Label: VELCADE- bortezomib injection, powder, lyophilized, for solution


- NDC Code(s): 63020-049-01
- Packager: Takeda Pharmaceuticals America, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 25, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VELCADE safely and effectively. See full prescribing information for VELCADE.
VELCADE® (bortezomib) for injection, for subcutaneous or intravenous use
Initial U.S. Approval: 2003INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- For subcutaneous or intravenous use only. Each route of administration has a different reconstituted concentration. Exercise caution when calculating the volume to be administered. (2.1, 2.10)
- The recommended starting dose of VELCADE is 1.3 mg/m2 administered either as a 3 to 5 second bolus intravenous injection or subcutaneous injection. (2.2, 2.4, 2.6)
- Retreatment for Multiple Myeloma: May retreat starting at the last tolerated dose. (2.6)
- Hepatic Impairment: Use a lower starting dose for patients with moderate or severe hepatic impairment. (2.8)
- Dose must be individualized to prevent overdose. (2.10)
DOSAGE FORMS AND STRENGTHS
For injection: Single-dose vial contains 3.5 mg of bortezomib as lyophilized powder for reconstitution and withdrawal of the appropriate individual patient dose. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Peripheral Neuropathy: Manage with dose modification or discontinuation. (2.7) Patients with pre-existing severe neuropathy should be treated with VELCADE only after careful risk-benefit assessment. (2.7, 5.1)
- Hypotension: Use caution when treating patients taking antihypertensives, with a history of syncope, or with dehydration. (5.2)
- Cardiac Toxicity: Worsening of and development of cardiac failure has occurred. Closely monitor patients with existing heart disease or risk factors for heart disease. (5.3)
- Pulmonary Toxicity: Acute respiratory syndromes have occurred. Monitor closely for new or worsening symptoms and consider interrupting VELCADE therapy. (5.4)
- Posterior Reversible Encephalopathy Syndrome: Consider MRI imaging for onset of visual or neurological symptoms; discontinue VELCADE if suspected. (5.5)
- Gastrointestinal Toxicity: Nausea, diarrhea, constipation, and vomiting may require use of antiemetic and antidiarrheal medications or fluid replacement. (5.6)
- Thrombocytopenia and Neutropenia: Monitor complete blood counts regularly throughout treatment. (5.7)
- Tumor Lysis Syndrome: Closely monitor patients with high tumor burden. (5.8)
- Hepatic Toxicity: Monitor hepatic enzymes during treatment. Interrupt VELCADE therapy to assess reversibility. (5.9)
- Thrombotic Microangiopathy: Monitor for signs and symptoms. Discontinue VELCADE if suspected. (5.10)
- Embryo-Fetal Toxicity: VELCADE can cause fetal harm. Advise females of reproductive potential and males with female partners of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.11)
ADVERSE REACTIONS
Most commonly reported adverse reactions (incidence ≥20%) in clinical studies include nausea, diarrhea, thrombocytopenia, neutropenia, peripheral neuropathy, fatigue, neuralgia, anemia, leukopenia, constipation, vomiting, lymphopenia, rash, pyrexia, and anorexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-866-VELCADE or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong CYP3A4 Inhibitors: Closely monitor patients with concomitant use. (7.1)
- Strong CYP3A4 Inducers: Avoid concomitant use. (7.3)
USE IN SPECIFIC POPULATIONS
Patients with diabetes may require close monitoring of blood glucose and adjustment of antidiabetic medication. (8.8)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Multiple Myeloma
1.2 Mantle Cell Lymphoma
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Guidelines
2.2 Dosage in Previously Untreated Multiple Myeloma
2.3 Dose Modification Guidelines for VELCADE When Given in Combination with Melphalan and Prednisone
2.4 Dosage in Previously Untreated Mantle Cell Lymphoma
2.5 Dose Modification Guidelines for VELCADE When Given in Combination with Rituximab, Cyclophosphamide, Doxorubicin and Prednisone
2.6 Dosage and Dose Modifications for Relapsed Multiple Myeloma and Relapsed Mantle Cell Lymphoma
2.7 Dose Modifications for Peripheral Neuropathy
2.8 Dosage in Patients with Hepatic Impairment
2.9 Administration Precautions
2.10 Reconstitution/Preparation for Intravenous and Subcutaneous Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Peripheral Neuropathy
5.2 Hypotension
5.3 Cardiac Toxicity
5.4 Pulmonary Toxicity
5.5 Posterior Reversible Encephalopathy Syndrome (PRES)
5.6 Gastrointestinal Toxicity
5.7 Thrombocytopenia/Neutropenia
5.8 Tumor Lysis Syndrome
5.9 Hepatic Toxicity
5.10 Thrombotic Microangiopathy
5.11 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on VELCADE
7.2 Drugs Without Clinically Significant Interactions with VELCADE
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Patients with Diabetes
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Multiple Myeloma
14.2 Mantle Cell Lymphoma
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Guidelines
VELCADE is for intravenous or subcutaneous use only. Do not administer VELCADE by any other route.
Because each route of administration has a different reconstituted concentration, use caution when calculating the volume to be administered.
The recommended starting dose of VELCADE is 1.3 mg/m2. VELCADE is administered intravenously at a concentration of 1 mg/mL, or subcutaneously at a concentration of 2.5 mg/mL [see Dosage and Administration (2.10)].
VELCADE retreatment may be considered for patients with multiple myeloma who had previously responded to treatment with VELCADE and who have relapsed at least six months after completing prior VELCADE treatment. Treatment may be started at the last tolerated dose [see Dosage and Administration (2.6)].
When administered intravenously, administer VELCADE as a 3 to 5 second bolus intravenous injection.
2.2 Dosage in Previously Untreated Multiple Myeloma
VELCADE is administered in combination with oral melphalan and oral prednisone for 9, six week treatment cycles as shown in Table 1. In Cycles 1 to 4, VELCADE is administered twice weekly (Days 1, 4, 8, 11, 22, 25, 29 and 32). In Cycles 5 to 9, VELCADE is administered once weekly (Days 1, 8, 22 and 29). At least 72 hours should elapse between consecutive doses of VELCADE.
Table 1: Dosage Regimen for Patients with Previously Untreated Multiple Myeloma Twice Weekly VELCADE (Cycles 1 to 4) Week 1 2 3 4 5 6 VELCADE
(1.3 mg/m2)Day 1 -- -- Day 4 Day 8 Day 11 rest period Day 22 Day 25 Day 29 Day 32 rest period Melphalan (9 mg/m2)
Prednisone (60 mg/m2)Day 1 Day 2 Day 3 Day 4 -- -- rest period -- -- -- -- rest period Once Weekly VELCADE (Cycles 5 to 9 when used in combination with Melphalan and Prednisone) Week 1 2 3 4 5 6 VELCADE
(1.3 mg/m2)Day 1 -- -- Day 8 rest period Day 22 Day 29 rest period Melphalan (9 mg/m2)
Prednisone (60 mg/m2)Day 1 Day 2 Day 3 Day 4 -- -- rest period -- -- -- -- rest period 2.3 Dose Modification Guidelines for VELCADE When Given in Combination with Melphalan and Prednisone
Prior to initiating any cycle of therapy with VELCADE in combination with melphalan and prednisone:
- Platelet count should be at least 70 × 109/L and the absolute neutrophil count (ANC) should be at least 1 × 109/L
- Nonhematological toxicities should have resolved to Grade 1 or baseline
Table 2: Dose Modifications During Cycles of Combination VELCADE, Melphalan and Prednisone Therapy Toxicity Dose Modification or Delay Hematological toxicity during a cycle: If prolonged Grade 4 neutropenia or thrombocytopenia, or thrombocytopenia with bleeding is observed in the previous cycle Consider reduction of the melphalan dose by 25% in the next cycle If platelet count is not above 30 × 109/L or ANC is not above 0.75 × 109/L on a VELCADE dosing day (other than Day 1) Withhold VELCADE dose If several VELCADE doses in consecutive cycles are withheld due to toxicity Reduce VELCADE dose by one dose level (from 1.3 mg/m2 to 1 mg/m2, or from 1 mg/m2 to 0.7 mg/m2) Grade 3 or higher nonhematological toxicities Withhold VELCADE therapy until symptoms of toxicity have resolved to Grade 1 or baseline. Then, VELCADE may be reinitiated with one dose level reduction (from 1.3 mg/m2 to 1 mg/m2, or from 1 mg/m2 to 0.7 mg/m2). For VELCADE-related neuropathic pain and/or peripheral neuropathy, hold or modify VELCADE as outlined in Table 5. For information concerning melphalan and prednisone, see manufacturer's prescribing information.
Dose modifications guidelines for peripheral neuropathy are provided [see Dosage and Administration (2.7)].
2.4 Dosage in Previously Untreated Mantle Cell Lymphoma
VELCADE (1.3 mg/m2) is administered intravenously in combination with intravenous rituximab, cyclophosphamide, doxorubicin and oral prednisone (VcR-CAP) for 6, three week treatment cycles as shown in Table 3. VELCADE is administered first followed by rituximab. VELCADE is administered twice weekly for two weeks (Days 1, 4, 8, and 11) followed by a ten day rest period on Days 12 to 21. For patients with a response first documented at Cycle 6, two additional VcR-CAP cycles are recommended. At least 72 hours should elapse between consecutive doses of VELCADE.
Table 3: Dosage Regimen for Patients with Previously Untreated Mantle Cell Lymphoma Twice Weekly VELCADE (6, Three Week Cycles)* Week 1 2 3 - *
- Dosing may continue for two more cycles (for a total of eight cycles) if response is first seen at Cycle 6.
VELCADE (1.3 mg/m2) Day 1 -- -- Day 4 -- Day 8 Day 11 rest period Rituximab (375 mg/m2)
Cyclophosphamide (750 mg/m2)
Doxorubicin (50 mg/m2)Day 1 -- -- -- -- rest period Prednisone (100 mg/m2) Day 1 Day 2 Day 3 Day 4 Day 5 -- -- rest period 2.5 Dose Modification Guidelines for VELCADE When Given in Combination with Rituximab, Cyclophosphamide, Doxorubicin and Prednisone
Prior to the first day of each cycle (other than Cycle 1):
- Platelet count should be at least 100 × 109/L and absolute neutrophil count (ANC) should be at least 1.5 × 109/L
- Hemoglobin should be at least 8 g/dL (at least 4.96 mmol/L)
- Nonhematologic toxicity should have recovered to Grade 1 or baseline
Interrupt VELCADE treatment at the onset of any Grade 3 hematologic or nonhematological toxicities, excluding neuropathy [see Table 5, Warnings and Precautions (5)]. For dose adjustments, see Table 4 below.
Table 4: Dose Modifications on Days 4, 8, and 11 During Cycles of Combination VELCADE, Rituximab, Cyclophosphamide, Doxorubicin and Prednisone Therapy Toxicity Dose Modification or Delay Hematological Toxicity - Grade 3 or higher neutropenia, or a platelet count not at or above 25 × 109/L
Withhold VELCADE therapy for up to 2 weeks until the patient has an ANC at or above 0.75 × 109/L and a platelet count at or above 25 × 109/L. - If, after VELCADE has been withheld, the toxicity does not resolve, discontinue VELCADE.
- If toxicity resolves such that the patient has an ANC at or above 0.75 × 109/L and a platelet count at or above 25 × 109/L, VELCADE dose should be reduced by 1 dose level (from 1.3 mg/m2 to 1 mg/m2, or from 1 mg/m2 to 0.7 mg/m2).
Grade 3 or higher nonhematological toxicities Withhold VELCADE therapy until symptoms of the toxicity have resolved to Grade 2 or better. Then, VELCADE may be reinitiated with one dose level reduction (from 1.3 mg/m2 to 1 mg/m2, or from 1 mg/m2 to 0.7 mg/m2).
For VELCADE-related neuropathic pain and/or peripheral neuropathy, hold or modify VELCADE as outlined in Table 5.For information concerning rituximab, cyclophosphamide, doxorubicin and prednisone, see manufacturer's prescribing information.
2.6 Dosage and Dose Modifications for Relapsed Multiple Myeloma and Relapsed Mantle Cell Lymphoma
VELCADE (1.3 mg/m2/dose) is administered twice weekly for two weeks (Days 1, 4, 8, and 11) followed by a ten day rest period (Days 12 to 21). For extended therapy of more than eight cycles, VELCADE may be administered on the standard schedule or, for relapsed multiple myeloma, on a maintenance schedule of once weekly for four weeks (Days 1, 8, 15, and 22) followed by a 13 day rest period (Days 23 to 35) [see Clinical Studies (14)]. At least 72 hours should elapse between consecutive doses of VELCADE.
Patients with multiple myeloma who have previously responded to treatment with VELCADE (either alone or in combination) and who have relapsed at least six months after their prior VELCADE therapy may be started on VELCADE at the last tolerated dose. Retreated patients are administered VELCADE twice weekly (Days 1, 4, 8, and 11) every three weeks for a maximum of eight cycles. At least 72 hours should elapse between consecutive doses of VELCADE. VELCADE may be administered either as a single agent or in combination with dexamethasone [see Clinical Studies (14.1)].
VELCADE therapy should be withheld at the onset of any Grade 3 nonhematological or Grade 4 hematological toxicities excluding neuropathy as discussed below [see Warnings and Precautions (5)]. Once the symptoms of the toxicity have resolved, VELCADE therapy may be reinitiated at a 25% reduced dose (1.3 mg/m2/dose reduced to 1 mg/m2/dose; 1 mg/m2/dose reduced to 0.7 mg/m2/dose).
For dose modifications guidelines for peripheral neuropathy, see section 2.7.
2.7 Dose Modifications for Peripheral Neuropathy
Starting VELCADE subcutaneously may be considered for patients with pre-existing or at high risk of peripheral neuropathy. Patients with pre-existing severe neuropathy should be treated with VELCADE only after careful risk-benefit assessment.
Patients experiencing new or worsening peripheral neuropathy during VELCADE therapy may require a decrease in the dose and/or a less dose-intense schedule.
For dose or schedule modification guidelines for patients who experience VELCADE-related neuropathic pain and/or peripheral neuropathy, see Table 5.
