Label: GABITRIL- tiagabine hydrochloride tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 16590-832-30, 16590-832-45, 16590-832-60, 16590-832-90 - Packager: STAT RX USA LLC
- This is a repackaged label.
- Source NDC Code(s): 63459-412
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated August 18, 2011
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
DESCRIPTION
GABITRIL® (tiagabine HCl) is an antiepilepsy drug available as 2 mg, 4 mg, 12 mg, and 16 mg tablets for oral administration. Its chemical name is (-)-(R)-1-[4,4-Bis(3-methyl-2-thienyl)-3-butenyl]nipecotic acid hydrochloride, its molecular formula is C20H25NO2S2 HCl, and its molecular weight is 412.0. Tiagabine HCl is a white to off-white, odorless, crystalline powder. It is insoluble in heptane, sparingly soluble in water, and soluble in aqueous base. The structural formula is:
Inactive Ingredients
GABITRIL tablets contain the following inactive ingredients: Ascorbic acid, colloidal silicon dioxide, crospovidone, hydrogenated vegetable oil wax, hydroxypropyl cellulose, hypromellose, lactose, magnesium stearate, microcrystalline cellulose, pregelatinized starch, stearic acid, and titanium dioxide.
In addition, individual tablets contain:
- 2 mg tablets: FD and C Yellow No. 6.
- 4 mg tablets: D and C Yellow No. 10.
- 12 mg tablets: D and C Yellow No. 10 and FD and C Blue No. 1.
- 16 mg tablets: FD and C Blue No. 2.
-
CLINICAL PHARMACOLOGY
Mechanism of ActionThe precise mechanism by which tiagabine exerts its antiseizure effect is unknown, although it is believed to be related to its ability, documented in in vitro experiments, to enhance the activity of gamma aminobutyric acid (GABA), the major inhibitory neurotransmitter in the central nervous system. These experiments have shown that tiagabine binds to recognition sites associated with the GABA uptake carrier. It is thought that, by this action, tiagabine blocks GABA uptake into presynaptic neurons, permitting more GABA to be available for receptor binding on the surfaces of post-synaptic cells. Inhibition of GABA uptake has been shown for synaptosomes, neuronal cell cultures, and glial cell cultures. In rat-derived hippocampal slices, tiagabine has been shown to prolong GABA-mediated inhibitory post-synaptic potentials. Tiagabine increases the amount of GABA available in the extracellular space of the globus pallidus, ventral palladum, and substantia nigra in rats at the ED50 and ED85 doses for inhibition of pentylenetetrazol (PTZ)-induced tonic seizures. This suggests that tiagabine prevents the propagation of neural impulses that contribute to seizures by a GABA-ergic action.
Tiagabine has shown efficacy in several animal models of seizures. It is effective against the tonic phase of subcutaneous PTZ-induced seizures in mice and rats, seizures induced by the proconvulsant DMCM in mice, audiogenic seizures in genetically epilepsy-prone rats (GEPR), and amygdala-kindled seizures in rats. Tiagabine has little efficacy against maximal electroshock seizures in rats and is only partially effective against subcutaneous PTZ-induced clonic seizures in mice, picrotoxin-induced tonic seizures in the mouse, bicuculline-induced seizures in the rat, and photic seizures in photosensitive baboons. Tiagabine produces a biphasic dose-response curve against PTZ- and DMCM-induced convulsions, with attenuated effectiveness at higher doses.
Based on in vitro binding studies, tiagabine does not significantly inhibit the uptake of dopamine, norepinephrine, serotonin, glutamate, or choline and shows little or no binding to dopamine D1 and D2, muscarinic, serotonin 5HT1A, 5HT2, and 5HT3, beta-1 and 2 adrenergic, alpha-1 and alpha-2 adrenergic, histamine H2 and H3, adenosine A1 and A2, opiate µ and K1, NMDA glutamate, and GABAA receptors at 100 µM. It also lacks significant affinity for sodium or calcium channels. Tiagabine binds to histamine H1, serotonin 5HT1B, benzodiazepine, and chloride channel receptors at concentrations 20 to 400 times those inhibiting the uptake of GABA.
PharmacokineticsTiagabine is well absorbed, with food slowing absorption rate but not altering the extent of absorption. The elimination half-life of tiagabine is 7 to 9 hours in normal volunteers. In epilepsy clinical trials, most patients were receiving hepatic enzyme-inducing agents (e.g., carbamazepine, phenytoin, primidone, and phenobarbital). The pharmacokinetic profile in induced patients is significantly different from the non-induced population (see PRECAUTIONS, General, Use in Non-Induced Patients). The systemic clearance of tiagabine in induced patients is approximately 60% greater resulting in considerably lower plasma concentrations and an elimination half-life of 2 to 5 hours. Given this difference in clearance, the systemic exposure after a dose of 32 mg/day in an induced population is expected to be comparable to the systemic exposure after a dose of 12 mg/day in a non-induced population. Similarly, the systemic exposure after a dose of 56 mg/day in an induced population is expected to be comparable to the systemic exposure after a dose of 22 mg/day in a non-induced population.
Absorption and DistributionAbsorption of tiagabine is rapid, with peak plasma concentrations occurring at approximately 45 minutes following an oral dose in the fasting state. Tiagabine is nearly completely absorbed (>95%), with an absolute oral bioavailability of about 90%. A high fat meal decreases the rate (mean Tmax was prolonged to 2.5 hours, and mean Cmax was reduced by about 40%) but not the extent (AUC) of tiagabine absorption. In all clinical trials, tiagabine was given with meals.
The pharmacokinetics of tiagabine are linear over the single dose range of 2 to 24 mg. Following multiple dosing, steady state is achieved within 2 days.
Tiagabine is 96% bound to human plasma proteins, mainly to serum albumin and α1-acid glycoprotein over the concentration range of 10 ng/mL to 10,000 ng/mL. While the relationship between tiagabine plasma concentrations and clinical response is not currently understood, trough plasma concentrations observed in controlled clinical trials at doses from 30 to 56 mg/day ranged from less than 1 ng/mL to 234 ng/mL.
Metabolism and EliminationAlthough the metabolism of tiagabine has not been fully elucidated, in vivo and in vitro studies suggest that at least two metabolic pathways for tiagabine have been identified in humans: 1) thiophene ring oxidation leading to the formation of 5-oxo-tiagabine; and 2) glucuronidation. The 5-oxo-tiagabine metabolite does not contribute to the pharmacologic activity of tiagabine.
Based on in vitro data, tiagabine is likely to be metabolized primarily by the 3A isoform subfamily of hepatic cytochrome P450 (CYP 3A), although contributions to the metabolism of tiagabine from CYP 1A2, CYP 2D6 or CYP 2C19 have not been excluded.
Approximately 2% of an oral dose of tiagabine is excreted unchanged, with 25% and 63% of the remaining dose excreted into the urine and feces, respectively, primarily as metabolites, at least 2 of which have not been identified. The mean systemic plasma clearance is 109 mL/min (CV = 23%) and the average elimination half-life for tiagabine in healthy subjects ranged from 7 to 9 hours. The elimination half-life decreased by 50 to 65% in hepatic enzyme-induced patients with epilepsy compared to uninduced patients with epilepsy.
A diurnal effect on the pharmacokinetics of tiagabine was observed. Mean steady-state Cminvalues were 40% lower in the evening than in the morning. Tiagabine steady-state AUC values were also found to be 15% lower following the evening tiagabine dose compared to the AUC following the morning dose.
Special Populations
Renal InsufficiencyThe pharmacokinetics of total and unbound tiagabine were similar in subjects with normal renal function (creatinine clearance >80 mL/min) and in subjects with mild (creatinine clearance 40 to 80 mL/min), moderate (creatinine clearance 20 to 39 mL/min), or severe (creatinine clearance 5 to 19 mL/min) renal impairment. The pharmacokinetics of total and unbound tiagabine were also unaffected in subjects with renal failure requiring hemodialysis.
Hepatic InsufficiencyIn patients with moderate hepatic impairment (Child-Pugh Class B), clearance of unbound tiagabine was reduced by about 60%. Patients with impaired liver function may require reduced initial and maintenance doses of tiagabine and/or longer dosing intervals compared to patients with normal hepatic function (see PRECAUTIONS).
