Label: XENLETA- lefamulin acetate injection, solution
CITRIC BUFFERED NORMAL SALINE- anhydrous citric acid injection, solution
XENLETA- lefamulin acetate tablet, coated






-
NDC Code(s):
72000-030-01,
72000-030-06,
72000-110-10,
72000-110-30, view more72000-120-01, 72000-120-06
- Packager: Nabriva Therapeutics US, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 11, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use XENLETA® safely and effectively. See full prescribing information for XENLETA.
XENLETA (lefamulin) injection, for intravenous use
XENLETA (lefamulin) tablets, for oral use
Initial U.S. Approval: 2019
INDICATIONS AND USAGE
XENLETA is a pleuromutilin antibacterial indicated for the treatment of adults with community-acquired bacterial pneumonia (CABP) caused by susceptible microorganisms. (1.1)
To reduce the development of drug resistant bacteria and maintain the effectiveness of XENLETA and other antibacterial drugs, XENLETA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1.2)
DOSAGE AND ADMINISTRATION
- For treatment of adults with CABP, the recommended dosage of XENLETA is as follows:
*With the option to switch to XENLETA Tablets 600 mg every 12 hours to complete the treatment course.
Dosage Treatment Duration 150 mg every 12 hours by intravenous infusion over 60 minutes* (2.1) 5 to 7 days 600 mg orally every 12 hours.(2.1) 5 days - Patients with Hepatic Impairment: Reduce the dosage of XENLETA Injection to 150 mg infused over 60 minutes every 24 hours in patients with severe hepatic impairment (Child-Pugh Class C). XENLETA Tablets have not been studied in and are not recommended for patients with moderate (Child-Pugh Class B) or severe hepatic impairment (2.2).
- Administration Instruction for XENLETA Tablets: Take at least 1 hour before a meal or 2 hours after a meal. Swallow XENLETA Tablets whole with water (6 to 8 ounces). (2.3)
- Administration Instruction for XENLETA Injection: Infuse over 60 minutes. (2.3)
- See Full Prescribing Information for additional information on the administration and preparation of XENLETA Tablets and Injection. (2.4)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- QT Prolongation: Avoid use in patients with known QT prolongation, ventricular arrhythmias including torsades de pointes, and patients receiving drugs that prolong the QT interval such as antiarrhythmic agents. (5.1)
- Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. (5.2, 8.1, 8.3)
- Clostridioides difficile-associated Diarrhea (CDAD): Evaluate patients who develop diarrhea. (5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥2%) are:
- XENLETA Injection: administration site reactions, hepatic enzyme elevation, nausea, hypokalemia, insomnia, headache. (6.1)
- XENLETA Tablets: diarrhea, nausea, vomiting, hepatic enzyme elevation. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Nabriva Therapeutics US, Inc. at 1-855-5NABRIVA or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
XENLETA Injection Strong or moderate CYP3A inducers or P-gp inducers Avoid XENLETA unless the benefit outweighs the risk. Monitor for reduced efficacy. (7.1) XENLETA Tablets Strong or moderate CYP3A inducers or P-gp inducers Avoid XENLETA unless the benefit outweighs the risk. Monitor for reduced efficacy. (7.1) Strong CYP3A inhibitors or P-gp inhibitors Avoid XENLETA. (7.1) Moderate CYP3A inhibitors or P-gp inhibitors Monitor for adverse reactions. (7.1) CYP3A substrates that prolong the QT interval Concomitant use is contraindicated. (4.2, 7.2) Midazolam and other sensitive CYP3A substrates Monitor for adverse reactions. (7.2) USE IN SPECIFIC POPULATIONS
Lactation: A lactating woman should pump and discard human milk for the duration of treatment with XENLETA and for 2 days after the final dose. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2021
- For treatment of adults with CABP, the recommended dosage of XENLETA is as follows:
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Community-Acquired Bacterial Pneumonia (CABP)
1.2 Usage
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Adjustment for Patients with Hepatic Impairment
2.3 Important Administration Instructions
2.4 Preparation of XENLETA Injection for Intravenous Infusion
2.5 Storage of XENLETA Injection After Dilution
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Hypersensitivity
4.2 CYP3A4 Substrates That Prolong the QT Interval
5 WARNINGS AND PRECAUTIONS
5.1 QT Prolongation
5.2 Embryo-Fetal Toxicity
5.3 Clostridioides difficile-associated Diarrhea
5.4 Development of Drug-Resistant Bacteria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on XENLETA
7.2 Effect of XENLETA on Other Drugs
7.3 Drugs that Prolong QT
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Community-Acquired Bacterial Pneumonia
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Community-Acquired Bacterial Pneumonia (CABP)
XENLETA is indicated for the treatment of adults with community-acquired bacterial pneumonia (CABP) caused by the following susceptible microorganisms: Streptococcus pneumoniae, Staphylococcus aureus (methicillin-susceptible isolates), Haemophilus influenzae, Legionella pneumophila, Mycoplasma pneumoniae, and Chlamydophila pneumoniae.