Table 5: Recommended Dose Modification for VELCADE-Related Neuropathic Pain and/or Peripheral Sensory or Motor Neuropathy Severity of Peripheral Neuropathy Signs and Symptoms* Modification of Dose and Regimen - *
- Grading based on NCI Common Terminology Criteria CTCAE v4.0
- †
- Instrumental ADL: refers to preparing meals, shopping for groceries or clothes, using telephone, managing money, etc.
- ‡
- Self care ADL: refers to bathing, dressing and undressing, feeding self, using the toilet, taking medications, and not bedridden
Grade 1 (asymptomatic; loss of deep tendon reflexes or paresthesia) without pain or loss of function No action Grade 1 with pain or Grade 2 (moderate symptoms; limiting instrumental Activities of Daily Living (ADL)†) Reduce VELCADE to 1 mg/m2 Grade 2 with pain or Grade 3 (severe symptoms; limiting self care ADL‡) Withhold VELCADE therapy until toxicity resolves. When toxicity resolves reinitiate with a reduced dose of VELCADE at 0.7 mg/m2 once per week. Grade 4 (life-threatening consequences; urgent intervention indicated) Discontinue VELCADE 2.8 Dosage in Patients with Hepatic Impairment
Do not adjust the starting dose for patients with mild hepatic impairment.
Start patients with moderate or severe hepatic impairment at a reduced dose of 0.7 mg/m2 per injection during the first cycle, and consider subsequent dose escalation to 1 mg/m2 or further dose reduction to 0.5 mg/m2 based on patient tolerance (see Table 6) [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
Table 6: Recommended Starting Dose Modification for VELCADE in Patients with Hepatic Impairment Bilirubin Level SGOT (AST) Levels Modification of Starting Dose Abbreviations: SGOT = serum glutamic oxaloacetic transaminase; AST = aspartate aminotransferase; ULN = upper limit of the normal range. Mild Less than or equal to 1× ULN More than ULN None More than 1× to 1.5× ULN Any None Moderate More than 1.5× to 3× ULN Any Reduce VELCADE to 0.7 mg/m2 in the first cycle. Consider dose escalation to 1 mg/m2 or further dose reduction to 0.5 mg/m2 in subsequent cycles based on patient tolerability. Severe More than 3× ULN Any 2.9 Administration Precautions
The drug quantity contained in one vial (3.5 mg) may exceed the usual dose required. Caution should be used in calculating the dose to prevent overdose [see Dosage and Administration (2.10)].
When administered subcutaneously, sites for each injection (thigh or abdomen) should be rotated. New injections should be given at least one inch from an old site and never into areas where the site is tender, bruised, erythematous, or indurated.
If local injection site reactions occur following VELCADE administration subcutaneously, a less concentrated VELCADE solution (1 mg/mL instead of 2.5 mg/mL) may be administered subcutaneously [see Dosage and Administration (2.10)]. Alternatively, consider use of the intravenous route of administration [see Dosage and Administration (2.10)].
VELCADE is a hazardous drug. Follow applicable special handling and disposal procedures.1
2.10 Reconstitution/Preparation for Intravenous and Subcutaneous Administration
Use proper aseptic technique. Reconstitute only with 0.9% sodium chloride. The reconstituted product should be a clear and colorless solution.
Different volumes of 0.9% sodium chloride are used to reconstitute the product for the different routes of administration. The reconstituted concentration of bortezomib for subcutaneous administration (2.5 mg/mL) is greater than the reconstituted concentration of bortezomib for intravenous administration (1 mg/mL). Because each route of administration has a different reconstituted concentration, use caution when calculating the volume to be administered [see Dosage and Administration (2.9)].
For each 3.5 mg single-dose vial of bortezomib, reconstitute with the following volume of 0.9% sodium chloride based on route of administration (Table 7):
Table 7: Reconstitution Volumes and Final Concentration for Intravenous and Subcutaneous Administration Route of Administration Bortezomib
(mg/vial)Diluent
(0.9% Sodium Chloride)Final Bortezomib Concentration
(mg/mL)Intravenous 3.5 mg 3.5 mL 1 mg/mL Subcutaneous 3.5 mg 1.4 mL 2.5 mg/mL Dose must be individualized to prevent overdosage. After determining patient body surface area (BSA) in square meters, use the following equations to calculate the total volume (mL) of reconstituted VELCADE to be administered:
-
Intravenous Administration [1 mg/mL concentration]
VELCADE dose (mg/m2) × patient BSA (m2) =Total VELCADE volume (mL) to be administered 1 mg/mL -
Subcutaneous Administration [2.5 mg/mL concentration]
VELCADE dose (mg/m2) × patient BSA (m2) =Total VELCADE volume (mL) to be administered 2.5 mg/mL
Stickers that indicate the route of administration are provided with each VELCADE vial. These stickers should be placed directly on the syringe of VELCADE once VELCADE is prepared to help alert practitioners of the correct route of administration for VELCADE.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. If any discoloration or particulate matter is observed, the reconstituted product should not be used.
Stability
Unopened vials of VELCADE are stable until the date indicated on the package when stored in the original package protected from light.
VELCADE contains no antimicrobial preservative. Administer reconstituted VELCADE within eight hours of preparation. When reconstituted as directed, VELCADE may be stored at 25°C (77°F). The reconstituted material may be stored in the original vial and/or the syringe prior to administration. The product may be stored for up to eight hours in a syringe; however, total storage time for the reconstituted material must not exceed eight hours when exposed to normal indoor lighting.
-
3 DOSAGE FORMS AND STRENGTHS
For injection: Each single-dose vial of VELCADE contains 3.5 mg of bortezomib as a sterile lyophilized white to off-white powder for reconstitution and withdrawal of the appropriate individual patient dose [see Dosage and Administration (2.10)].
-
4 CONTRAINDICATIONS
VELCADE is contraindicated in patients with hypersensitivity (not including local reactions) to bortezomib, boron, or mannitol. Reactions have included anaphylactic reactions [see Adverse Reactions (6.1)].
VELCADE is contraindicated for intrathecal administration. Fatal events have occurred with intrathecal administration of VELCADE.
-
5 WARNINGS AND PRECAUTIONS
5.1 Peripheral Neuropathy
VELCADE treatment causes a peripheral neuropathy that is predominantly sensory; however, cases of severe sensory and motor peripheral neuropathy have been reported. Patients with pre-existing symptoms (numbness, pain or a burning feeling in the feet or hands) and/or signs of peripheral neuropathy may experience worsening peripheral neuropathy (including ≥Grade 3) during treatment with VELCADE. Patients should be monitored for symptoms of neuropathy, such as a burning sensation, hyperesthesia, hypoesthesia, paresthesia, discomfort, neuropathic pain or weakness. In the Phase 3 relapsed multiple myeloma trial comparing VELCADE subcutaneous vs intravenous, the incidence of Grade ≥2 peripheral neuropathy was 24% for subcutaneous and 39% for intravenous. Grade ≥3 peripheral neuropathy occurred in 6% of patients in the subcutaneous treatment group, compared with 15% in the intravenous treatment group [see Adverse Reactions (6.1)]. Starting VELCADE subcutaneously may be considered for patients with pre-existing or at high risk of peripheral neuropathy.
Patients experiencing new or worsening peripheral neuropathy during VELCADE therapy may require a decrease in the dose and/or a less dose-intense schedule [see Dosage and Administration (2.7)]. In the VELCADE vs dexamethasone Phase 3 relapsed multiple myeloma study, improvement in or resolution of peripheral neuropathy was reported in 48% of patients with ≥Grade 2 peripheral neuropathy following dose adjustment or interruption. Improvement in or resolution of peripheral neuropathy was reported in 73% of patients who discontinued due to Grade 2 neuropathy or who had ≥Grade 3 peripheral neuropathy in the Phase 2 multiple myeloma studies. The long-term outcome of peripheral neuropathy has not been studied in mantle cell lymphoma.
5.2 Hypotension
The incidence of hypotension (postural, orthostatic, and hypotension NOS) was 8% [see Adverse Reactions (6.1)]. These events are observed throughout therapy. Patients with a history of syncope, patients receiving medications known to be associated with hypotension, and patients who are dehydrated may be at increased risk of hypotension. Management of orthostatic/postural hypotension may include adjustment of antihypertensive medications, hydration, and administration of mineralocorticoids and/or sympathomimetics.
5.3 Cardiac Toxicity
Acute development or exacerbation of congestive heart failure and new onset of decreased left ventricular ejection fraction have occurred during VELCADE therapy, including reports in patients with no risk factors for decreased left ventricular ejection fraction [see Adverse Reactions (6.1)]. Patients with risk factors for, or existing heart disease should be frequently monitored. In the relapsed multiple myeloma study of VELCADE vs dexamethasone, the incidence of any treatment-related cardiac disorder was 8% and 5% in the VELCADE and dexamethasone groups, respectively. The incidence of adverse reactions suggestive of heart failure (acute pulmonary edema, pulmonary edema, cardiac failure, congestive cardiac failure, cardiogenic shock) was ≤1% for each individual reaction in the VELCADE group. In the dexamethasone group the incidence was ≤1% for cardiac failure and congestive cardiac failure; there were no reported reactions of acute pulmonary edema, pulmonary edema, or cardiogenic shock. There have been isolated cases of QT-interval prolongation in clinical studies; causality has not been established.
5.4 Pulmonary Toxicity
Acute Respiratory Distress Syndrome (ARDS) and acute diffuse infiltrative pulmonary disease of unknown etiology such as pneumonitis, interstitial pneumonia, lung infiltration have occurred in patients receiving VELCADE. Some of these events have been fatal.
In a clinical trial, the first two patients given high-dose cytarabine (2 g/m2 per day) by continuous infusion with daunorubicin and VELCADE for relapsed acute myelogenous leukemia died of ARDS early in the course of therapy.
There have been reports of pulmonary hypertension associated with VELCADE administration in the absence of left heart failure or significant pulmonary disease.
In the event of new or worsening cardiopulmonary symptoms, consider interrupting VELCADE until a prompt and comprehensive diagnostic evaluation is conducted.
5.5 Posterior Reversible Encephalopathy Syndrome (PRES)
Posterior Reversible Encephalopathy Syndrome (PRES; formerly termed Reversible Posterior Leukoencephalopathy Syndrome (RPLS)) has occurred in patients receiving VELCADE. PRES is a rare, reversible, neurological disorder which can present with seizure, hypertension, headache, lethargy, confusion, blindness, and other visual and neurological disturbances. Brain imaging, preferably MRI (Magnetic Resonance Imaging), is used to confirm the diagnosis. In patients developing PRES, discontinue VELCADE. The safety of reinitiating VELCADE therapy in patients previously experiencing PRES is not known.
5.6 Gastrointestinal Toxicity
VELCADE treatment can cause nausea, diarrhea, constipation, and vomiting [see Adverse Reactions (6.1)] sometimes requiring use of antiemetic and antidiarrheal medications. Ileus can occur. Fluid and electrolyte replacement should be administered to prevent dehydration. Interrupt VELCADE for severe symptoms.
5.7 Thrombocytopenia/Neutropenia
VELCADE is associated with thrombocytopenia and neutropenia that follow a cyclical pattern with nadirs occurring following the last dose of each cycle and typically recovering prior to initiation of the subsequent cycle. The cyclical pattern of platelet and neutrophil decreases and recovery remain consistent in the studies of multiple myeloma and mantle cell lymphoma, with no evidence of cumulative thrombocytopenia or neutropenia in the treatment regimens studied.
Monitor complete blood counts (CBC) frequently during treatment with VELCADE. Measure platelet counts prior to each dose of VELCADE. Adjust dose/schedule for thrombocytopenia [see Dosage and Administration (2.6)]. Gastrointestinal and intracerebral hemorrhage has occurred during thrombocytopenia in association with VELCADE. Support with transfusions and supportive care, according to published guidelines.
In the single agent, relapsed multiple myeloma study of VELCADE vs dexamethasone, the mean platelet count nadir measured was approximately 40% of baseline. The severity of thrombocytopenia related to pretreatment platelet count is shown in Table 8. The incidence of bleeding (≥Grade 3) was 2% on the VELCADE arm and was <1% in the dexamethasone arm.
Table 8: Severity of Thrombocytopenia Related to Pretreatment Platelet Count in the Relapsed Multiple Myeloma Study of VELCADE vs Dexamethasone Pretreatment Platelet Count* Number of Patients
(N=331)†Number (%) of Patients with Platelet Count <10,000/µL Number (%) of Patients with Platelet Count 10,000 to 25,000/µL ≥75,000/µL 309 8 (3%) 36 (12%) ≥50,000/µL to <75,000/µL 14 2 (14%) 11 (79%) ≥10,000/µL to <50,000/µL 7 1 (14%) 5 (71%) In the combination study of VELCADE with rituximab, cyclophosphamide, doxorubicin and prednisone (VcR-CAP) in previously untreated mantle cell lymphoma patients, the incidence of thrombocytopenia (≥Grade 4) was 32% vs 1% for the rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) arm as shown in Table 12. The incidence of bleeding events (≥Grade 3) was 1.7% in the VcR-CAP arm (four patients) and was 1.2% in the R-CHOP arm (three patients).
Platelet transfusions were given to 23% of the patients in the VcR-CAP arm and 3% of the patients in the R-CHOP arm.
The incidence of neutropenia (≥Grade 4) was 70% in the VcR-CAP arm and was 52% in the R-CHOP arm. The incidence of febrile neutropenia (≥Grade 4) was 5% in the VcR-CAP arm and was 6% in the R-CHOP arm. Myeloid growth factor support was provided at a rate of 78% in the VcR-CAP arm and 61% in the R-CHOP arm.
5.8 Tumor Lysis Syndrome
Tumor lysis syndrome has been reported with VELCADE therapy. Patients at risk of tumor lysis syndrome are those with high tumor burden prior to treatment. Monitor patients closely and take appropriate precautions.
5.9 Hepatic Toxicity
Cases of acute liver failure have been reported in patients receiving multiple concomitant medications and with serious underlying medical conditions. Other reported hepatic reactions include hepatitis, increases in liver enzymes, and hyperbilirubinemia. Interrupt VELCADE therapy to assess reversibility. There is limited rechallenge information in these patients.
