” in patients without a prior history of insulin-dependent diabetes mellitus or non-insulin dependent diabetes mellitus. Data are presented in Tables 11 to 14. PTDM was reported in 20% of Tacrolimus /Azathioprine (AZA)-treated kidney transplant patients without pre-transplant history of diabetes mellitus in a Phase 3 trial ( Table 11). The median time to onset of PTDM was 68 days. Insulin dependence was reversible in 15% of these PTDM patients at one year and in 50% at 2 years post-transplant. African-American and Hispanic kidney transplant patients were at an increased risk of development of PTDM ( Table 12).
Table 11. Incidence of Post-Transplant Diabetes Mellitus and Insulin Use at 2 Years in Kidney Transplant Recipients in a Phase 3 Trial using Azathioprine (AZA)
Status of PTDM* Tacrolimus/AZA CsA/AZA Patients without pretransplant history of diabetes mellitus. 151 151 New onset PTDM *, 1 st Year 30/151 (20%) 6/151 (4%) Still insulin dependent at one year in those without prior history of diabetes. 25/151 (17%) 5/151 (3%) New onset PTDM * post 1 year 1 0 Patients with PTDM * at 2 years 16/151 (11%) 5/151 (3%) *Use of insulin for 30 or more consecutive days, with < 5 day gap, without a prior history of insulin-dependent diabetes mellitus or non-insulin dependent diabetes mellitus.
Table 12. Development of Post-Transplant Diabetes Mellitus by Race or Ethnicity and by Treatment Group During First Year Post Kidney Transplantation in a Phase 3 Trial
Patient Race Patients Who Developed PTDM * Tacrolimus Cyclosporine African-American 15/41 (37%) 3 (8%) Hispanic 5/17 (29%) 1 (6%) Caucasian 10/82 (12%) 1 (1%) Other 0/11 (0%) 1 (10%) Total 30/151 (20%) 6 (4%) * Use of insulin for 30 or more consecutive days, with < 5 day gap, without a prior history of insulin-dependent diabetes mellitus or non-insulin dependent diabetes mellitus.
Liver TransplantationInsulin-dependent PTDM was reported in 18% and 11% of tacrolimus treated liver transplant patients and was reversible in 45% and 31% of these patients at 1 year post-transplant, in the U.S. and European randomized trials, respectively ( Table 13). Hyperglycemia was associated with the use of tacrolimus in 47% and 33% of liver transplant recipients in the U.S. and European randomized trials, respectively, and may require treatment [see Adverse Reactions ( 6.1)].
Table 13. Incidence of Post-Transplant Diabetes Mellitus and Insulin Use at 1 Year in Liver Transplant RecipientsStatus of PTDM * US Trial European Trial Tacrolimus Cyclosporine Tacrolimus Cyclosporine Patients at risk † 239 236 239 249 New Onset PTDM * 42 (18%) 30 (13%) 26 (11%) 12 (5%) Patients still on insulin at 1 year 23 (10%) 19 (8%) 18 (8%) 6 (2%) *Use of insulin for 30 or more consecutive days, with < 5 day gap, without a prior history of insulin-dependent diabetes mellitus or non-insulin dependent diabetes mellitus.
† Patients without pre-transplant history of dibetes mellitus.
Heart Transplantation
Insulin-dependent PTDM was reported in 13% and 22% of tacrolimus treated heart transplant patients receiving mycophenolate mofetil (MMF) or azathioprine (AZA) and was reversible in 30% and 17% of these patients at one year post-transplant, in the U.S. and European randomized trials, respectively ( Table 14). Hyperglycemia defined as two fasting plasma glucose levels ≥ 126 mg/dL was reported with the use of tacrolimus plus MMF or AZA in 32% and 35% of heart transplant recipients in the U.S. and European randomized trials, respectively, and may require treatment [see Adverse Reactions ( 6.1)].
Table 14. Incidence of Post-Transplant Diabetes Mellitus and Insulin Use at 1 Year in Heart Transplant Recipients
Status of PTDM* US Trial European Trial Tacrolimus MMF Cyclosporine/MMF Tacrolimus/AZA Cyclosporine/AZA Patients at risk † 75 83 132 138 New Onset PTDM* 10 (13%) 6 (7%) 29 (22%) 5 (4%) Patients still on insulin at 1 year ‡ 7 (9%) 1 (1%) 24 (18%) 4 (3%) * Use of insulin for 30 or more consecutive days without a prior history of insulin-dependent diabetes mellitus or non-insulin dependent diabetes mellitus.
† Patients without pre-transplant history of diabetes mellitus.
‡ 7-12 months for the U.S. trial.
Less Frequently Reported Adverse Reactions (> 3% and < 15%) in Liver, Kidney, and Heart Transplant Studies
The following adverse reactions were reported in either liver, kidney, and/or heart transplant recipients who were treated with tacrolimus in clinical trials.
- Nervous System [see Warnings and Precautions (5.6)]: Abnormal dreams, agitation, amnesia, anxiety, confusion, convulsion, crying, depression, elevated mood, emotional lability, encephalopathy, hemorrhagic stroke, hallucinations, hypertonia, incoordination, monoparesis, myoclonus, nerve compression, nervousness, neuralgia, neuropathy, paralysis flaccid, psychomotor skills impaired, psychosis, quadriparesis, somnolence, thinking abnormal, vertigo, writing impaired
- Special Senses: Abnormal vision, amblyopia, ear pain, otitis media, tinnitus
- Gastrointestinal: Cholangitis, cholestatic jaundice, duodenitis, dysphagia, esophagitis, flatulence, gastritis, gastroesophagitis, gastrointestinal hemorrhage, GGT increase, GI disorder, GI perforation, hepatitis, hepatitis granulomatous, ileus, increased appetite, jaundice, liver damage, esophagitis ulcerative, oral moniliasis, pancreatic pseudocyst, stomatitis
- Cardiovascular: Abnormal ECG, angina pectoris, arrhythmia, atrial fibrillation, atrial flutter, bradycardia, cardiac fibrillation, cardiopulmonary failure, congestive heart failure, deep thrombophlebitis, echocardiogram abnormal, electrocardiogram QRS complex abnormal, electrocardiogram ST segment abnormal, heart failure, heart rate decreased, hemorrhage, hypotension, phlebitis, postural hypotension, syncope, tachycardia, thrombosis, vasodilatation
- Urogenital: Acute kidney failure [see Warnings and Precautions ( 5.5)] , albuminuria, BK nephropathy, bladder spasm, cystitis, dysuria, hematuria, hydronephrosis, kidney failure, kidney tubular necrosis, nocturia, pyuria, toxic nephropathy, urge incontinence, urinary frequency, urinary incontinence, urinary retention, vaginitis
- Metabolic/Nutritional: Acidosis, alkaline phosphatase increased, alkalosis, ALT (SGPT) increased, AST (SGOT) increased, bicarbonate decreased, bilirubinemia, dehydration, GGT increased, gout, healing abnormal, hypercalcemia, hypercholesterolemia, hyperphosphatemia, hyperuricemia, hypervolemia, hypocalcemia, hypoglycemia, hyponatremia, hypoproteinemia, lactic dehydrogenase increase, weight gain
- Endocrine: Cushing’s syndrome
- Hemic/Lymphatic: Coagulation disorder, ecchymosis, hematocrit increased, hypochromic anemia, leukocytosis, polycythemia, prothrombin decreased, serum iron decreased
- Miscellaneous: Abdomen enlarged, abscess, accidental injury, allergic reaction, cellulitis, chills, fall, flu syndrome, generalized edema, hernia, mobility decreased, peritonitis, photosensitivity reaction, sepsis, temperature intolerance, ulcer
- Musculoskeletal: Arthralgia, cramps, generalized spasm, leg cramps, myalgia, myasthenia, osteoporosis
- Respiratory: Asthma, emphysema, hiccups, lung function decreased, pharyngitis, pneumonia, pneumothorax, pulmonary edema, rhinitis, sinusitis, voice alteration
- Skin: Acne, alopecia, exfoliative dermatitis, fungal dermatitis, herpes simplex, herpes zoster, hirsutism, neoplasm skin benign, skin discoloration, skin ulcer, sweating
6.2 Postmarketing Adverse Reactions
The following adverse reactions have been reported from worldwide marketing experience with tacrolimus. Because these reactions are reported voluntarily from a population of uncertain size it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. Decisions to include these reactions in labeling are typically based on one or more of the following factors: (1) seriousness of the reaction, (2) frequency of the reporting, or (3) strength of causal connection to the drug.
Other reactions include:
- Cardiovascular: Atrial fibrillation, atrial flutter, cardiac arrhythmia, cardiac arrest, electrocardiogram T wave abnormal, flushing, myocardial infarction, myocardial ischemia, pericardial effusion, QT prolongation, Torsade de Pointes, venous thrombosis deep limb, ventricular extrasystoles, ventricular fibrillation, myocardial hypertrophy [see Warnings and Precautions ( 5.13)]
- Gastrointestinal: Bile duct stenosis, colitis, enterocolitis, gastroenteritis, gastroesophageal reflux disease, hepatic cytolysis, hepatic necrosis, hepatotoxicity, impaired gastric emptying, liver fatty, mouth ulceration, pancreatitis hemorrhagic, pancreatitis necrotizing, stomach ulcer, veno-occlusive liver disease
- Hemic/Lymphatic: Agranulocytosis, disseminated intravascular coagulation, hemolytic anemia, neutropenia, pancytopenia, thrombocytopenic purpura, thrombotic thrombocytopenic purpura, pure red cell aplasia [see Warnings and Precautions ( 5.15)]
- Infections: Cases of progressive multifocal leukoencephalopathy (PML), sometimes fatal; polyoma virus- associated nephropathy, (PVAN) including graft loss [see Warnings and Precautions ( 5.2)]
- Miscellaneous: Feeling hot and cold, feeling jittery, hot flushes, multi-organ failure, primary graft dysfunction
- Musculoskeletal and Connective Tissue Disorders: Pain in extremity including Calcineurin-Inhibitor Induced Pain Syndrome (CIPS)
- Nervous System: Carpal tunnel syndrome, cerebral infarction, hemiparesis, leukoencephalopathy, mental disorder, mutism, posterior reversible encephalopathy syndrome (PRES) [see Warnings and Precautions (5.6)] , progressive multifocal leukoencephalopathy (PML) [see Warnings and Precautions ( 5.2)] , quadriplegia, speech disorder, syncope
-
7 DRUG INTERACTIONS
7.1 Mycophenolic Acid
When tacrolimus is prescribed with a given dose of a mycophenolic acid (MPA) product, exposure to MPA is higher with tacrolimus co-administration than with cyclosporine co-administration because cyclosporine interrupts the enterohepatic recirculation of MPA while tacrolimus does not. Monitor for MPA associated adverse reactions and reduce the dose of concomitantly administered mycophenolic acid products as needed.
7.2 Effects of Other Drugs on Tacrolimus
Table 15 displays the effects of other drugs on tacrolimus.