1.2 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of XENLETA and other antibacterial drugs, XENLETA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
For treatment of adults with CABP, the recommended dosage of XENLETA is described in Table 1 below. For patients with severe hepatic impairment, dosage adjustment is required [see Dosage and Administration (2.2)].
Table 1: Dosage of XENLETA in Adult CABP Patients *With the option to switch to XENLETA Tablets 600 mg every 12 hours to complete the treatment course.
Dosage Treatment Duration 150 mg every 12 hours by intravenous infusion over 60 minutes* 5 to 7 days 600 mg orally every 12 hours 5 days 2.2 Dosage Adjustment for Patients with Hepatic Impairment
Monitor patients with hepatic impairment for adverse reactions associated with XENLETA Injection and Tablets throughout the treatment period [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
XENLETA Injection
Reduce the dosage of XENLETA Injection to 150 mg infused intravenously over 60 minutes every 24 hours for patients with severe hepatic impairment (Child-Pugh Class C). No dosage adjustment of XENLETA Injection is needed for patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment.
XENLETA Tablets
XENLETA Tablets have not been studied in and are not recommended for patients with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment. No dosage adjustment of XENLETA Tablets is needed for patients with mild hepatic impairment (Child-Pugh Class A).
2.3 Important Administration Instructions
XENLETA Injection
Administer XENLETA Injection by intravenous infusion over 60 minutes. Must dilute in a 250 mL solution of 10 mM citrate buffered 0.9% sodium chloride for injection supplied with XENLETA Injection before use [see Dosage and Administration (2.4)].
XENLETA Tablets
Take XENLETA Tablets at least 1 hour before a meal or 2 hours after a meal. Swallow XENLETA Tablets whole with water (6 to 8 ounces). Do not crush or divide XENLETA Tablets [see Clinical Pharmacology (12.3)].
Missed Dose
If a dose is missed, the patient should take the dose as soon as possible and anytime up to 8 hours prior to the next scheduled dose. If less than 8 hours remain before the next scheduled dose, do not take the missed dose, and resume dosing at the next scheduled dose.
2.4 Preparation of XENLETA Injection for Intravenous Infusion
- Dilute the entire 15 mL vial of XENLETA Injection into the diluent bag supplied with XENLETA injection that contains 250 mL of 10 mM citrate buffered 0.9% sodium chloride.
- Use aseptic technique when adding XENLETA Injection into the diluent bag. Mix thoroughly.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Use the diluent bag only if the solution is clear and the container is undamaged.
- Do not use the diluent bag in series connections.
- Do not add other additives to the diluent bag because their compatibilities with XENLETA Injection have not been established.
-
3 DOSAGE FORMS AND STRENGTHS
XENLETA Injection
Clear, colorless solution in a single-dose clear glass vial. Each vial contains 150 mg of lefamulin in 15 mL of 0.9% sodium chloride for further dilution [see Dosage and Administration (2.4)].
XENLETA Tablets
Blue, oval, film-coated tablet with ‘LEF 600’ printed in black on one side. Each tablet contains 600 mg of lefamulin.
-
4 CONTRAINDICATIONS
4.1 Hypersensitivity
XENLETA is contraindicated in patients with known hypersensitivity to lefamulin, pleuromutilin class drugs, or any of the components of XENLETA.