5.10 Thrombotic Microangiopathy
Cases, sometimes fatal, of thrombotic microangiopathy, including thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS), have been reported in the postmarketing setting in patients who received VELCADE. Monitor for signs and symptoms of TTP/HUS. If the diagnosis is suspected, stop VELCADE and evaluate. If the diagnosis of TTP/HUS is excluded, consider restarting VELCADE. The safety of reinitiating VELCADE therapy in patients previously experiencing TTP/HUS is not known.
5.11 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, VELCADE can cause fetal harm when administered to a pregnant woman. Bortezomib administered to rabbits during organogenesis at a dose approximately 0.5 times the clinical dose of 1.3 mg/m2 based on body surface area caused postimplantation loss and a decreased number of live fetuses [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with VELCADE and for seven months following treatment. Advise males with female partners of reproductive potential to use effective contraception during treatment with VELCADE and for four months following treatment. If VELCADE is used during pregnancy or if the patient becomes pregnant during VELCADE treatment, the patient should be apprised of the potential risk to the fetus [see Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are also discussed in other sections of the labeling:
- Peripheral Neuropathy [see Warnings and Precautions (5.1)]
- Hypotension [see Warnings and Precautions (5.2)]
- Cardiac Toxicity [see Warnings and Precautions (5.3)]
- Pulmonary Toxicity [see Warnings and Precautions (5.4)]
- Posterior Reversible Encephalopathy Syndrome (PRES) [see Warnings and Precautions (5.5)]
- Gastrointestinal Toxicity [see Warnings and Precautions (5.6)]
- Thrombocytopenia/Neutropenia [see Warnings and Precautions (5.7)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.8)]
- Hepatic Toxicity [see Warnings and Precautions (5.9)]
- Thrombotic Microangiopathy [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Summary of Clinical Trial in Patients with Previously Untreated Multiple Myeloma
Table 9 describes safety data from 340 patients with previously untreated multiple myeloma who received VELCADE (1.3 mg/m2) administered intravenously in combination with melphalan (9 mg/m2) and prednisone (60 mg/m2) in a prospective randomized study.
The safety profile of VELCADE in combination with melphalan/prednisone is consistent with the known safety profiles of both VELCADE and melphalan/prednisone.
Table 9: Most Commonly Reported Adverse Reactions (≥10% in the VELCADE, Melphalan and Prednisone Arm) with Grades 3 and ≥4 Intensity in the Previously Untreated Multiple Myeloma Study VELCADE, Melphalan and Prednisone Melphalan and Prednisone (n=340) (n=337) Body System Total Toxicity Grade, n (%) Total Toxicity Grade, n (%) Adverse Reaction n (%) 3 ≥4 n (%) 3 ≥4 - *
- Represents High Level Term Peripheral Neuropathies NEC
Blood and Lymphatic System Disorders Thrombocytopenia 164 (48) 60 (18) 57 (17) 140 (42) 48 (14) 39 (12) Neutropenia 160 (47) 101 (30) 33 (10) 143 (42) 77 (23) 42 (12) Anemia 109 (32) 41 (12) 4 (1) 156 (46) 61 (18) 18 (5) Leukopenia 108 (32) 64 (19) 8 (2) 93 (28) 53 (16) 11 (3) Lymphopenia 78 (23) 46 (14) 17 (5) 51 (15) 26 (8) 7 (2) Gastrointestinal Disorders Nausea 134 (39) 10 (3) 0 70 (21) 1 (<1) 0 Diarrhea 119 (35) 19 (6) 2 (1) 20 (6) 1 (<1) 0 Vomiting 87 (26) 13 (4) 0 41 (12) 2 (1) 0 Constipation 77 (23) 2 (1) 0 14 (4) 0 0 Abdominal pain upper 34 (10) 1 (<1) 0 20 (6) 0 0 Nervous System Disorders Peripheral neuropathy* 156 (46) 42 (12) 2 (1) 4 (1) 0 0 Neuralgia 117 (34) 27 (8) 2 (1) 1 (<1) 0 0 Paresthesia 42 (12) 6 (2) 0 4 (1) 0 0 General Disorders and Administration Site Conditions Fatigue 85 (25) 19 (6) 2 (1) 48 (14) 4 (1) 0 Asthenia 54 (16) 18 (5) 0 23 (7) 3 (1) 0 Pyrexia 53 (16) 4 (1) 0 19 (6) 1 (<1) 1 (<1) Infections and Infestations Herpes Zoster 39 (11) 11 (3) 0 9 (3) 4 (1) 0 Metabolism and Nutrition Disorders Anorexia 64 (19) 6 (2) 0 19 (6) 0 0 Skin and Subcutaneous Tissue Disorders Rash 38 (11) 2 (1) 0 7 (2) 0 0 Psychiatric Disorders Insomnia 35 (10) 1 (<1) 0 21 (6) 0 0 Relapsed Multiple Myeloma Randomized Study of VELCADE vs Dexamethasone
The safety data described below and in Table 10 reflect exposure to either VELCADE (n=331) or dexamethasone (n=332) in a study of patients with relapsed multiple myeloma. VELCADE was administered intravenously at doses of 1.3 mg/m2 twice weekly for two out of three weeks (21 day cycle). After eight, 21 day cycles patients continued therapy for three, 35 day cycles on a weekly schedule. Duration of treatment was up to 11 cycles (nine months) with a median duration of six cycles (4.1 months). For inclusion in the trial, patients must have had measurable disease and one to three prior therapies. There was no upper age limit for entry. Creatinine clearance could be as low as 20 mL/min and bilirubin levels as high as 1.5 times the upper limit of normal. The overall frequency of adverse reactions was similar in men and women, and in patients <65 and ≥65 years of age. Most patients were Caucasian [see Clinical Studies (14.1)].
Among the 331 VELCADE-treated patients, the most commonly reported (>20%) adverse reactions overall were nausea (52%), diarrhea (52%), fatigue (39%), peripheral neuropathies (35%), thrombocytopenia (33%), constipation (30%), vomiting (29%), and anorexia (21%). The most commonly reported (>20%) adverse reaction reported among the 332 patients in the dexamethasone group was fatigue (25%). Eight percent (8%) of patients in the VELCADE-treated arm experienced a Grade 4 adverse reaction; the most common reactions were thrombocytopenia (4%) and neutropenia (2%). Nine percent (9%) of dexamethasone-treated patients experienced a Grade 4 adverse reaction. All individual dexamethasone-related Grade 4 adverse reactions were less than 1%.
Serious Adverse Reactions and Adverse Reactions Leading to Treatment Discontinuation in the Relapsed Multiple Myeloma Study of VELCADE vs Dexamethasone
Serious adverse reactions are defined as any reaction that results in death, is life-threatening, requires hospitalization or prolongs a current hospitalization, results in a significant disability, or is deemed to be an important medical event. A total of 80 (24%) patients from the VELCADE treatment arm experienced a serious adverse reaction during the study, as did 83 (25%) dexamethasone-treated patients. The most commonly reported serious adverse reactions in the VELCADE treatment arm were diarrhea (3%), dehydration, herpes zoster, pyrexia, nausea, vomiting, dyspnea, and thrombocytopenia (2% each). In the dexamethasone treatment group, the most commonly reported serious adverse reactions were pneumonia (4%), hyperglycemia (3%), pyrexia, and psychotic disorder (2% each).
A total of 145 patients, including 84 (25%) of 331 patients in the VELCADE treatment group and 61 (18%) of 332 patients in the dexamethasone treatment group were discontinued from treatment due to adverse reactions. Among the 331 VELCADE-treated patients, the most commonly reported adverse reaction leading to discontinuation was peripheral neuropathy (8%). Among the 332 patients in the dexamethasone group, the most commonly reported adverse reactions leading to treatment discontinuation were psychotic disorder and hyperglycemia (2% each).
Four deaths were considered to be VELCADE-related in this relapsed multiple myeloma study: one case each of cardiogenic shock, respiratory insufficiency, congestive heart failure and cardiac arrest. Four deaths were considered dexamethasone-related: two cases of sepsis, one case of bacterial meningitis, and one case of sudden death at home.
Most Commonly Reported Adverse Reactions in the Relapsed Multiple Myeloma Study of VELCADE vs Dexamethasone
The most common adverse reactions from the relapsed multiple myeloma study are shown in Table 10. All adverse reactions with incidence ≥10% in the VELCADE arm are included.
Table 10: Most Commonly Reported Adverse Reactions (≥10% in VELCADE Arm), with Grades 3 and 4 Intensity in the Relapsed Multiple Myeloma Study of VELCADE vs Dexamethasone (N=663) VELCADE
(N=331)Dexamethasone
(N=332)Adverse Reactions All Grade 3 Grade 4 All Grade 3 Grade 4 - *
- Represents High Level Term Peripheral Neuropathies NEC
Any Adverse Reactions 324 (98) 193 (58) 28 (8) 297 (89) 110 (33) 29 (9) Nausea 172 (52) 8 (2) 0 31 (9) 0 0 Diarrhea NOS 171 (52) 22 (7) 0 36 (11) 2 (<1) 0 Fatigue 130 (39) 15 (5) 0 82 (25) 8 (2) 0 Peripheral neuropathies* 115 (35) 23 (7) 2 (<1) 14 (4) 0 1 (<1) Thrombocytopenia 109 (33) 80 (24) 12 (4) 11 (3) 5 (2) 1 (<1) Constipation 99 (30) 6 (2) 0 27 (8) 1 (<1) 0 Vomiting NOS 96 (29) 8 (2) 0 10 (3) 1 (<1) 0 Anorexia 68 (21) 8 (2) 0 8 (2) 1 (<1) 0 Pyrexia 66 (20) 2 (<1) 0 21 (6) 3 (<1) 1 (<1) Paresthesia 64 (19) 5 (2) 0 24 (7) 0 0 Anemia NOS 63 (19) 20 (6) 1 (<1) 21 (6) 8 (2) 0 Headache NOS 62 (19) 3 (<1) 0 23 (7) 1 (<1) 0 Neutropenia 58 (18) 37 (11) 8 (2) 1 (<1) 1 (<1) 0 Rash NOS 43 (13) 3 (<1) 0 7 (2) 0 0 Appetite decreased NOS 36 (11) 0 0 12 (4) 0 0 Dyspnea NOS 35 (11) 11 (3) 1 (<1) 37 (11) 7 (2) 1 (<1) Abdominal pain NOS 35 (11) 5 (2) 0 7 (2) 0 0 Weakness 34 (10) 10 (3) 0 28 (8) 8 (2) 0 Safety Experience from the Phase 2 Open-Label Extension Study in Relapsed Multiple Myeloma
In the Phase 2 extension study of 63 patients, no new cumulative or new long-term toxicities were observed with prolonged VELCADE treatment. These patients were treated for a total of 5.3 to 23 months, including time on VELCADE in the prior VELCADE study [see Clinical Studies (14.1)].
Safety Experience from the Phase 3 Open-Label Study of VELCADE Subcutaneous vs Intravenous in Relapsed Multiple Myeloma
The safety and efficacy of VELCADE administered subcutaneously were evaluated in one Phase 3 study at the recommended dose of 1.3 mg/m2. This was a randomized, comparative study of VELCADE subcutaneous vs intravenous in 222 patients with relapsed multiple myeloma. The safety data described below and in Table 11 reflect exposure to either VELCADE subcutaneous (N=147) or VELCADE intravenous (N=74) [see Clinical Studies (14.1)].
Table 11: Most Commonly Reported Adverse Reactions (≥10%), with Grade 3 and ≥4 Intensity in the Relapsed Multiple Myeloma Study (N=221) of VELCADE Subcutaneous vs Intravenous Subcutaneous Intravenous (N=147) (N=74) Body System Total Toxicity Grade, n (%) Total Toxicity Grade, n (%) Adverse Reaction n (%) 3 ≥4 n (%) 3 ≥4 Note: Safety population: 147 patients in the subcutaneous treatment group and 74 patients in the intravenous treatment group who received at least one dose of study medication - *
- Represents High Level Term Peripheral Neuropathies NEC
Blood and Lymphatic System Disorders Anemia 28 (19) 8 (5) 0 17 (23) 3 (4) 0 Leukopenia 26 (18) 8 (5) 0 15 (20) 4 (5) 1 (1) Neutropenia 34 (23) 15 (10) 4 (3) 20 (27) 10 (14) 3 (4) Thrombocytopenia 44 (30) 7 (5) 5 (3) 25 (34) 7 (9) 5 (7) Gastrointestinal Disorders Diarrhea 28 (19) 1 (1) 0 21 (28) 3 (4) 0 Nausea 24 (16) 0 0 10 (14) 0 0 Vomiting 13 (9) 3 (2) 0 8 (11) 0 0 General Disorders and Administration Site Conditions Asthenia 10 (7) 1 (1) 0 12 (16) 4 (5) 0 Fatigue 11 (7) 3 (2) 0 11 (15) 3 (4) 0 Pyrexia 18 (12) 0 0 6 (8) 0 0 Nervous System Disorders Neuralgia 34 (23) 5 (3) 0 17 (23) 7 (9) 0 Peripheral neuropathies* 55 (37) 8 (5) 1 (1) 37 (50) 10 (14) 1 (1) In general, safety data were similar for the subcutaneous and intravenous treatment groups. Differences were observed in the rates of some Grade ≥3 adverse reactions. Differences of ≥5% were reported in neuralgia (3% subcutaneous vs 9% intravenous), peripheral neuropathies (6% subcutaneous vs 15% intravenous), neutropenia (13% subcutaneous vs 18% intravenous), and thrombocytopenia (8% subcutaneous vs 16% intravenous).
A local reaction was reported in 6% of patients in the subcutaneous group, mostly redness. Only two (1%) patients were reported as having severe reactions, one case of pruritus and one case of redness. Local reactions led to reduction in injection concentration in one patient and drug discontinuation in one patient. Local reactions resolved in a median of six days.