Table 15: Effects of Other Drugs/Substances on Tacrolimus* Drug/Substance Class or Name Drug Interaction Effect Recommendations Grapefruit or grapefruit juice † May increase tacrolimus whole blood trough concentrations and increase the risk of serious adverse reactions (e.g., neurotoxicity, QT prolongation) [see Warnings and Precautions ( 5.6, 5.11, 5.12)]. Avoid grapefruit or grapefruit juice. Strong CYP3A Inducers #:
Antimycobacterials (e.g., rifampin, rifabutin),anticonvulsants (e.g., phenytoin, carbamazepine and phenobarbital), St John's Wort
May decrease tacrolimus whole blood trough concentrations and increase the risk of rejection [see Warnings and Precautions ( 5.11)]. Increase tacrolimus dose and monitor tacrolimus whole blood trough concentrations [see Dosage and Administration (2.2, 2.6) and Clinical Pharmacology ( 12.3)]. Strong CYP3A Inhibitors #:
Protease inhibitors (e.g, nelfinavir, telaprevir, boceprevir, ritonavir), azole antifungals (e.g., voriconazole, posaconazole, itraconazole, ketoconazole), antibiotics (e.g., clarithromycin, troleandomycin, chloramphenicol), nefazodone, schisandra sphenanthera extracts
May increase tacrolimus whole blood trough concentrations and increase the risk of serious adverse reactions (e.g., neurotoxicity, QT prolongation) [see Warnings and Precautions ( 5.6, 5.11, 5.12)]. Reduce tacrolimus dose (for voriconazole and posaconazole, give one-third of the original dose) and adjust dose based on tacrolimus whole blood trough concentrations [see Dosage and Administration ( 2.2, 2.6) and Clinical Pharmacology ( 12.3)]. Mild or Moderate CYP3A Inhibitors:
Clotrimazole, antibiotics (e.g., erythromycin, fluconazole), calcium channel blockers (e.g., verapamil, diltiazem, nifedipine, nicardipine), amiodarone, danazol, ethinyl estradiol, cimetidine, lansoprazole and omeprazole
May increase tacrolimus whole blood trough concentrations and increase the risk of serious adverse reactions (e.g., neurotoxicity, QT prolongation) [see Warnings and Precautions ( 5.6, 5.11, 5.12)]. Monitor tacrolimus whole blood trough concentrations and reduce tacrolimus dose if needed [see Dosage andAdministration ( 2.2, 2.6) and Clinical Pharmacology ( 12.3)]. Other drugs, such as:
Magnesium and aluminum hydroxideantacids Metoclopramide
May increase tacrolimus whole blood trough concentrations and increase the risk of serious adverse reactions (e.g., neurotoxicity, QT prolongation) [see Warnings and Precautions ( 5.6, 5.11, 5.12)]. Monitor tacrolimus whole blood trough concentrations and reduce tacrolimus dose if needed [see Dosage and Administration ( 2.2, 2.6) and Clinical Pharmacology ( 12.3)]. Mild or Moderate CYP3A Inducers Methylprednisolone, prednisone May decrease tacrolimus concentrations Monitor tacrolimus whole blood trough concentrations and adjust tacrolimus dose if needed [see Dosage and Administration ( 2.2, 2.6)]. * Tacrolimus dosage adjustment recommendation based on observed effect of coadministered drug on tacrolimus exposures [see Clinical Pharmacology ( 12.3)] , literature reports of altered tacrolimus exposures, or the other drug's known CYP3A inhibitor/inducer status.
† High dose or double strength grapefruit juice is a strong CYP3A inhibitor; low dose or single strength grapefruit juice is a moderate CYP3A inhibitor.
# Strong CYP3A inhibitor/inducer, based on reported effect on exposures to tacrolimus along with supporting in vitro CYP3A inhibitor/inducer data, or based on drug-drug interaction studies with midazolam (sensitive CYP3A probe substrate).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy registry that monitors pregnancy outcomes in women exposed to tacrolimus during pregnancy. The Transplantation Pregnancy Registry International (TPRI) is a voluntary pregnancy exposure registry that monitors outcomes of pregnancy in female transplant recipients and those fathered by male transplant recipients exposed to immunosuppressants including tacrolimus. Healthcare providers are encouraged to advise their patients to register by contacting the Transplantation Pregnancy Registry International at 1-877-955-6877 or https://www.transplantpregnancyregistry.org/.
Risk Summary
Tacrolimus can cause fetal harm when administered to a pregnant woman. Data from postmarketing surveillance and TPRI suggest that infants exposed to tacrolimus in utero are at a risk of prematurity, birth defects/congenital anomalies, low birth weight, and fetal distress [see Human Data]. Advise pregnant women of the potential risk to the fetus. Administration of oral tacrolimus to pregnant rabbits and rats throughout the period of organogenesis was associated with maternal toxicity/lethality, and an increased incidence of abortion, malformation and embryofetal death at clinically relevant doses (0.5 to 6.9 times the recommended clinical dose range [0.2 to 0.075 mg/kg/day], on a mg/m 2 basis). Administration of oral tacrolimus to pregnant rats after organogenesis and throughout lactation produced maternal toxicity, effects on parturition, reduced pup viability and reduced pup weight at clinically relevant doses (0.8 to 6.9 times the recommended clinical dose range, on a mg/m 2 basis). Administration of oral tacrolimus to rats prior to mating, and throughout gestation and lactation produced maternal toxicity/lethality, marked effects on parturition, embryofetal loss, malformations, and reduced pup viability at clinically relevant doses (0.8 to 6.9 times the recommended clinical dose range, on a mg/m 2 basis). Interventricular septal defects, hydronephrosis, craniofacial malformations and skeletal effects were observed in offspring that died [see Animal Data].
The background risk of major birth defects and miscarriage in the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo-Fetal Risk
Risks during pregnancy are increased in organ transplant recipients.
The risk of premature delivery following transplantation is increased. Pre-existing hypertension and diabetes confer additional risk to the pregnancy of an organ transplant recipient. Pre-gestational and gestational diabetes are associated with birth defects/congenital anomalies, hypertension, low birth weight and fetal death.
Cholestasis of pregnancy (COP) was reported in 7% of liver or liver-kidney (LK) transplant recipients, compared with approximately 1% of pregnancies in the general population. However, COP symptoms resolved postpartum and no long term effects on the offspring were reported.
Maternal Adverse Reactions
Tacrolimus may increase hyperglycemia in pregnant women with diabetes (including gestational diabetes). Monitor maternal blood glucose levels regularly [see Warnings and Precautions ( 5.4)].
Tacrolimus may exacerbate hypertension in pregnant women and increase pre-eclampsia. Monitor and control blood pressure [see Warnings and Precautions ( 5.7, 5.8)].
Fetal/Neonatal Adverse Reactions
Renal dysfunction, transient neonatal hyperkalemia and low birth weight have been reported at the time of delivery in infants of mothers taking Tacrolimus.
Labor or Delivery
There is an increased risk for premature delivery (< 37 weeks) following transplantation and maternal exposure to Tacrolimus
Data
Human Data
There are no adequate and well controlled studies on the effects of tacrolimus in human pregnancy.Safety data from the TPRI and postmarketing surveillance suggest infants exposed to tacrolimus in utero have an increased risk for miscarriage, pre-term delivery (< 37 weeks), low birth weight (< 2500 g), birth defects/congenital anomalies and fetal distress.
TPRI reported 450 and 241 total pregnancies in kidney and liver transplant recipients exposed to tacrolimus, respectively. The TPRI pregnancy outcomes are summarized in Table 16. In the table below, the number of recipients exposed to tacrolimus concomitantly with mycophenolic acid (MPA) products during the preconception and first trimester periods is high (27% and 29% for renal and liver transplant recipients, respectively). Because MPA products may also cause birth defects, the birth defect rate may be confounded and this should be taken into consideration when reviewing the data, particularly for birth defects. Birth defects observed include cardiac malformations, craniofacial malformations, renal/urogenital disorders, skeletal abnormalities, neurological abnormalities and multiple malformations.
Table 16. TPRI Reported Pregnancy Outcomes in Transplant Recipients with Exposure to Tacrolimus
Kidney Liver Pregnancy Outcomes* 462 253 Miscarriage 24.50% 25% Live births 331 180 Pre-term delivery (< 37 weeks) 49% 42% Low birth weight (< 2500 g) 42% 30% Birth defects † 8% † 5% * Includes multiple births and terminations.
† Birth defect rate confounded by concomitant MPA products exposure in over half of offspring with birth defects.
Additional information reported by TPRI in pregnant transplant patients receiving tacrolimus included diabetes during pregnancy in 9% of kidney recipients and 13% of liver recipients and hypertension during pregnancy in 53% of kidney recipients and 16.2% of liver recipients.
Animal Data
Administration of oral tacrolimus to pregnant rabbits throughout organogenesis produced maternal toxicity and abortion at 0.32 mg/kg (0.5 to 1.4 times the recommended clinical dose range [0.2 to 0.075 mg/kg/day], on a mg/m 2 basis). At 1 mg/kg (1.6 to 4.3 times the recommended clinical dose range), embryofetal lethality and fetal malformations (ventricular hypoplasia, interventricular septal defect, bulbous aortic arch, stenosis of ductus arteriosus, omphalocele, gallbladder agenesis, skeletal anomalies) were observed. Administration of 3.2 mg/kg oral tacrolimus (2.6 to 6.9 times the recommended clinical dose range) to pregnant rats throughout organogenesis produced maternal toxicity/lethality, embryofetal lethality and decreased fetal body weight in the offspring of C-sectioned dams; and decreased pup viability and interventricular septal defect in offspring of dams that delivered.
In a peri-/postnatal development study, oral administration of tacrolimus to pregnant rats during late gestation (after organogenesis) and throughout lactation produced maternal toxicity, effects on parturition, and reduced pup viability at 3.2 mg/kg (2.6 to 6.9 times the recommended clinical dose range); among these pups that died early, an increased incidence of kidney hydronephrosis was observed. Reduced pup weight was observed at 1.0 mg/kg (0.8 to 2.2 times the recommended clinical dose range).
Administration of oral tacrolimus to rats prior to mating, and throughout gestation and lactation produced maternal toxicity/lethality, embryofetal loss and reduced pup viability at 3.2 mg/kg (2.6 to 6.9 times the recommended clinical dose range). Interventricular septal defects, hydronephrosis, craniofacial malformations and skeletal effects were observed in offspring that died. Effects on parturition (incomplete delivery of nonviable pups) were observed at 1 mg/kg (0.8 to 2.2 times the recommended clinical dose range) [see Nonclinical Toxicology ( 13.1)].
8.2 Lactation
Risk Summary
Controlled lactation studies have not been conducted in humans; however tacrolimus has been reported to be present in human milk. The effects of tacrolimus on the breastfed infant, or on milk production have not been assessed. Tacrolimus is excreted in rat milk and in peri-/postnatal rat studies, exposure to tacrolimus during the postnatal period was associated with developmental toxicity in the offspring at clinically relevant doses [see Pregnancy ( 8.1), Nonclinical Toxicology ( 13.1)].