4.2 CYP3A4 Substrates That Prolong the QT Interval
XENLETA Tablets are contraindicated with sensitive CYP3A4 substrates that prolong the QT interval (for example, pimozide). Concomitant administration of oral XENLETA with sensitive CYP3A4 substrates may result in increased plasma concentrations of these drugs, leading to QT prolongation and cases of torsades de pointes [see Warnings and Precautions (5.1), Drug Interactions (7.2), and Clinical Pharmacology (12.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 QT Prolongation
XENLETA has the potential to prolong the QT interval of the electrocardiogram (ECG) in some patients. Avoid XENLETA use in the following patients:
- Patients with known prolongation of the QT interval
- Patients with ventricular arrhythmias including torsades de pointes
- Patients receiving Class IA (for example, quinidine, procainamide) or Class III (for example, amiodarone, sotalol) antiarrhythmic agents
- Patients receiving other drugs that prolong the QT interval, such as antipsychotics, erythromycin, pimozide, moxifloxacin, and tricyclic antidepressants
In patients with renal failure who require dialysis, metabolic disturbances associated with renal failure may lead to QT prolongation.
In patients with mild, moderate, or severe hepatic impairment, metabolic disturbances associated with hepatic impairment may lead to QT prolongation.
If use with XENLETA cannot be avoided in specific populations predisposed to QT prolongation or those receiving another drug that prolongs the QT interval, ECG monitoring is recommended during treatment.
The magnitude of QT prolongation may increase with increasing concentrations of XENLETA or increasing the rate of infusion of the intravenous formulation. Therefore, the recommended dose and infusion rate should not be exceeded.
5.2 Embryo-Fetal Toxicity
Based on findings from animal studies, lefamulin may cause fetal harm when administered to pregnant women. Animal studies indicate that administration of lefamulin resulted in an increased incidence of post-implantation fetal loss and stillbirths in rats and rabbits treated during the period of organogenesis or in rats treated from the beginning of organogenesis through the time of weaning. Additional rat pup deaths were observed during early lactation that were likely related to maternal treatment with lefamulin. Decreased fetal body weights and ossification in rats and rabbits, and apparent delay in sexual maturation in rats may indicate treatment-related developmental delay, while other findings such as malformations in rats at systemic exposures lower than the systemic exposure in CABP patients may indicate a risk for embryo-fetal toxicity.
Verify pregnancy status in females of reproductive potential prior to initiating XENLETA. Advise females of reproductive potential to use effective contraception during treatment with XENLETA and for 2 days after the final dose. Advise pregnant women and females of reproductive potential of the potential risk to a fetus [see Use in Specific Populations (8.1, 8.3)].
5.3 Clostridioides difficile-associated Diarrhea
Clostridioides difficile-associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including XENLETA, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin-producing isolates of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial drug use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial drug use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial drug treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- QT Prolongation [see Warnings and Precautions (5.1)].
- Clostridioides difficile-associated Diarrhea [see Warnings and Precautions (5.3)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
XENLETA was evaluated in two clinical trials in CABP patients (Trial 1 and Trial 2). Across the two trials, a total of 641 patients were treated with XENLETA. Trial 1 (intravenous [IV] to oral dosing switch trial) enrolled 551 adult patients, 276 randomized to XENLETA (273 received at least one dose of XENLETA) and 275 randomized to moxifloxacin (273 received at least one dose of moxifloxacin). Trial 2 (oral dosing only trial) enrolled 738 adult patients, 370 randomized to XENLETA (368 received at least one dose of XENLETA) and 368 randomized to moxifloxacin (all 368 received at least one dose of moxifloxacin).
Trial 1 enrolled patients with Pneumonia Outcomes Research Team (PORT) Risk Class III-V. The mean duration of intravenous treatment was 6 days; the mean total duration of treatment was 7 days. Trial 2 enrolled patients with PORT Risk Class II-IV. The mean duration of treatment was 5 days for XENLETA and 7 days for moxifloxacin.
