Label: TRISENOX- arsenic trioxide injection, solution

- NDC Code(s): 63459-601-06, 63459-601-11
- Packager: Cephalon, LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 28, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TRISENOX safely and effectively. See full prescribing information for TRISENOX.
TRISENOX® (arsenic trioxide) injection, for intravenous use
Initial U.S. Approval: 2000WARNING: DIFFERENTIATION SYNDROME, CARDIAC CONDUCTION ABNORMALITIES, and ENCEPHALOPATHY INCLUDING WERNICKE'S
See full prescribing information for complete boxed warning.
- Patients with acute promyelocytic leukemia (APL) treated with TRISENOX have experienced symptoms of differentiation syndrome, which may be life-threatening or fatal. If differentiation syndrome is suspected, immediately initiate high-dose corticosteroids and hemodynamic monitoring until resolution. Temporarily withhold TRISENOX. (2.3, 5.1)
- TRISENOX can cause QTc interval prolongation, complete atrioventricular block and torsade de pointes, which can be fatal. Before administering TRISENOX, assess the QTc interval, correct electrolyte abnormalities, and consider discontinuing drugs known to prolong QTc interval. Do not administer TRISENOX to patients with ventricular arrhythmia or prolonged QTc interval. Withhold TRISENOX until resolution and resume at reduced dose for QTc prolongation. (2.3, 5.2)
- Serious encephalopathy, including Wernicke's, has occurred with TRISENOX. If Wernicke's encephalopathy is suspected, immediately interrupt TRISENOX and initiate parenteral thiamine. Monitor until symptoms resolve or improve and thiamine levels normalize. (5.3)
INDICATIONS AND USAGE
TRISENOX is an arsenical indicated:
- In combination with tretinoin for treatment of adults with newly-diagnosed low-risk acute promyelocytic leukemia (APL) whose APL is characterized by the presence of the t(15;17) translocation or PML/RAR-alpha gene expression. (1.1)
- For induction of remission and consolidation in patients with APL who are refractory to, or have relapsed from, retinoid and anthracycline chemotherapy, and whose APL is characterized by the presence of the t(15;17) translocation or PML/RAR-alpha gene expression. (1.2)
DOSAGE AND ADMINISTRATION
Newly-diagnosed low-risk APL:
- Induction: Administer 0.15 mg/kg/day intravenously daily in combination with tretinoin until bone marrow remission. Do not exceed 60 days. (2.1)
- Consolidation: Administer 0.15 mg/kg/day intravenously daily for 5 days per week during weeks 1-4 of each 8-week cycle for a total of 4 cycles in combination with tretinoin. (2.1)
Relapsed or refractory APL:
DOSAGE FORMS AND STRENGTHS
Injection: 12 mg/6 mL (2 mg/mL) arsenic trioxide in single-dose vial. (3)
CONTRAINDICATIONS
Hypersensitivity to arsenic. (4)
WARNINGS AND PRECAUTIONS
- Hepatotoxicity: Elevated aspartate aminotransferase (AST), alkaline phosphatase and serum bilirubin have occurred in patients with newly-diagnosed low-risk APL treated with TRISENOX in combination with tretinoin. Monitor hepatic function tests at least twice weekly during induction and at least once weekly during consolidation. Withhold TRISENOX for certain elevations in AST, alkaline phosphatase and bilirubin and resume at reduced dose upon resolution.(2.3, 5.4)
- Carcinogenesis: Arsenic trioxide is a human carcinogen. Monitor patients for the development of second primary malignancies. (5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise of potential risk to a fetus and use of effective contraception. (5.6, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (> 30%) are nausea, cough, fatigue, pyrexia, headache, abdominal pain, vomiting, tachycardia, diarrhea, dyspnea, hypokalemia, leukocytosis, hyperglycemia, hypomagnesemia, insomnia, dermatitis, edema, QTc prolongation, rigors, sore throat, arthralgia, paresthesia, and pruritus (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Teva Pharmaceuticals at 1-888-483-8279 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. (8.2)
- Renal Impairment: Monitor patients with severe renal impairment (creatinine clearance less than 30 mL/min) for toxicity when treated with TRISENOX; dose reduction may be warranted. (8.6)
- Hepatic Impairment: Monitor patients with severe hepatic impairment (Child-Pugh Class C) for toxicity when treated with TRISENOX. (8.7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DIFFERENTIATION SYNDROME, CARDIAC CONDUCTION ABNORMALITIES AND ENCEPHALOPATHY INCLUDING WERNICKE'S
1 INDICATIONS AND USAGE
1.1 Newly-Diagnosed Low-Risk APL
1.2 Relapsed or Refractory APL
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Newly-Diagnosed Low-Risk Acute Promyelocytic Leukemia (APL)
2.2 Recommended Dosage for Relapsed or Refractory APL
2.3 Monitoring and Dosage Modifications for Adverse Reactions
2.4 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome
5.2 Cardiac Conduction Abnormalities
5.3 Encephalopathy
5.4 Hepatotoxicity
5.5 Carcinogenesis
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Newly-Diagnosed Low-Risk APL
14.2 Relapsed or Refractory APL
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DIFFERENTIATION SYNDROME, CARDIAC CONDUCTION ABNORMALITIES AND ENCEPHALOPATHY INCLUDING WERNICKE'S
Differentiation Syndrome: Patients with acute promyelocytic leukemia (APL) treated with TRISENOX have experienced differentiation syndrome, which may be life-threatening or fatal. Signs and symptoms may include unexplained fever, dyspnea, hypoxia, acute respiratory distress, pulmonary infiltrates, pleural or pericardial effusions, weight gain, peripheral edema, hypotension, renal insufficiency, hepatopathy, and multi-organ dysfunction, in the presence or absence of leukocytosis. If differentiation syndrome is suspected, immediately initiate high-dose corticosteroids and hemodynamic monitoring until resolution. Temporarily withhold TRISENOX [see Dosage and Administration (2.3), Warnings and Precautions (5.1)].
Cardiac Conduction Abnormalities: TRISENOX can cause QTc interval prolongation, complete atrioventricular block and torsade de pointes, which can be fatal. Before administering TRISENOX, assess the QTc interval, correct electrolyte abnormalities, and consider discontinuing drugs known to prolong QTc interval. Do not administer TRISENOX to patients with a ventricular arrhythmia or prolonged QTc interval. Withhold TRISENOX until resolution and resume at reduced dose for QTc prolongation [see Dosage and Administration (2.3), Warnings and Precautions (5.2)].
Encephalopathy: Serious encephalopathy, including Wernicke's, has occurred with TRISENOX. Wernicke's is a neurologic emergency. Consider testing thiamine levels in patients at risk for thiamine deficiency. Administer parenteral thiamine in patients with or at risk for thiamine deficiency. Monitor patients for neurological symptoms and nutritional status while receiving TRISENOX. If Wernicke's encephalopathy is suspected, immediately interrupt TRISENOX and initiate parenteral thiamine. Monitor until symptoms resolve or improve and thiamine levels normalize [see Warnings and Precautions (5.3)].