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for tacrolimus and any potential adverse effects on the breastfed child from tacrolimus or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Tacrolimus can cause fetal harm when administered to pregnant women. Advise female and male patients of reproductive potential to speak to their healthcare provider on family planning options including appropriate contraception prior to starting treatment with tacrolimus [see Use in Specific Populations ( 8.1), Nonclinical Toxicology ( 13.1)].Infertility
Based on findings in animals, male and female fertility may be compromised by treatment with tacrolimus [see Nonclinical Toxicology ( 13.1)].
8.4 Pediatric Use
Safety and effectiveness have been established in pediatric liver, kidney, and heart transplant patients.
Liver transplant:
Safety and efficacy using tacrolimus Granules in pediatric de novo liver transplant patients less than 16 years of age are based on evidence from active controlled studies that included 56 pediatric patients, 31 of which received tacrolimus and supported by two pharmacokinetic and safety studies in 151 children who received tacrolimus.
Additionally, 122 pediatric patients were studied in an uncontrolled trial of tacrolimus in living related donor liver transplantation. Dose adjustments were made in the PK studies based on clinical status and whole blood concentrations. Pediatric patients generally required higher doses of tacrolimus to maintain blood trough concentrations of tacrolimus similar to adult patients [see Dosage and Administration ( 2.3), Adverse Reactions ( 6.1), Clinical Pharmacology ( 12.3) and Clinical Studies ( 14.2)].
Kidney and heart transplant:
Use of Tacrolimus capsules and tacrolimus Granules in pediatric kidney and heart transplant patients is supported by adequate and well-controlled studies and pharmacokinetic data in adult kidney and heart transplant patients with additional pharmacokinetic data in pediatric kidney and heart transplant patients and safety data in pediatric liver transplant patients [see Dosage and Administration ( 2.3) and Clinical Pharmacology ( 12.3)].
8.5 Geriatric Use
Clinical trials of tacrolimus did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Use in Renal Impairment
The pharmacokinetics of tacrolimus in patients with renal impairment was similar to that in healthy volunteers with normal renal function. However, consideration should be given to dosing tacrolimus at the lower end of the therapeutic dosing range in patients who have received a liver or heart transplant and have pre-existing renal impairment. Further reductions in dose below the targeted range may be required [see Dosage and Administration ( 2.4) and Clinical Pharmacology ( 12.3)].
8.7 Hepatic Impairment
The mean clearance of tacrolimus was substantially lower in patients with severe hepatic impairment (mean Child-Pugh score: > 10) compared to healthy volunteers with normal hepatic function. Close monitoring of tacrolimus trough concentrations is warranted in patients with hepatic impairment [see Clinical Pharmacology ( 12.3)].
The use of tacrolimus in liver transplant recipients experiencing post-transplant hepatic impairment may be associated with increased risk of developing renal insufficiency related to high whole blood trough concentrations of tacrolimus. These patients should be monitored closely and dosage adjustments should be considered. Some evidence suggests that lower doses should be used in these patients [see Dosage and Administration ( 2.5) and Clinical Pharmacology ( 12.3)].
8.8 Race or Ethnicity
African-American patients may need to be titrated to higher dosages to attain comparable trough concentrations compared to Caucasian patients [see Dosage and Administration ( 2.2) and Clinical Pharmacology ( 12.3)].
African-American and Hispanic patients are at increased risk for new onset diabetes after transplant. Monitor blood glucose concentrations and treat appropriately [see Warnings and Precautions ( 5.4)].
-
10 OVERDOSAGE
Limited overdosage experience is available. Acute overdosages of up to 30 times the intended dose have been reported. Almost all cases have been asymptomatic and all patients recovered with no sequelae. Acute overdosage was sometimes followed by adverse reactions consistent with those listed in Adverse Reactions ( 6) (including tremors, abnormal renal function, hypertension, and peripheral edema); in one case of acute overdosage, transient urticaria and lethargy were observed. Based on the poor aqueous solubility and extensive erythrocyte and plasma protein binding, it is anticipated that tacrolimus is not dialyzable to any significant extent; there is no experience with charcoal hemoperfusion. The oral use of activated charcoal has been reported in treating acute overdoses, but experience has not been sufficient to warrant recommending its use. General supportive measures and treatment of specific symptoms should be followed in all cases of overdosage.
In acute oral and IV toxicity studies, mortalities were seen at or above the following doses: in adult rats, 52 times the recommended human oral dose; in immature rats, 16 times the recommended oral dose; and in adult rats, 16 times the recommended human IV dose (all based on body surface area corrections).
-
11 DESCRIPTION
Tacrolimus, previously known as FK506, is the active ingredient in Tacrolimus Capsules. Tacrolimus is a calcineurin-inhibitor immunosuppressant produced by Streptomyces tsukubaensis. Chemically, tacrolimus is designated as [3S [3R*[E(1S*, 3S*,4S*)],4S*,5R*,8S*,9E,12R*,14R*,15S*,16R*,18S*,19S*,26aR*]] 5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[2-(4-hydroxy-3-methoxycyclohexyl)-1methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone, monohydrate.
The chemical structure of tacrolimus USP is:
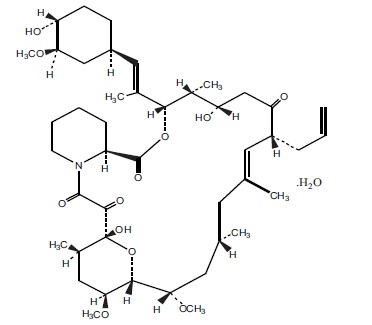
Tacrolimus USP has an empirical formula of C 44H 69NO 12H 2O and a formula weight of 822.03. Tacrolimus appears as white to off white powder. It is soluble in acetone, chloroform, ethyl acetate and insoluble in water. Tacrolimus Capsules, USP are available for oral administration as capsules (tacrolimus capsules) containing the equivalent of 0.5 mg, 1 mg or 5 mg of tacrolimus USP. Inactive ingredients include croscarmellose sodium NF, hypromellose USP, lactose anhydrous USNF, and magnesium stearate USNF. The 0.5 mg capsule shell contains ferric oxide, gelatin and titanium dioxide. The 1 mg capsule shell contains gelatin and titanium dioxide . The 5 mg capsule shell contains ferric oxide, gelatin, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tacrolimus binds to an intracellular protein, FKBP-12. A complex of tacrolimus-FKBP-12, calcium, calmodulin, and calcineurin (a ubiquitous mammalian intracellular enzyme) is then formed, after which the phosphatase activity of calcineurin inhibited. Such inhibition prevents the dephosphorylation and translocation of various factors such as the nuclear factor of activated T-cells (NF-AT), and nuclear factor kappa-light-chain enhancer of activated B-cells (NF-κB).
Tacrolimus inhibits the expression and/or production of several cytokines that include interleukin (IL)-1 beta, IL-2, IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, gamma interferon, tumor necrosis factor-alpha, and granulocyte macrophage colony stimulating factor. Tacrolimus also inhibits IL-2 receptor expression and nitric oxide release, induces apoptosis and production of transforming growth factor beta that can lead to immunosuppressive activity. The net result is the inhibition of T-lymphocyte activation and proliferation as well as T-helper-cell-dependent B-cell response (i.e., immunosuppression).
12.3 Pharmacokinetics
Tacrolimus activity is primarily due to the parent drug. The pharmacokinetic parameters (mean ± S.D.) of tacrolimus have been determined following intravenous (IV) and/or oral (PO) administration in healthy volunteers, and in kidney transplant, liver transplant, and heart transplant patients ( Table 17).
Table 17. Pharmacokinetics Parameters (mean ± S.D.) of Tacrolimus in Healthy Volunteers and Patients
Population N Route (Dose) Parameters C max (ng/mL) T max (hr) AUC (ng•hr/mL) t 1/2 (hr) CI (L/hr/kg) V (L/kg) Healthy Volunteers 8 IV (0.025 mg/kg/4 hr) * * 652 † ± 156 34.2 ± 7.7 0.040 ± 0.009 1.91 ± 0.31 30 PO (5 mg) (granules) 35.6 ± 10.9 1.3 ± 0.5 320 † ± 164 32.1 ± 5.9 ‡ ‡ PO (5 mg) (capsules) 28.8 ± 8.9 1.5 ± 0.7 266 † ± 95 32.3 ± 8.8 ‡ ‡ Kidney Transplant Patients 26 IV (0.02 mg/kg/12 hr) * * 294 † ± 262 18.8 ± 16.7 0.083 ± 0.05 1.41 ± 0.66 PO (0.2 mg/kg/day) 19.2 ± 10.3 3 203 † ± 42 ‡ ‡ ‡ PO (0.3 mg/kg/day) 24.2 ± 15.8 1.5 288 † ± 93 ‡ ‡ ‡ Liver Transplant Patients 17 IV (0.05 mg/kg/12 hr) * * 3300 † ± 2130 11.7 ± 3.9 0.053 ± 0.017 0.85 ± .30 PO (0.3 mg/kg/day) 68.5 ± 30.0 2.3 ± 1.5 519 † ± 179 ‡ ‡ ‡ Heart Transplant Patients 11 IV (0.01 mg/kg/day as a continuous infusion) * * 954 § ± 334 23.6 ± 9.22 0.051 ± 0.015 ‡ 11 PO (0.075 mg/kg/day)¶ 14.7 ± 7.79 2.1 [0.5-6.0] # 82.7 Þ ± 63.2 * ‡ ‡ 14 PO (0.15 mg/kg/day)¶ 24.5 ± 13.7 1.5 [0.4-4.0] # 142 Þ ± 116 * ‡ ‡ * not applicable
† AUC0-inf
‡ not available
§ AUC0-t
¶ Determined after the first dose
# Median [range]
Þ AUC0-12Due to intersubject variability in tacrolimus pharmacokinetics, individualization of dosing regimen is necessary for optimal therapy [see Dosage and Administration ( 2.6)] . Pharmacokinetic data indicate that whole blood concentrations rather than plasma concentrations serve as the more appropriate sampling compartment to describe tacrolimus pharmacokinetics.
Absorption
Absorption of tacrolimus from the gastrointestinal tract after oral administration is incomplete and variable. The absolute bioavailability of tacrolimus was 17 ± 10% in adult kidney transplant patients (N = 26), 22 ± 6% in adult liver transplant patients (N = 17), 23 ± 9% in adult heart transplant patients (N = 11) and 18 ± 5% in healthy volunteers (N = 16).
A single dose trial conducted in 32 healthy volunteers established the bioequivalence of the 1 mg and 5 mg capsules. Another single dose trial in 32 healthy volunteers established the bioequivalence of the 0.5 mg and 1 mg capsules. Tacrolimus maximum blood concentrations (C max) and area under the curve (AUC) appeared to increase in a dose- proportional fashion in 18 fasted healthy volunteers receiving a single oral dose of 3, 7, and 10 mg.