Dose reductions occurred due to adverse reactions in 31% of patients in the subcutaneous treatment group compared with 43% of the intravenously-treated patients. The most common adverse reactions leading to a dose reduction included peripheral sensory neuropathy (17% in the subcutaneous treatment group compared with 31% in the intravenous treatment group); and neuralgia (11% in the subcutaneous treatment group compared with 19% in the intravenous treatment group).
Serious Adverse Reactions and Adverse Reactions Leading to Treatment Discontinuation in the Relapsed Multiple Myeloma Study of VELCADE Subcutaneous vs Intravenous
The incidence of serious adverse reactions was similar for the subcutaneous treatment group (20%) and the intravenous treatment group (19%). The most commonly reported serious adverse reactions in the subcutaneous treatment arm were pneumonia and pyrexia (2% each). In the intravenous treatment group, the most commonly reported serious adverse reactions were pneumonia, diarrhea, and peripheral sensory neuropathy (3% each).
In the subcutaneous treatment group, 27 patients (18%) discontinued study treatment due to an adverse reaction compared with 17 patients (23%) in the intravenous treatment group. Among the 147 subcutaneously-treated patients, the most commonly reported adverse reactions leading to discontinuation were peripheral sensory neuropathy (5%) and neuralgia (5%). Among the 74 patients in the intravenous treatment group, the most commonly reported adverse reactions leading to treatment discontinuation were peripheral sensory neuropathy (9%) and neuralgia (9%).
Two patients (1%) in the subcutaneous treatment group and one (1%) patient in the intravenous treatment group died due to an adverse reaction during treatment. In the subcutaneous group the causes of death were one case of pneumonia and one case of sudden death. In the intravenous group the cause of death was coronary artery insufficiency.
Safety Experience from the Clinical Trial in Patients with Previously Untreated Mantle Cell Lymphoma
Table 12 describes safety data from 240 patients with previously untreated mantle cell lymphoma who received VELCADE (1.3 mg/m2) administered intravenously in combination with rituximab (375 mg/m2), cyclophosphamide (750 mg/m2), doxorubicin (50 mg/m2), and prednisone (100 mg/m2) (VcR-CAP) in a prospective randomized study.
Infections were reported for 31% of patients in the VcR-CAP arm and 23% of the patients in the comparator (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone [R-CHOP]) arm, including the predominant preferred term of pneumonia (VcR-CAP 8% vs R-CHOP 5%).
Table 12: Most Commonly Reported Adverse Reactions (≥5%) with Grades 3 and ≥4 Intensity in the Previously Untreated Mantle Cell Lymphoma Study VcR-CAP
(n=240)R-CHOP
(n=242)Body System
Adverse ReactionsAll
n (%)Toxicity Grade 3
n (%)Toxicity Grade ≥4
n (%)All
n (%)Toxicity Grade 3
n (%)Toxicity Grade ≥4
n (%)Key: R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; VcR-CAP = VELCADE, rituximab, cyclophosphamide, doxorubicin, and prednisone. - *
- Represents High Level Term Peripheral Neuropathies NEC
Blood and Lymphatic System Disorders Neutropenia 209 (87) 32 (13) 168 (70) 172 (71) 31 (13) 125 (52) Leukopenia 116 (48) 34 (14) 69 (29) 87 (36) 39 (16) 27 (11) Anemia 106 (44) 27 (11) 4 (2) 71 (29) 23 (10) 4 (2) Thrombocytopenia 172 (72) 59 (25) 76 (32) 42 (17) 9 (4) 3 (1) Febrile neutropenia 41 (17) 24 (10) 12 (5) 33 (14) 17 (7) 15 (6) Lymphopenia 68 (28) 25 (10) 36 (15) 28 (12) 15 (6) 2 (1) Nervous System Disorders Peripheral neuropathy* 71 (30) 17 (7) 1 (<1) 65 (27) 10 (4) 0 Hypoesthesia 14 (6) 3 (1) 0 13 (5) 0 0 Paresthesia 14 (6) 2 (1) 0 11 (5) 0 0 Neuralgia 25 (10) 9 (4) 0 1 (<1) 0 0 General Disorders and Administration Site Conditions Fatigue 43 (18) 11 (5) 1 (<1) 38 (16) 5 (2) 0 Pyrexia 48 (20) 7 (3) 0 23 (10) 5 (2) 0 Asthenia 29 (12) 4 (2) 1 (<1) 18 (7) 1 (<1) 0 Edema peripheral 16 (7) 1 (<1) 0 13 (5) 0 0 Gastrointestinal Disorders Nausea 54 (23) 1 (<1) 0 28 (12) 0 0 Constipation 42 (18) 1 (<1) 0 22 (9) 2 (1) 0 Stomatitis 20 (8) 2 (1) 0 19 (8) 0 1 (<1) Diarrhea 59 (25) 11 (5) 0 11 (5) 3 (1) 1 (<1) Vomiting 24 (10) 1 (<1) 0 8 (3) 0 0 Abdominal distension 13 (5) 0 0 4 (2) 0 0 Infections and Infestations Pneumonia 20 (8) 8 (3) 5 (2) 11 (5) 5 (2) 3 (1) Skin and Subcutaneous Tissue Disorders Alopecia 31 (13) 1 (<1) 1 (<1) 33 (14) 4 (2) 0 Metabolism and Nutrition Disorders Hyperglycemia 10 (4) 1 (<1) 0 17 (7) 10 (4) 0 Decreased appetite 36 (15) 2 (1) 0 15 (6) 1 (<1) 0 Vascular Disorders Hypertension 15 (6) 1 (<1) 0 3 (1) 0 0 Psychiatric Disorders Insomnia 16 (7) 1 (<1) 0 8 (3) 0 0 The incidence of herpes zoster reactivation was 4.6% in the VcR-CAP arm and 0.8% in the R-CHOP arm. Antiviral prophylaxis was mandated by protocol amendment.
The incidences of Grade ≥3 bleeding events were similar between the two arms (four patients in the VcR-CAP arm and three patients in the R-CHOP arm). All of the Grade ≥3 bleeding events resolved without sequelae in the VcR-CAP arm.
Adverse reactions leading to discontinuation occurred in 8% of patients in VcR-CAP group and 6% of patients in R-CHOP group. In the VcR-CAP group, the most commonly reported adverse reaction leading to discontinuation was peripheral sensory neuropathy (1%; three patients). The most commonly reported adverse reaction leading to discontinuation in the R-CHOP group was febrile neutropenia (<1%; two patients).
Integrated Summary of Safety (Relapsed Multiple Myeloma and Relapsed Mantle Cell Lymphoma)
Safety data from Phase 2 and 3 studies of single agent VELCADE 1.3 mg/m2/dose twice weekly for two weeks followed by a ten day rest period in 1163 patients with previously-treated multiple myeloma (N=1008) and previously-treated mantle cell lymphoma (N=155) were integrated and tabulated. This analysis does not include data from the Phase 3 open-label study of VELCADE subcutaneous vs intravenous in relapsed multiple myeloma. In the integrated studies, the safety profile of VELCADE was similar in patients with multiple myeloma and mantle cell lymphoma.
In the integrated analysis, the most commonly reported (>20%) adverse reactions were nausea (49%), diarrhea (46%), asthenic conditions including fatigue (41%) and weakness (11%), peripheral neuropathies (38%), thrombocytopenia (32%), vomiting (28%), constipation (25%), and pyrexia (21%). Eleven percent (11%) of patients experienced at least one episode of ≥Grade 4 toxicity, most commonly thrombocytopenia (4%) and neutropenia (2%).
In the Phase 2 relapsed multiple myeloma clinical trials of VELCADE administered intravenously, local skin irritation was reported in 5% of patients, but extravasation of VELCADE was not associated with tissue damage.
Serious Adverse Reactions and Adverse Reactions Leading to Treatment Discontinuation in the Integrated Summary of Safety
A total of 26% of patients experienced a serious adverse reaction during the studies. The most commonly reported serious adverse reactions included diarrhea, vomiting and pyrexia (3% each), nausea, dehydration, and thrombocytopenia (2% each) and pneumonia, dyspnea, peripheral neuropathies, and herpes zoster (1% each).
Adverse reactions leading to discontinuation occurred in 22% of patients. The reasons for discontinuation included peripheral neuropathy (8%), and fatigue, thrombocytopenia, and diarrhea (2% each).
In total, 2% of the patients died and the cause of death was considered by the investigator to be possibly related to study drug: including reports of cardiac arrest, congestive heart failure, respiratory failure, renal failure, pneumonia and sepsis.
Most Commonly Reported Adverse Reactions in the Integrated Summary of Safety
The most common adverse reactions are shown in Table 13. All adverse reactions occurring at ≥10% are included. In the absence of a randomized comparator arm, it is often not possible to distinguish between adverse events that are drug-caused and those that reflect the patient's underlying disease. Please see the discussion of specific adverse reactions that follows.
Table 13: Most Commonly Reported (≥10% Overall) Adverse Reactions in Integrated Analyses of Relapsed Multiple Myeloma and Relapsed Mantle Cell Lymphoma Studies Using the 1.3 mg/m2 Dose (N=1163) All Patients
(N=1163)Multiple Myeloma
(N=1008)Mantle Cell Lymphoma
(N=155)Adverse Reactions All ≥Grade 3 All ≥Grade 3 All ≥Grade 3 - *
- Represents High Level Term Peripheral Neuropathies NEC
Nausea 567 (49) 36 (3) 511 (51) 32 (3) 56 (36) 4 (3) Diarrhea NOS 530 (46) 83 (7) 470 (47) 72 (7) 60 (39) 11 (7) Fatigue 477 (41) 86 (7) 396 (39) 71 (7) 81 (52) 15 (10) Peripheral neuropathies* 443 (38) 129 (11) 359 (36) 110 (11) 84 (54) 19 (12) Thrombocytopenia 369 (32) 295 (25) 344 (34) 283 (28) 25 (16) 12 (8) Vomiting NOS 321 (28) 44 (4) 286 (28) 40 (4) 35 (23) 4 (3) Constipation 296 (25) 17 (1) 244 (24) 14 (1) 52 (34) 3 (2) Pyrexia 249 (21) 16 (1) 233 (23) 15 (1) 16 (10) 1 (<1) Anorexia 227 (20) 19 (2) 205 (20) 16 (2) 22 (14) 3 (2) Anemia NOS 209 (18) 65 (6) 190 (19) 63 (6) 19 (12) 2 (1) Headache NOS 175 (15) 8 (<1) 160 (16) 8 (<1) 15 (10) 0 Neutropenia 172 (15) 121 (10) 164 (16) 117 (12) 8 (5) 4 (3) Rash NOS 156 (13) 8 (<1) 120 (12) 4 (<1) 36 (23) 4 (3) Paresthesia 147 (13) 9 (<1) 136 (13) 8 (<1) 11 (7) 1 (<1) Dizziness (excl vertigo) 129 (11) 13 (1) 101 (10) 9 (<1) 28 (18) 4 (3) Weakness 124 (11) 31 (3) 106 (11) 28 (3) 18 (12) 3 (2) Description of Selected Adverse Reactions from the Integrated Phase 2 and 3 Relapsed Multiple Myeloma and Phase 2 Relapsed Mantle Cell Lymphoma Studies
Gastrointestinal Toxicity
A total of 75% of patients experienced at least one gastrointestinal disorder. The most common gastrointestinal disorders included nausea, diarrhea, constipation, vomiting, and appetite decreased. Other gastrointestinal disorders included dyspepsia and dysgeusia. Grade 3 adverse reactions occurred in 14% of patients; ≥Grade 4 adverse reactions were ≤1%. Gastrointestinal adverse reactions were considered serious in 7% of patients. Four percent (4%) of patients discontinued due to a gastrointestinal adverse reaction. Nausea was reported more often in patients with multiple myeloma (51%) compared to patients with mantle cell lymphoma (36%).
Thrombocytopenia
Across the studies, VELCADE-associated thrombocytopenia was characterized by a decrease in platelet count during the dosing period (Days 1 to 11) and a return toward baseline during the ten day rest period during each treatment cycle. Overall, thrombocytopenia was reported in 32% of patients. Thrombocytopenia was Grade 3 in 22%, ≥Grade 4 in 4%, and serious in 2% of patients, and the reaction resulted in VELCADE discontinuation in 2% of patients [see Warnings and Precautions (5.7)]. Thrombocytopenia was reported more often in patients with multiple myeloma (34%) compared to patients with mantle cell lymphoma (16%). The incidence of ≥Grade 3 thrombocytopenia also was higher in patients with multiple myeloma (28%) compared to patients with mantle cell lymphoma (8%).
Peripheral Neuropathy
Overall, peripheral neuropathies occurred in 38% of patients. Peripheral neuropathy was Grade 3 for 11% of patients and ≥Grade 4 for <1% of patients. Eight percent (8%) of patients discontinued VELCADE due to peripheral neuropathy. The incidence of peripheral neuropathy was higher among patients with mantle cell lymphoma (54%) compared to patients with multiple myeloma (36%).
In the VELCADE vs dexamethasone Phase 3 relapsed multiple myeloma study, among the 62 VELCADE-treated patients who experienced ≥Grade 2 peripheral neuropathy and had dose adjustments, 48% had improved or resolved with a median of 3.8 months from first onset.
In the Phase 2 relapsed multiple myeloma studies, among the 30 patients who experienced Grade 2 peripheral neuropathy resulting in discontinuation or who experienced ≥Grade 3 peripheral neuropathy, 73% reported improvement or resolution with a median time of 47 days to improvement of one grade or more from the last dose of VELCADE.
Hypotension
The incidence of hypotension (postural, orthostatic and hypotension NOS) was 8% in patients treated with VELCADE. Hypotension was Grade 1 or 2 in the majority of patients and Grade 3 in 2% and ≥Grade 4 in <1%. Two percent (2%) of patients had hypotension reported as a serious adverse reaction, and 1% discontinued due to hypotension. The incidence of hypotension was similar in patients with multiple myeloma (8%) and those with mantle cell lymphoma (9%). In addition, <1% of patients experienced hypotension associated with a syncopal reaction.