-
1 INDICATIONS AND USAGE
1.1 Newly-Diagnosed Low-Risk APL
TRISENOX is indicated in combination with tretinoin for treatment of adults with newly-diagnosed low-risk acute promyelocytic leukemia (APL) whose APL is characterized by the presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
1.2 Relapsed or Refractory APL
TRISENOX is indicated for induction of remission and consolidation in patients with APL who are refractory to, or have relapsed from, retinoid and anthracycline chemotherapy, and whose APL is characterized by the presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Newly-Diagnosed Low-Risk Acute Promyelocytic Leukemia (APL)
A treatment course for patients with newly-diagnosed low-risk APL consists of 1 induction cycle and 4 consolidation cycles.
- For the induction cycle, the recommended dosage of TRISENOX is 0.15 mg/kg/day intravenously daily in combination with tretinoin until bone marrow remission but not to exceed 60 days (see Table 1).
- For the consolidation cycles, the recommended dosage of TRISENOX is 0.15 mg/kg/day intravenously daily 5 days per week during weeks 1-4 of each 8-week cycle for a total of 4 cycles in combination with tretinoin (see Table 1). Omit tretinoin during weeks 5-6 of the fourth cycle of consolidation.
Table 1: Recommended Dosage of TRISENOX in Combination with Tretinoin
Induction (1 cycle)
TRISENOX
0.15 mg/kg once daily intravenously
until marrow remission but not to exceed 60 days
Tretinoina
22.5 mg/m2 twice daily orally
until marrow remission but not to exceed 60 days
Consolidation (4 cycles)
Week 1
Week 2
Week 3
Week 4
Week 5
Week 6
Week 7
Week 8
TRISENOX
0.15 mg/kg once
daily intravenously
Days
1-5Days
1-5Days
1-5Days
1-5--
--
--
--
Tretinoina
22.5 mg/m2 twice daily orally
Days
1-7Days
1-7--
--
Daysb
1-7Daysb
1-7--
--
a Rounded to the nearest 10 mg increment
b Omitted during the 4th cycle of consolidation
Differentiation syndrome prophylaxis consisting of prednisone 0.5 mg/kg daily from day 1 until the end of induction cycle with TRISENOX and tretinoin is recommended.
2.2 Recommended Dosage for Relapsed or Refractory APL
A treatment course for patients with relapsed or refractory APL consists of 1 induction cycle and 1 consolidation cycle [see Clinical Studies (14.2)].
- For the induction cycle, the recommended dosage of TRISENOX is 0.15 mg/kg/day intravenously daily until bone marrow remission or up to a maximum of 60 days.
- For the consolidation cycle, the recommended dosage of TRISENOX is 0.15 mg/kg/day intravenously daily for 25 doses over a period of up to 5 weeks. Begin consolidation 3 to 6 weeks after completion of induction cycle.
2.3 Monitoring and Dosage Modifications for Adverse Reactions
During induction, monitor coagulation studies, blood counts, and chemistries at least 2-3 times per week through recovery. During consolidation, monitor coagulation studies, blood counts, and chemistries at least weekly.
Table 2 shows the dosage modifications for adverse reactions due to TRISENOX when used alone or in combination with tretinoin.
Table 2: Dosage Modifications for Adverse Reactions of TRISENOX
Adverse Reaction Dosage Modification Differentiation syndrome, defined by the presence of 2 or more of the following:
- Unexplained fever
- Dyspnea
- Pleural and/or pericardial effusion
- Pulmonary infiltrates
- Renal failure
- Hypotension
- Weight gain greater than 5 kg
[see Warnings and Precautions (5.1)]
- Temporarily withhold TRISENOX. Consider holding tretinoin if symptoms are severe.
- Administer dexamethasone 10 mg intravenously every 12 hours until the resolution of signs and symptoms for a minimum of 3 days.
- Resume treatment when the clinical condition improves and reduce the dose of the withheld drug(s) by 50%.
- Increase the dose of the withheld drug(s) to the recommended dosage after one week in the absence of recurrence of symptoms of differentiation syndrome.
- If symptoms re-appear, decrease TRISENOX and/or tretinoin to the previous dose.
QTc (Framingham formula) Prolongation greater than 450 msec for men or greater than 460 msec for women [see Warnings and Precautions (5.2)] - Withhold TRISENOX and any medication known to prolong the QTc interval.
- Correct electrolyte abnormalities.
- After the QTc normalizes and electrolyte abnormalities are corrected, resume treatment with TRISENOX at a 50% reduced dose (0.075 mg/kg/day daily) for one week after resolution.
- If the 50% reduced dose is tolerated for one week (in the absence of QTc prolongation), increase the dose of TRISENOX to 0.11 mg/kg/day daily for the next week [see Dosage and Administration (2.1)].
- The dose of TRISENOX can be increased to 0.15 mg/kg/day in the absence of QTc prolongation during that 14-day dose-escalation period.
Hepatotoxicity, defined by 1 or more of the following:
- Total bilirubin (TB) greater than 3 times the upper limit of normal (ULN)
- Aspartate aminotransferase (AST) greater than 5 times the ULN
- Alkaline phosphatase (AP) greater than 5 times the ULN
[see Warnings and Precautions (5.4)]
- Withhold TRISENOX and/or tretinoin.
- Resume treatment at a 50% reduced dose of the withheld drug(s) when TB is less than 1.5 times the ULN and AP/AST are less than 3 times the ULN.
- Increase the dose of the withheld drug(s) back to the recommended dosage after one week on the reduced dose in the absence of worsening of hepatotoxicity.
- Discontinue the withheld drug(s) permanently if hepatotoxicity recurs.
Other severe or life-threatening (grade 3-4) nonhematologic reactions [see Adverse Reactions (6)] - Temporarily withhold TRISENOX and tretinoin.
- When the adverse reaction resolves to no more than mild (grade 1), resume TRISENOX and tretinoin reduced by 2 dose levels (see Table 3 below).
Moderate (grade 2) nonhematologic reactions
[see Adverse Reactions (6)]
- Reduce the dose of TRISENOX and/or tretinoin by 1 dose level (see Table 3 below).
Leukocytosis (WBC count greater than 10 Gi/L)
[see Adverse Reactions (6.1)]
- Administer hydroxyurea.
- Hydroxyurea may be discontinued when the WBC declines below 10 Gi/L.
Myelosuppression, defined by 1 or more of the following: - absolute neutrophil count less than 1 Gi/L
- platelets less than 50 Gi/L lasting more than 5 weeks
[see Adverse Reactions (6)]
- Consider reducing the dose of TRISENOX and tretinoin by 1 dose level (see Table 3 below).
- If myelosuppression lasts ≥ 50 days or occurs on 2 consecutive cycles, assess a marrow aspirate for remission status. In the case of molecular remission, resume TRISENOX and tretinoin at 1 dose level lower (see Table 3 below).
Table 3: Dose Reduction Levels for Hematologic and Nonhematologic Toxicities
Dose Level TRISENOX
mg/kg intravenously
once dailyTretinoin*
mg/m2 orally
twice dailyStarting level 0.15 22.5 -1 0.11 18.75 -2 0.10 12.5 -3 0.075 10 *Rounded to the nearest 10 mg increment
2.4 Preparation and Administration
Reconstitution
Dilute TRISENOX with 100 to 250 mL 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP, using proper aseptic technique, immediately after withdrawal from the vial. Do not save any unused portions for later administration.