In 18 kidney transplant patients, tacrolimus trough concentrations from 3 to 30 ng/mL measured at 10-12 hours post-dose (C min) correlated well with the AUC (correlation coefficient 0.93). In 24 liver transplant patients over a concentration range of 10 to 60 ng/mL, the correlation coefficient was 0.94. In 25 heart transplant patients over a concentration range of 2 to 24 ng/mL, the correlation coefficient was 0.89 after an oral dose of 0.075 or 0.15 mg/kg/day at steady-state.
In a healthy volunteer adult study, the systemic exposure to tacrolimus (AUC) for tacrolimus Granules was approximately 16% higher than that for tacrolimus capsules when administered as single doses. If pediatric patients are converted between formulations, therapeutic drug monitoring must be performed and dose adjustments made to ensure that systemic exposure to tacrolimus is maintained.
Food Effects
The rate and extent of tacrolimus absorption were greatest under fasted conditions. The presence and composition of food decreased both the rate and extent of tacrolimus absorption when administered to 15 healthy volunteers.
The effect was most pronounced with a high-fat meal (848 kcal, 46% fat): mean AUC and C max were decreased 37% and 77%, respectively; T max was lengthened 5-fold. A high-carbohydrate meal (668 kcal, 85% carbohydrate) decreased mean AUC and mean C max by 28% and 65%, respectively.
In healthy volunteers (N = 16), the time of the meal also affected tacrolimus bioavailability. When given immediately following the meal, mean C max was reduced 71%, and mean AUC was reduced 39%, relative to the fasted condition.
When administered 1.5 hours following the meal, mean C max was reduced 63%, and mean AUC was reduced 39%, relative to the fasted condition.
In 11 liver transplant patients, tacrolimus administered 15 minutes after a high fat (400 kcal, 34% fat) breakfast, resulted in decreased AUC (27 ± 18%) and C max (50 ± 19%), as compared to a fasted state.
Tacrolimus capsules, USP should be taken consistently every day either with or without food because the presence and composition of food decreases the bioavailability of tacrolimus [see Dosage and Administration ( 2.1)].
Distribution
The plasma protein binding of tacrolimus is approximately 99% and is independent of concentration over a range of 5-50 ng/mL. Tacrolimus is bound mainly to albumin and alpha-1-acid glycoprotein, and has a high level of association with erythrocytes. The distribution of tacrolimus between whole blood and plasma depends on several factors, such as hematocrit, temperature at the time of plasma separation, drug concentration, and plasma protein concentration. In a U.S. trial, the ratio of whole blood concentration to plasma concentration averaged 35 (range 12 to 67).
Elimination
Metabolism
Tacrolimus is extensively metabolized by the mixed-function oxidase system, primarily the cytochrome P-450 system (CYP3A). A metabolic pathway leading to the formation of 8 possible metabolites has been proposed. Demethylation and hydroxylation were identified as the primary mechanisms of biotransformation in vitro. The major metabolite identified in incubations with human liver microsomes is 13-demethyl tacrolimus. In in vitro studies, a 31-demethyl metabolite has been reported to have the same activity as tacrolimus.
Excretion
The mean clearance following IV administration of tacrolimus is 0.040, 0.083, and 0.053, and 0.051 L/hr/kg in healthy volunteers, adult kidney transplant patients, adult liver transplant patients, and adult heart transplant patients, respectively. In man, less than 1% of the dose administered is excreted unchanged in urine.
In a mass balance study of IV-administered radiolabeled tacrolimus to 6 healthy volunteers, the mean recovery of radiolabel was 77.8 ± 12.7%. Fecal elimination accounted for 92.4 ± 1.0% and the elimination half-life based on radioactivity was 48.1 ± 15.9 hours whereas it was 43.5 ± 11.6 hours based on tacrolimus concentrations. The mean clearance of radiolabel was 0.029 ± 0.015 L/hr/kg and clearance of tacrolimus was 0.029 ± 0.009 L/hr/kg. When administered PO, the mean recovery of the radiolabel was 94.9 ± 30.7%. Fecal elimination accounted for 92.6 ± 30.7%, urinary elimination accounted for 2.3 ± 1.1% and the elimination half-life based on radioactivity was 31.9 ± 10.5 hours whereas it was 48.4 ± 12.3 hours based on tacrolimus concentrations. The mean clearance of radiolabel was 0.226 ± 0.116 L/hr/kg and clearance of tacrolimus 0.172 ± 0.088 L/hr/kg.
Specific Populations
Pediatric Patients
Tacrolimus Pharmacokinetics in Pediatric Patients
Pharmacokinetics of tacrolimus have been studied in liver transplantation patients, 0.7 to 13.2 years of age. Following IV administration of a 0.037 mg/kg/day dose to 12 pediatric patients, mean terminal half-life, volume of distribution and clearance were 11.5 ± 3.8 hours, 2.6 ± 2.1 L/kg and 0.138 ± 0.071 L/hr/kg, respectively. Following oral administration to 9 patients, mean AUC and Cmax were 337 ± 167 ng·hr/mL and 48.4 ± 27.9 ng/mL, respectively. The absolute bioavailability was 31 ± 24%.
Pharmacokinetics of tacrolimus have also been studied in kidney transplantation patients, 8.2 ± 2.4 years of age. Following IV infusion of a 0.06 mg/kg/day to 12 pediatric patients (8 male and 4 female), mean terminal half-life and clearance were 10.2 ± 5.0 hours and 0.12 ± 0.04 L/hr/kg, respectively. Following oral administration to the same patients, mean AUC and C max were 181 ± 65 ng·hr/mL and 30 ± 11 ng/mL, respectively. The absolute bioavailability was 19 ± 14%.
Whole blood trough concentrations from 31 patients less than 12 years old showed that pediatric patients needed higher doses than adults to achieve similar tacrolimus trough concentrations [see Dosage and Administration ( 2.3)].
Tacrolimus Granules Pharmacokinetics in Pediatric Patients
A multicenter, open-label, single arm, pharmacokinetic study (OPTION, NCT01371331) was conducted using tacrolimus granules for oral suspension in pediatric patients undergoing de novo liver, kidney, or heart transplant. After an initial 24 hour continuous IV infusion of tacrolimus (0.025 mg/kg/hour) for 12 hours to 4 days, oral Tacrolimus Granules were dosed at 0.3 mg/kg/day in divided doses twice daily. Tacrolimus whole blood trough concentrations ranged from 5-15 ng/mL for the first month post-transplant, and 5-10 ng/mL thereafter. Two pharmacokinetic (PK) profiles, AUC, C max, T max and C trough, were taken after the first oral dose (Day 1) and at steady state (Day 7). Subsequent oral doses of Tacrolimus Granules were adjusted based on clinical evidence of efficacy, the whole-blood trough levels, and/or occurrence of adverse events. Of 52 patients enrolled, thirty-eight (38) had evaluable PK profile. The mean pediatric age was 6.1 years for heart transplant, 1.1 years for liver transplant and 3.6 years for kidney transplant. Summary results of PK parameters are presented in Table 18.
Table 18. Summary of Whole Blood PK Parameters of Tacrolimus after Administration of Tacrolimus Granules in Pediatric Patients
Population N (age range) Parameters AUC tau [hr*ng/mL]
mean ± SDC max [ng/mL]
mean ± SDT max [hr]
mean ± S DCtrough [ng/mL]
mean ± SDHeart Transplant Patients 12 (0.58-13 years) Day 1
Day 7224.13 ± 114.30
165.17 ± 39.1245.61 ± 19.55
32.69 ± 9.782.95 ± 4.33
0.84 ± 0.4412.60 ± 13.40
7.57 ± 1.80Liver Transplant Patients 14 (0.33-12 years) Day 1
Day 7210.56 ± 84.01
195.08 ± 94.6325.11 ± 10.78
30.52 ± 19.352.73 ± 1.84
1.71 ± 1.1213.41 ± 7.11
9.71 ± 4.03Kidney Transplant Patients 12 (2.42-11 years) Day 1
Day 797.40 ± 36.77
208.32 ± 68.7518.04 ± 8.10
36.63 ± 13.971.78 ± 0.88
1.09 ± 0.613.54 ± 1.45
8.92 ± 3.59Renal and Hepatic Impaired Patients
The mean pharmacokinetic parameters for tacrolimus following single administrations to adult patients with renal and hepatic impairment are given in Table 19.
Table 19. Pharmacokinetics in Renal and Hepatic Impaired Adult Patients
Population (No. of Patients) Dose AUC 0-t (ng-hr/mL) t 1/2 (hr) V (L/kg) CI (L/hr/kg) Renal Impairment (n=12) 0.02 mg/kg/4hr IV 393±123 (t=60 hr) 26.3±9.2 1.07±0.20 0.038±0.014 Mild Hepatic Impairment (n=6) 0.02 mg/kg/4hr IV 367±107 (t=72 hr) 60.6±43.8 Range: 27.8-141 3.1±1.6 0.042±0.02 7.7 mg PO 488±320 (t=72 hr) 66.1±44.8 Range: 29.5-138 3.7±4.7* 0.034±0.019* Severe Hepatic Impairment (n=6, IV) 0.02 mg/kg/4hr IV (n=2) 762±204 (t=120hr) 198±158 Range: 81-436 3.9±1.0 0.017±0.013 0.01 mg/kg/8hr IV (n=4) 289±117 (t=144 hr) (n=5, PO)† 8 mg PO (n=1) 658 (t=120 hr) 119±35 Range: 85-178 3.1±3.4* 0.016±0.011* 5 mg PO (n=4) 533±156 (t=144 hr) 4 mg PO (n=1) *corrected for bioavailability
†1 patient did not receive the PO dose
Patients with Renal Impairment
Tacrolimus pharmacokinetics following a single IV administration were determined in 12 patients (7 not on dialysis and 5 on dialysis, serum creatinine of 3.9 ± 1.6 and 12.0 ± 2.4 mg/dL, respectively) prior to their kidney transplant. The pharmacokinetic parameters obtained were similar for both groups. The mean clearance of tacrolimus in patients with renal dysfunction was similar to that in normal volunteers (Table 19) [see Dosage and Administration (2.2) and Use in Specific Populations ( 8.6)].
Patients with Hepatic Impairment
Tacrolimus pharmacokinetics have been determined in six patients with mild hepatic dysfunction (mean Pugh score: 6.2) following single IV and oral administrations. The mean clearance of tacrolimus in patients with mild hepatic dysfunction was not substantially different from that in normal volunteers (see previous table). Tacrolimus pharmacokinetics were studied in 6 patients with severe hepatic dysfunction (mean Pugh score: > 10). The mean clearance was substantially lower in patients with severe hepatic dysfunction, irrespective of the route of administration [see Dosage and Administration ( 2.5) and Use in Specific Populations (8.7)].