Neutropenia
Neutrophil counts decreased during the VELCADE dosing period (Days 1 to 11) and returned toward baseline during the ten day rest period during each treatment cycle. Overall, neutropenia occurred in 15% of patients and was Grade 3 in 8% of patients and ≥Grade 4 in 2%. Neutropenia was reported as a serious adverse reaction in <1% of patients and <1% of patients discontinued due to neutropenia. The incidence of neutropenia was higher in patients with multiple myeloma (16%) compared to patients with mantle cell lymphoma (5%). The incidence of ≥Grade 3 neutropenia also was higher in patients with multiple myeloma (12%) compared to patients with mantle cell lymphoma (3%).
Asthenic Conditions (Fatigue, Malaise, Weakness, Asthenia)
Asthenic conditions were reported in 54% of patients. Fatigue was reported as Grade 3 in 7% and ≥Grade 4 in <1% of patients. Asthenia was reported as Grade 3 in 2% and ≥Grade 4 in <1% of patients. Two percent (2%) of patients discontinued treatment due to fatigue and <1% due to weakness and asthenia. Asthenic conditions were reported in 53% of patients with multiple myeloma and 59% of patients with mantle cell lymphoma.
Pyrexia
Pyrexia (>38°C) was reported as an adverse reaction for 21% of patients. The reaction was Grade 3 in 1% and ≥Grade 4 in <1%. Pyrexia was reported as a serious adverse reaction in 3% of patients and led to VELCADE discontinuation in <1% of patients. The incidence of pyrexia was higher among patients with multiple myeloma (23%) compared to patients with mantle cell lymphoma (10%). The incidence of ≥Grade 3 pyrexia was 1% in patients with multiple myeloma and <1% in patients with mantle cell lymphoma.
Herpes Virus Infection
Consider using antiviral prophylaxis in subjects being treated with VELCADE. In the randomized studies in previously untreated and relapsed multiple myeloma, herpes zoster reactivation was more common in subjects treated with VELCADE (ranging between 6 to 11%) than in the control groups (3 to 4%). Herpes simplex was seen in 1 to 3% in subjects treated with VELCADE and 1 to 3% in the control groups. In the previously untreated multiple myeloma study, herpes zoster virus reactivation in the VELCADE, melphalan and prednisone arm was less common in subjects receiving prophylactic antiviral therapy (3%) than in subjects who did not receive prophylactic antiviral therapy (17%).
Retreatment in Relapsed Multiple Myeloma
A single-arm trial was conducted in 130 patients with relapsed multiple myeloma to determine the efficacy and safety of retreatment with intravenous VELCADE. The safety profile of patients in this trial is consistent with the known safety profile of VELCADE-treated patients with relapsed multiple myeloma as demonstrated in Tables 10, 11, and 13; no cumulative toxicities were observed upon retreatment. The most common adverse drug reaction was thrombocytopenia which occurred in 52% of the patients. The incidence of ≥Grade 3 thrombocytopenia was 24%. Peripheral neuropathy occurred in 28% of patients, with the incidence of ≥Grade 3 peripheral neuropathy reported at 6%. The incidence of serious adverse reactions was 12.3%. The most commonly reported serious adverse reactions were thrombocytopenia (3.8%), diarrhea (2.3%), and herpes zoster and pneumonia (1.5% each).
Adverse reactions leading to discontinuation occurred in 13% of patients. The reasons for discontinuation included peripheral neuropathy (5%) and diarrhea (3%).
Two deaths considered to be VELCADE-related occurred within 30 days of the last VELCADE dose; one in a patient with cerebrovascular accident and one in a patient with sepsis.
Additional Adverse Reactions from Clinical Studies
The following clinically important serious adverse reactions that are not described above have been reported in clinical trials in patients treated with VELCADE administered as monotherapy or in combination with other chemotherapeutics. These studies were conducted in patients with hematological malignancies and in solid tumors.
Blood and Lymphatic System Disorders: Anemia, disseminated intravascular coagulation, febrile neutropenia, lymphopenia, leukopenia
Cardiac Disorders: Angina pectoris, atrial fibrillation aggravated, atrial flutter, bradycardia, sinus arrest, cardiac amyloidosis, complete atrioventricular block, myocardial ischemia, myocardial infarction, pericarditis, pericardial effusion, Torsades de pointes, ventricular tachycardia
Ear and Labyrinth Disorders: Hearing impaired, vertigo
Eye Disorders: Diplopia and blurred vision, conjunctival infection, irritation
Gastrointestinal Disorders: Abdominal pain, ascites, dysphagia, fecal impaction, gastroenteritis, gastritis hemorrhagic, hematemesis, hemorrhagic duodenitis, ileus paralytic, large intestinal obstruction, paralytic intestinal obstruction, peritonitis, small intestinal obstruction, large intestinal perforation, stomatitis, melena, pancreatitis acute, oral mucosal petechiae, gastroesophageal reflux
General Disorders and Administration Site Conditions: Chills, edema, edema peripheral, injection site erythema, neuralgia, injection site pain, irritation, malaise, phlebitis
Hepatobiliary Disorders: Cholestasis, hepatic hemorrhage, hyperbilirubinemia, portal vein thrombosis, hepatitis, liver failure
Immune System Disorders: Anaphylactic reaction, drug hypersensitivity, immune complex mediated hypersensitivity, angioedema, laryngeal edema
Infections and Infestations: Aspergillosis, bacteremia, bronchitis, urinary tract infection, herpes viral infection, listeriosis, nasopharyngitis, pneumonia, respiratory tract infection, septic shock, toxoplasmosis, oral candidiasis, sinusitis, catheter-related infection
Injury, Poisoning and Procedural Complications: Catheter-related complication, skeletal fracture, subdural hematoma
Investigations: Weight decreased
Metabolism and Nutrition Disorders: Dehydration, hypocalcemia, hyperuricemia, hypokalemia, hyperkalemia, hyponatremia, hypernatremia
Musculoskeletal and Connective Tissue Disorders: Arthralgia, back pain, bone pain, myalgia, pain in extremity
Nervous System Disorders: Ataxia, coma, dizziness, dysarthria, dysesthesia, dysautonomia, encephalopathy, cranial palsy, grand mal convulsion, headache, hemorrhagic stroke, motor dysfunction, neuralgia, spinal cord compression, paralysis, postherpetic neuralgia, transient ischemic attack
Psychiatric Disorders: Agitation, anxiety, confusion, insomnia, mental status change, psychotic disorder, suicidal ideation
Renal and Urinary Disorders: Calculus renal, bilateral hydronephrosis, bladder spasm, hematuria, hemorrhagic cystitis, urinary incontinence, urinary retention, renal failure (acute and chronic), glomerular nephritis proliferative
Respiratory, Thoracic and Mediastinal Disorders: Acute respiratory distress syndrome, aspiration pneumonia, atelectasis, chronic obstructive airways disease exacerbated, cough, dysphagia, dyspnea, dyspnea exertional, epistaxis, hemoptysis, hypoxia, lung infiltration, pleural effusion, pneumonitis, respiratory distress, pulmonary hypertension
Skin and Subcutaneous Tissue Disorders: Urticaria, face edema, rash (which may be pruritic), leukocytoclastic vasculitis, pruritus
Vascular Disorders: Cerebrovascular accident, cerebral hemorrhage, deep venous thrombosis, hypertension, peripheral embolism, pulmonary embolism, pulmonary hypertension
6.2 Postmarketing Experience
The following adverse reactions have been identified from the worldwide postmarketing experience with VELCADE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Cardiac Disorders: Cardiac tamponade
Ear and Labyrinth Disorders: Deafness bilateral
Eye Disorders: Optic neuropathy, blindness, chalazion/blepharitis
Gastrointestinal Disorders: Ischemic colitis
Infections and Infestations: Progressive multifocal leukoencephalopathy (PML), ophthalmic herpes, herpes meningoencephalitis
Nervous System Disorders: Posterior reversible encephalopathy syndrome (PRES, formerly RPLS), Guillain-Barré syndrome, demyelinating polyneuropathy
Respiratory, Thoracic and Mediastinal Disorders: Acute diffuse infiltrative pulmonary disease
Skin and Subcutaneous Tissue Disorders: Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), acute febrile neutrophilic dermatosis (Sweet's syndrome)
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on VELCADE
Strong CYP3A4 Inducers
Coadministration with a strong CYP3A4 inducer decreases the exposure of bortezomib [see Clinical Pharmacology (12.3)] which may decrease VELCADE efficacy. Avoid coadministration with strong CYP3A4 inducers.
Strong CYP3A4 Inhibitors
Coadministration with a strong CYP3A4 inhibitor increases the exposure of bortezomib [see Clinical Pharmacology (12.3)] which may increase the risk of VELCADE toxicities. Monitor patients for signs of bortezomib toxicity and consider a bortezomib dose reduction if bortezomib must be given in combination with strong CYP3A4 inhibitors.
7.2 Drugs Without Clinically Significant Interactions with VELCADE
No clinically significant drug interactions have been observed when VELCADE was coadministered with dexamethasone, omeprazole, or melphalan in combination with prednisone [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action [see Clinical Pharmacology (12.1)] and findings in animals, VELCADE can cause fetal harm when administered to a pregnant woman. There are no studies with the use of VELCADE in pregnant women to inform drug-associated risks. Bortezomib caused embryo-fetal lethality in rabbits at doses lower than the clinical dose (see Data). Advise pregnant women of the potential risk to the fetus.
Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Bortezomib was not teratogenic in nonclinical developmental toxicity studies in rats and rabbits at the highest dose tested (0.075 mg/kg; 0.5 mg/m2 in the rat and 0.05 mg/kg; 0.6 mg/m2 in the rabbit) when administered during organogenesis. These dosages are approximately 0.5 times the clinical dose of 1.3 mg/m2 based on body surface area.
Bortezomib caused embryo-fetal lethality in rabbits at doses lower than the clinical dose (approximately 0.5 times the clinical dose of 1.3 mg/m2 based on body surface area). Pregnant rabbits given bortezomib during organogenesis at a dose of 0.05 mg/kg (0.6 mg/m2) experienced significant postimplantation loss and decreased number of live fetuses. Live fetuses from these litters also showed significant decreases in fetal weight.
8.2 Lactation
Risk Summary
There are no data on the presence of bortezomib or its metabolites in human milk, the effects of the drug on the breastfed child, or the effects of the drug on milk production. Because many drugs are excreted in human milk and because the potential for serious adverse reactions in a breastfed child from VELCADE is unknown, advise nursing women not to breastfeed during treatment with VELCADE and for two months after treatment.
8.3 Females and Males of Reproductive Potential
Based on its mechanism of action and findings in animals, VELCADE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Conduct pregnancy testing in females of reproductive potential prior to initiating VELCADE treatment.
Contraception
Infertility
Based on the mechanism of action and findings in animals, VELCADE may have an effect on either male or female fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness have not been established in pediatric patients.
The activity and safety of VELCADE in combination with intensive reinduction chemotherapy was evaluated in pediatric and young adult patients with lymphoid malignancies (pre-B cell ALL 77%, 16% with T-cell ALL, and 7% T-cell lymphoblastic lymphoma (LL)), all of whom relapsed within 36 months of initial diagnosis in a single-arm multicenter, nonrandomized cooperative group trial. An effective reinduction multiagent chemotherapy regimen was administered in three blocks. Block 1 included vincristine, prednisone, doxorubicin and pegaspargase; Block 2 included cyclophosphamide, etoposide and methotrexate; Block 3 included high-dose cytosine arabinoside and asparaginase. VELCADE was administered at a dose of 1.3 mg/m2 as a bolus intravenous injection on Days 1, 4, 8, and 11 of Block 1 and Days 1, 4, and 8 of Block 2. There were 140 patients with ALL or LL enrolled and evaluated for safety. The median age was ten years (range: 1 to 26), 57% were male, 70% were white, 14% were black, 4% were Asian, 2% were American Indian/Alaska Native, 1% were Pacific Islander.
The activity was evaluated in a prespecified subset of the first 60 evaluable patients enrolled on the study with pre-B ALL ≤21 years and relapsed <36 months from diagnosis. The complete remission (CR) rate at day 36 was compared to that in a historical control set of patients who had received the identical backbone therapy without VELCADE. There was no evidence that the addition of VELCADE had any impact on the CR rate.
No new safety concerns were observed when VELCADE was added to a chemotherapy backbone regimen as compared with a historical control group in which the backbone regimen was given without VELCADE.
The BSA-normalized clearance of bortezomib in pediatric patients was similar to that observed in adults.
8.5 Geriatric Use
Of the 669 patients enrolled in the relapsed multiple myeloma study, 245 (37%) were 65 years of age or older: 125 (38%) on the VELCADE arm and 120 (36%) on the dexamethasone arm. Median time to progression and median duration of response for patients ≥65 were longer on VELCADE compared to dexamethasone [5.5 mo vs 4.3 mo, and 8.0 mo vs 4.9 mo, respectively]. On the VELCADE arm, 40% (n=46) of evaluable patients aged ≥65 experienced response (CR + PR) vs 18% (n=21) on the dexamethasone arm. The incidence of Grade 3 and 4 events was 64%, 78% and 75% for VELCADE patients ≤50, 51 to 64 and ≥65 years old, respectively [see Adverse Reactions (6.1), Clinical Studies (14.1)].
No overall differences in safety or effectiveness were observed between patients ≥age 65 and younger patients receiving VELCADE; but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No starting dosage adjustment of VELCADE is recommended for patients with renal impairment. In patients requiring dialysis, VELCADE should be administered after the dialysis procedure [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No starting dosage adjustment of VELCADE is recommended for patients with mild hepatic impairment (total bilirubin ≤1× ULN and AST >ULN, or total bilirubin >1 to 1.5× ULN and any AST). The exposure of bortezomib is increased in patients with moderate (total bilirubin ≥1.5 to 3× ULN and any AST) and severe (total bilirubin >3× ULN and any AST) hepatic impairment. Reduce the starting dose in patients with moderate or severe hepatic impairment [see Dosage and Administration (2.8), Clinical Pharmacology (12.3)].