After dilution, store TRISENOX for no more than 24 hours at room temperature and 48 hours when refrigerated.
Administration
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Administer TRISENOX as an intravenous infusion over 2 hours. The infusion duration may be extended up to 4 hours if acute vasomotor reactions are observed. A central venous catheter is not required.
The TRISENOX vial is single-dose and does not contain any preservatives. Discard unused portions of each vial properly. Do not mix TRISENOX with other medications.
Safe Handling Procedures
TRISENOX is a hazardous drug. Follow applicable special handling and disposal procedures.1
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome
Differentiation syndrome, which may be life-threatening or fatal, has been observed in patients with acute promyelocytic leukemia (APL) treated with TRISENOX. In clinical trials, 16-23% of patients treated with TRISENOX for APL developed differentiation syndrome. Signs and symptoms include unexplained fever, dyspnea, hypoxia, acute respiratory distress, pulmonary infiltrates, pleural or pericardial effusion, weight gain, peripheral edema, hypotension, renal insufficiency, hepatopathy and multi-organ dysfunction. Differentiation syndrome has been observed with and without concomitant leukocytosis, and it has occurred as early as day 1 of induction to as late as the second month induction therapy.
When TRISENOX is used in combination with tretinoin, prophylaxis with prednisone is recommended during the induction cycle [see Dosage and Administration (2.1)].
If differentiation syndrome is suspected, temporarily withhold TRISENOX and immediately initiate dexamethasone 10 mg intravenously every 12 hours and hemodynamic monitoring until resolution of signs and symptoms for a minimum of 3 days [see Dosage and Administration (2.3)].
5.2 Cardiac Conduction Abnormalities
Patients treated with TRISENOX can develop QTc prolongation, torsade de pointes, and complete atrioventricular block. In the clinical trials of patients with newly-diagnosed low-risk APL treated with TRISENOX in combination with tretinoin, 11% experienced QTc (Framingham formula) prolongation > 450 msec for men and > 460 msec for women throughout the treatment cycles. In the clinical trial of patients with relapsed or refractory APL treated with TRISENOX monotherapy, 40% had at least one ECG tracing with a QTc interval greater than 500 msec. A prolonged QTc was observed between 1 and 5 weeks after start of TRISENOX infusion, and it usually resolved by 8 weeks after TRISENOX infusion. There are no data on the effect of TRISENOX on the QTc interval during the infusion of the drug.
The risk of torsade de pointes is related to the extent of QTc prolongation, concomitant administration of QTc prolonging drugs, a history of torsade de pointes, pre-existing QTc interval prolongation, congestive heart failure, administration of potassium-wasting diuretics, or other conditions that result in hypokalemia or hypomagnesemia. The risk may be increased when TRISENOX is coadministered with medications that can lead to electrolyte abnormalities (such as diuretics or amphotericin B) [see Drug Interactions (7)].
Prior to initiating therapy with TRISENOX, assess the QTc interval by electrocardiogram, correct pre-existing electrolyte abnormalities, and consider discontinuing drugs known to prolong QTc interval. Do not administer TRISENOX to patients with a ventricular arrhythmia or prolonged QTc. If possible, discontinue drugs that are known to prolong the QTc interval. If it is not possible to discontinue the interacting drug, perform cardiac monitoring frequently [see Drug Interactions (7)]. During TRISENOX therapy, maintain potassium concentrations above 4 mEq/L and magnesium concentrations above 1.8 mg/dL. Monitor ECG weekly and more frequently for clinically unstable patients.
For patients who develop a QTc Framingham greater than 450 msec for men or greater than 460 msec for women, withhold TRISENOX and any medication known to prolong the QTc interval. Correct electrolyte abnormalities. When the QTc normalizes and electrolyte abnormalities are corrected, resume TRISENOX at a reduced dose [see Dosage and Administration (2.3)].
5.3 Encephalopathy
Serious encephalopathies were reported in patients receiving TRISENOX. Monitor patients for neurological symptoms, such as confusion, decreased level of consciousness, seizures, cognitive deficits, ataxia, visual symptoms and ocular motor dysfunction. Advise patients and caregivers of the need for close observation.
Wernicke’s Encephalopathy
Wernicke’s encephalopathy occurred in patients receiving TRISENOX. Wernicke’s encephalopathy is a neurologic emergency that can be prevented and treated with thiamine. Consider testing thiamine levels in patients at risk for thiamine deficiency (e.g., chronic alcohol use, malabsorption, nutritional deficiency, concomitant use of furosemide). Administer parenteral thiamine in patients with or at risk for thiamine deficiency. Monitor patients for neurological symptoms and nutritional status while receiving TRISENOX. If Wernicke’s encephalopathy is suspected, immediately interrupt TRISENOX and initiate parenteral thiamine. Monitor until symptoms resolve or improve and thiamine levels normalize.
5.4 Hepatotoxicity
In the clinical trials, 44% of patients with newly-diagnosed low-risk APL treated with TRISENOX in combination with tretinoin experienced elevated aspartate aminotransferase (AST), alkaline phosphatase, and/or serum bilirubin. These abnormalities resolved with temporary discontinuation of TRISENOX and/or tretinoin.
Long-term liver abnormalities can occur in patients with APL treated with TRISENOX in combination with tretinoin. In a published series, mild liver dysfunction and hepatic steatosis were seen in 15% and 43%, respectively, of patients at a median of 7 years (range 0 to 14 years) after treatment with arsenic trioxide in combination with tretinoin.
During treatment with TRISENOX, monitor hepatic function tests at least twice weekly during induction and at least once weekly during consolidation. Withhold TRISENOX and/or tretinoin if elevations in AST or alkaline phosphatase occur to greater than 5 times the upper limit of normal and/or elevation in serum total bilirubin occurs to greater than 3 times the upper limit of normal and resume at reduced dose upon resolution [see Dosage and Administration (2.3)].
5.5 Carcinogenesis
The active ingredient of TRISENOX, arsenic trioxide, is a human carcinogen. Monitor patients for the development of second primary malignancies.
5.6 Embryo-Fetal Toxicity
TRISENOX can cause fetal harm when administered to a pregnant woman. Arsenic trioxide was embryolethal and teratogenic in rats when administered on gestation day 9 at a dose approximately 10 times the recommended human daily dose on a mg/m2 basis. A related trivalent arsenic, sodium arsenite, produced teratogenicity when administered during gestation in mice at a dose approximately 5 times the projected human dose on a mg/m2 basis and in hamsters at an intravenous dose approximately equivalent to the projected human daily dose on a mg/m2 basis.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRISENOX and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with TRISENOX and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Differentiation Syndrome [see Warnings and Precautions (5.1)]
- Cardiac Conduction Abnormalities [see Warnings and Precautions (5.2)]
- Encephalopathy [see Warnings and Precautions (5.3)]
- Hepatotoxicity [see Warnings and Precautions (5.4)]
- Carcinogenesis [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Newly-Diagnosed Low-Risk APL
The safety of TRISENOX in combination with tretinoin was evaluated in Study APL0406, a randomized trial comparing TRISENOX plus tretinoin (n=129) versus chemotherapy plus tretinoin (n=137) in patients with newly-diagnosed APL [see Clinical Studies (14.1)]. In the TRISENOX/tretinoin group, 98% of patients completed induction therapy and 89% completed at least three consolidation cycles. In the chemotherapy/tretinoin group, 96% completed induction therapy and 87% patients completed all three courses of consolidation therapy.