Racial or Ethnic Groups
The pharmacokinetics of tacrolimus have been studied following single IV and oral administration of tacrolimus to 10 African-American, 12 Latino-American, and 12 Caucasian healthy volunteers. There were no significant pharmacokinetic differences among the three ethnic groups following a 4-hour IV infusion of 0.015 mg/kg. However, after single oral administration of 5 mg, mean (± SD) tacrolimus C max in African-Americans (23.6 ± 12.1 ng/mL) was significantly lower than in Caucasians (40.2 ± 12.6 ng/mL) and the Latino-Americans (36.2 ± 15.8 ng/mL) (p < 0.01). Mean AUC 0-inf tended to be lower in African-Americans (203 ± 115 ng·hr/mL) than Caucasians (344 ± 186 ng·hr/mL) and Latino-Americans (274 ± 150 ng·hr/mL). The mean (± SD) absolute oral bioavailability (F) in African-Americans (12 ± 4.5%) and Latino-Americans (14 ± 7.4%) was significantly lower than in Caucasians (19 ± 5.8%, p = 0.011). There was no significant difference in mean terminal T1/2 among the three ethnic groups (range from approximately 25 to 30 hours). A retrospective comparison of African-American and Caucasian kidney transplant patients indicated that African-American patients required higher tacrolimus doses to attain similar trough concentrations [see Dosage and Administration ( 2.2)].
Male and Female Patients
A formal trial to evaluate the effect of gender on tacrolimus pharmacokinetics has not been conducted, however, there was no difference in dosing by gender in the kidney transplant trial. A retrospective comparison of pharmacokinetics in healthy volunteers, and in kidney, liver, and heart transplant patients indicated no gender-based differences.
Drug Interaction Studies
Frequent monitoring of whole blood concentrations and appropriate dosage adjustments of tacrolimus are recommended when concomitant use of the following drugs with tacrolimus is initiated or discontinued [see Drug Interactions ( 7)].
- Telaprevir: In a single-dose study in 9 healthy volunteers, co-administration of tacrolimus (0.5 mg single dose) with telaprevir (750 mg three times daily for 13 days) increased the tacrolimus dose-normalized C max by 9.3-fold and AUC by 70-fold compared to tacrolimus alone [see Drug Interactions ( 7.2)].
- Boceprevir: In a single-dose study in 12 subjects, co-administration of tacrolimus (0.5 mg single dose) with boceprevir (800 mg three times daily for 11 days) increased tacrolimus C max by 9.9-fold and AUC by 17-fold compared to tacrolimus alone [see Drug Interactions ( 7.2)].
- Nelfinavir: Based on a clinical study of 5 liver transplant recipients, co-administration of tacrolimus with nelfinavir increased blood concentrations of tacrolimus significantly and, as a result, a reduction in the tacrolimus dose by an average of 16-fold was needed to maintain mean trough tacrolimus blood concentrations of 9.7 ng/mL. It is recommended to avoid concomitant use of Tacrolimus capsules, USP and nelfinavir unless the benefits outweigh the risks [see Drug Interactions ( 7.2)].
- Rifampin: In a study of 6 normal volunteers, a significant decrease in tacrolimus oral bioavailability (14 ± 6% vs.7 ± 3%) was observed with concomitant rifampin administration (600 mg). In addition, there was a significant increase in tacrolimus clearance (0.036 ± 0.008 L/hr/kg vs. 0.053 ± 0.010 L/hr/kg) with concomitant rifampin administration [see Drug Interactions ( 7.2)].
- Magnesium and Aluminum-hydroxide: In a single-dose crossover study in healthy volunteers, co-administration of tacrolimus and magnesium-aluminum-hydroxide resulted in a 21% increase in the mean tacrolimus AUC and a 10% decrease in the mean tacrolimus C max relative to tacrolimus administration alone [see Drug Interactions ( 7.2)].
- Ketoconazole: In a study of 6 normal volunteers, a significant increase in tacrolimus oral bioavailability (14 ± 5% vs. 30 ± 8%) was observed with concomitant ketoconazole administration (200 mg). The apparent oral clearance of tacrolimus during ketoconazole administration was significantly decreased compared to tacrolimus alone (0.430 ± 0.129 L/hr/kg vs. 0.148 ± 0.043 L/hr/kg). Overall, IV clearance of tacrolimus was not significantly changed by ketoconazole co-administration, although it was highly variable between patients [see Drug Interactions ( 7.2)].
- Voriconazole (see complete prescribing information for VFEND) : Repeat oral dose administration of voriconazole (400 mg every 12 hours for one day, then 200 mg every 12 hours for 6 days) increased tacrolimus (0.1 mg/kg single dose) C max and AUC τ in healthy subjects by an average of 2-fold (90% CI: 1.9, 2.5) and 3-fold (90% CI: 2.7, 3.8), respectively [see Drug Interactions ( 7.2)].
- Posaconazole (see complete prescribing information for Noxafil) : Repeat oral administration of posaconazole (400 mg twice daily for 7 days) increased tacrolimus (0.05 mg/kg single dose) C max and AUC in healthy subjects by an average of 2-fold (90% CI: 2.01, 2.42) and 4.5-fold (90% CI 4.03, 5.19), respectively [see Drug Interactions ( 7.2)].
- Caspofungin (see complete prescribing information for CANCIDAS) : Caspofungin reduced the blood AUC0-12 of tacrolimus by approximately 20%, peak blood concentration (C max) by 16%, and 12-hour blood concentration(C 12hr) by 26% in healthy adult subjects when tacrolimus (2 doses of 0.1 mg/kg 12 hours apart) was administered on the 10th day of CANCIDAS 70 mg daily, as compared to results from a control period in which tacrolimus was administered alone [see Drug Interactions ( 7.2)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies were conducted in male and female rats and mice. In the 80-week mouse oral study and in the 104-week rat oral study, no relationship of tumor incidence to tacrolimus dosage was found. The highest dose used in the mouse was 3.0 mg/kg/day (0.9 to 2.2 times the AUC at clinical doses of 0.075 to 0.2 mg/kg/day) and in the rat was 5.0 mg/kg/day (0.265 to 0.65 times the AUC at clinical doses of 0.075 to 0.2 mg/kg/day) [see Boxed Warning and Warnings and Precautions ( 5.1)] .
A 104-week dermal carcinogenicity study was performed in mice with tacrolimus ointment (0.03% - 3%), equivalent to tacrolimus doses of 1.1-118 mg/kg/day or 3.3-354 mg/m 2/day. In the study, the incidence of skin tumors was minimal and the topical application of tacrolimus was not associated with skin tumor formation under ambient room lighting. However, a statistically significant elevation in the incidence of pleomorphic lymphoma in high-dose male (25/50) and female animals (27/50) and in the incidence of undifferentiated lymphoma in high-dose female animals (13/50) was noted in the mouse dermal carcinogenicity study. Lymphomas were noted in the mouse dermal carcinogenicity study at a daily dose of 3.5 mg/kg (0.1% tacrolimus ointment). No drug-related tumors were noted in the mouse dermal carcinogenicity study at a daily dose of 1.1 mg/kg (0.03% tacrolimus ointment). The relevance of topical administration of tacrolimus in the setting of systemic tacrolimus use is unknown.
The implications of these carcinogenicity studies to the human condition are limited; doses of tacrolimus were administered that likely induced immunosuppression in these animals, impairing their immune system’s ability to inhibit unrelated carcinogenesis.
Mutagenesis
No evidence of genotoxicity was seen in bacterial ( Salmonella and E. coli) or mammalian (Chinese hamster lung-derived cells) in vitro assays of mutagenicity, the in vitro CHO/HGPRT assay of mutagenicity, or in vivo clastogenicity assays performed in mice; tacrolimus did not cause unscheduled DNA synthesis in rodent hepatocytes.
Impairment of Fertility
Tacrolimus subcutaneously administered to male rats at paternally toxic doses of 2 mg/kg/day (1.6 to 4.3 times the recommended clinical dose range [0.2 to 0.075 mg/kg/day] on a mg/m 2 basis) or 3 mg/kg/day (2.4 to 6.4 times the recommended clinical dose range) resulted in a dose-related decrease in sperm count. Tacrolimus administered orally at 1.0 mg/kg (0.8 to 2.2 times the clinical dose range) to male and female rats, prior to and during mating, as well as to dams during gestation and lactation, was associated with embryolethality and adverse effects on female reproduction. Effects on female reproductive function (parturition) and embryolethal effects were indicated by a higher rate of pre-and post- implantation loss and increased numbers of undelivered and nonviable pups. When administered at 3.2 mg/kg (2.6 to 6.9 times the clinical dose range based on body surface area), tacrolimus was associated with maternal and paternal toxicity as well as reproductive toxicity including marked adverse effects on estrus cycles, parturition, pup viability, and pup malformations.
-
14 CLINICAL STUDIES
14.1 Kidney Transplantation
Tacrolimus /Azathioprine (AZA)
Tacrolimus-based immunosuppression in conjunction with azathioprine and corticosteroids following kidney transplantation was assessed in a randomized, multicenter, non-blinded, prospective trial. There were 412 kidney transplant patients enrolled at 19 clinical sites in the United States. Study therapy was initiated when renal function was stable as indicated by a serum creatinine ≤ 4 mg/dL (median of 4 days after transplantation, range 1 to 14 days). Patients less than 6 years of age were excluded.
There were 205 patients randomized to tacrolimus-based immunosuppression and 207 patients were randomized to cyclosporine-based immunosuppression. All patients received prophylactic induction therapy consisting of an antilymphocyte antibody preparation, corticosteroids, and azathioprine. Overall 1-year patient and graft survival was 96.1% and 89.6%, respectively.
Data from this trial of tacrolimus in conjunction with azathioprine indicate that during the first 3 months of that trial, 80% of the patients maintained trough concentrations between 7-20 ng/mL, and then between 5-15 ng/mL, through 1 year.
Tacrolimus /Mycophenolate Mofetil (MMF)
Tacrolimus-based immunosuppression in conjunction with MMF, corticosteroids, and induction has been studied. In a randomized, open-label, multicenter trial (Study 1), 1589 kidney transplant patients received Tacrolimus (Group C, n = 401), sirolimus (Group D, n = 399), or one of two cyclosporine (CsA) regimens (Group A, n = 390 and Group B, n = 399) in combination with MMF and corticosteroids; all patients, except those in one of the two cyclosporine groups, also received induction with daclizumab. The trial was conducted outside the United States; the trial population was 93% Caucasian. In this trial, mortality at 12 months in patients receiving Tacrolimus/MMF was similar (3%) compared to patients receiving cyclosporine/MMF (3% and 2%) or sirolimus/MMF (3%). Patients in the Tacrolimus group exhibited higher estimated creatinine clearance rates (eCL cr) using the Cockcroft-Gault formula ( Table 20) and experienced fewer efficacy failures, defined as biopsy-proven acute rejection (BPAR), graft loss, death, and/or lost to follow-up ( Table 21) in comparison to each of the other three groups. Patients randomized to Tacrolimus/MMF were more likely to develop diarrhea and diabetes after the transplantation and experienced similar rates of infections compared to patients randomized to either cyclosporine/MMF regimen [see Adverse Reactions ( 6.1)].