GeriatricThe pharmacokinetic profile of tiagabine was similar in healthy elderly and healthy young adults.
PediatricTiagabine has not been investigated in adequate and well-controlled clinical trials in patients below the age of 12. The apparent clearance and volume of distribution of tiagabine per unit body surface area or per kg were fairly similar in 25 children (age: 3 to 10 years) and in adults taking enzyme-inducing antiepilepsy drugs ([AEDs] e.g., carbamazepine or phenytoin). In children who were taking a non-inducing AED (e.g., valproate), the clearance of tiagabine based upon body weight and body surface area was 2 and 1.5-fold higher, respectively, than in non-induced adults with epilepsy.
Gender, Race and Cigarette SmokingNo specific pharmacokinetic studies were conducted to investigate the effect of gender, race and cigarette smoking on the disposition of tiagabine. Retrospective pharmacokinetic analyses, however, suggest that there is no clinically important difference between the clearance of tiagabine in males and females, when adjusted for body weight. Population pharmacokinetic analyses indicated that tiagabine clearance values were not significantly different in Caucasian (N=463), Black (N=23), or Hispanic (N=17) patients with epilepsy, and that tiagabine clearance values were not significantly affected by tobacco use.
Interactions with other Antiepilepsy DrugsThe clearance of tiagabine is affected by the co-administration of hepatic enzyme-inducing antiepilepsy drugs. Tiagabine is eliminated more rapidly in patients who have been taking hepatic enzyme-inducing drugs, e.g., carbamazepine, phenytoin, primidone and phenobarbital than in patients not receiving such treatment (see PRECAUTIONS, Drug Interactions).
Interactions with Other Drugs -
CLINICAL STUDIES
The effectiveness of GABITRIL as adjunctive therapy (added to other antiepilepsy drugs) was examined in three multi-center, double-blind, placebo-controlled, parallel-group, clinical trials in 769 patients with refractory partial seizures who were taking at least one hepatic enzyme-inducing antiepilepsy drug (AED), and two placebo-controlled cross-over studies in 90 patients. In the parallel-group trials, patients had a history of at least six complex partial seizures (Study 1 and Study 2, U.S. studies), or six partial seizures of any type (Study 3, European study), occurring alone or in combination with any other seizure type within the 8-week period preceding the first study visit in spite of receiving one or more AEDs at therapeutic concentrations.
In the first two studies, the primary protocol-specified outcome measure was the median reduction from baseline in the 4-week complex partial seizure (CPS) rates during treatment. In the third study, the protocol-specified primary outcome measure was the proportion of patients achieving a 50% or greater reduction from baseline in the 4-week seizure rate of all partial seizures during treatment. The results given below include data for complex partial seizures and all partial seizures for the intent-to-treat population (all patients who received at least one dose of treatment and at least one seizure evaluation) in each study.
Study 1 was a double-blind, placebo-controlled, parallel-group trial comparing GABITRIL 16 mg/day, GABITRIL 32 mg/day, GABITRIL 56 mg/day, and placebo. Study drug was given as a four times a day regimen. After a prospective Baseline Phase of 12 weeks, patients were randomized to one of the four treatment groups described above. The 16-week Treatment Phase consisted of a 4-week Titration Period, followed by a 12-week Fixed-Dose Period, during which concomitant AED doses were held constant. The primary outcome was assessed for the combined 32 and 56 mg/day groups compared to placebo.
Study 2 was a double-blind, placebo-controlled, parallel-group trial consisting of an 8-week Baseline Phase and a 12-week Treatment Phase, the first 4 weeks of which constituted a Titration Period and the last 8 weeks a Fixed-Dose Period. This study compared GABITRIL 16 mg BID and 8 mg QID to placebo. The protocol-specified primary outcome measure was assessed separately for each group treated with GABITRIL.
The following tables display the results of the analyses of these two trials.
Table 1: Median Reduction and Median Percent Reduction from Baseline in 4-Week Seizure Rates in Study 1
Placebo (N=91) GABITRIL 16 mg/day (N=61) GABITRIL 32 mg/day (N=87) GABITRIL 56mg/day (N=56) Combined 32 and 56 mg/day (N=143)
Complex Partial Median Reduction 0.6 0.8 2.2 * 2.9 * 2.6 *
Median % Reduction † 9% 13% 25% 32% 29%
All Partial Median Reduction 0.2 1.2 2.7 * 3.5 * 2.9 *
Median % Reduction † 3% 12% 24% 36% 27% * p less than 0.05
† Statistical significance was not assessed for median % reduction.
Table 2: Median Reduction and Median Percent Reduction from Baseline in 4-Week Seizure Rates in Study 2
* p less than 0.027 necessary for statistical significance due to multiple comparisons.
Placebo (N=107) GABITRIL 16 mg BID (N=106) GABITRIL 8 mg QID (N=104)
Complex Partial Median Reduction 0.3 1.6 1.3 *
Median % Reduction† 4% 22% 15%
All Partial Median Reduction 0.5 1.6 1.3
Median % Reduction† 5% 19% 13%
† Statistical significance was not assessed for median % reduction.
-
SPL UNCLASSIFIED SECTION
Figures 1 to 4 present the proportion of patients (X-axis) whose percent reduction from baseline in the all partial seizure rate was at least as great as that indicated on the Y axis in the three placebo-controlled adjunctive studies (Studies 1, 2, and 3). A positive value on the Y axis indicates an improvement from baseline (i.e., a decrease in seizure rate), while a negative value indicates a worsening from baseline (i.e., an increase in seizure rate). Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo.
Figure 1 indicates that the proportion of patients achieving any particular level of reduction in seizure rate was consistently higher for the combined GABITRIL 32 mg and 56 mg groups compared to the placebo group in Study 1. For example, Figure 1 indicates that approximately 24% of patients treated with GABITRIL experienced a 50% or greater reduction, compared to 4% in the placebo group.
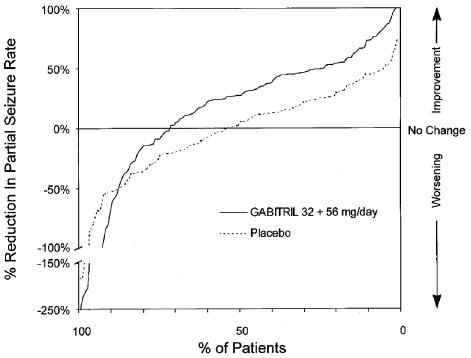
Figure 2 also displays the results for Study 1, which was a dose-response study, by treatment group, without combining GABITRIL dosage groups. Figure 2 indicates a dose-response relationship across the three GABITRIL groups. The proportion of patients achieving any particular level of reduction in all partial seizure rates was consistently higher as the dose of GABITRIL was increased. For example, Figure 2 indicates that approximately 4% of patients in the placebo group experienced a 50% or greater reduction in all partial seizure rate, compared to approximately 10% of the GABITRIL 16 mg/day group, 21% of the GABITRIL 32 mg/day group, and 30% of the GABITRIL 56 mg/day group.
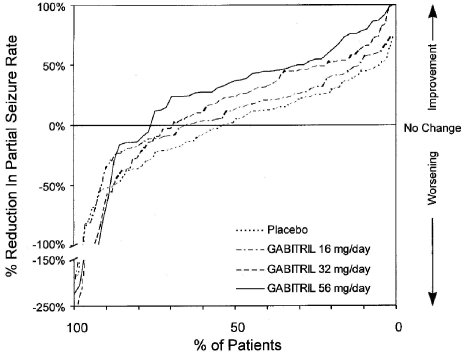
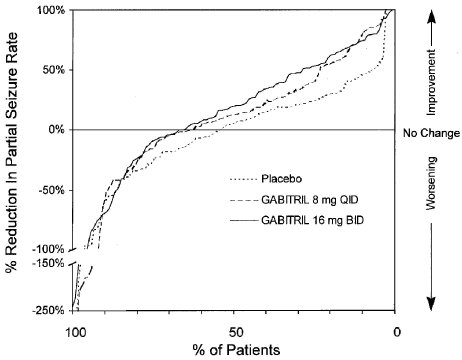
Figure 3 indicates that the proportion of patients achieving any particular level of reduction in partial seizure rate was consistently greater in patients taking GABITRIL than in those taking placebo in Study 2. (Study 2 compared placebo to GABITRIL 32 mg/day; one of the GABITRIL groups received 8 mg QID, while the other GABITRIL group received 16 mg BID). For example, Figure 3 indicates that approximately 7% of patients in the placebo group experienced a 50% or greater reduction in their partial seizure rate, compared to approximately 23% of patients in the GABITRIL 8 mg QID group and 28% of patients in the GABITRIL 16 mg BID group.