Serious adverse reactions were reported in 25% of patients on the TRISENOX/tretinoin arm and 24% on the chemotherapy/tretinoin arm. The serious adverse reactions reported in ≥ 2% of patients who received TRISENOX/tretinoin were abnormal liver tests, differentiation syndrome, dyspnea, pneumonia, and other infections. Fatal adverse reactions were reported in 1 (1%) patient on the TRISENOX/tretinoin arm and 8 (6%) patients on the chemotherapy/tretinoin arm.
TRISENOX/tretinoin was discontinued due to toxicity in 1 patient during induction and in 4 patients during the first three consolidation courses, whereas chemotherapy/tretinoin was discontinued due to toxicity in 4 patients during induction and in 6 patients during consolidation.
Selected hematologic and nonhematologic toxicities that occurred during induction or consolidation are presented in Table 4.
Table 4: Select Adverse Reactions of Trisenox in Combination with Tretinoin in Patients with Newly-Diagnosed APL in Study APL0406
Induction
First Consolidation
Second Consolidation
Third Consolidation
Adverse Reaction
n (%)
n (%)
n (%)
n (%)
Thrombocytopenia > 15 days (Grade 3-4)
TRISENOX/tretinoin
74 (58%)
6 (5%)
6 (5%)
8 (7%)
Chemotherapy/tretinoin
120 (88%)
17 (14%)
77 (63%)
26 (22%)
Neutropenia >15 days (Grade 3-4)
TRISENOX/tretinoin
61 (48%)
8 (7%)
7 (6%)
5 (4%)
Chemotherapy/tretinoin
109 (80%)
40 (32%)
90 (73%)
28 (24%)
Hepatic toxicity (Grade 3-4)
TRISENOX/tretinoin
51 (40%)
5 (4%)
1 (1%)
0 (0%)
Chemotherapy/tretinoin
4 (3%)
1 (1%)
0 (0%)
0 (0%)
Infection and fever of unknown origin
TRISENOX/tretinoin
30 (23%)
10 (8%)
4 (3%)
2 (2%)
Chemotherapy/tretinoin
75 (55%)
8 (6%)
46 (38%)
2 (2%)
Hypertriglyceridemia
TRISENOX/tretinoin
29 (22%)
22 (18%)
17 (14%)
16 (14%)
Chemotherapy/tretinoin
29 (22%)
19 (15%)
10 (8%)
13 (11%)
Hypercholesterolemia
TRISENOX/tretinoin
14 (10%)
19 (16%)
19 (16%)
16 (14%)
Chemotherapy/tretinoin
12 (9%)
12 (10%)
12 (10%)
11 (9%)
QT prolongation
TRISENOX/tretinoin
11 (9%)
3 (2%)
3 (2%)
2 (2%)
Chemotherapy/tretinoin
1 (1%)
0 (0%)
0 (0%)
0 (0%)
Gastrointestinal toxicity (Grade 3-4)
TRISENOX/tretinoin
3 (2%)
0 (0%)
0 (0%)
0 (0%)
Chemotherapy/tretinoin
25 (18%)
1 (1%)
6 (5%)
0 (0%)
Neurotoxicity*
TRISENOX/tretinoin
1 (1%)
5 (4%)
6 (5%)
7 (6%)
Chemotherapy/tretinoin
0 (0%)
0 (0%)
0 (0%)
0 (0%)
Cardiac function (Grade 3-4)
TRISENOX/tretinoin
0 (0%)
0 (0%)
0 (0%)
0 (0%)
Chemotherapy/tretinoin
5 (4%)
0 (0%)
0 (0%)
0 (0%)
*Mostly cases of reversible peripheral neuropathy
Relapsed or Refractory APL
Safety information was available for 52 patients with relapsed or refractory APL who participated in clinical trials of TRISENOX. Forty patients in the Study PLRXAS01 received the recommended dose of 0.15 mg/kg, of whom 28 completed both induction and consolidation cycles. An additional 12 patients with relapsed or refractory APL received doses generally similar to the recommended dose.
Serious adverse reactions observed in the 40 patients with refractory or relapsed APL enrolled in Study PLRXAS01 included differentiation syndrome (n=3), hyperleukocytosis (n=3), QTc interval ≥ 500 msec (n=16, 1 with torsade de pointes), atrial dysrhythmias (n=2), and hyperglycemia (n=2).
The most common adverse reactions (> 30%) were nausea, cough, fatigue, pyrexia, headache, abdominal pain, vomiting, tachycardia, diarrhea, dyspnea, hypokalemia, leukocytosis, hyperglycemia, hypomagnesemia, insomnia, dermatitis, edema, QTc prolongation, rigors, sore throat, arthralgia, paresthesia, and pruritus.
Table 5 describes the adverse reactions in patients aged 5 to 73 years with APL who received TRISENOX at the recommended dose. Similar adverse reactions profiles were seen in the other patient populations who received TRISENOX.