Table 20. Estimated Creatinine Clearance at 12 Months (Study 1)
Group eCLcr[mL/min] at Month 12* N MEAN SD MEDIAN Treatment Difference with Group C (99.2%CI†) (A) CsA/MMF/CS 390 56.5 25.8 56.9 - 8.6 (-13.7, -3.7) (B) CsA/MMF/CS/Daclizumab 399 58.9 25.6 60.9 - 6.2 (-11.2, -1.2) (C) Tac/MMF/CS/Daclizumab 401 65.1 27.4 66.2 - (D) Siro/MMF/CS/Daclizumab 399 56.2 27.4 57.3 - 8.9 (-14.1, -3.9) Total 1589 59.2 26.8 60.5 Key: CsA=Cyclosporine, CS=Corticosteroids, Tac=Tacrolimus, Siro=Sirolimus * All death/graft loss (n = 41, 27, 23, and 42 in Groups A, B, C, and D) and patients whose last recorded creatinine values were prior to month 3 visit (n = 10, 9, 7, and 9 in Groups A, B, C, and D, respectively) were imputed with Glomerular Filtration Rate (GFR) of 10 mL/min; a subject's last observed creatinine value from month 3 on was used for the remainder of subjects with missing creatinine at month 12 (n = 11, 12, 15, and 19 for Groups A, B, C, and D, respectively). Weight was also imputed in the calculation of estimated GFR, if missing.
† Adjusted for multiple (6) pairwise comparisons using Bonferroni corrections.
Table 21. Incidence of BPAR, Graft Loss, Death, or Loss to Follow-up at 12 Months (Study 1)
Group A N=390 Group B N=399 Group C N=401 Group D N=399 Overall Failure 141 (36.2%) 126 (31.6%) 82 (20.4%) 185 (46.4%) Components of efficacy failure BPAR 113 (29.0%) 106 (26.6%) 60 (15.0%) 152 (38.1%) Graft loss excluding death 28 (7.2%) 20 (5.0%) 12 (3.0%) 30 (7.5%) Mortality 13 (3.3%) 7 (1.8%) 11 (2.7%) 12 (3.0%) Lost to follow-up 5 (1.3%) 7 (1.8%) 5 (1.3%) 6 (1.5%) Treatment Difference of efficacy failure compared to Group C 15.8% 11.2% - 26.00% (99.2% CI*) (7.1%, 24.3%) (2.7%, 19.5%) (17.2%, 34.7%) Key: Group A =CsA/MMF/CS, B =CsA/MMF/CS/Daclizumab, C=Tac/MMF/CS/Daclizumab, and D=Siro/MMF/CS/Daclizumab *Adjusted for multiple (6) pairwise comparisons using Bonferroni corrections.
The protocol-specified target tacrolimus trough concentrations (C trough,Tac) were 3-7 ng/mL; however, the observed median C troughs,Tac approximated 7 ng/mL throughout the 12-month trial ( Table 22). Approximately 80% of patients maintained tacrolimus whole blood concentrations between 4-11 ng/mL through 1 year post-transplant.
Table 22. Tacrolimus Whole Blood Trough Concentration Range (Study 1)
Time Median (P10-P90*) tacrolimus whole blood trough concentrations range (ng/mL) Day 30 (N=366) 6.9 (4.4 – 11.3) Day 90 (N=351) 6.8 (4.1 – 10.7) Day 180 (N=355) 6.5 (4.0 – 9.6) Day 365 (N=346) 6.5 (3.8 – 10.0) *10 to 90th Percentile: range of Ctrough,Tac that excludes lowest 10% and highest 10% of Ctrough,Tac
The protocol-specified target cyclosporine trough concentrations (C trough,CsA) for Group B were 50-100 ng/mL; however, the observed median C troughs,CsA approximated 100 ng/mL throughout the 12 month trial. The protocol-specified target C troughs,CsA for Group A were 150-300 ng/mL for the first 3 months and 100-200 ng/mL from month 4 to month 12; the observed median C troughs,CsA approximated 225 ng/mL for the first 3 months and 140 ng/mL from month 4 to month 12.
While patients in all groups started MMF at 1gram twice daily, the MMF dose was reduced to less than 2 g per day in 63% of patients in the tacrolimus treatment arm by month 12 ( Table 23); approximately 50% of these MMF dose reductions were due to adverse reactions. By comparison, the MMF dose was reduced to less than 2 g per day in 49% and 45% of patients in the two cyclosporine arms (Group A and Group B, respectively), by month 12 and approximately 40% of MMF dose reductions were due to adverse reactions.Table 23: MMF Dose Over Time in Tacrolimus/MMF (Group C) (Study 1)
Time period (Days) Time-averaged MMF dose (grams per day)* Less than 2.0 2 Greater than 2.0 0-30 (N=364) 37% 60% 2% 0-90 (N=373) 47% 51% 2% 0-180 (N=377) 56% 42% 2% 0-365 (N=380) 63% 36% 1% Key: Time-averaged MMF dose = (total MMF dose) / (duration of treatment) * Percentage of patients for each time-averaged MMF dose range during various treatment periods. Administration of 2 g per day of time-averaged MMF dose means that MMF dose was not reduced in those patients during the treatment periods.
In a second randomized, open-label, multicenter trial (Study 2), 424 kidney transplant patients received Tacrolimus (N = 212) or cyclosporine (N = 212) in combination with MMF 1 gram twice daily, basiliximab induction, and corticosteroids. In this trial, the rate for the combined endpoint of BPAR, graft failure, death, and/or lost to follow-up at 12 months in theTacrolimus /MMF group was similar to the rate in the cyclosporine/MMF group. There was, however, an imbalance in mortality at 12 months in those patients receiving Tacrolimus /MMF (4%) compared to those receiving cyclosporine/MMF (2%), including cases attributed to over-immunosuppression ( Table 24).
Table 24. Incidence of BPAR, Graft Loss, Death, or Loss to Follow-up at 12 Months (Study 2)
Tacrolimus /MMF (n=212) Cyclosporine/MMF (n=212) Overall Failure 32 (15.1%) 36 (17.0%) Components of efficacy failure BPAR 16 (7.5%) 29 (13.7%) Graft loss excluding death 6 (2.8%) 4 (1.9%) Mortality 9 (4.2%) 5 (2.4%) Lost to follow-up 4 (1.9%) 1 (0.5%) Treatment Difference of efficacy failure compared to Tacrolimus/MMF group (95% CI*) - 1.9% (-5.2%, 9.0%) * 95% confidence interval calculated using Fisher's Exact Test
The protocol-specified target tacrolimus whole blood trough concentrations (C trough,Tac) in Study 2 were 7-16 ng/mL for the first three months and 5-15 ng/mL thereafter. The observed median C trough,Tac approximated 10 ng/mL during the first three months and 8 ng/mL from month 4 to month 12 ( Table 25). Approximately 80% of patients maintained tacrolimus whole blood trough concentrations between 6 to 16 ng/mL during months 1 through 3 and, then, between 5 to 12 ng/mL from month 4 through 1 year.
Table 25. Tacrolimus Whole Blood Trough Concentration Range (Study 2)
Time Median (P10-P90*) tacrolimus whole blood trough concentrations range (ng/mL) Day 30 (N=174) 10.5 (6.3 - 16.8) Day 60 (N=179) 9.2 (5.9 - 15.3) Day 120 (N=176) 8.3 (4.6 - 13.3) Day 180 (N=171) 7.8 (5.5 - 13.2) Day 365 (N=178) 7.1 (4.2 - 12.4) *a) 10 to 90th Percentile: range of Ctrough,Tac that excludes lowest 10% and highest 10% of Ctrough,Tac
The protocol-specified target cyclosporine whole blood concentrations (C trough,CsA) were 125 to 400 ng/mL for the first three months, and 100 to 300 ng/mL thereafter. The observed median C troughs, C sA approximated 280 ng/mL during the first three months and 190 ng/mL from month 4 to month 12.
Patients in both groups started MMF at 1 gram twice daily. The MMF dose was reduced to less than 2 grams per day by month 12 in 62% of patients in the Tacrolimus/ MMF group (Table 26) and in 47% of patients in the cyclosporine/MMF group. Approximately 63% and 55% of these MMF dose reductions were because of adverse reactions in the Tacrolimus/MMF group and the cyclosporine/MMF group, respectively [see Adverse Reactions ( 6.1)].
Table 26. MMF Dose Over Time in the Tacrolimus/MMF Group (Study 2)
Time Period (Days) Time-averaged MMF dose (g/day)* Less than 2.0 2 Greater than 2.0 0-30 (N=212) 25% 69% 6% 0-90 (N=212) 41% 53% 6% 0-180 (N=212) 52% 41% 7% 0-365 (N=212) 62% 34% 4% Key: Time-averaged MMF dose = (total MMF dose) / (duration of treatment) * Percentage of patients for each time-averaged MMF dose range during various treatment periods. Two grams per day of time- averaged MMF dose means that MMF dose was not reduced in those patients during the treatment periods.
14.2 Liver Transplantation
The safety and efficacy of tacrolimus-based immunosuppression following orthotopic liver transplantation were assessed in two prospective, randomized, non-blinded multicenter trials. The active control groups were treated with a cyclosporine-based immunosuppressive regimen (CsA/AZA). Both trials used concomitant adrenal corticosteroids as part of the immunosuppressive regimens. These trials compared patient and graft survival rates at 12 months following transplantation.
In one trial, 529 patients were enrolled at 12 clinical sites in the United States; prior to surgery, 263 were randomized to the tacrolimus-based immunosuppressive regimen and 266 to the CsA/AZA. In 10 of the 12 sites, the same CsA/AZA protocol was used, while 2 sites used different control protocols. This trial excluded patients with renal dysfunction, fulminant hepatic failure with Stage IV encephalopathy, and cancers; pediatric patients (≤12 years old) were allowed.
In the second trial, 545 patients were enrolled at 8 clinical sites in Europe; prior to surgery, 270 were randomized to the tacrolimus-based immunosuppressive regimen and 275 to CsA/AZA. In this trial, each center used its local standard CsA/AZA protocol in the active-control arm. This trial excluded pediatric patients, but did allow enrollment of subjects with renal dysfunction, fulminant hepatic failure in Stage IV encephalopathy, and cancers other than primary hepatic with metastases.
One-year patient survival and graft survival in the tacrolimus-based treatment groups were similar to those in the CsA/AZA treatment groups in both trials.The overall 1-year patient survival (CsA/AZA and tacrolimus-based treatment groups combined) was 88% in the U.S. trial and 78% in the European trial. The overall 1-year graft survival (CsA/AZA and tacrolimus-based treatment groups combined) was 81% in the U.S. trial and 73% in the European trial. In both trials, the median time to convert from IV to oral tacrolimus capsules dosing was 2 days.