-
SPL UNCLASSIFIED SECTION
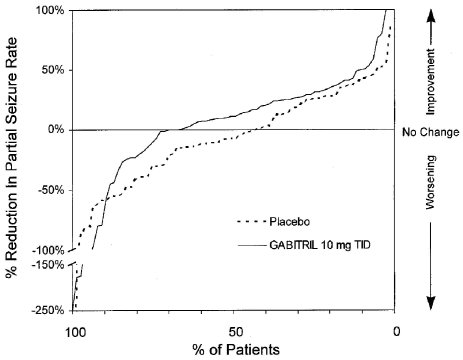
Study 3 was a double-blind, placebo-controlled, parallel-group trial that compared GABITRIL 10 mg TID (N=77) with placebo (N=77). In this trial, patients were followed prospectively during a 12-week Baseline Phase and then randomized to receive study drug during an 18-week Treatment Phase. During the first 6 weeks of treatment (Titration Period), patients were titrated to 30 mg/day, after which they were maintained on this dose during the 12-week Fixed-Dose Period. The protocol-specified primary outcome measure (proportion of patients who achieved at least a 50% reduction from baseline in partial seizure rate) did not reach statistical significance. However, analyses of the median reduction from baseline in 4-week partial seizure rate (the analyses presented above for Study 1 and Study 2) were performed and showed a statistically significant improvement compared to placebo in all partial and complex partial seizure rates (Table 3):
Table 3: Median Reduction and Median Percent Reduction from Baseline in 4-Week Seizure Rates in Study 3
Placebo (N=77) GABITRIL 30 mg/day (N=77)
Complex Partial‡ Median Reduction -0.1 1.3*
Median % Reduction† -1% 14%
All Partial Median Reduction -0.5 1.1*
Median % Reduction† -7% 11% * p less than 0.05
†Statistical significance was not assessed for median % reduction.
‡ N=72 and 75 for placebo and GABITRIL, respectively.
Figure 4 indicates that the proportion of patients achieving any particular level of reduction in seizure activity was consistently higher in those taking GABITRIL than those taking placebo in Study 3. For example, Figure 4 indicates that approximately 5% of patients in the placebo group experienced a 50% or greater reduction in their partial seizure rate compared to approximately 10% of patients in the GABITRIL group.
The two other placebo-controlled trials that examined the effectiveness of GABITRIL were small cross-over trials (N=46 and 44). Both trials included an open Screening Phase during which patients were titrated to an optimal dose and then treated with this dose for an additional 4 weeks. After this Open Phase, patients were randomized to one of two blinded treatment sequences (GABITRIL followed by placebo or placebo followed by GABITRIL). The Double-Blind Phase consisted of two Treatment Periods, each lasting 7 weeks (with a 3 week washout between periods). The outcome measures were median with-in patient differences between placebo and GABITRIL Treatment Periods in 4-week complex partial and all partial seizure rates. The reductions in seizure rates were statistically significant in both studies.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Seizures in Patients Without Epilepsy: Post-marketing reports have shown that GABITRIL use has been associated with new onset seizures and status epilepticus in patients without epilepsy. Dose may be an important predisposing factor in the development of seizures, although seizures have been reported in patients taking daily doses of GABITRIL as low as 4 mg/day. In most cases, patients were using concomitant medications (antidepressants, antipsychotics, stimulants, narcotics) that are thought to lower the seizure threshold. Some seizures occurred near the time of a dose increase, even after periods of prior stable dosing.
The GABITRIL dosing recommendations in current labeling for treatment of epilepsy were based on use in patients with partial seizures 12 years of age and older, most of whom were taking enzyme-inducing antiepileptic drugs (AEDs; e.g., carbamazepine, phenytoin, primidone and phenobarbital) which lower plasma levels of GABITRIL by inducing its metabolism. Use of GABITRIL without enzyme-inducing antiepileptic drugs results in blood levels about twice those attained in the studies on which current dosing recommendations are based (see DOSAGE AND ADMINISTRATION).
Safety and effectiveness of GABITRIL have not been established for any indication other than as adjunctive therapy for partial seizures in adults and children 12 years and older.
In nonepileptic patients who develop seizures while on GABITRIL treatment, GABITRIL should be discontinued and patients should be evaluated for an underlying seizure disorder.
Seizures and status epilepticus are known to occur with GABITRIL overdosage (see OVERDOSAGE).
Suicidal Behavior and IdeationAntiepileptic drugs (AEDs), including GABITRIL, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed.
Table 4 shows absolute and relative risk by indication for all evaluated AEDs.
Table 4: Risk by Indication for Antiepileptic Drugs in the Pooled Analysis
Indication
Placebo Patients with Events per 1000 Patients
Drug Patients with Events per 1000 Patients
Relative Risk: Incidence of Events in Drug Patients/Incidence in Placebo Patients
Risk Difference: Additional Drug Patients with Events per 1000 Patients
Epilepsy
1.0
3.4
3.5
2.4 Psychiatric
5.7
8.5
1.5
2.9 Other
1.0
1.8
1.9
0.9
Total
2.4
4.3
1.8
1.9 The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing GABITRIL or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
Withdrawal SeizuresAs a rule, antiepilepsy drugs should not be abruptly discontinued because of the possibility of increasing seizure frequency. In a placebo-controlled, double-blind, dose-response study (Study 1 described in CLINICAL STUDIES) designed, in part, to investigate the capacity of GABITRIL to induce withdrawal seizures, study drug was tapered over a 4-week period after 16 weeks of treatment. Patients’ seizure frequency during this 4-week withdrawal period was compared to their baseline seizure frequency (before study drug). For each partial seizure type, for all partial seizure types combined, and for secondarily generalized tonic-clonic seizures, more patients experienced increases in their seizure frequencies during the withdrawal period in the three GABITRIL groups than in the placebo group. The increase in seizure frequency was not affected by dose. GABITRIL should be withdrawn gradually to minimize the potential of increased seizure frequency, unless safety concerns require a more rapid withdrawal.
Cognitive/Neuropsychiatric Adverse EventsAdverse events most often associated with the use of GABITRIL were related to the central nervous system. The most significant of these can be classified into 2 general categories: 1) impaired concentration, speech or language problems, and confusion (effects on thought processes); and 2) somnolence and fatigue (effects on level of consciousness). The majority of these events were mild to moderate. In controlled clinical trials, these events led to discontinuation of treatment with GABITRIL in 6% (31 of 494) of patients compared to 2% (5 of 275) of the placebo-treated patients. A total of 1.6% (8 of 494) of the GABITRIL treated patients in the controlled trials were hospitalized secondary to the occurrence of these events compared to 0% of the placebo treated patients. Some of these events were dose related and usually began during initial titration.
Patients with a history of spike and wave discharges on EEG have been reported to have exacerbations of their EEG abnormalities associated with these cognitive/neuropsychiatric events. This raises the possibility that these clinical events may, in some cases, be a manifestation of underlying seizure activity (see PRECAUTIONS, Laboratory Tests, EEG). In the documented cases of spike and wave discharges on EEG with cognitive/neuropsychiatric events, patients usually continued tiagabine, but required dosage adjustment.
Additionally, there have been postmarketing reports of patients who have experienced cognitive/neuropsychiatric symptoms, some accompanied by EEG abnormalities such as generalized spike and wave activity, that have been reported as nonconvulsant status epilepticus. Some reports describe recovery following reduction of dose or discontinuation of GABITRIL.