Table 5: Adverse Reactions (≥ 5%) in Patients with Relapsed or Refractory APL Who Received TRISENOX in Study PLRXAS01
Body System Adverse reaction Any Grade Adverse Reactions Grade ≥3 Adverse Reactions n % n % Gastrointestinal disorders
Nausea
30
75
Abdominal pain (lower & upper)
23
58
4
10
Vomiting
23
58
Diarrhea
21
53
Sore throat
14
35
Constipation
11
28
1
3
Anorexia
9
23
Appetite decreased
6
15
Loose stools
4
10
Dyspepsia
4
10
Oral blistering
3
8
Fecal incontinence
3
8
Gastrointestinal hemorrhage
3
8
Dry mouth
3
8
Abdominal tenderness
3
8
Diarrhea hemorrhagic
3
8
Abdominal distension
3
8
Respiratory
Cough
26
65
Dyspnea
21
53
4
10
Epistaxis
10
25
Hypoxia
9
23
4
10
Pleural effusion
8
20
1
3
Post nasal drip
5
13
Wheezing
5
13
Decreased breath sounds
4
10
Crepitations
4
10
Rales
4
10
Hemoptysis
3
8
Tachypnea
3
8
Rhonchi
3
8
General disorders and administration site conditions
Fatigue
25
63
2
5
Pyrexia (fever)
25
63
2
5
Edema - non-specific
16
40
Rigors
15
38
Chest pain
10
25
2
5
Injection site pain
8
20
Pain - non-specific
6
15
1
3
Injection site erythema
5
13
Weight gain
5
13
Injection site edema
4
10
Weakness
4
10
2
5
Hemorrhage
3
8
Weight loss
3
8
Drug hypersensitivity
2
5
1
3
Nervous system disorders
Headache
24
60
1
3
Insomnia
17
43
1
3
Paresthesia
13
33
2
5
Dizziness (excluding vertigo)
9
23
Tremor
5
13
Convulsion
3
8
2
5
Somnolence
3
8
Coma
2
5
2
5
Cardiac disorders
Tachycardia
22
55
ECG QT corrected interval prolonged
> 500 msec
16
40
Palpitations
4
10
ECG abnormal other than QT interval prolongation
3
8
Metabolism and nutrition disorders
Hypokalemia
20
50
5
13
Hypomagnesemia
18
45
5
13
Hyperglycemia
18
45
5
13
ALT increased
8
20
2
5
Hyperkalemia
7
18
2
5
AST increased
5
13
1
3
Hypocalcemia
4
10
Hypoglycemia
3
8
Acidosis
2
5
Hematologic disorders
Leukocytosis
20
50
1
3
Anemia
8
20
2
5
Thrombocytopenia
7
18
5
13
Febrile neutropenia
5
13
3
8
Neutropenia
4
10
4
10
Disseminated intravascular coagulation
3
8
3
8
Lymphadenopathy
3
8
Skin and subcutaneous tissue disorders
Dermatitis
17
43
Pruritus
13
33
1
3
Ecchymosis
8
20
Dry skin
6
15
Erythema - non-specific
5
13
Increased sweating
5
13
Facial edema
3
8
Night sweats
3
8
Petechiae
3
8
Hyperpigmentation
3
8
Non-specific skin lesions
3
8
Urticaria
3
8
Local exfoliation
2
5
Eyelid edema
2
5
Musculoskeletal, connective tissue, and bone disorders
Arthralgia
13
33
3
8
Myalgia
10
25
2
5
Bone pain
9
23
4
10
Back pain
7
18
1
3
Neck pain
5
13
Pain in limb
5
13
2
5
Psychiatric disorders
Anxiety
12
30
Depression
8
20
Agitation
2
5
Confusion
2
5
Vascular disorders
Hypotension
10
25
2
5
Flushing
4
10
Hypertension
4
10
Pallor
4
10
Infections and infestations
Sinusitis
8
20
Herpes simplex
5
13
Upper respiratory tract infection
5
13
1
3
Bacterial infection - non-specific
3
8
1
3
Herpes zoster
3
8
Nasopharyngitis
2
5
Oral candidiasis
2
5
Sepsis
2
5
2
5
Reproductive system disorders
Vaginal hemorrhage
5
13
Intermenstrual bleeding
3
8
Ocular disorders
Eye irritation
4
10
Blurred vision
4
10
Dry eye
3
8
Painful red eye
2
5
Renal and urinary disorders
Renal failure
3
8
1
3
Renal impairment
3
8
Oliguria
2
5
Incontinence
2
5
Ear disorders
Earache
3
8
Tinnitus
2
5
Other Clinically Relevant Adverse Reactions
Leukocytosis
TRISENOX can induce proliferation of leukemic promyelocytes resulting in a rapid increase in white blood cell count. Leukocytosis greater than 10 Gi/L developed during induction therapy in 43% patients receiving TRISENOX/tretinoin for newly-diagnosed low-risk APL and in 50% of patients receiving TRISENOX monotherapy for relapsed/refractory APL. In the relapsed/refractory setting, a relationship did not exist between baseline WBC counts and development of hyperleukocytosis nor baseline WBC counts and peak WBC counts.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of TRISENOX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Cardiac disorders: Ventricular extrasystoles in association with QT prolongation, ventricular tachycardia in association with QT prolongation, including torsade de pointes, atrioventricular block, and congestive heart failure
Ear and labyrinth disorders: Deafness
Hematologic disorders: Pancytopenia, bone marrow necrosis
Infections: Herpes zoster
Investigations: Gamma-glutamyltransferase increased
Musculoskeletal and connective tissue disorders: Bone pain, myalgia, rhabdomyolysis
Neoplasms benign, malignant and unspecified: Melanoma, pancreatic cancer, squamous cell carcinoma
Nervous system disorders: Peripheral neuropathy, paresis, seizures, confusion, encephalopathy, Wernicke's encephalopathy, posterior reversible encephalopathy syndrome
Skin and subcutaneous tissue disorders: Toxic epidermal necrolysis
-
7 DRUG INTERACTIONS
Drugs That Can Prolong the QT/QTc Interval
Concomitant use of these drugs and TRISENOX may increase the risk of serious QT/QTc interval prolongation [see Warnings and Precautions (5.1)]. Discontinue or replace with an alternative drug that does not prolong the QT/QTc interval while the patient is using TRISENOX. Monitor ECGs more frequently in patients when it is not feasible to avoid concomitant use.
Drugs That Can Lead to Electrolyte Abnormalities
Electrolyte abnormalities increase the risk of serious QT/QTc interval prolongation [see Warnings and Precautions (5.1)]. Avoid concomitant use of drugs that can lead to electrolyte abnormalities. Monitor electrolytes more frequently in patients who must receive concomitant use of these drugs and TRISENOX.
Drugs That Can Lead to Hepatotoxicity
Concomitant use of these drugs and TRISENOX, particularly when given in combination with tretinoin, may increase the risk of serious hepatotoxicity [see Warnings and Precautions (5.4)]. Discontinue or replace with an alternative drug that does not cause hepatotoxicity while the patient is using TRISENOX. Monitor liver function tests more frequently in patients when it is not feasible to avoid concomitant use.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on the mechanism of action [see Clinical Pharmacology (12.1)] and findings in animal studies, TRISENOX can cause fetal harm when administered to a pregnant woman. Arsenic trioxide was embryolethal and teratogenic in rats when administered on gestation day 9 at a dose approximately 10 times the recommended human daily dose on a mg/m2 basis (see Data). A related trivalent arsenic, sodium arsenite, produced teratogenicity when administered during gestation in mice at a dose approximately 5 times the projected human dose on a mg/m2 basis and in hamsters at an intravenous dose approximately equivalent to the projected human daily dose on a mg/m2 basis. There are no studies with the use of TRISENOX in pregnant women, and limited published data on arsenic trioxide use during pregnancy are insufficient to inform a drug-associated risk of major birth defects and miscarriage. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
One patient was reported to deliver a live infant with no reported congenital anomalies after receiving arsenic trioxide during the first five months of pregnancy. A second patient became pregnant three months after discontinuing arsenic trioxide and was reported to have a normal pregnancy outcome. A third patient was a pregnant healthcare provider who experienced dermal contact with liquid arsenic trioxide and had a normal pregnancy outcome after treatment and monitoring. A fourth patient who became pregnant while receiving arsenic trioxide had a miscarriage.
Animal Data
Studies in pregnant mice, rats, hamsters, and primates have shown that inorganic arsenicals cross the placental barrier when given orally or by injection. An increase in resorptions, neural-tube defects, anophthalmia and microphthalmia were observed in rats administered 10 mg/kg of arsenic trioxide on gestation day 9 (approximately 10 times the recommended human daily dose on a mg/m2 basis). Similar findings occurred in mice administered a 10 mg/kg dose of a related trivalent arsenic, sodium arsenite (approximately 5 times the projected human dose on a mg/m2 basis), on gestation days 6, 7, 8, or 9. Intravenous injection of 2 mg/kg sodium arsenite (approximately equivalent to the projected human daily dose on a mg/m2 basis) on gestation day 7 (the lowest dose tested) resulted in neural-tube defects in hamsters.