8.8 Patients with Diabetes
During clinical trials, hypoglycemia and hyperglycemia were reported in diabetic patients receiving oral hypoglycemics. Patients on oral antidiabetic agents receiving VELCADE treatment may require close monitoring of their blood glucose levels and adjustment of the dose of their antidiabetic medication.
-
10 OVERDOSAGE
There is no known specific antidote for VELCADE overdosage. In humans, fatal outcomes following the administration of more than twice the recommended therapeutic dose have been reported, which were associated with the acute onset of symptomatic hypotension (5.2) and thrombocytopenia (5.7). In the event of an overdosage, the patient's vital signs should be monitored and appropriate supportive care given.
Studies in monkeys and dogs showed that intravenous bortezomib doses as low as two times the recommended clinical dose on a mg/m2 basis were associated with increases in heart rate, decreases in contractility, hypotension, and death. In dog studies, a slight increase in the corrected QT interval was observed at doses resulting in death. In monkeys, doses of 3.0 mg/m2 and greater (approximately twice the recommended clinical dose) resulted in hypotension starting at one hour postadministration, with progression to death in 12 to 14 hours following drug administration.
-
11 DESCRIPTION
VELCADE® for Injection, a proteasome inhibitor, contains bortezomib which is an antineoplastic agent. Bortezomib is a modified dipeptidyl boronic acid. The chemical name for bortezomib, the monomeric boronic acid, is [(1R)-3-methyl-1-[[(2S)-1-oxo-3-phenyl-2-[(pyrazinylcarbonyl) amino]propyl]amino]butyl] boronic acid.
Bortezomib has the following chemical structure:
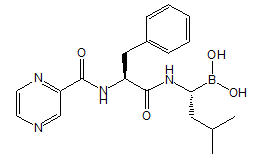
The molecular weight is 384.24. The molecular formula is C19H25BN4O4. The solubility of bortezomib, as the monomeric boronic acid, in water is 3.3 to 3.8 mg/mL in a pH range of 2 to 6.5.
VELCADE is available for intravenous injection or subcutaneous use. Each single-dose vial contains 3.5 mg of bortezomib as a sterile lyophilized powder. It also contains the inactive ingredient: 35 mg mannitol, USP. The product is provided as a mannitol boronic ester which, in reconstituted form, consists of the mannitol ester in equilibrium with its hydrolysis product, the monomeric boronic acid. The drug substance exists in its cyclic anhydride form as a trimeric boroxine.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bortezomib is a reversible inhibitor of the chymotrypsin-like activity of the 26S proteasome in mammalian cells. The 26S proteasome is a large protein complex that degrades ubiquitinated proteins. The ubiquitin-proteasome pathway plays an essential role in regulating the intracellular concentration of specific proteins, thereby maintaining homeostasis within cells. Inhibition of the 26S proteasome prevents this targeted proteolysis, which can affect multiple signaling cascades within the cell. This disruption of normal homeostatic mechanisms can lead to cell death. Experiments have demonstrated that bortezomib is cytotoxic to a variety of cancer cell types in vitro. Bortezomib causes a delay in tumor growth in vivo in nonclinical tumor models, including multiple myeloma.
12.2 Pharmacodynamics
Following twice weekly administration of 1 mg/m2 and 1.3 mg/m2 bortezomib doses, the maximum inhibition of 20S proteasome activity (relative to baseline) in whole blood was observed five minutes after drug administration. Comparable maximum inhibition of 20S proteasome activity was observed between 1 and 1.3 mg/m2 doses. Maximal inhibition ranged from 70% to 84% and from 73% to 83% for the 1 mg/m2 and 1.3 mg/m2 dose regimens, respectively.
12.3 Pharmacokinetics
Following intravenous administration of 1 mg/m2 and 1.3 mg/m2 doses, the mean maximum plasma concentrations of bortezomib (Cmax) after the first dose (Day 1) were 57 and 112 ng/mL, respectively. When administered twice weekly, the mean maximum observed plasma concentrations ranged from 67 to 106 ng/mL for the 1 mg/m2 dose and 89 to 120 ng/mL for the 1.3 mg/m2 dose.
Following an intravenous bolus or subcutaneous injection of a 1.3 mg/m2 dose to patients with multiple myeloma, the total systemic exposure after repeat dose administration (AUClast) was equivalent for subcutaneous and intravenous administration. The AUClast geometric mean ratio (90% confidence interval) was 0.99 (0.80 to 1.23). The Cmax after subcutaneous administration (20.4 ng/mL) was lower than after intravenous administration (223 ng/mL) with repeat dose administration.
Distribution
The mean distribution volume of bortezomib ranged from approximately 498 to 1884 L/m2 following single- or repeat-dose administration of 1 mg/m2 or 1.3 mg/m2 to patients with multiple myeloma. The binding of bortezomib to human plasma proteins averaged 83% over the concentration range of 100 to 1000 ng/mL.
Elimination
The mean elimination half-life of bortezomib upon multiple dosing ranged from 40 to 193 hours after the 1 mg/m2 dose and 76 to 108 hours after the 1.3 mg/m2 dose. The mean total body clearances were 102 and 112 L/h following the first dose for doses of 1 mg/m2 and 1.3 mg/m2, respectively, and ranged from 15 to 32 L/h following subsequent doses for doses of 1 and 1.3 mg/m2, respectively.
Specific Populations
No clinically significant differences in the pharmacokinetics of bortezomib were observed based on age, sex, or renal impairment (including patients administered VELCADE after dialysis). The effect of race on bortezomib pharmacokinetics is unknown.
Patients with Hepatic Impairment
Following administration of bortezomib doses ranging from 0.5 to 1.3 mg/m2, mild (total bilirubin ≤1× ULN and AST >ULN, or total bilirubin >1 to 1.5× ULN and any AST) hepatic impairment did not alter dose-normalized bortezomib AUC when compared to patients with normal hepatic function. Dose normalized mean bortezomib AUC increased by approximately 60% in patients with moderate (total bilirubin >1.5 to 3× ULN and any AST) or severe (total bilirubin >3× ULN and any AST) hepatic impairment. A lower starting dose is recommended in patients with moderate or severe hepatic impairment.
Drug Interaction Studies
Clinical Studies
No clinically significant differences in bortezomib pharmacokinetics were observed when coadministered with dexamethasone (weak CYP3A4 inducer), omeprazole (strong CYP2C19 inhibitor), or melphalan in combination with prednisone.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with bortezomib.
Bortezomib showed clastogenic activity (structural chromosomal aberrations) in the in vitro chromosomal aberration assay using Chinese hamster ovary cells. Bortezomib was not genotoxic when tested in the in vitro mutagenicity assay (Ames test) and in vivo micronucleus assay in mice.
Fertility studies with bortezomib were not performed but evaluation of reproductive tissues has been performed in the general toxicity studies. In the six month rat toxicity study, degenerative effects in the ovary were observed at doses ≥0.3 mg/m2 (one-fourth of the recommended clinical dose), and degenerative changes in the testes occurred at 1.2 mg/m2.
13.2 Animal Toxicology and/or Pharmacology
Cardiovascular Toxicity
Studies in monkeys showed that administration of dosages approximately twice the recommended clinical dose resulted in heart rate elevations, followed by profound progressive hypotension, bradycardia, and death 12 to 14 hours postdose. Doses ≥1.2 mg/m2 induced dose-proportional changes in cardiac parameters. Bortezomib has been shown to distribute to most tissues in the body, including the myocardium. In a repeated dosing toxicity study in the monkey, myocardial hemorrhage, inflammation, and necrosis were also observed.
Chronic Administration
In animal studies at a dose and schedule similar to that recommended for patients (twice weekly dosing for two weeks followed by one week rest), toxicities observed included severe anemia and thrombocytopenia, and gastrointestinal, neurological and lymphoid system toxicities. Neurotoxic effects of bortezomib in animal studies included axonal swelling and degeneration in peripheral nerves, dorsal spinal roots, and tracts of the spinal cord. Additionally, multifocal hemorrhage and necrosis in the brain, eye, and heart were observed.
-
14 CLINICAL STUDIES
14.1 Multiple Myeloma
Randomized, Open-Label Clinical Study in Patients with Previously Untreated Multiple Myeloma
A prospective, international, randomized (1:1), open-label clinical study (NCT00111319) of 682 patients was conducted to determine whether VELCADE administered intravenously (1.3 mg/m2) in combination with melphalan (9 mg/m2) and prednisone (60 mg/m2) resulted in improvement in time to progression (TTP) when compared to melphalan (9 mg/m2) and prednisone (60 mg/m2) in patients with previously untreated multiple myeloma. Treatment was administered for a maximum of nine cycles (approximately 54 weeks) and was discontinued early for disease progression or unacceptable toxicity. Antiviral prophylaxis was recommended for patients on the VELCADE study arm.
The median age of the patients in the study was 71 years (48;91), 50% were male, 88% were Caucasian and the median Karnofsky performance status score for the patients was 80 (60;100). Patients had IgG/IgA/Light chain myeloma in 63%/25%/8% instances, a median hemoglobin of 105 g/L (64;165), and a median platelet count of 221,500/microliter (33,000;587,000).
Efficacy results for the trial are presented in Table 14. At a prespecified interim analysis (with median follow-up of 16.3 months), the combination of VELCADE, melphalan and prednisone therapy resulted in significantly superior results for time to progression, progression-free survival, overall survival and response rate. Further enrollment was halted, and patients receiving melphalan and prednisone were offered VELCADE in addition. A later, prespecified analysis of overall survival (with median follow-up of 36.7 months with a hazard ratio of 0.65, 95% CI: 0.51, 0.84) resulted in a statistically significant survival benefit for the VELCADE, melphalan and prednisone treatment arm despite subsequent therapies including VELCADE based regimens. In an updated analysis of overall survival based on 387 deaths (median follow-up 60.1 months), the median overall survival for the VELCADE, melphalan and prednisone treatment arm was 56.4 months and for the melphalan and prednisone treatment arm was 43.1 months, with a hazard ratio of 0.695 (95% CI: 0.57, 0.85).
Table 14: Summary of Efficacy Analyses in the Previously Untreated Multiple Myeloma Study Efficacy Endpoint VELCADE, Melphalan and Prednisone
(n=344)Melphalan and Prednisone
(n=338)Note: All results are based on the analysis performed at a median follow-up duration of 16.3 months except for the overall survival analysis. - *
- Kaplan-Meier estimate
- †
- Hazard ratio estimate is based on a Cox proportional-hazard model adjusted for stratification factors: beta2-microglobulin, albumin, and region. A hazard ratio less than one indicates an advantage for VELCADE, melphalan and prednisone
- ‡
- p-value based on the stratified log-rank test adjusted for stratification factors: beta2-microglobulin, albumin, and region
- §
- EBMT criteria
- ¶
- p-value for Response Rate (CR + PR) from the Cochran-Mantel-Haenszel chi-square test adjusted for the stratification factors
Time to Progression Events n (%) 101 (29) 152 (45) Median* (months)
(95% CI)20.7
(17.6, 24.7)15.0
(14.1, 17.9)Hazard ratio†
(95% CI)0.54
(0.42, 0.70)p-value‡ 0.000002 Progression-Free Survival Events n (%) 135 (39) 190 (56) Median* (months)
(95% CI)18.3
(16.6, 21.7)14.0
(11.1, 15.0)Hazard ratio†
(95% CI)0.61
(0.49, 0.76)p-value ‡ 0.00001 Response Rate CR§ n (%) 102 (30) 12 (4) PR§ n (%) 136 (40) 103 (30) nCR n (%) 5 (1) 0 CR + PR§ n (%) 238 (69) 115 (34) p-value¶ <10-10 Overall Survival at Median Follow-Up of 36.7 Months Events (deaths) n (%) 109 (32) 148 (44) Median* (months)
(95% CI)Not Reached
(46.2, NR)43.1
(34.8, NR)Hazard ratio†
(95% CI)0.65
(0.51, 0.84)p-value‡ 0.00084 TTP was statistically significantly longer on the VELCADE, melphalan and prednisone arm (see Figure 1). (median follow-up 16.3 months)
Figure 1: Time to Progression VELCADE, Melphalan and Prednisone vs Melphalan and Prednisone
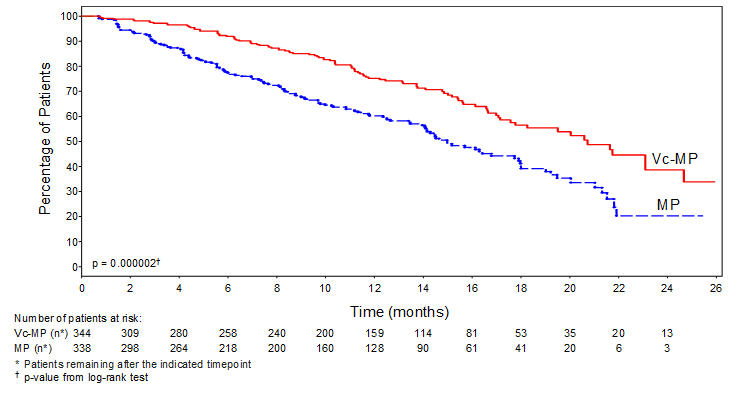
Overall survival was statistically significantly longer on the VELCADE, melphalan and prednisone arm (see Figure 2). (median follow-up 60.1 months)
Figure 2: Overall Survival VELCADE, Melphalan and Prednisone vs Melphalan and Prednisone
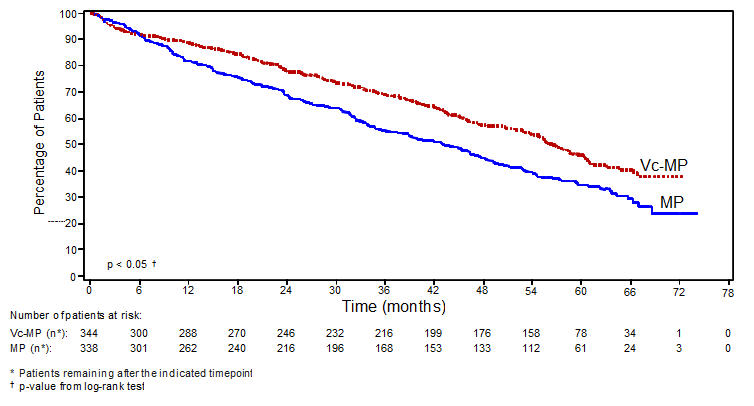
Randomized, Clinical Study in Relapsed Multiple Myeloma of VELCADE vs Dexamethasone
A prospective Phase 3, international, randomized (1:1), stratified, open-label clinical study (NCT00048230) enrolling 669 patients was designed to determine whether VELCADE resulted in improvement in time to progression (TTP) compared to high-dose dexamethasone in patients with progressive multiple myeloma following 1 to 3 prior therapies. Patients considered to be refractory to prior high-dose dexamethasone were excluded as were those with baseline Grade ≥2 peripheral neuropathy or platelet counts <50,000/µL. A total of 627 patients were evaluable for response.