Status EpilepticusIn the three double-blind, placebo-controlled, parallel-group studies (Studies 1, 2, and 3), the incidence of any type of status epilepticus (simple, complex, or generalized tonic-clonic) in patients receiving GABITRIL was 0.8% (4 of 494 patients) versus 0.7% (2 of 275 patients) receiving placebo. Among the patients treated with GABITRIL across all epilepsy studies (controlled and uncontrolled), 5% had some form of status epilepticus. Of the 5%, 57% of patients experienced complex partial status epilepticus. A critical risk factor for status epilepticus was the presence of a previous history; 33% of patients with a history of status epilepticus had recurrence during GABITRIL treatment. Because adequate information about the incidence of status epilepticus in a similar population of patients with epilepsy who have not received treatment with GABITRIL is not available, it is impossible to state whether or not treatment with GABITRIL is associated with a higher or lower rate of status epilepticus than would be expected to occur in a similar population not treated with GABITRIL.
Sudden Unexpected Death In Epilepsy (SUDEP)There have been as many as 10 cases of sudden unexpected deaths during the clinical development of tiagabine among 2531 patients with epilepsy (3831 patient-years of exposure).
This represents an estimated incidence of 0.0026 deaths per patient-year. This rate is within the range of estimates for the incidence of sudden and unexpected deaths in patients with epilepsy not receiving GABITRIL (ranging from 0.0005 for the general population with epilepsy, 0.003 to 0.004 for clinical trial populations similar to that in the clinical development program for GABITRIL, to 0.005 for patients with refractory epilepsy). The estimated SUDEP rates in patients receiving GABITRIL are also similar to those observed in patients receiving other antiepilepsy drugs, chemically unrelated to GABITRIL, that underwent clinical testing in similar populations at about the same time. This evidence suggests that the SUDEP rates reflect population rates, not a drug effect.
-
PRECAUTIONS
General
Use in Non-Induced PatientsVirtually all experience with GABITRIL has been obtained in patients with epilepsy receiving at least one concomitant enzyme-inducing antiepilepsy drug (AED), which lowers the plasma levels of tiagabine. Use in non-induced patients requires lower doses of GABITRIL. These patients may also require a slower titration of GABITRIL compared to that of induced patients (see DOSAGE AND ADMINISTRATION). Patients taking a combination of inducing and non-inducing agents (e.g., carbamazepine and valproate) should be considered to be induced. Patients not receiving hepatic enzyme-inducing agents are referred to as non-induced patients.
Generalized WeaknessModerately severe to incapacitating generalized weakness has been reported following administration of GABITRIL in 28 of 2531 (approximately 1%) patients with epilepsy. The weakness resolved in all cases after a reduction in dose or discontinuation of GABITRIL.
Binding in the Eye and Other Melanin-Containing TissuesWhen dogs received a single dose of radiolabeled tiagabine, there was evidence of residual binding in the retina and uvea after 3 weeks (the latest time point measured). Although not directly measured, melanin binding is suggested. The ability of available tests to detect potentially adverse consequences, if any, of the binding of tiagabine to melanin-containing tissue is unknown and there was no systematic monitoring for relevant ophthalmological changes during the clinical development of GABITRIL. However, long term (up to one year) toxicological studies of tiagabine in dogs showed no treatment-related ophthalmoscopic changes and macro- and microscopic examinations of the eye were unremarkable. Accordingly, although there are no specific recommendations for periodic ophthalmologic monitoring, prescribers should be aware of the possibility of long-term ophthalmologic effects.
Use in Hepatically-Impaired PatientsBecause the clearance of tiagabine is reduced in patients with liver disease, dosage reduction may be necessary in these patients.
Serious RashFour patients treated with tiagabine during the product’s premarketing clinical testing developed what were considered to be serious rashes. In two patients, the rash was described as maculopapular; in one it was described as vesiculobullous; and in the 4th case, a diagnosis of Stevens Johnson Syndrome was made. In none of the 4 cases is it certain that tiagabine was the primary, or even a contributory, cause of the rash. Nevertheless, drug associated rash can, if extensive and serious, cause irreversible morbidity, even death.
Information for PatientsPatients should be informed of the availability of a Medication Guide, and they should be instructed to read it prior to taking GABITRIL. The complete text of the Medication Guide is provided at the end of this labeling.
Suicidal Thinking and BehaviorPatients, their caregivers, and families should be counseled that AEDs, including GABITRIL, may increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
Patients should be advised that GABITRIL may cause dizziness, somnolence, and other symptoms and signs of CNS depression. Accordingly, patients should be advised neither to drive nor to operate other complex machinery until they have gained sufficient experience on GABITRIL to gauge whether or not it affects their mental and/or motor performance adversely. Because of the possible additive depressive effects, caution should also be used when patients are taking other CNS depressants in combination with GABITRIL.
Because teratogenic effects were seen in the offspring of rats exposed to maternally toxic doses of tiagabine and because experience in humans is limited, patients should be advised to notify their physicians if they become pregnant or intend to become pregnant during therapy.
Because of the possibility that tiagabine may be excreted in breast milk, patients should be advised to notify those providing care to themselves and their children if they intend to breast-feed or are breast-feeding an infant.
Patients should be encouraged to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy. To enroll, patients can call the toll free number 1-888-233-2334 (see PRECAUTIONS, Pregnancy).
Laboratory Tests
Therapeutic Monitoring of Plasma Concentrations of TiagabineA therapeutic range for tiagabine plasma concentrations has not been established. In controlled trials, trough plasma concentrations observed among patients randomized to doses of tiagabine that were statistically significantly more effective than placebo ranged from less than 1 ng/mL to 234 ng/mL (median, 10th and 90th percentiles are 23.7 ng/mL, 5.4 ng/mL, and 69.8 ng/mL, respectively). Because of the potential for pharmacokinetic interactions between GABITRIL and drugs that induce or inhibit hepatic metabolizing enzymes, it may be useful to obtain plasma levels of tiagabine before and after changes are made in the therapeutic regimen.
Clinical Chemistry and HematologyDuring the development of GABITRIL, no systematic abnormalities on routine laboratory testing were noted. Therefore, no specific guidance is offered regarding routine monitoring; the practitioner retains responsibility for determining how best to monitor the patient in his/her care.
EEGPatients with a history of spike and wave discharges on EEG have been reported to have exacerbations of their EEG abnormalities associated with cognitive/neuropsychiatric events. This raises the possibility that these clinical events may, in some cases, be a manifestation of underlying seizure activity (see WARNINGS, Cognitive/Neuropsychiatric Adverse Events). In the documented cases of spike and wave discharges on EEG with cognitive/neuropsychiatric events, patients usually continued tiagabine, but required dosage adjustment.
Drug InteractionsIn evaluating the potential for interactions among co-administered antiepilepsy drugs (AEDs), whether or not an AED induces or does not induce metabolic enzymes is an important consideration. Carbamazepine, phenytoin, primidone, and phenobarbital are generally classified as enzyme inducers; valproate and gabapentin are not. GABITRIL is considered to be a non-enzyme inducing AED (see PRECAUTIONS, General, Use in Non-Induced Patients).
The drug interaction data described in this section were obtained from studies involving either healthy subjects or patients with epilepsy.
Effects of GABITRIL on other Antiepilepsy Drugs (AEDs):Phenytoin: Tiagabine had no effect on the steady-state plasma concentrations of phenytoin in patients with epilepsy.
Carbamazepine: Tiagabine had no effect on the steady-state plasma concentrations of carbamazepine or its epoxide metabolite in patients with epilepsy.
Valproate: Tiagabine causes a slight decrease (about 10%) in steady-state valproate concentrations.
Phenobarbital or Primidone: No formal pharmacokinetic studies have been performed examining the addition of tiagabine to regimens containing phenobarbital or primidone. The addition of tiagabine in a limited number of patients in three well-controlled studies caused no systematic changes in phenobarbital or primidone concentrations when compared to placebo.
Effects of other Antiepilepsy Drugs (AEDs) on GABITRIL:Carbamazepine: Population pharmacokinetic analyses indicate that tiagabine clearance is 60% greater in patients taking carbamazepine with or without other enzyme-inducing AEDs.