In Trial 1 and Trial 2 (pooled), the median age of patients treated with XENLETA was 61 (range 19-97) years; 42% of patients were 65 years or older and 18% were 75 years or older. Patients were predominantly male (58%) and white (79%) and had a median body mass index (BMI) of 26.0 (range 13.0-56.8) kg/m2. Approximately 52% of XENLETA-treated patients had creatinine clearance (CrCl) <90 mL/min.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In Trial 1 and Trial 2 (pooled), serious adverse reactions occurred in 36/641 (5.6%) patients treated with XENLETA and 31/641 (4.8%) patients treated with moxifloxacin. Treatment was discontinued due to an adverse reaction in 21/641 (3.3%) patients treated with XENLETA and 21/641 (3.3%) patients treated with moxifloxacin. Death within 28 days occurred in 8/641 (1.2%) patients treated with XENLETA and 7/641 (1.1%) patients treated with moxifloxacin.
Most Common Adverse Reactions
Table 2 and Table 3 include adverse reactions occurring in ≥2% of patients receiving XENLETA in Trials 1 and 2.
Table 2: Adverse Reactions Occurring in ≥2% of Patients Receiving XENLETA in Trial 1 *Administration site reactions include infusion site pain, infusion site phlebitis, and injection site reaction.
**Hepatic enzyme elevation includes alanine aminotransferase increased, aspartate aminotransferase increased, and liver function test increased.
Adverse Reaction Trial 1
IV ± Oral DosingXENLETA
N=273Moxifloxacin
N=273Administration site reactions* 7% 3% Hepatic enzyme elevation** 3% 3% Nausea 3% 2% Hypokalemia 3% 2% Insomnia 3% 2% Headache 2% 2% Table 3: Adverse Reactions Occurring in ≥2% of Patients Receiving XENLETA in Trial 2 **Hepatic enzyme elevation includes alanine aminotransferase increased, aspartate aminotransferase increased, and liver function test increased.
Adverse Reaction Trial 2
Oral DosingXENLETA
N=368Moxifloxacin
N=368Diarrhea 12% 1% Nausea 5% 2% Vomiting 3% 1% Hepatic enzyme elevation** 2% 2% Selected Adverse Reactions Occurring in Less Than 2% of Patients Receiving XENLETA in Trials 1 and 2
Blood and Lymphatic System Disorders: anemia, thrombocytopenia
Cardiac Disorders: atrial fibrillation, palpitations
Gastrointestinal Disorders: abdominal pain, constipation, dyspepsia, epigastric discomfort, erosive gastritis
Infections and Infestations: Clostridioides difficile colitis, oropharyngeal candidiasis, vulvovaginal candidiasis
Investigations: alkaline phosphatase increased, creatine phosphokinase increased, electrocardiogram QT prolonged, gamma-glutamyl transferase increased
Nervous System Disorders: somnolence
Psychiatric Disorders: anxiety
Renal and Urinary Disorders: urinary retention
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on XENLETA
Strong and Moderate CYP3A Inducers or P-gp Inducers
Concomitant use of oral or intravenous XENLETA with strong CYP3A4 inducers or P-gp inducers decreases lefamulin AUC and Cmax [see Clinical Pharmacology (12.3)] ,which may reduce the efficacy of XENLETA. Avoid concomitant use of XENLETA Injection and XENLETA Tablets with strong and moderate CYP3A4 inducers or P-gp inducers unless the benefit outweighs the risks.
Strong and Moderate CYP3A Inhibitors or P-gp Inhibitors
Concomitant use of XENLETA Tablets with strong CYP3A inhibitors or P-gp inhibitors increases lefamulin AUC [see Clinical Pharmacology (12.3)] , which may increase the risk of adverse reactions with XENLETA Tablets. Avoid concomitant use of XENLETA Tablets with strong CYP3A inhibitors or P-gp inhibitors. Monitor for adverse effects of XENLETA Tablets when administered concomitantly with moderate CYP3A inhibitors or P-gp inhibitors.