Stratification factors were based on the number of lines of prior therapy the patient had previously received (one previous line vs more than one line of therapy), time of progression relative to prior treatment (progression during or within six months of stopping their most recent therapy vs relapse >6 months after receiving their most recent therapy), and screening beta2-microglobulin levels (≤2.5 mg/L vs >2.5 mg/L).
Baseline patient and disease characteristics are summarized in Table 15.
Table 15: Summary of Baseline Patient and Disease Characteristics in the Relapsed Multiple Myeloma Study Patient Characteristics VELCADE
(N=333)Dexamethasone
(N=336)Median age in years (range) 62.0 (33, 84) 61.0 (27, 86) Gender: Male/female 56%/44% 60%/40% Race: Caucasian/black/other 90%/6%/4% 88%/7%/5% Karnofsky performance status score ≤70 13% 17% Hemoglobin <100 g/L 32% 28% Platelet count <75 × 109/L 6% 4% Disease Characteristics Type of myeloma (%): IgG/IgA/Light chain 60%/23%/12% 59%/24%/13% Median beta2-microglobulin (mg/L) 3.7 3.6 Median albumin (g/L) 39.0 39.0 Creatinine clearance ≤30 mL/min [n (%)] 17 (5%) 11 (3%) Median Duration of Multiple Myeloma Since Diagnosis (Years) 3.5 3.1 Number of Prior Therapeutic Lines of Treatment Median 2 2 1 prior line 40% 35% >1 prior line 60% 65% Previous Therapy Any prior steroids, e.g., dexamethasone, VAD 98% 99% Any prior anthracyclines, e.g., VAD, mitoxantrone 77% 76% Any prior alkylating agents, e.g., MP, VBMCP 91% 92% Any prior thalidomide therapy 48% 50% Vinca alkaloids 74% 72% Prior stem cell transplant/other high-dose therapy 67% 68% Prior experimental or other types of therapy 3% 2% Patients in the VELCADE treatment group were to receive 8, three week treatment cycles followed by 3, five week treatment cycles of VELCADE. Patients achieving a CR were treated for four cycles beyond first evidence of CR. Within each three week treatment cycle, VELCADE 1.3 mg/m2/dose alone was administered by intravenous bolus twice weekly for two weeks on Days 1, 4, 8, and 11 followed by a ten day rest period (Days 12 to 21). Within each five week treatment cycle, VELCADE 1.3 mg/m2/dose alone was administered by intravenous bolus once weekly for four weeks on Days 1, 8, 15, and 22 followed by a 13 day rest period (Days 23 to 35) [see Dosage and Administration (2.2)].
Patients in the dexamethasone treatment group were to receive 4, five week treatment cycles followed by 5, four week treatment cycles. Within each five week treatment cycle, dexamethasone 40 mg/day PO was administered once daily on Days 1 to 4, 9 to 12, and 17 to 20 followed by a 15 day rest period (Days 21 to 35). Within each four week treatment cycle, dexamethasone 40 mg/day PO was administered once daily on Days 1 to 4 followed by a 24 day rest period (Days 5 to 28). Patients with documented progressive disease on dexamethasone were offered VELCADE at a standard dose and schedule on a companion study. Following a preplanned interim analysis of time to progression, the dexamethasone arm was halted and all patients randomized to dexamethasone were offered VELCADE, regardless of disease status.
In the VELCADE arm, 34% of patients received at least one VELCADE dose in all eight of the three week cycles of therapy, and 13% received at least one dose in all 11 cycles. The average number of VELCADE doses during the study was 22, with a range of 1 to 44. In the dexamethasone arm, 40% of patients received at least one dose in all four of the five week treatment cycles of therapy, and 6% received at least one dose in all nine cycles.
The time to event analyses and response rates from the relapsed multiple myeloma study are presented in Table 16. Response and progression were assessed using the European Group for Blood and Marrow Transplantation (EBMT) criteria. Complete response (CR) required <5% plasma cells in the marrow, 100% reduction in M-protein, and a negative immunofixation test (IF-). Partial response (PR) requires ≥50% reduction in serum myeloma protein and ≥90% reduction of urine myeloma protein on at least two occasions for a minimum of at least six weeks along with stable bone disease and normal calcium. Near complete response (nCR) was defined as meeting all the criteria for complete response including 100% reduction in M-protein by protein electrophoresis; however, M-protein was still detectable by immunofixation (IF+).
Table 16: Summary of Efficacy Analyses in the Relapsed Multiple Myeloma Study Efficacy Endpoint All Patients 1 Prior Line of Therapy >1 Prior Line of Therapy VELCADE Dex VELCADE Dex VELCADE Dex (n=333) (n=336) (n=132) (n=119) (n=200) (n=217) - *
- Kaplan-Meier estimate
- †
- Hazard ratio is based on Cox proportional-hazard model with the treatment as single independent variable. A hazard ratio less than one indicates an advantage for VELCADE
- ‡
- p-value based on the stratified log-rank test including randomization stratification factors
- §
- Precise p-value cannot be rendered
- ¶
- Response population includes patients who had measurable disease at baseline and received at least one dose of study drug
- #
- EBMT criteria; nCR meets all EBMT criteria for CR but has positive IF. Under EBMT criteria nCR is in the PR category
- Þ
- In two patients, the IF was unknown
- ß
- p-value for Response Rate (CR + PR) from the Cochran-Mantel-Haenszel chi-square test adjusted for the stratification factors
Time to Progression
Events n (%)147 (44) 196 (58) 55 (42) 64 (54) 92 (46) 132 (61) Median*
(95% CI)6.2 mo
(4.9, 6.9)3.5 mo
(2.9, 4.2)7.0 mo
(6.2, 8.8)5.6 mo
(3.4, 6.3)4.9 mo
(4.2, 6.3)2.9 mo
(2.8, 3.5)Hazard ratio†
(95% CI)0.55
(0.44, 0.69)0.55
(0.38, 0.81)0.54
(0.41, 0.72)p-value‡ <0.0001 0.0019 <0.0001 Overall Survival
Events (deaths) n (%)51 (15) 84 (25) 12 (9) 24 (20) 39 (20) 60 (28) Hazard ratio†
(95% CI)0.57
(0.40, 0.81)0.39
(0.19, 0.81)0.65
(0.43, 0.97)p-value‡, § <0.05 <0.05 <0.05 Response Rate
Population¶ n=627n=315 n=312 n=128 n=110 n=187 n=202 CR# n (%) 20 (6) 2 (<1) 8 (6) 2 (2) 12 (6) 0 (0) PR# n(%) 101 (32) 54 (17) 49 (38) 27 (25) 52 (28) 27 (13) nCR#, Þ n(%) 21 (7) 3 (<1) 8 (6) 2 (2) 13 (7) 1 (<1) CR + PR# n (%) 121 (38) 56 (18) 57 (45) 29 (26) 64 (34) 27 (13) p-value ß <0.0001 0.0035 <0.0001 TTP was statistically significantly longer on the VELCADE arm (see Figure 3).
Figure 3: Time to Progression Bortezomib vs Dexamethasone (Relapsed Multiple Myeloma Study)
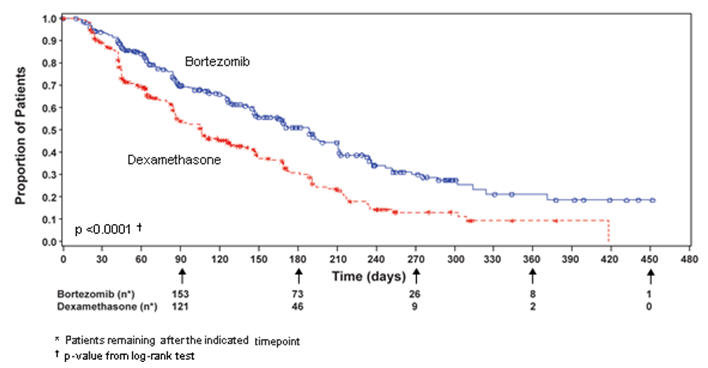
As shown in Figure 4, VELCADE had a significant survival advantage relative to dexamethasone (p <0.05). The median follow-up was 8.3 months.
Figure 4: Overall Survival Bortezomib vs Dexamethasone (Relapsed Multiple Myeloma Study)
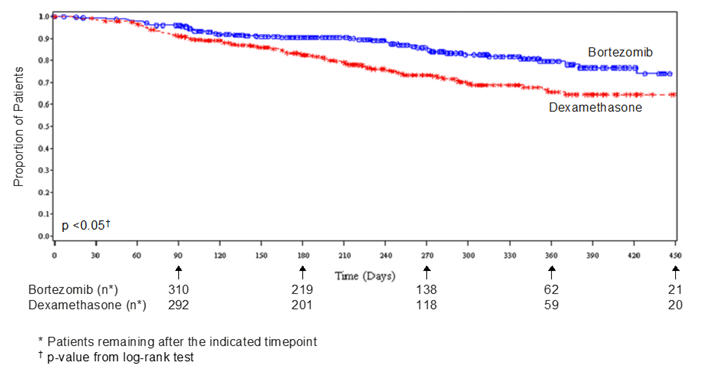
For the 121 patients achieving a response (CR or PR) on the VELCADE arm, the median duration was 8.0 months (95% CI: 6.9, 11.5 months) compared to 5.6 months (95% CI: 4.8, 9.2 months) for the 56 responders on the dexamethasone arm. The response rate was significantly higher on the VELCADE arm regardless of beta2-microglobulin levels at baseline.
Randomized, Open-Label Clinical Study of VELCADE Subcutaneous vs Intravenous in Relapsed Multiple Myeloma
An open-label, randomized, Phase 3 noninferiority study (NCT00722566) compared the efficacy and safety of the subcutaneous administration of VELCADE vs the intravenous administration. This study included 222 bortezomib naïve patients with relapsed multiple myeloma, who were randomized in a 2:1 ratio to receive 1.3 mg/m2 of VELCADE by either the subcutaneous (n=148) or intravenous (n=74) route for eight cycles. Patients who did not obtain an optimal response (less than Complete Response (CR)) to therapy with VELCADE alone after four cycles were allowed to receive oral dexamethasone 20 mg daily on the day of and after VELCADE administration (82 patients in subcutaneous treatment group and 39 patients in the intravenous treatment group). Patients with baseline Grade ≥2 peripheral neuropathy or neuropathic pain, or platelet counts <50,000/µL were excluded. A total of 218 patients were evaluable for response.
Stratification factors were based on the number of lines of prior therapy the patient had received (one previous line vs more than one line of therapy), and international staging system (ISS) stage (incorporating beta2-microglobulin and albumin levels; Stages I, II, or III).
The baseline demographic and other characteristics of the two treatment groups are summarized as follows: the median age of the patient population was approximately 64 years of age (range: 38 to 88 years), primarily male (subcutaneous: 50%, intravenous: 64%); the primary type of myeloma is IgG (subcutaneous: 65% IgG, 26% IgA, 8% light chain; intravenous: 72% IgG, 19% IgA, 8% light chain), ISS staging I/II/III (%) was 27, 41, 32 for both subcutaneous and intravenous, Karnofsky performance status score was ≤70% in 22% of subcutaneous and 16% of intravenous, creatinine clearance was 67.5 mL/min in subcutaneous and 73 mL/min in intravenous, the median years from diagnosis was 2.68 and 2.93 in subcutaneous and intravenous respectively and the proportion of patients with more than one prior line of therapy was 38% in subcutaneous and 35% in intravenous.
This study met its primary (noninferiority) objective that single agent subcutaneous VELCADE retains at least 60% of the overall response rate after four cycles relative to single agent intravenous VELCADE. The results are provided in Table 17.
Table 17: Summary of Efficacy Analyses in the Relapsed Multiple Myeloma Study of VELCADE Subcutaneous vs Intravenous Subcutaneous VELCADE Intravenous VELCADE Intent to Treat Population (n=148) (n=74) - *
- Median duration of follow-up is 11.8 months
Primary Endpoint Response Rate at 4 Cycles ORR (CR + PR) n(%) 63 (43) 31 (42) Ratio of Response Rates (95% CI) 1.01 (0.73, 1.40) CR n (%) 11 (7) 6 (8) PR n (%) 52 (35) 25 (34) nCR n (%) 9 (6) 4 (5) Secondary Endpoints Response Rate at 8 Cycles ORR (CR + PR) 78 (53) 38 (51) CR n (%) 17 (11) 9 (12) PR n (%) 61 (41) 29 (39) nCR n (%) 14 (9) 7 (9) Median Time to Progression, months 10.4 9.4 Median Progression-Free Survival, months 10.2 8.0 1 Year Overall Survival (%)* 72.6 76.7 A Randomized, Phase 2 Dose-Response Study in Relapsed Multiple Myeloma
An open-label, multicenter study randomized 54 patients with multiple myeloma who had progressed or relapsed on or after front-line therapy to receive VELCADE 1 mg/m2 or 1.3 mg/m2 intravenous bolus twice weekly for two weeks on Days 1, 4, 8, and 11 followed by a ten day rest period (Days 12 to 21). The median duration of time between diagnosis of multiple myeloma and first dose of VELCADE on this trial was two years, and patients had received a median of one prior line of treatment (median of three prior therapies). A single complete response was seen at each dose. The overall response rates (CR + PR) were 30% (8/27) at 1 mg/m2 and 38% (10/26) at 1.3 mg/m2.