Phenytoin: Population pharmacokinetic analyses indicate that tiagabine clearance is 60% greater in patients taking phenytoin with or without other enzyme-inducing AEDs.
Phenobarbital (Primidone): Population pharmacokinetic analyses indicate that tiagabine clearance is 60% greater in patients taking phenobarbital (primidone) with or without other enzyme-inducing AEDs.
Valproate: The addition of tiagabine to patients taking valproate chronically had no effect on tiagabine pharmacokinetics, but valproate significantly decreased tiagabine binding in vitro from 96.3 to 94.8%, which resulted in an increase of approximately 40% in the free tiagabine concentration. The clinical relevance of this in vitro finding is unknown.
Interaction of GABITRIL with Other Drugs:Cimetidine: Co-administration of cimetidine (800 mg/day) to patients taking tiagabine chronically had no effect on tiagabine pharmacokinetics.
Theophylline: A single 10 mg dose of tiagabine did not affect the pharmacokinetics of theophylline at steady state.
Warfarin: No significant differences were observed in the steady-state pharmacokinetics of R-warfarin or S-warfarin with the addition of tiagabine given as a single dose. Prothrombin times were not affected by tiagabine.
Digoxin: Concomitant administration of tiagabine did not affect the steady-state pharmacokinetics of digoxin or the mean daily trough serum level of digoxin.
Ethanol or Triazolam: No significant differences were observed in the pharmacokinetics of triazolam (0.125 mg) and tiagabine (10 mg) when given together as a single dose. The pharmacokinetics of ethanol were not affected by multiple-dose administration of tiagabine. Tiagabine has shown no clinically important potentiation of the pharmacodynamic effects of triazolam or alcohol. Because of the possible additive effects of drugs that may depress the nervous system, ethanol or triazolam should be used cautiously in combination with tiagabine.
Oral Contraceptives: Multiple dose administration of tiagabine (8 mg/day monotherapy) did not alter the pharmacokinetics of oral contraceptives in healthy women of child-bearing age.
Antipyrine: Antipyrine pharmacokinetics were not significantly different before and after tiagabine multiple-dose regimens. This indicates that tiagabine does not cause induction or inhibition of the hepatic microsomal enzyme systems responsible for the metabolism of antipyrine.
Interaction of GABITRIL with Highly Protein Bound Drugs:In vitro data showed that tiagabine is 96% bound to human plasma protein and therefore has the potential to interact with other highly protein bound compounds. Such an interaction can potentially lead to higher free fractions of either tiagabine or the competing drug.
Carcinogenesis, Mutagenesis, Impairment of FertilityCarcinogenesis: In rats, a study of the potential carcinogenicity associated with tiagabine HCl administration showed that 200 mg/kg/day (plasma exposure [AUC] 36 to 100 times that at the maximum recommended human dosage [MRHD] of 56 mg/day) for 2 years resulted in small, but statistically significant increases in the incidences of hepatocellular adenomas in females and Leydig cell tumors of the testis in males. The significance of these findings relative to the use of GABITRIL in humans is unknown. The no effect dosage for induction of tumors in this study was 100 mg/kg/day (17 to 50 times the exposure at the MRHD). No statistically significant increases in tumor formation were noted in mice at dosages up to 250 mg/kg/day (20 times the MRHD on a mg/m2 basis).
Mutagenesis: Tiagabine produced an increase in structural chromosome aberration frequency in human lymphocytes in vitro in the absence of metabolic activation. No increase in chromosomal aberration frequencies was demonstrated in this assay in the presence of metabolic activation. No evidence of genetic toxicity was found in the in vitro bacterial gene mutation assays, the in vitro HGPRT forward mutation assay in Chinese hamster lung cells, the in vivo mouse micronucleus test, or an unscheduled DNA synthesis assay.
Impairment of Fertility: Studies of male and female rats administered dosages of tiagabine HCl prior to and during mating, gestation, and lactation have shown no impairment of fertility at doses up to 100 mg/kg/day. This dose represents approximately 16 times the maximum recommended human dose (MRHD) of 56 mg/day, based on body surface area (mg/m2). Lowered maternal weight gain and decreased viability and growth in the rat pups were found at 100 mg/kg, but not at 20 mg/kg/day (3 times the MRHD on a mg/m2 basis).
Pregnancy:Pregnancy Category C: Tiagabine has been shown to have adverse effects on embryo-fetal development, including teratogenic effects, when administered to pregnant rats and rabbits at doses greater than the human therapeutic dose.
An increased incidence of malformed fetuses (various craniofacial, appendicular, and visceral defects) and decreased fetal weights were observed following oral administration of 100 mg/kg/day to pregnant rats during the period of organogenesis. This dose is approximately 16 times the maximum recommended human dose (MRHD) of 56 mg/day, based on body surface area (mg/m2). Maternal toxicity (transient weight loss/reduced maternal weight gain during gestation) was associated with this dose, but there is no evidence to suggest that the teratogenic effects were secondary to the maternal effects. No adverse maternal or embryo-fetal effects were seen at a dose of 20 mg/kg/day (3 times the MRHD on a mg/m2 basis).
Decreased maternal weight gain, increased resorption of embryos and increased incidences of fetal variations, but not malformations, were observed when pregnant rabbits were given 25 mg/kg/day (8 times the MRHD on a mg/m2 basis) during organogenesis. The no effect level for maternal and embryo-fetal toxicity in rabbits was 5 mg/kg/day (equivalent to the MRHD on a mg/m2 basis).
When female rats were given tiagabine 100 mg/kg/day during late gestation and throughout parturition and lactation, decreased maternal weight gain during gestation, an increase in stillbirths, and decreased postnatal offspring viability and growth were found. There are no adequate and well-controlled studies in pregnant women. Tiagabine should be used during pregnancy only if clearly needed.
To provide additional information regarding the effects of in utero exposure to GABITRIL, physicians are advised to recommend that pregnant patients taking GABITRIL enroll in the NAAED Pregnancy Registry. This can be done by calling the toll free number 1-888-233-2334, and must be done by patients themselves. Information on the registry can also be found at the website http://www.aedpregnancyregistry.org/.
Use in Nursing Mothers:Studies in rats have shown that tiagabine HCl and/or its metabolites are excreted in the milk of that species. Levels of excretion of tiagabine and/or its metabolites in human milk have not been determined and effects on the nursing infant are unknown. GABITRIL should be used in women who are nursing only if the benefits clearly outweigh the risks.
Pediatric Use:Safety and effectiveness in pediatric patients below the age of 12 have not been established. The pharmacokinetics of tiagabine were evaluated in pediatric patients age 3 to 10 years (see CLINICAL PHARMACOLOGY, Special Populations, Pediatric).
Geriatric Use:Because few patients over the age of 65 (approximately 20) were exposed to GABITRIL during its clinical evaluation, no specific statements about the safety or effectiveness of GABITRIL in this age group could be made.
-
ADVERSE REACTIONS
The most commonly observed adverse events in placebo-controlled, parallel-group, add-on epilepsy trials associated with the use of GABITRIL in combination with other antiepilepsy drugs not seen at an equivalent frequency among placebo-treated patients were dizziness/light-headedness, asthenia/lack of energy, somnolence, nausea, nervousness/irritability, tremor, abdominal pain, and thinking abnormal/difficulty with concentration or attention.
Approximately 21% of the 2531 patients who received GABITRIL in clinical trials of epilepsy discontinued treatment because of an adverse event. The adverse events most commonly associated with discontinuation were dizziness (1.7%), somnolence (1.6%), depression (1.3%), confusion (1.1%), and asthenia (1.1%).
In Studies 1 and 2 (U.S. studies), the double-blind, placebo-controlled, parallel-group, add-on studies, the proportion of patients who discontinued treatment because of adverse events was 11% for the group treated with GABITRIL and 6% for the placebo group. The most common adverse events considered the primary reason for discontinuation were confusion (1.2%), somnolence (1.0%), and ataxia (1.0%).