Although there is a lack of direct correlation between tacrolimus concentrations and drug efficacy, data from clinical trials of liver transplant patients have shown an increasing incidence of adverse reactions with increasing trough blood concentrations. Most patients are stable when trough whole blood concentrations are maintained between 5 to 20 ng/mL. Long-term post-transplant patients often are maintained at the low end of this target range. Data from the U.S. clinical trial show that the median trough blood concentrations, measured at intervals from the second week to one year post-transplantation ranged from 9.8 ng/mL to 19.4 ng/mL.
Pediatric Liver Transplantation Using Tacrolimus Granules
The efficacy and safety of Tacrolimus Granules plus corticosteroids were compared with a triple regimen of cyclosporine/corticosteroids/azathioprine in a randomized, open-label study, in de novo pediatric liver transplant patients. The study was conducted outside the United States and enrolled patients aged 16 years or younger. The distribution of pediatric patients by age was similar in both treatment groups, with a majority < 5 years. Patients were randomized to either tacrolimus for oral suspension 0.3 mg/kg/day (N = 91) or cyclosporine 10 mg/kg/day orally (N = 90) initiated 6 hours after completion of transplant surgery. Doses throughout the 1-year study period were adjusted to maintain whole blood trough levels within 5-20 ng/mL [see Dosage and Administration ( 2.3)] . Based on trough levels, doses of tacrolimus were adjusted to 0.17 mg/kg/day and 0.14 mg/kg/day by days 2 and 3, respectively. At 12 months, the incidence rate of BPAR, graft loss, death, or lost to follow-up was 52.7% in the tacrolimus group and 61.1% in the cyclosporine group ( Table 27).
Table 27. Key Efficacy Results at 12 Months in Pediatric Liver Transplant Recipients Receiving Tacrolimus Granules or Cyclosporine
Tacrolimus Granules (N = 91) Cyclosporine (N = 90) Overall Failure 48 (52.7%) 55 (61.1%) Components of efficacy failure BPAR 40 (44.0%) 49 (54.4%) Graft loss 7 (7.7%) 13 (14.4%) Graft loss excluding death 1 (1.1%) 6 (6.7%) Mortality 6 (6.6%) 7 (7.8%) Lost to follow-up 2 (2.2%) 0 Treatment Difference of efficacy failure compared to Cyclosporine (95% CI*) -8.4% (-22.7%, 6.0%) * 95% confidence interval calculated using normal approximation.
14.3 Heart Transplantation
Two open-label, randomized, comparative trials evaluated the safety and efficacy of tacrolimus-based and cyclosporine based immunosuppression in primary orthotopic heart transplantation. In a trial conducted in Europe, 314 patients received a regimen of antibody induction, corticosteroids, and azathioprine in combination with tacrolimus or cyclosporine modified for 18 months. In a 3-arm trial conducted in the US, 331 patients received corticosteroids and tacrolimus plus sirolimus, tacrolimus plus mycophenolate mofetil (MMF) or cyclosporine modified plus MMF for 1 year.
In the European trial, patient/graft survival at 18 months post-transplant was similar between treatment arms, 92% in the tacrolimus group and 90% in the cyclosporine group. In the U.S. trial, patient and graft survival at 12 months was similar with 93% survival in the tacrolimus plus MMF group and 86% survival in the cyclosporine modified plus MMF group. In the European trial, the cyclosporine trough concentrations were above the pre-defined target range (i.e., 100 to 200 ng/mL) at Day 122 and beyond in 32% to 68% of the patients in the cyclosporine treatment arm, whereas the tacrolimus trough concentrations were within the pre-defined target range (i.e., 5 to 15 ng/mL) in 74% to 86% of the patients in the tacrolimus treatment arm. Data from this European trial indicate that from 1 week to 3 months post-transplant, approximately 80% of patients maintained trough concentrations between 8 to 20 ng/mL and, from 3 months through 18 months post-transplant, approximately 80% of patients maintained trough concentrations between 6 to 18 ng/mL.
The U.S. trial contained a third arm of a combination regimen of sirolimus, 2 mg per day, and full-dose tacrolimus; however, this regimen was associated with increased risk of wound-healing complications, renal function impairment, and insulin-dependent post-transplant diabetes mellitus, and is not recommended [see Warnings and Precautions ( 5.10)].
- REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Tacrolimus Capsules, USP
Tacrolimus Capsules, USP, 0.5 mg: Light yellow color, oblong shape, size “5” hard gelatin capsules printed with “PBT” and “0.5” in red ink on cap and body respectively.
Capsules are supplied as follows:
NDC 69452- 153-20 Bottle of 100Tacrolimus Capsules, USP, 1 mg: White color, oblong shape, size “5” hard gelatin capsules printed with “PBT” and “1.0” in red ink on cap and body respectively.
Capsules are supplied as follows:
NDC 69452- 154-20 Bottle of 100Tacrolimus Capsules, USP, 5 mg: Pink color, oblong shape, size “4” hard gelatin capsules printed with “PBT” and “5.0” in red ink on cap and body respectively.
Capsules are supplied as follows:
NDC 69452- 155-20 Bottle of 100Note: Tacrolimus capsule are not filled to maximum capsule capacity. Capsule contains labeled amount.
Store and Dispense
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [See USP Controlled Room Temperature].16.4 Handling and Disposal
Tacrolimus can cause fetal harm. Tacrolimus capsules should not be opened or crushed. Avoid inhalation or direct contact with skin or mucous membranes of the powder contained in tacrolimus capsules. If such contact occurs wash the skin thoroughly with soap and water; if ocular contact occurs, rinse eyes with water. In case a spill occurs, wipe the surface with a wet paper towel. Follow applicable special handling and disposal procedures 1.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
17.1 Administration
Advise the patient or caregiver to:
- Inspect their tacrolimus capsules medicine when they receive a new prescription and before taking it. If the appearance of the capsule is not the same as usual, or if dosage instructions have changed, advise patients to contact their healthcare provider as soon as possible to make sure that they have the right medicine. Other tacrolimus products cannot be substituted for tacrolimus capsules.
- Take tacrolimus capsules at the same 12-hour intervals every day to achieve consistent blood concentrations.
- Take tacrolimus Ccapsules consistently either with or without food because the presence and composition of food decreases the bioavailability of Tacrolimus Capsules.
- Not to eat grapefruit or drink grapefruit juice in combination with tacrolimus capsules [see Drug Interactions ( 7.2)].
17.2 Development of Lymphoma and Other Malignancies
Inform patients they are at increased risk of developing lymphomas and other malignancies, particularly of the skin, due to immunosuppression. Advise patients to limit exposure to sunlight and ultraviolet (UV) light by wearing protective clothing and using a broad spectrum with a high protection factor [see Boxed Warnings and Warnings and Precautions ( 5.1)].
17.3 Increased Risk of Infection
Inform patients they are at increased risk of developing a variety of infections, including opportunistic infections, due to immunosuppression and to contact their physician if they develop any symptoms of infection such as fever, sweats or chills, cough or flu-like symptoms, muscle aches, or warm, red, painful areas on the skin [see Boxed Warnings and Warnings and Precautions ( 5.2)]
17.4 New Onset Diabetes After Transplant
Inform patients that tacrolimus capsules can cause diabetes mellitus and should be advised to contact their physician if they develop frequent urination, increased thirst, or hunger [see Warnings and Precautions ( 5.4)].
17.5 Nephrotoxicity
Inform patients that Tacrolimus capsules can have toxic effects on the kidney that should be monitored. Advise patients to attend all visits and complete all blood tests ordered by their medical team [see Warnings and Precautions ( 5.5)].
17.6 Neurotoxicity
Inform patients that they are at risk of developing adverse neurologic reactions including seizure, altered mental status, and tremor. Advise patients to contact their physician should they develop vision changes, deliriums, or tremors [see Warnings and Precautions ( 5.6)].
17.7 Hyperkalemia
Inform patients that tacrolimus capsules can cause hyperkalemia. Monitoring of potassium levels may be necessary, especially with concomitant use of other drugs known to cause hyperkalemia [see Warnings and Precautions ( 5.7)].
17.8 Hypertension
Inform patients that tacrolimus capsules can cause high blood pressure which may require treatment with anti-hypertensive therapy. Advise patients to monitor their blood pressure [see Warnings and Precautions ( 5.8)].
17.9 Drug Interactions
Instruct patients to tell their healthcare providers when they start or stop taking all the medicines, including prescription medicines and non-prescription medicines, natural or herbal remedies, nutritional supplements, and vitamins. Advise patients to avoid grapefruit and grapefruit juice [see Drug Interactions ( 7)].
17.10 Pregnancy, Lactation and Infertility
Inform women of childbearing potential that tacrolimus capsules can harm the fetus. Instruct male and female patients to discuss with their healthcare provider family planning options including appropriate contraception. Also, discuss with pregnant patients the risks and benefits of breastfeeding their infant [see Use in Specific Populations ( 8.1, 8.2, 8.3)].
Encourage female transplant patients who become pregnant and male patients who have fathered a pregnancy, exposed to immunosuppressants including tacrolimus, to enroll in the voluntary Transplantation Pregnancy Registry International. To enroll or register, patients can call the toll free number 1-877-955-6877 or https://www.transplantpregnancyregistry.org/[see Use in Specific Populations ( 8.1)].
Based on animal studies, tacrolimus capsules may affect fertility in males and females [see Nonclinical Toxicology ( 13.1)].
17.11 Myocardial Hypertrophy
Inform patients to report symptoms of tiredness, swelling, and/or shortness of breath (heart failure).
17.12 Immunizations
Inform patients that tacrolimus capsules can interfere with the usual response to immunizations and that they should avoid live vaccines [see Warnings and Precautions ( 5.14)].
-
SPL UNCLASSIFIED SECTION
Rx only
Manufactured by:
Panacea Biotec Pharma Limited
Malpur, Baddi,
Distt. Solan (H.P.) – 173205, IndiaMfg.Lic.No: MB/05/203
Distributed by:
Bionpharma Inc., 600 Alexander Road,
Princeton, NJ 08540, USA.All other trademarks and registered trademarks are the property of their respective owners.
Resource Number: L5678Item Code: PPIT081C
Revised Date: October 2020 -
Patient Information
Tacrolimus Capsules, USP
(ta-KROE-li-mus)Read this Patient Information before you start taking tacrolimus capsules and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment. If you have any questions about tacrolimus capsules ask your healthcare provider or pharmacist.
What is the most important information I should know about tacrolimus capsules?
Tacrolimus capsules can cause serious side effects, including:
Increased risk of cancer. People who take tacrolimus capsules have an increased risk of getting some kinds of cancer, including skin and lymph gland cancer (lymphoma).