8.2 Lactation
Risk Summary
Arsenic trioxide is excreted in human milk. There are no data on the effects of arsenic trioxide on the breastfed child or on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with TRISENOX and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
TRISENOX can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Conduct pregnancy testing in females of reproductive potential prior to initiation of TRISENOX.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with TRISENOX and for 6 months after the final dose.
Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with TRISENOX and for 3 months after the final dose.
Infertility
Males
Based on testicular toxicities including decreased testicular weight and impaired spermatogenesis observed in animal studies, TRISENOX may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of TRISENOX in combination with tretinoin in pediatric patients has not been established.
The safety and efficacy of TRISENOX as a single agent for treatment of pediatric patients with relapsed or refractory APL is supported by the pivotal phase 2 study in 40 patients with relapsed or refractory APL. Five patients below the age of 18 years (age range: 5 to 16 years) were treated with TRISENOX at the recommended dose of 0.15 mg/kg/day. A literature review included an additional 17 patients treated with arsenic trioxide for relapsed or refractory APL, with ages ranging from 4 to 21 years. No differences in efficacy and safety were observed by age.
8.5 Geriatric Use
Use of TRISENOX in combination with tretinoin in newly-diagnosed adult patients with low-risk APL is supported by a randomized, controlled trial that included 16 patients between the ages of 60 and 70 years. No overall differences in safety or effectiveness were observed between these patients and younger patients. A literature review included an additional 77 patients aged 60 to 84 years who were treated with arsenic trioxide in combination with tretinoin as part of induction and consolidation therapy for low- and high-risk APL. These studies showed lower survival rates in older patients. Monitor elderly patients frequently during treatment with TRISENOX.
Use of TRISENOX as monotherapy in patients with relapsed or refractory APL is supported by the open-label, single-arm trial that included 6 patients aged 65 and older (range: 65 to 73 years). A literature review included an additional 4 patients aged 69 to 72 years who were treated with arsenic trioxide for relapsed or refractory APL. No overall differences in safety or effectiveness were observed between these patients and younger patients.
8.6 Renal Impairment
Exposure of arsenic trioxide may be higher in patients with severe renal impairment [see Clinical Pharmacology (12.3)]. Monitor patients with severe renal impairment (creatinine clearance [CLcr] less than 30 mL/min) frequently for toxicity; a dose reduction may be warranted.
The use of TRISENOX in patients on dialysis has not been studied.
8.7 Hepatic Impairment
Since limited data are available across all hepatic impairment groups, caution is advised in the use of TRISENOX in patients with hepatic impairment [see Clinical Pharmacology (12.3)]. Monitor patients with severe hepatic impairment (Child-Pugh Class C) frequently for toxicity.
-
10 OVERDOSAGE
Manifestations
Manifestations of TRISENOX (arsenic trioxide) overdosage include convulsions, muscle weakness, and confusion.
Management
For symptoms of TRISENOX (arsenic trioxide) overdosage, immediately discontinue TRISENOX and consider chelation therapy.
A conventional protocol for acute arsenic intoxication includes dimercaprol administered at a dose of 3 mg/kg intramuscularly every 4 hours until immediate life-threatening toxicity has subsided. Thereafter, penicillamine at a dose of 250 mg orally, up to a maximum frequency of four times per day (≤ 1 g per day), may be given.
-
11 DESCRIPTION
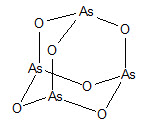
TRISENOX is a sterile injectable solution of arsenic trioxide. The molecular formula of arsenic trioxide in the solid state is As2O3, with a molecular weight of 197.8 and the following structural formula:
TRISENOX is available in 10 mL, single-dose vials containing 12 mg of arsenic trioxide. TRISENOX is formulated as a sterile, nonpyrogenic, clear solution of arsenic trioxide in water for injection using sodium hydroxide and dilute hydrochloric acid to adjust to pH 8. TRISENOX is preservative-free. Arsenic trioxide, the active ingredient, is present at a concentration of 2 mg/mL. Inactive ingredients and their respective approximate concentrations are sodium hydroxide (1.2 mg/mL) for solubilization, and sodium hydroxide and hydrochloric acid for pH adjustment to pH 8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of TRISENOX is not completely understood. Arsenic trioxide causes morphological changes and DNA fragmentation characteristic of apoptosis in NB4 human promyelocytic leukemia cells in vitro. Arsenic trioxide also causes damage or degradation of the fusion protein promyelocytic leukemia (PML)-retinoic acid receptor (RAR)-alpha.
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a single-arm trial of TRISENOX (0.15 mg/kg daily), 16 of 40 patients (40%) had a QTc interval greater than 500 msec. Prolongation of the QTc was observed between 1 and 5 weeks after TRISENOX infusion, and then returned towards baseline by the end of 8 weeks after TRISENOX infusion.
12.3 Pharmacokinetics
The inorganic, lyophilized form of arsenic trioxide, when placed into solution, immediately forms the hydrolysis product arsenious acid (AsIII). AsIII is the pharmacologically active species of arsenic trioxide. Monomethylarsonic acid (MMAV), and dimethylarsinic acid (DMAV) are the main pentavalent metabolites formed during metabolism, in addition to arsenic acid (AsV) a product of AsIII oxidation.
The pharmacokinetics of arsenical species ([AsIII], [AsV], [MMAV], [DMAV]) were determined in 6 APL patients following once-daily doses of 0.15 mg/kg for 5 days per week. Over the total single-dose range of 7 to 32 mg (administered as 0.15 mg/kg), systemic exposure (AUC) appears to be linear.
Peak plasma concentrations of arsenious acid (AsIII), the primary active arsenical species were reached at the end of infusion (2 hours). Plasma concentration of AsIII declined in a biphasic manner with a mean elimination half-life of 10 to 14 hours and is characterized by an initial rapid distribution phase followed by a slower terminal elimination phase. The daily exposure to AsIII (mean AUC0-24h) was 194 ng·hr/mL (n=5) on Day 1 of Cycle 1 and 332 ng·hr/mL (n=6) on Day 25 of Cycle 1, which represents an approximate 2-fold accumulation.
The primary pentavalent metabolites, MMAV and DMAV, are slow to appear in plasma (approximately 10 to 24 hours after first administration of arsenic trioxide), but, due to their longer half-life, accumulate more upon multiple dosing than does AsIII. The mean estimated terminal elimination half-lives of the metabolites MMAV and DMAV are 32 hours and 72 hours, respectively. Approximate accumulation ranged from 1.4- to 8-fold following multiple dosing as compared to single-dose administration. AsV is present in plasma only at relatively low levels.
Distribution
The volume of distribution (Vss) for AsIII is large (mean 562 L, N=10) indicating that AsIII is widely distributed throughout body tissues. Vss is also dependent on body weight and increases as body weight increases.
Elimination
Metabolism
Much of the AsIII is distributed to the tissues where it is methylated to the less cytotoxic metabolites, monomethylarsonic acid (MMAV) and dimethylarsinic acid (DMAV) by methyltransferases primarily in the liver. The metabolism of arsenic trioxide also involves oxidation of AsIII to AsV, which may occur in numerous tissues via enzymatic or nonenzymatic processes. AsV is present in plasma only at relatively low levels following administration of arsenic trioxide.