Adverse Event Incidence in Controlled Clinical TrialsTable 5 lists treatment-emergent signs and symptoms that occurred in at least 1% of patients treated with GABITRIL for epilepsy participating in parallel-group, placebo-controlled trials and were numerically more common in the GABITRIL group. In these studies, either GABITRIL or placebo was added to the patient’s current antiepilepsy drug therapy. Adverse events were usually mild or moderate in intensity.
The prescriber should be aware that these figures, obtained when GABITRIL was added to concurrent antiepilepsy drug therapy, cannot be used to predict the frequency of adverse events in the course of usual medical practice when patient characteristics and other factors may differ from those prevailing during clinical studies. Similarly, the cited frequencies cannot be directly compared with figures obtained from other clinical investigations involving different treatments, uses, or investigators. An inspection of these frequencies, however, does provide the prescribing physician with one basis to estimate the relative contribution of drug and non-drug factors to the adverse event incidences in the population studied.
Table 5: Treatment-Emergent Adverse Event1 Incidence in Parallel-Group,
Placebo-Controlled, Add-On Trials (events in at least 1% of patients treated with GABITRIL and numerically more frequent than in the placebo group)
Body System/COSTART
GABITRIL N=494
Placebo N=275
%
% BODY AS A WHOLE
Abdominal Pain
7
3 Pain (unspecified)
5
3
CARDIOVASCULAR
Vasodilation
2
1
DIGESTIVE
Nausea
11
9 Diarrhea
7
3 Vomiting
7
4 Increased Appetite
2
0 Mouth Ulceration
1
0
MUSCULOSKELETAL
Myasthenia
1
0
NERVOUS SYSTEM
Dizziness
27
15 Asthenia
20
14 Somnolence
18
15 Nervousness
10
3 Tremor
9
3 Difficulty with Concentration/Attention*
6
2 Insomnia
6
4 Ataxia
5
3 Confusion
5
3 Speech Disorder
4
2 Difficulty with Memory*
4
3 Paresthesia
4
2 Depression
3
1 Emotional Lability
3
2 Abnormal Gait
3
2 Hostility
2
1 Nystagmus
2
1 Language Problems*
2
0 Agitation
1
0
RESPIRATORY SYSTEM
Pharyngitis
7
4 Cough Increased
4
3
SKIN AND APPENDAGES
Rash
5
4 Pruritus
2
0 1Patients in these add-on studies were receiving one to three concomitant enzyme-inducing antiepilepsy drugs in addition to GABITRIL or placebo. Patients may have reported multiple adverse experiences; thus, patients may be included in more than one category.
*COSTART term substituted with a more clinically descriptive term.
Other events reported by 1% or more of patients treated with GABITRIL but equally or more frequent in the placebo group were: accidental injury, chest pain, constipation, flu syndrome, rhinitis, anorexia, back pain, dry mouth, flatulence, ecchymosis, twitching, fever, amblyopia, conjunctivitis, urinary tract infection, urinary frequency, infection, dyspepsia, gastroenteritis, nausea and vomiting, myalgia, diplopia, headache, anxiety, acne, sinusitis, and incoordination.
Study 1 was a dose-response study including doses of 32 mg and 56 mg. Table 6 shows adverse events reported at a rate of ≥ 5% in at least one GABITRIL group and more frequent than in the placebo group. Among these events, depression, tremor, nervousness, difficulty with concentration/attention, and perhaps asthenia exhibited a positive relationship to dose.
-
SPL UNCLASSIFIED SECTION
Table 6: Treatment-Emergent Adverse Event Incidence in Study 1† (events in at least 5% of patients treated with GABITRIL 32 or 56 mg and numerically more frequent than in the placebo group)
Body System/COSTART Term
GABITRIL 56 MG
GABITRIL 32 MG
Placebo
(N = 57)
(N = 88)
(N = 91)
%
%
% BODY AS A WHOLE
Accidental Injury
21
15
20 Infection
19
10
12 Flu Syndrome
9
6
3 Pain
7
2
3 Abdominal Pain
5
7
4
DIGESTIVE SYSTEM
Diarrhea
2
10
6
HEMIC AND LYMPHATIC SYSTEM
Ecchymosis
0
6
1
MUSCULOSKELETAL SYSTEM
Myalgia
5
2
3
NERVOUS SYSTEM
Dizziness
28
31
12 Asthenia
23
18
15 Tremor
21
14
1 Somnolence
19
21
17 Nervousness
14
11
6 Difficulty with Concentration/Attention*
14
7
3 Ataxia
9
6
6 Depression
7
1
0 Insomnia
5
6
3 Abnormal Gait
5
5
3 Hostility
5
5
2
RESPIRATORY SYSTEM
Pharyngitis
7
8
6
SPECIAL SENSES
Amblyopia
4
9
8
UROGENITAL SYSTEM
Urinary Tract Infection
5
0
2
† Patients in this study were receiving one to three concomitant enzyme-inducing antiepilepsy drugs in addition to GABITRIL or placebo. Patients may have reported multiple adverse experiences; thus, patients may be included in more than one category.
* COSTART term substituted with a more clinically descriptive term.
The effects of GABITRIL in relation to those of placebo on the incidence of adverse events and the types of adverse events reported were independent of age, weight, and gender. Because only 10% of patients were non-Caucasian in parallel-group, placebo-controlled trials, there is insufficient data to support a statement regarding the distribution of adverse experience reports by race.
Other Adverse Events Observed During All Clinical TrialsGABITRIL has been administered to 2531 patients during all phase 2/3 clinical trials, only some of which were placebo-controlled. During these trials, all adverse events were recorded by the clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals having adverse events, similar types of events were grouped into a smaller number of standardized categories using modified COSTART dictionary terminology. These categories are used in the listing below. The frequencies presented represent the proportion of the 2531 patients exposed to GABITRIL who experienced events of the type cited on at least one occasion while receiving GABITRIL. All reported events are included except those already listed above, events seen only three times or fewer (unless potentially important), events very unlikely to be drug-related, and those too general to be informative. Events are included without regard to determination of a causal relationship to tiagabine.
Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in at least 1/100 patients; infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare events are those occurring in fewer than 1/1000 patients.
Body as a Whole: Frequent: Allergic reaction, chest pain, chills, cyst, neck pain, and malaise. Infrequent: Abscess, cellulitis, facial edema, halitosis, hernia, neck rigidity, neoplasm, pelvic pain, photosensitivity reaction, sepsis, sudden death, and suicide attempt.
Cardiovascular System: Frequent: Hypertension, palpitation, syncope, and tachycardia. Infrequent: Angina pectoris, cerebral ischemia, electrocardiogram abnormal, hemorrhage, hypotension, myocardial infarct, pallor, peripheral vascular disorder, phlebitis, postural hypotension, and thrombophlebitis.
Digestive System: Frequent: Gingivitis and stomatitis. Infrequent: Abnormal stools, cholecystitis, cholelithiasis, dysphagia, eructation, esophagitis, fecal incontinence, gastritis, gastrointestinal hemorrhage, glossitis, gum hyperplasia, hepatomegaly, increased salivation, liver function tests abnormal, melena, periodontal abscess, rectal hemorrhage, thirst, tooth caries, and ulcerative stomatitis.
Endocrine System: Infrequent: Goiter and hypothyroidism.
Hemic and Lymphatic System: Frequent: Lymphadenopathy. Infrequent: Anemia, erythrocytes abnormal, leukopenia, petechia, and thrombocytopenia.
Metabolic and Nutritional: Frequent: Edema, peripheral edema, weight gain, and weight loss. Infrequent: Dehydration, hypercholesteremia, hyperglycemia, hyperlipemia, hypoglycemia, hypokalemia, and hyponatremia.
Musculoskeletal System: Frequent: Arthralgia. Infrequent: Arthritis, arthrosis, bursitis, generalized spasm, and tendinous contracture.
Nervous System: Frequent: Depersonalization, dysarthria, euphoria, hallucination, hyperkinesia, hypertonia, hypesthesia, hypokinesia, hypotonia, migraine, myoclonus, paranoid reaction, personality disorder, reflexes decreased, stupor, twitching, and vertigo. Infrequent: Abnormal dreams, apathy, choreoathetosis, circumoral paresthesia, CNS neoplasm, coma, delusions, dry mouth, dystonia, encephalopathy, hemiplegia, leg cramps, libido increased, libido decreased, movement disorder, neuritis, neurosis, paralysis, peripheral neuritis, psychosis, reflexes increased, and urinary retention.