Increased risk of infection. Tacrolimus capsules is a medicine that affects your immune system. Tacrolimus capsules can lower the ability of your immune system to fight infections. Serious infections can happen in people receiving tacrolimus capsules that can cause death. Call your healthcare provider right away if you have any symptoms of an infection, including:- fever
- muscle aches
- sweats or chills
- warm, red, or painful areas on your skin
- cough or flu-like symptoms
What is tacrolimus capsules?
- Tacrolimus capsules is a prescription medicine used with other medicines to help prevent organ rejection in people who have had a kidney, liver, or heart transplant.
- Tacrolimus capsules are types of tacrolimus immediate-release drugs and they are not the same as tacrolimus extended-release tablets or tacrolimus extended-release capsules. Your healthcare provider should decide what medicine is right for you.
Who should not take tacrolimus Capsules?
Do not take tacrolimus Capsules if you are allergic to tacrolimus or any of the ingredients in tacrolimus capsules. See the end of this leaflet for a complete list of ingredients in tacrolimus capsules.
What should I tell my healthcare provider before taking tacrolimus Capsules?
Before you take tacrolimus capsules, tell your healthcare provider about all of your medical conditions, including if you:
- plan to receive any live vaccines. People taking tacrolimus capsules should not receive live vaccines
- have or have had liver, kidney, or heart problems.
- are pregnant or plan to become pregnant. Tacrolimus capsules can harm your unborn baby.
- If you are able to become pregnant, you should use effective birth control before and during treatment with tacrolimus capsules. Talk to your healthcare provider before starting treatment with tacrolimus capsules about birth control methods that may be right for you.
- Males who have female partners who are able to become pregnant should also use effective birth control before and during treatment with tacrolimus capsules. Talk to your healthcare provider before starting treatment with tacrolimus capsules about birth control methods that may be right for you.
- There is a pregnancy registry for females who become pregnant and males who have fathered a pregnancy during treatment with tacrolimus capsules, USP. The purpose of this registry is to collect information about the health of you and your baby. To enroll in this voluntary registry, call 1-877-955-6877 or go to https://www.transplantpregnancyregistry.org/.
- are breastfeeding or plan to breastfeed. Tacrolimus Capsules passes into your breast milk. You and your healthcare provider should decide if you will breastfeed while taking tacrolimus Capsules.
Tell your healthcare provider about all the medicines you take, and when you start a new medicine or stop taking a medicine, including prescription and over-the-counter medicines, vitamins, natural, herbal or nutritional supplements.
Especially tell your healthcare provider if you take:
- sirolimus (RAPAMUNE)
- cyclosporine (GENGRAF, NEORAL, and SANDIMMUNE)
- medicines called aminoglycosides that are used to treat bacterial infections
- ganciclovir (CYTOVENE IV, VALCYTE)
- amphotericin B (ABELCET, AMBISOME)
- cisplatin
- antiviral medicines called nucleoside reverse transcriptase inhibitors
- antiviral medicines called protease inhibitors
- water pill (diuretic)
- medicine to treat high blood pressure
- nelfinavir (VIRACEPT)
- telaprevir (INCIVEK)
- boceprevir
- ritonavir (KALETRA, NORVIR, TECHNIVIE, VIEKIRA PAK, VIEKIRA XR)
- letermovir (PREVYMIS)
- ketoconazole
- itraconazole (ONMEL, SPORANOX)
- voriconazole (VFEND)
- clarithromycin (BIAXIN, BIAXIN XL, PREVPAC)
- rifampin (RIFADIN, RIFAMATE, RIFATER, RIMACTANE)
- rifabutin (MYCOBUTIN)
- amiodarone (NEXTERONE, PACERONE)
Ask your healthcare provider or pharmacist if you are not sure if you take any of the medicines listed above. Tacrolimus capsules may affect the way other medicines work, and other medicines may affect how tacrolimus capsules works.
Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take tacrolimus capsules?
- Take tacrolimus capsules exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much tacrolimus capsules to take and when to take it.
- Your healthcare provider may change your tacrolimus capsules dose if needed. Do not stop taking or change your dose of tacrolimus capsules without talking to your healthcare provider.
- Take tacrolimus capsules with or without food.
- Take tacrolimus capsules the same way every day. For example, if you choose to take tacrolimus capsules with food, you should always take tacrolimus capsules with food.
- Take tacrolimus capsules at the same time each day, 12 hours apart. For example, if you take your first dose at 7:00 a.m., you should take your second dose at 7:00 p.m.
- Taking tacrolimus capsules at the same time each day helps to keep the amount of medicine in your body at a steady level.
- Do not eat graperuit or drink grapefruit juice while taking tacrolimus capsules.
- If you take too much tacrolimus capsules, call your healthcare provider or go to the nearest hospital emergency room right away.
Tacrolimus capsules:
• Do not open or crush tacrolimus capsules.
What should I avoid while taking tacrolimus capsules?
While you take tacrolimus capsules you should not receive any live vaccines.
Limit the amount of time you spend in sunlight and avoid exposure to ultraviolet (UV) light, such as tanning machines. Wear protective clothing and use a sunscreen with a high sun protection factor (SPF).What are the possible side effects of tacrolimus Capsules?
Tacrolimus capsules may cause serious side effects, including:
- See “What is the most important information I should know about tacrolimus Capsules?”
- problems from medicine errors. People who take tacrolimus capsules have sometimes been given the wrong type of tacrolimus product. Tacrolimus extended-release medicines are not the same as tacrolimus capsules and cannot be substituted for each other, unless specifically prescribed by your healthcare provider, who will send you to get blood tacrolimus levels at a lab. Check your tacrolimus capsules when you get a new prescription and before you take it to make sure you have received tacrolimus capsules.
- Check with the pharmacist and call your healthcare provider if you think you were given the wrong medicine.
- high blood sugar (diabetes). Your healthcare provider may do blood tests to check for diabetes while you take tacrolimus capsules. Call your healthcare provider right away if you have any symptoms of high blood sugar, including:
- frequent urination
- drowsiness
- increased thirst or hunger
- loss of appetite
- blurred vision
- fruity smell on your breath
- confusion
- nausea, vomiting, or stomach pain
- kidney problems. Kidney problems are a serious and common side effect of tacrolimus capsules. Your healthcare provider may do blood tests to check your kidney function while you take tacrolimus capsules.
- nervous system problems. Nervous system problems are a serious and common side effect of tacrolimus capsules. Call your healthcare provider right away if you get any of these symptoms while taking tacrolimus capsules. These could be signs of a serious nervous system problem:
- headache
- changes in behavior
- confusion
- coma
- seizures
- tremors
- changes in your vision
- numbness and tingling
- high levels of potassium in your blood. Your healthcare provider may do blood tests to check your potassium level while you take tacrolimus capsules.
- high blood pressure. High blood pressure is a serious and common side effect of tacrolimus capsules. Your healthcare provider will monitor your blood pressure while you take tacrolimus capsules and may prescribe blood pressure medicine for you, if needed. Your healthcare provider may instruct you to check your blood pressure at home.
- changes in the electrical activity of your heart (QT prolongation).
- heart problems (myocardial hypertrophy). Tell your healthcare provider right away if you get any of these symptoms of heart problems while taking tacrolimus capsules:
- shortness of breath
- feel lightheaded
- chest pain
- feel faint
-
severe low red blood cell count (anemia).
The most common side effects of tacrolimus Capsules in people who have received a kidney, liver or heart transplant are: - infections in general, including cytomegalovirus (CMV) infection
- swelling of the hands, legs, ankles, or feet
- weakness
- tremors(shaking of the body)
- pain
- constipation
- high levels of fat in your blood
- diarrhea
- high levels of potassium in your blood
- headache
- low red blood cell count (anemia)
- stomach pain
- low white blood cell count
- trouble sleeping
- fever
- nausea
- numbness or tingling in your hands and feet
- high blood sugar (diabetes)
- inflammation of your airway (bronchitis)
- low levels of magnesium in your blood
- fluid around your heart
- low levels of phosphate in your blood
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of tacrolimus capsules. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA1088.
How should I store tacrolimus capsules?
tacrolimus capsules
- Store Tacrolimus Capsules, USP at room temperature between 68°F to 77°F (20°C to 25°C).
Keep tacrolimus capsules and all medicines out of the reach of children.
General information about the safe and effective use of tacrolimus capsules.
- Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use tacrolimus capsules for a condition for which it was not prescribed. Do not give tacrolimus capsules to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about tacrolimus capsules that is written for health professionals.
- This Patient Information leaflet summarizes the most important information about tacrolimus capsules. If you would like more information, talk to your healthcare provider.
What are the ingredients in tacrolimus capsules?
Active ingredient: Tacrolimus USPInactive ingredients:
Tacrolimus capsules: croscarmellose sodium, hypromellose, lactose anhydrous, and magnesium stearate. The 0.5 mg capsule shell contains ferric oxide, gelatin, and titanium dioxide. The 1 mg capsule shell contains gelatin and titanium dioxide. The 5 mg capsule shell contains ferric oxide, gelatin, and titanium dioxide.
Manufactured by:
Panacea Biotec Pharma Limited, Malpur, Baddi,
Distt. Solan (H.P.) – 173205, IndiaAll other trademarks and registered trademarks are the property of their respective owners.
Mfg.Lic.No: MB/05/203
Distributed by:
Bionpharma Inc., 600 Alexander Road,
Princeton, NJ 08540, USA.Item Code: PPIT082B
For more information, call Bionpharma, Inc. at 1-888-235-BION or 1-888-235-2466.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised Date: October 2020
- 0.5 mg bottle
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TACROLIMUS
tacrolimus capsule, gelatin coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69452-153 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TACROLIMUS (UNII: WM0HAQ4WNM) (TACROLIMUS ANHYDROUS - UNII:Y5L2157C4J) TACROLIMUS ANHYDROUS 0.5 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color yellow Score no score Shape CAPSULE Size 5mm Flavor Imprint Code PBT;0;5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69452-153-20 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/26/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090802 02/26/2016 TACROLIMUS
tacrolimus capsule, gelatin coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69452-154 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TACROLIMUS (UNII: WM0HAQ4WNM) (TACROLIMUS ANHYDROUS - UNII:Y5L2157C4J) TACROLIMUS ANHYDROUS 1 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white Score no score Shape CAPSULE Size 5mm Flavor Imprint Code PBT;1;0 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69452-154-20 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/26/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090802 02/26/2016 TACROLIMUS
tacrolimus capsule, gelatin coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69452-155 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TACROLIMUS (UNII: WM0HAQ4WNM) (TACROLIMUS ANHYDROUS - UNII:Y5L2157C4J) TACROLIMUS ANHYDROUS 5 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color pink Score no score Shape CAPSULE Size 4mm Flavor Imprint Code PBT;5;0 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69452-155-20 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/26/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090802 02/26/2016 Labeler - BionPharma Inc., (079637826) Registrant - Panacea Biotec Pharma Limited (878533279) Establishment Name Address ID/FEI Business Operations Panacea Biotec Pharma Limited 857979552 manufacture(69452-153, 69452-154, 69452-155)