Excretion
Approximately 15% of the administered TRISENOX dose is excreted in the urine as unchanged AsIII. The methylated metabolites of AsIII (MMAV, DMAV) are primarily excreted in the urine. The total clearance of AsIII is 49 L/h and the renal clearance is 9 L/h. Clearance is not dependent on body weight or dose administered over the range of 7 to 32 mg.
Specific Populations
Patients with Renal Impairment
The effect of renal impairment on the pharmacokinetics of AsIII, AsV, and the pentavalent metabolites MMAV and DMAV was evaluated in 20 patients with advanced malignancies. Patients were classified as having normal renal function (creatinine clearance [CLcr] > 80 mL/min, n=6), mild renal impairment (CLcr 50 to 80 mL/min, n=5), moderate renal impairment (CLcr 30 to 49 mL/min, n=6), or severe renal impairment (CLcr < 30 mL/min, n=3). Following twice-weekly administration of 0.15 mg/kg over a 2-hour infusion, the mean AUC0-INF for AsIII was comparable among the normal, mild and moderate renal impairment groups. However, in the severe renal impairment group, the mean AUC0-INF for AsIII was approximately 48% higher than that in the normal group.
Systemic exposure to MMAV and DMAV tended to be larger in patients with renal impairment; however, the clinical consequences of this increased exposure are not known. AsV plasma levels were generally below the limit of assay quantitation in patients with impaired renal function [see Use in Specific Populations (8.6)]. The use of arsenic trioxide in patients on dialysis has not been studied.
Patients with Hepatic Impairment
The effect of pharmacokinetics of AsIII, AsV, and the pentavalent metabolites MMAV and DMAV was evaluated following administration of 0.25 to 0.50 mg/kg of arsenic trioxide in patients with hepatocellular carcinoma. Patients were classified as having normal hepatic function (n=4), mild hepatic impairment (Child-Pugh class A, n=12), moderate hepatic impairment (Child-Pugh class B, n=3), or severe hepatic impairment (Child-Pugh class C, n=1). No clear trend toward an increase in systemic exposure to AsIII, AsV, MMAV or DMAV was observed with decreasing level of hepatic function as assessed by dose-normalized (per mg dose) AUC in the mild and moderate hepatic impairment groups. However, the one patient with severe hepatic impairment had mean dose-normalized AUC0‑24h and Cmax values 40% and 70% higher, respectively, than those patients with normal hepatic function. The mean dose-normalized trough plasma levels for both MMAV and DMAV in this severely hepatically impaired patient were 2.2-fold and 4.7-fold higher, respectively, than those in the patients with normal hepatic function [see Use in Specific Populations (8.7)].
Pediatric Patients
Following intravenous administration of 0.15 mg/kg/day of arsenic trioxide in 10 APL patients (median age = 13.5 years, range 4-20 years), the daily exposure to AsIII (mean AUC0-24h) was 317 ng·hr/mL on Day 1 of Cycle 1 [see Use in Specific Populations (8.4)].
Drug Interaction Studies
No formal assessments of pharmacokinetic drug-drug interactions between TRISENOX and other drugs have been conducted. The methyltransferases responsible for metabolizing arsenic trioxide are not members of the cytochrome P450 family of isoenzymes. In vitro incubation of arsenic trioxide with human liver microsomes showed no inhibitory activity on substrates of the major cytochrome P450 (CYP) enzymes such as 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, and 4A9/11. The pharmacokinetics of drugs that are substrates for these CYP enzymes are not expected to be affected by concomitant treatment with arsenic trioxide.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with TRISENOX [see Warnings and Precautions (5.6)].
Arsenic trioxide and trivalent arsenite salts have not been demonstrated to be mutagenic to bacteria, yeast, or mammalian cells. Arsenite salts are clastogenic in vitro (human fibroblast, human lymphocytes, Chinese hamster ovary cells, Chinese hamster V79 lung cells). Trivalent arsenic was genotoxic in the chromosome aberrations assay and micronucleus bone marrow assay in mice.
The effect of arsenic on fertility has not been adequately studied in humans. Decreased testicular weight and impaired spermatogenesis have been reported in animal studies. Male Wistar rat pups were administered 1.5 mg/kg sodium arsenite solution via the intraperitoneal route from postnatal days 1 to 14 and testes were collected for evaluation on postnatal days 15, 21, and 50. Results of this study revealed an altered morphology of the seminiferous tubules along with degeneration of spermatogenic cells, increased number of sperm with abnormal morphology, and decreased sperm counts. In beagle dogs administered intravenous arsenic trioxide for 90 days, reduced inner cell layers within seminiferous tubules and significantly decreased numbers of spermatocytes, spermatozoa, and sperm cells were observed at doses of 1 mg/kg/day and higher. The 1 mg/kg/day dose is approximately 3 times the recommended human daily dose on a mg/m² basis.
-
14 CLINICAL STUDIES
14.1 Newly-Diagnosed Low-Risk APL
TRISENOX in combination with tretinoin was investigated in Study APL0406 (NCT00482833), a multicenter, randomized, open-label trial in patients with newly-diagnosed low-risk APL (white blood cell count at diagnosis ≤10 Gi/L). The patients were randomized 1:1 to receive TRISENOX/tretinoin for induction and consolidation or chemotherapy/tretinoin for induction, consolidation, and maintenance.
Patients in the TRISENOX/tretinoin group received induction treatment with TRISENOX 0.15 mg/kg intravenously once daily in combination with tretinoin 22.5 mg/m2 (rounded to the nearest 10 mg increment) orally twice daily until hematologic complete remission (CR) or for a maximum of 60 days. Patients in this group who achieved a CR during induction received four 8-week cycles of consolidation treatment with TRISENOX 0.15 mg/kg intravenously once daily for 5 days every week during weeks 1 to 4 of the 8-week cycle, in combination with tretinoin 22.5 mg/m2 (rounded to the nearest 10 mg increment) orally twice daily during weeks 1 to 2 and 5 to 6 of the 8-week cycle. Tretinoin was omitted during weeks 5 to 6 of the last cycle.
Patients in the chemotherapy/tretinoin group received idarubicin 12 mg/m2 intravenously once daily on days 2, 4, 6, and 8 in combination with tretinoin 22.5 mg/m2 (rounded to the nearest 10 mg increment) orally twice daily, starting on day 1, until hematologic CR or for a maximum of 60 days. Patients in this group who achieved a CR during induction received consolidation and maintenance treatment with tretinoin in combination with chemotherapy.
The trial enrolled 162 patients with a morphologic diagnosis of APL. The median age of patients was 45 years in the TRISENOX/tretinoin arm and 47 years in the chemotherapy/tretinoin arm, and 52% and 46% were male in the TRISENOX/tretinoin and chemotherapy/tretinoin arms, respectively. Baseline characteristics were balanced between treatment arms, including median WBC count, platelet count, PML-RARA isoform, and FLT3-ITD status.