Respiratory System: Frequent: Bronchitis, dyspnea, epistaxis, and pneumonia. lnfrequent: Apnea, asthma, hemoptysis, hiccups, hyperventilation, laryngitis, respiratory disorder, and voice alteration.
Skin and Appendages: Frequent: Alopecia, dry skin, and sweating. Infrequent: Contact dermatitis, eczema, exfoliative dermatitis, furunculosis, herpes simplex, herpes zoster, hirsutism, maculopapular rash, psoriasis, skin benign neoplasm, skin carcinoma, skin discolorations, skin nodules, skin ulcer, subcutaneous nodule, urticaria, and vesiculobullous rash.
Special Senses: Frequent: Abnormal vision, ear pain, otitis media, and tinnitus. Infrequent: Blepharitis, blindness, deafness, eye pain, hyperacusis, keratoconjunctivitis, otitis externa, parosmia, photophobia, taste loss, taste perversion, and visual field defect.
Urogenital System: Frequent: Dysmenorrhea, dysuria, metrorrhagia, urinary incontinence, and vaginitis. Infrequent: Abortion, amenorrhea, breast enlargement, breast pain, cystitis, fibrocystic breast, hematuria, impotence, kidney failure, menorrhagia, nocturia, papanicolaou smear suspicious, polyuria, pyelonephritis, salpingitis, urethritis, urinary urgency, and vaginal hemorrhage.
- DRUG ABUSE AND DEPENDENCE
-
OVERDOSAGE
Human Overdose Experience: Human experience of acute overdose with GABITRIL is limited. Eleven patients in clinical trials took single doses of GABITRIL up to 800 mg. All patients fully recovered, usually within one day. The most common symptoms reported after overdose included somnolence, impaired consciousness, agitation, confusion, speech difficulty, hostility, depression, weakness, and myoclonus. One patient who ingested a single dose of 400 mg experienced generalized tonic-clonic status epilepticus, which responded to intravenous phenobarbital.
From post-marketing experience, there have been no reports of fatal overdoses involving GABITRIL alone (doses up to 720 mg), although a number of patients required intubation and ventilatory support as part of the management of their status epilepticus. Overdoses involving multiple drugs, including GABITRIL, have resulted in fatal outcomes. Symptoms most often accompanying GABITRIL overdose, alone or in combination with other drugs, have included: seizures including status epilepticus in patients with and without underlying seizure disorders, nonconvulsive status epilepticus, coma, ataxia, confusion, somnolence, drowsiness, impaired speech, agitation, lethargy, myoclonus, spike wave stupor, tremors, disorientation, vomiting, hostility, and temporary paralysis. Respiratory depression was seen in a number of patients, including children, in the context of seizures.
Management of Overdose: There is no specific antidote for overdose with GABITRIL. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage; usual precautions should be observed to maintain the airway. General supportive care of the patient is indicated including monitoring of vital signs and observation of clinical status of the patient. Since tiagabine is mostly metabolized by the liver and is highly protein bound, dialysis is unlikely to be beneficial. A Certified Poison Control Center should be consulted for up to date information on the management of overdose with GABITRIL.
-
DOSAGE AND ADMINISTRATION
General:
The blood level of tiagabine obtained after a given dose depends on whether the patient also is receiving a drug that induces the metabolism of tiagabine. The presence of an inducer means that the attained blood level will be substantially reduced. Dosing should take the presence of concomitant medications into account.
GABITRIL (tiagabine HCl) is recommended as adjunctive therapy for the treatment of partial seizures in patients 12 years and older.
The following dosing recommendations apply to all patients taking GABITRIL:
- GABITRIL is given orally and should be taken with food.
- Do not use a loading dose of GABITRIL.
- Dose titration: Rapid escalation and/or large dose increments of GABITRIL should not be used.
- Missed dose(s): If the patient forgets to take the prescribed dose of GABITRIL at the scheduled time, the patient should not attempt to make up for the missed dose by increasing the next dose. If a patient has missed multiple doses, patient should refer back to his or her physician for possible re-titration as clinically indicated.
- Dosage adjustment of GABITRIL should be considered whenever a change in patient’s enzyme-inducing status occurs as a result of the addition, discontinuation, or dose change of the enzyme-inducing agent.
The following dosing recommendations apply to patients who are already taking enzyme-inducing antiepilepsy drugs (AEDs) (e.g., carbamazepine, phenytoin, primidone, and phenobarbital). Such patients are considered induced patients when administering GABITRIL.
In adolescents 12 to 18 years old, GABITRIL should be initiated at 4 mg once daily. Modification of concomitant antiepilepsy drugs is not necessary, unless clinically indicated. The total daily dose of GABITRIL may be increased by 4 mg at the beginning of Week 2. Thereafter, the total daily dose may be increased by 4 to 8 mg at weekly intervals until clinical response is achieved or up to 32 mg/day. The total daily dose should be given in divided doses two to four times daily. Doses above 32 mg/day have been tolerated in a small number of adolescent patients for a relatively short duration.
In adults, GABITRIL should be initiated at 4 mg once daily. Modification of concomitant antiepilepsy drugs is not necessary, unless clinically indicated. The total daily dose of GABITRIL may be increased by 4 to 8 mg at weekly intervals until clinical response is achieved or, up to 56 mg/day. The total daily dose should be given in divided doses two to four times daily. Doses above 56 mg/day have not been systematically evaluated in adequate and well-controlled clinical trials.
Experience is limited in patients taking total daily doses above 32 mg/day using twice daily dosing. A typical dosing titration regimen for patients taking enzyme-inducing AEDs (induced patients) is provided in Table 7.
Table 7: Typical Dosing Titration Regimen for Patients Already Taking Enzyme-Inducing AEDs
Usual Adult Maintenance Dose in Induced Patients: 32 to 56 mg / day in two to four divided doses
INITIATION AND TITRATION SCHEDULE
TOTAL DAILY DOSE
WEEK 1
Initiate at 4 mg once daily
4 mg / day
WEEK 2
Increase total daily dose by 4 mg
8 mg / day (in two divided doses)
WEEK 3
Increase total daily dose by 4 mg
12 mg / day (in three divided doses)
WEEK 4
Increase total daily dose by 4 mg
16 mg / day (in two to four divided doses)
WEEK 5
Increase total daily dose by 4 to 8 mg
20 to 24 mg / day (in two to four divided doses)
WEEK 6
Increase total daily dose by 4 to 8 mg
24 to 32 mg / day (in two to four divided doses)
Non-Induced Adults and Adolescents 12 Years or Older:The following dosing recommendations apply to patients who are taking only non-enzyme-inducing AEDs. Such patients are considered non-induced patients:
Following a given dose of GABITRIL, the estimated plasma concentration in the non-induced patients is more than twice that in patients receiving enzyme-inducing agents. Use in non-induced patients requires lower doses of GABITRIL. These patients may also require a slower titration of GABITRIL compared to that of induced patients (see CLINICAL PHARMACOLOGY, Pharmacokinetics and PRECAUTIONS, General, Use in Non-Induced Patients).
-
HOW SUPPLIED
GABITRIL tablets are available in four dosage strengths.
• 2 mg orange-peach, round tablets, debossed with
 on one side and 402 on the opposite side, are available in bottles of 30 (NDC 63459-402-30).
on one side and 402 on the opposite side, are available in bottles of 30 (NDC 63459-402-30).• 4 mg yellow, round tablets, debossed with
on one side and 404 on the opposite side, are available in bottles of 30 (NDC 63459-404-30).• 12 mg green, ovaloid tablets, debossed with
on one side and 412 on the opposite side, are available in bottles of 30 (NDC 63459-412-30).• 16 mg blue, ovaloid tablets, debossed with
on one side and 416 on the opposite side, are available in bottles of 30 (NDC 63459-416-30).Recommended Storage: Store tablets at controlled room temperature, between 20-25°C (68-77°F). See USP. Protect from light and moisture.