A Phase 2 Open-Label Extension Study in Relapsed Multiple Myeloma
Patients from the two Phase 2 studies, who in the investigators' opinion would experience additional clinical benefit, continued to receive VELCADE beyond 8 cycles on an extension study. Sixty-three (63) patients from the Phase 2 multiple myeloma studies were enrolled and received a median of seven additional cycles of VELCADE therapy for a total median of 14 cycles (range: 7 to 32). The overall median dosing intensity was the same in both the parent protocol and extension study. Sixty-seven percent (67%) of patients initiated the extension study at the same or higher dose intensity at which they completed the parent protocol, and 89% of patients maintained the standard three week dosing schedule during the extension study. No new cumulative or new long-term toxicities were observed with prolonged VELCADE treatment [see Adverse Reactions (6.1)].
A Single-Arm Trial of Retreatment in Relapsed Multiple Myeloma
A single-arm, open-label trial (NCT00431769) was conducted to determine the efficacy and safety of retreatment with VELCADE. One hundred and thirty patients (≥18 years of age) with multiple myeloma who previously had at least partial response on a VELCADE-containing regimen (median of two prior lines of therapy [range: 1 to 7]) were retreated upon progression with VELCADE administered intravenously. Patients were excluded from trial participation if they had peripheral neuropathy or neuropathic pain of Grade ≥2. At least six months after prior VELCADE therapy, VELCADE was restarted at the last tolerated dose of 1.3 mg/m2 (n=93) or ≤1 mg/m2 (n=37) and given on Days 1, 4, 8 and 11 every three weeks for maximum of eight cycles either as single agent or in combination with dexamethasone in accordance with the standard of care. Dexamethasone was administered in combination with VELCADE to 83 patients in Cycle 1 with an additional 11 patients receiving dexamethasone during the course of VELCADE retreatment cycles.
The primary endpoint was best confirmed response to retreatment as assessed by European Group for Blood and Marrow Transplantation (EBMT) criteria. Fifty of the 130 patients achieved a best confirmed response of Partial Response or better for an overall response rate of 38.5% (95% CI: 30.1, 47.4). One patient achieved a Complete Response and 49 achieved Partial Response. In the 50 responding patients, the median duration of response was 6.5 months and the range was 0.6 to 19.3 months.
14.2 Mantle Cell Lymphoma
A Randomized, Open-Label Clinical Study in Patients with Previously Untreated Mantle Cell Lymphoma
A randomized, open-label, Phase 3 study (NCT00722137) was conducted in 487 adult patients with previously untreated mantle cell lymphoma (Stage II, III or IV) who were ineligible or not considered for bone marrow transplantation to determine whether VELCADE administered in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (VcR-CAP) resulted in improvement in progression-free survival (PFS) when compared to the combination of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP). This clinical study utilized independent pathology confirmation and independent radiologic response assessment.
Patients in the VcR-CAP treatment arm received VELCADE (1.3 mg/m2) administered intravenously on Days 1, 4, 8, and 11 (rest period Days 12 to 21); rituximab (375 mg/m2) on Day 1; cyclophosphamide (750 mg/m2) on Day 1; doxorubicin (50 mg/m2) on Day 1; and prednisone (100 mg/m2) on Day 1 through Day 5 of the 21 day treatment cycle. For patients with a response first documented at Cycle 6, two additional treatment cycles were allowed.
Median patient age was 66 years, 74% were male, 66% were Caucasian and 32% were Asian. Sixty-nine percent of patients had a positive bone marrow aspirate and/or a positive bone marrow biopsy for MCL, 54% of patients had an International Prognostic Index (IPI) score of three (high-intermediate) or higher and 76% had Stage IV disease.
The majority of the patients in both groups received six or more cycles of treatment, 84% in the VcR-CAP group and 83% in the R-CHOP group. Median number of cycles received by patients in both treatment arms was six with 17% of patients in the R-CHOP group and 14% of subjects in the VcR-CAP group receiving up to two additional cycles.
The efficacy results for PFS, CR and ORR with a median follow-up of 40 months are presented in Table 18. The response criteria used to assess efficacy were based on the International Workshop to Standardize Response Criteria for Non-Hodgkin's Lymphoma (IWRC). Final overall survival results at a median follow-up of 78.5 months are also presented in Table 18 and Figure 6. The combination of VcR-CAP resulted in statistically significant prolongation of PFS compared with R-CHOP (see Table 18, Figure 5).
Table 18: Summary of Efficacy Analyses in the Previously Untreated Mantle Cell Lymphoma Study Efficacy Endpoint
n: Intent to Treat patientsVcR-CAP
(n=243)R-CHOP
(n=244)Note: All results are based on the analysis performed at a median follow-up duration of 40 months except for the overall survival analysis, which was performed at a median follow-up of 78.5 months. CI = Confidence Interval; IPI = International Prognostic Index; LDH = Lactate dehydrogenase - *
- Based on Kaplan-Meier product limit estimates.
- †
- Hazard ratio estimate is based on a Cox's model stratified by IPI risk and stage of disease. A hazard ratio <1 indicates an advantage for VcR-CAP.
- ‡
- Based on Log rank test stratified with IPI risk and stage of disease.
- §
- Includes CR by independent radiographic assessment, bone marrow, and LDH using ITT population.
- ¶
- Includes CR + CRu + PR by independent radiographic assessment, regardless of the verification by bone marrow and LDH, using ITT population.
Progression-Free Survival (by independent radiographic assessment) Events n (%) 133 (55) 165 (68) Median* (months)
(95% CI)25
(20, 32)14
(12, 17)Hazard ratio†
(95% CI)0.63
(0.50, 0.79)p-value‡ <0.001 Complete Response Rate (CR)§ n (%)
(95% CI)108 (44)
(38, 51)82 (34)
(28, 40)Overall Response Rate (CR + CRu + PR)¶ n (%) 214 (88) 208 (85) (95% CI) (83, 92) (80, 89) Overall Survival Events n (%) 103 (42) 138 (57) Median* (months)
(95% CI)91
(71, NE)56
(47, 69)Hazard Ratio†
(95% CI)0.66
(0.51, 0.85)Figure 5: Progression-Free Survival VcR-CAP vs R-CHOP (previously Untreated Mantle Cell Lymphoma Study)
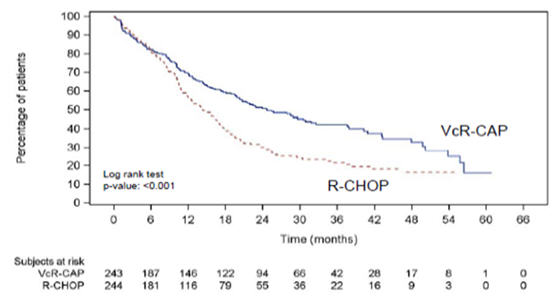
Key: R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; VcR-CAP = VELCADE, rituximab, cyclophosphamide, doxorubicin, and prednisone.
Figure 6: Overall Survival VcR-CAP vs R-CHOP (previously Untreated Mantle Cell Lymphoma Study)
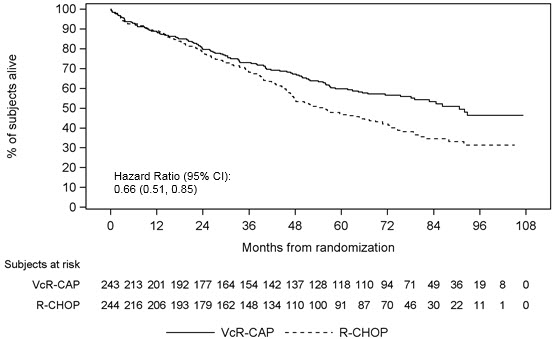
Key: R-CHOP = rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; VcR-CAP = VELCADE, rituximab, cyclophosphamide, doxorubicin, and prednisone.
A Phase 2 Single-Arm Clinical Study in Relapsed Mantle Cell Lymphoma after Prior Therapy
The safety and efficacy of VELCADE in relapsed or refractory mantle cell lymphoma were evaluated in an open-label, single-arm, multicenter study (NCT00063713) of 155 patients with progressive disease who had received at least one prior therapy. The median age of the patients was 65 years (42, 89), 81% were male, and 92% were Caucasian. Of the total, 75% had one or more extra-nodal sites of disease, and 77% were Stage 4. In 91% of the patients, prior therapy included all of the following: an anthracycline or mitoxantrone, cyclophosphamide, and rituximab. A total of thirty-seven percent (37%) of patients were refractory to their last prior therapy. An intravenous bolus injection of VELCADE 1.3 mg/m2/dose was administered twice weekly for two weeks on Days 1, 4, 8, and 11 followed by a ten day rest period (Days 12 to 21) for a maximum of 17 treatment cycles. Patients achieving a CR or CRu were treated for four cycles beyond first evidence of CR or CRu. The study employed dose modifications for toxicity [see Dosage and Administration (2.6, 2.7)].
Responses to VELCADE are shown in Table 19. Response rates to VELCADE were determined according to the International Workshop Response Criteria (IWRC) based on independent radiologic review of CT scans. The median number of cycles administered across all patients was four; in responding patients the median number of cycles was eight. The median time to response was 40 days (range: 31 to 204 days). The median duration of follow-up was more than 13 months.
Table 19: Response Outcomes in a Phase 2 Relapsed Mantle Cell Lymphoma Study Response Analyses (N=155) N (%) 95% CI Overall Response Rate (IWRC) (CR + CRu + PR) 48 (31) (24, 39) Complete Response (CR + CRu) 12 (8) (4, 13) CR 10 (6) (3, 12) CRu 2 (1) (0, 5) Partial Response (PR) 36 (23) (17, 31) Duration of Response Median 95% CI CR + CRu + PR (N=48) 9.3 months (5.4, 13.8) CR + CRu (N=12) 15.4 months (13.4, 15.4) PR (N=36) 6.1 months (4.2, 9.3) - 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VELCADE® (bortezomib) for Injection is supplied as individually cartoned 10 mL vials containing 3.5 mg of bortezomib as a white to off-white cake or powder.
NDC 63020-049-01
3.5 mg single-dose vialUnopened vials may be stored at controlled room temperature 25°C (77°F); excursions permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Retain in original package to protect from light.
Follow guidelines for handling and disposal for hazardous drugs, including the use of gloves and other protective clothing to prevent skin contact1.
-
17 PATIENT COUNSELING INFORMATION
Discuss the following with patients prior to treatment with VELCADE:
Peripheral Neuropathy
Advise patients to report the development or worsening of sensory and motor peripheral neuropathy to their healthcare provider [see Warnings and Precautions (5.1)].
Hypotension
Advise patients to drink adequate fluids to avoid dehydration and to report symptoms of hypotension to their healthcare provider [see Warnings and Precautions (5.2)].
Instruct patients to seek medical advice if they experience symptoms of dizziness, light headedness or fainting spells, or muscle cramps.
Cardiac Toxicity
Advise patients to report signs or symptoms of heart failure to their healthcare provider [see Warnings and Precautions (5.3)].
Pulmonary Toxicity
Advise patients to report symptoms of ARDS, pulmonary hypertension, pneumonitis, and pneumonia immediately to their healthcare provider [see Warnings and Precautions (5.4)].
Posterior Reversible Encephalopathy Syndrome (PRES)
Advise patients to seek immediate medical attention for signs or symptoms of PRES [see Warnings and Precautions (5.5)].
Gastrointestinal Toxicity
Advise patients to report symptoms of gastrointestinal toxicity to their healthcare provider and to drink adequate fluids to avoid dehydration. Instruct patients to seek medical advice if they experience symptoms of dizziness, light headedness or fainting spells, or muscle cramps [see Warnings and Precautions (5.6)].
Thrombocytopenia/Neutropenia
Advise patients to report signs or symptoms of bleeding or infection immediately to their healthcare provider [see Warnings and Precautions (5.7)].
Tumor Lysis Syndrome
Advise patients of the risk of tumor lysis syndrome and to drink adequate fluids to avoid dehydration [see Warnings and Precautions (5.8)].
Hepatic Toxicity
Advise patients to report signs or symptoms of hepatic toxicity to their healthcare provider [see Warnings and Precautions (5.9)].
Thrombotic Microangiopathy
Advise patients to seek immediate medical attention if any signs or symptoms of thrombotic microangiopathy occur [see Warnings and Precautions (5.10)].
Ability to Drive or Operate Machinery or Impairment of Mental Ability
VELCADE may cause fatigue, dizziness, syncope, orthostatic/postural hypotension. Advise patients not to drive or operate machinery if they experience any of these symptoms [see Warnings and Precautions (5.2, 5.5)].
Embryo-Fetal Toxicity
Advise females of the potential risk to the fetus and to use effective contraception during treatment with VELCADE and for seven months following the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with VELCADE and for four months following the last dose. Instruct patients to report pregnancy to their physicians immediately if they or their female partner becomes pregnant during treatment or within seven months following last dose [see Warnings and Precautions (5.11)].
Lactation
Advise women not to breastfeed while receiving VELCADE and for two months after last dose [see Use in Specific Populations (8.2)].
Concomitant Medications
Advise patients to speak with their physicians about any other medication they are currently taking.
Diabetic Patients
Advise patients to check their blood sugar frequently if using an oral antidiabetic medication and to notify their physicians of any changes in blood sugar level.
Dermal
Advise patients to contact their physicians if they experience rash, severe injection site reactions [see Dosage and Administration (2.9)], or skin pain. Discuss with patients the option for antiviral prophylaxis for herpes virus infection [see Adverse Reactions (6.1)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 3.5 mg Vial Label
- PRINCIPAL DISPLAY PANEL - 3.5 mg Vial Carton
-
INGREDIENTS AND APPEARANCE
VELCADE
bortezomib injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63020-049 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength bortezomib (UNII: 69G8BD63PP) (bortezomib - UNII:69G8BD63PP) bortezomib 3.5 mg Inactive Ingredients Ingredient Name Strength mannitol (UNII: 3OWL53L36A) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63020-049-01 1 in 1 CARTON 05/13/2003 1 1 in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021602 05/13/2003 Labeler - Takeda Pharmaceuticals America, Inc. (039997266)