Efficacy was based on event-free survival (EFS) rate at 2 years. EFS was defined as the time from randomization to the occurrence of treatment failure, defined as no achievement of CR or CRi after induction therapy, no achievement of molecular remission after 3 consolidation courses, molecular relapse, hematologic relapse, or death. The primary analysis of EFS was based on the difference between the two treatment arms in patients achieving EFS at 2 years. With a median follow-up of 34.4 months, the 2 year EFS rate of the modified ITT (mITT) population (patients who received at least one dose of the assigned treatment) was 94% in the TRISENOX/tretinoin arm (n=77) versus 82% in the chemotherapy/tretinoin arm (n=79), a treatment difference of 11% (95% CI: 1, 22; p-value 0.048). Overall survival (OS) for the mITT population was 99% (95% CI: 93, 100) in the TRISENOX/tretinoin arm versus 91% (95% CI: 86, 97) in the chemotherapy/tretinoin arm. The difference in 2-year OS rate between the arms was 8% (95% CI: 0, 16).
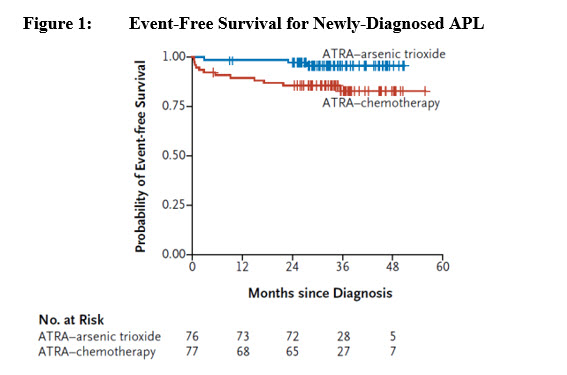
The number of patients in the plot is based on the mITT population.
14.2 Relapsed or Refractory APL
TRISENOX was investigated in Study PLRXAS01, an open-label, single-arm trial in 40 patients with relapsed or refractory APL who were previously treated with an anthracycline and a retinoid regimen. Patients received TRISENOX 0.15 mg/kg/day intravenously over 1 to 2 hours until the bone marrow was cleared of leukemic cells or for a maximum of 60 days. The CR (absence of visible leukemic cells in bone marrow and peripheral recovery of platelets and white blood cells with a confirmatory bone marrow ≥ 30 days later) rate in this population of previously treated patients was 28 of 40 (70%). Among the 22 patients who had relapsed less than one year after treatment with tretinoin, there were 18 complete responders (82%). Of the 18 patients receiving TRISENOX ≥ one year from tretinoin treatment, there were 10 complete responders (55%). The median time to bone marrow remission was 44 days and to onset of CR was 53 days. Three of 5 children, 5 years or older, achieved CR. No children less than 5 years old were treated.
Three to six weeks following bone marrow remission, 31 patients received consolidation therapy with TRISENOX, at the same dose, for 25 additional days over a period up to 5 weeks. In follow-up treatment, 18 patients received further TRISENOX as a maintenance course. Fifteen patients had bone marrow transplants. At last follow-up, 27 of 40 patients were alive with a median follow-up time of 484 days (range 280 to 755) and 23 of 40 patients remained in complete response with a median follow-up time of 483 days (range 280 to 755).
Cytogenetic conversion to no detection of the APL chromosome rearrangement was observed in 24 of 28 (86%) patients who met the response criteria defined above, in 5 of 5 (100%) patients who met some, but not all, of the response criteria, and 3 of 7 (43%) of patients who did not respond. RT-PCR conversions to no detection of the APL gene rearrangement were demonstrated in 22 of 28 (79%) of patients who met the response criteria, in 3 of 5 (60%) of patients who met some, but not all, of the response criteria, and in 2 of 7 (29%) of patients who did not respond.
Responses were seen across all age groups tested, ranging from 6 to 72 years. The ability to achieve a CR was similar for both sexes. There were insufficient patients of Black, Hispanic, or Asian ancestry to estimate relative response rates in these groups, but responses were seen in each group.
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
TRISENOX (arsenic trioxide) injection is supplied as a sterile, clear, colorless solution in 10 mL glass, single-dose vials.
NDC 63459-601-06: 12 mg/6 mL (2 mg/mL) vial in packages of ten vials.
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [See USP Controlled Room Temperature]. Do not freeze.
TRISENOX is a hazardous drug. Follow applicable special handling and disposal procedures.1
-
17 PATIENT COUNSELING INFORMATION
Differentiation Syndrome
Advise patients that symptoms of APL differentiation syndrome include fever, sudden weight gain, dizziness/lightheadedness, labored breathing, and accumulation of fluid in the lungs, heart, and chest. This syndrome is managed by immediate treatment with high-dose corticosteroids. Advise patients to immediately report any of these symptoms [see Warnings and Precautions (5.1)].Cardiac Conduction Abnormalities
Advise patients that TRISENOX may cause ECG abnormalities, including QT prolongation. If extreme, this prolongation has the potential to cause fainting, irregular heartbeat, or more serious side effects. Advise patients to immediately report any of these symptoms. Advise patients to provide a complete list of current medications as caution should be taken when TRISENOX is coadministered with other medications that can cause QT prolongation or lead to electrolyte abnormalities [see Warnings and Precautions (5.2) and Drug Interactions (7)].Encephalopathy and Wernicke's Encephalopathy (WE)
Advise patients that symptoms of encephalopathies include neurological symptoms such as confusion, decreased level of consciousness, seizures, cognitive deficits, ataxia, visual symptoms and ocular motor dysfunction. Advise patients and caregivers to closely monitor for neurological symptoms and immediately report them to their healthcare provider [see Warnings and Precautions (5.3)].
Advise patients at risk for thiamine deficiency (e.g., chronic alcohol use, malabsorption, nutritional deficiency, concomitant use of furosemide) that Wernicke’s encephalopathy is a neurologic emergency that can be prevented and treated with thiamine supplementation, and to immediately report any neurological symptoms to their healthcare provider [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.5) and Use in Specific Populations 8.1)].Advise females of reproductive potential to use effective contraception during treatment with TRISENOX and for 6 months after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with TRISENOX for 3 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with TRISENOX and for 2 weeks after the last dose [see Use in Specific Populations (8.2)].Infertility
Advise males of reproductive potential that TRISENOX may impair fertility [see Use in Specific Population (8.3)].
Other Adverse Reactions
Advise patients of the expected adverse reactions of TRISENOX. Most patients in clinical trials experienced some drug-related toxicity, most commonly leukocytosis, gastrointestinal symptoms (nausea, vomiting, diarrhea, and abdominal pain), fatigue, edema, hyperglycemia, dyspnea, cough, rash or itching, headaches, and dizziness. Advise patients to call their healthcare provider at the onset of any adverse reactions [see Adverse Reactions (6.1)].Manufactured for:
Teva Pharmaceuticals
Parsippany, NJ 07054©2022 Teva Pharmaceuticals, Inc.
Rev. 6/2022
TRI-015
- Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
TRISENOX
arsenic trioxide injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:63459-601 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ARSENIC TRIOXIDE (UNII: S7V92P67HO) (ARSENIC CATION (3+) - UNII:C96613F5AV) ARSENIC TRIOXIDE 2 mg in 1 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:63459-601-06 10 in 1 CARTON 12/22/2017 1 NDC:63459-601-11 6 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021248 12/22/2017 Labeler - Cephalon, LLC (183236314)