-
ANIMAL TOXICOLOGY
In repeat dose toxicology studies, dogs receiving daily oral doses of 5 mg/kg/day or greater experienced unexpected CNS effects throughout the study. These effects occurred acutely and included marked sedation and apparent visual impairment which was characterized by a lack of awareness of objects, failure to fix on and follow moving objects, and absence of a blink reaction. Plasma exposures (AUCs) at 5 mg/kg/day were equal to those in humans receiving the maximum recommended daily human dose of 56 mg/day. The effects were reversible upon cessation of treatment and were not associated with any observed structural abnormality. The implications of these findings for humans are unknown.
GAB-013
Revised: September 2010Distributed by:
Cephalon, Inc.
Frazer, PA 19355GABITRIL is a trademark of Cephalon, Inc., or its affiliates.
©1997-2010 Cephalon, Inc., or its affiliates. All rights reserved.
U.S. Patent Nos. 5,354,760; 5,866,590; 5,958,951
Printed in U.S.A.
-
Medication Guide
GABITRIL® (găb-ĭ-trĭl)
(tiagabine hydrochloride)
Tablets
Read this Medication Guide before you start taking GABITRIL and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
What is the most important information I should know about GABITRIL?
Do not stop taking GABITRIL without first talking to your healthcare provider.
Stopping GABITRIL suddenly can cause serious problems.
GABITRIL can cause serious side effects, including:
1. GABITRIL may cause seizures in people who do not have epilepsy. If you do not have a seizure disorder and you take GABITRIL, you may have a seizure or seizures that do not stop (status epilepticus). Call your healthcare provider right away if you have a seizure and you are not taking GABITRIL for epilepsy.
2. Like other antiepileptic drugs, GABITRIL may cause suicidal thoughts or actions in a very small number of people, about 1 in 500. Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions?
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
- Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
Do not stop GABITRIL without first talking to a healthcare provider.
Stopping GABITRIL suddenly can cause serious problems. If you have epilepsy and stop a seizure medicine suddenly, you may have more frequent seizures or seizures that will not stop (status epilepticus).
What is GABITRIL?
GABITRIL is a prescription medicine used with other medicines to treat partial seizures in adults and children age 12 and older.
Who should not take GABITRIL?
Do not take GABITRIL if you are allergic to tiagabine hydrochloride or any of the other ingredients in GABITRIL. See the end of this Medication Guide for a complete list of ingredients in GABITRIL.
What should I tell my healthcare provider before taking GABITRIL?
Before taking GABITRIL, tell your healthcare provider if you:
- have or have had depression, mood problems, or suicidal thoughts or behavior
- have liver problems
- have a history of seizures that do not stop (status epilepticus)
- have any other medical conditions
- are pregnant or plan to become pregnant. It is not known if GABITRIL can
harm your unborn baby. Tell your healthcare provider right away if you become
pregnant while taking GABITRIL. You and your healthcare provider will decide if
you should take GABITRIL while you are pregnant.
- If you become pregnant while taking GABITRIL, talk to your healthcare provider about registering with the North American Antiepileptic Drug (NAAED) Pregnancy Registry. The purpose of this registry is to collect information about the safety of antiepileptic medicines during pregnancy. You can enroll in this registry by calling 1-888-233-2334.
- are breastfeeding or plan to breastfeed. It is not known if GABITRIL passes into breast milk or if it can harm your baby. Talk to your healthcare provider about the best way to feed your baby if you take GABITRIL.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Taking GABITRIL with certain other medicines can cause side effects or affect how well they work. Do not start or stop other medicines without talking to your healthcare provider.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine. Always tell your healthcare provider if there are any changes in any other medicines that you take.
How should I take GABITRIL?
- Take GABITRIL exactly as your healthcare provider tells you.
- Your healthcare provider may change your dose.
- GABITRIL should be taken with food.
- Do not stop taking GABITRIL without talking to your healthcare provider. Stopping GABITRIL suddenly can increase your chances of having a seizure or cause seizures that will not stop.
- If you miss a dose of GABITRIL, do not take 2 doses of GABITRIL at the same time. Contact your healthcare provider if you miss more than one dose.
If you take too much GABITRIL, call your healthcare provider or local Poison Control Center right away.
What should I avoid while taking GABITRIL?
- Do not drink alcohol or take other medicines that make you sleepy or dizzy while taking GABITRIL without first talking to your healthcare provider. Taking GABITRIL with alcohol or drugs that cause sleepiness or dizziness may make your sleepiness or dizziness worse.
- Do not drive, operate heavy machinery, or do other dangerous activities until you know how GABITRIL affects you. GABITRIL can slow your thinking and motor skills.
What are possible side effects of GABITRIL?
See “What is the most important information I should know about GABITRIL?”
GABITRIL may cause other serious side effects including:
- seizures that can happen more often or become worse
- trouble concentrating, problems with speech and language, feeling confused, feeling sleepy and tired, and problems thinking
- weakness all over your body
- eye and vision problems
- serious rash
Call your healthcare provider right away if you have any of the serious side effects listed above.
The most common side effects of GABITRIL include:
- dizziness
- lack of energy
- drowsiness
- nausea
- nervousness
- tremor
- stomach pain
- abnormal thinking
- difficulty with concentration or attention
These are not all the possible side effects of GABITRIL. For more information, ask your healthcare provider or pharmacist. Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store GABITRIL?
- Store GABITRIL between 68°F to 77°F (20°C to 25°C)
- Keep GABITRIL out of light
- Keep GABITRIL tablets dry
Keep GABITRIL and all medicines out of the reach of children.
General Information about GABITRIL
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use GABITRIL for a condition for which it was not prescribed. Do not give GABITRIL to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about GABITRIL. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about GABITRIL that is written for health professionals.
For more information, go to www.GABITRIL.com or call 1-800-896-5855.
What are the ingredients in GABITRIL?
Active Ingredient: tiagabine hydrochloride
Inactive Ingredients: ascorbic acid, colloidal silicon dioxide, crospovidone, hydrogenated vegetable oil wax, hydroxypropyl cellulose, hypromellose, lactose, magnesium stearate, microcrystalline cellulose, pregelatinized starch, stearic acid and titanium dioxide.
In addition:
- the 2 mg tablets contain FD and C Yellow No. 6
- the 4 mg tablets contain D and C Yellow No. 10
- the 12 mg tablets contain D and C Yellow No. 10 and FD and C Blue No. 1
- the 16 mg tablets contain FD and C Blue No. 2
This Medication Guide has been approved by the U.S. Food and Drug Administration.
GABMG-001
September 2010
Rx only
Distributed by:
Cephalon, Inc.
Frazer, PA 19355
U.S. Patent Nos. 5,354,760; 5,866,590; 5,958,951
©2010 Cephalon, Inc., or its affiliates. All rights reserved.
Printed in U.S.A.
Label for 63459-402-30
- PACKAGE LABEL - GABITRIL 12 MG TABLET
-
INGREDIENTS AND APPEARANCE
GABITRIL
tiagabine hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:16590-832(NDC:63459-412) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TIAGABINE HYDROCHLORIDE (UNII: DQH6T6D8OY) (TIAGABINE - UNII:Z80I64HMNP) TIAGABINE 12 mg Inactive Ingredients Ingredient Name Strength ASCORBIC ACID (UNII: PQ6CK8PD0R) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 68401960MK) HYDROXYPROPYL CELLULOSE (UNII: RFW2ET671P) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE (UNII: J2B2A4N98G) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) STEARIC ACID (UNII: 4ELV7Z65AP) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) Product Characteristics Color green Score no score Shape OVAL Size 12mm Flavor Imprint Code 412 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:16590-832-30 30 in 1 BOTTLE 2 NDC:16590-832-45 45 in 1 BOTTLE 3 NDC:16590-832-60 60 in 1 BOTTLE 4 NDC:16590-832-90 90 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020646 11/03/1997 Labeler - STAT RX USA LLC (786036330) Establishment Name Address ID/FEI Business Operations STAT RX USA LLC 786036330 relabel, repack