7.2 Effect of XENLETA on Other Drugs
CYP3A4 Substrates
Concomitant use of XENLETA Tablets with sensitive CYP3A4 substrates increases the AUC and Cmax of CYP3A4 substrates [see Clinical Pharmacology (12.3)] , which may increase the risk of toxicities associated with cardiac conduction. Concomitant use with CYP3A substrates known to prolong the QT interval is contraindicated [see Contraindications (4.2)]. Concomitant use of sensitive CYP3A substrates with XENLETA Tablets requires close monitoring for adverse effects of these drugs (for example, alprazolam, diltiazem, verapamil, simvastatin, vardenafil). Concomitant use of XENLETA Injection with CYP3A4 substrates does not affect the exposure of CYP3A4 substrates.
7.3 Drugs that Prolong QT
The pharmacodynamic interaction potential to prolong the QT interval of the electrocardiogram between XENLETA and other drugs that effect cardiac conduction is unknown. Therefore, avoid concomitant use of XENLETA Injection and XENLETA Tablets with such drugs (for example, Class IA and III antiarrhythmics, antipsychotics, erythromycin, moxifloxacin, tricyclic antidepressants).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies, lefamulin may cause fetal harm when administered to pregnant women. There are no available data on the use of XENLETA in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes.
Animal studies indicate that intravenous administration of lefamulin during organogenesis resulted in an increased incidence of prenatal mortality at mean maternal exposures 0.9 times the mean exposure in clinical patients (based on AUC0-24h), decreased fetal body weights, apparent delay in sexual maturation that suggest treatment-related developmental delay, and malformations in rats at maternal exposures greater than 0.4 times the mean exposure in CABP patients for which the litter incidence was nonexistent in concurrent controls and rare (0 to approximately 0.3%) in historical controls. Decreased ossification was seen in fetuses at all doses in a dose-related manner, suggestive of developmental delay (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
There is a pregnancy pharmacovigilance program for XENLETA. If XENLETA is inadvertently administered during pregnancy or if a patient becomes pregnant while receiving XENLETA, healthcare providers should report XENLETA exposure by calling 1-855-5NABRIVA to enroll.
Animal Data
In a prenatal and postnatal development study in rats treated from the beginning of organogenesis through lactation (Gestation Day [GD] 6 through lactation day 21), the percent of live births was reduced (87.4% compared with the concurrent control of 98.7%) in the high dose group of 100 mg/kg/day (0.9 times the mean exposure in CABP patients treated IV). Equivocal findings in that study were indicative of early post-natal mortality and apparent developmental delay that may be related to pre-natal effects.
In the rat embryo-fetal development study of IV lefamulin during organogenesis (GD 6-17), findings included late resorptions in the high-dose group and malformations (cleft palate/jaw/vertebral malformations at the mid and high doses and enlarged ventricular heart chamber with a thin ventricular wall at the high dose) for which the litter incidence was nonexistent in concurrent controls and rare in historical controls (0 to approximately 0.3%). Decreased or no ossification in a number of skeletal elements in all treated groups may indicate treatment-related developmental delay at all doses. The mean exposure at the lowest dose was approximately 0.4 times the mean exposure in CABP patients treated IV. The main human metabolite, 2R-hydroxy lefamulin, was evaluated in an embryo-fetal development study in rats after IV administration and was also associated with the same cardiac malformation seen in the above study, enlarged ventricular heart chamber with or without a thin ventricular wall (which could be associated with undetected valve or great vessel anomalies).
In the rabbit embryo-fetal development study of IV lefamulin during organogenesis (GD 6-18), low numbers of live fetuses in utero in treated groups limited evaluation of the study. Additional findings at the high dose included decreased fetal weight and decreased or no ossification of skeletal elements, which may be indicative of developmental delay. A NOAEL was not determined. The lowest dose (not fully evaluated due to fetal mortality) would correspond to a mean exposure approximately 0.1 times the mean exposure in CABP patients.
Results of animal studies indicate that lefamulin crosses the placenta and is found in fetal tissues. Following a single intravenous administration of 30 mg/kg radio-labelled lefamulin to pregnant female rats on Day 17 of gestation, radioactivity was visible in fetal tissue, with greatest concentrations measured in the placenta and fetal liver (34.3 and 8.26 mcg equivalents/g, respectively) compared to 96.6 mcg equivalents/g in the maternal liver. Radioactivity in fetal tissues generally declined rapidly, and radioactivity associated with the fetus itself was below the limit of quantification by 12 hours post-dose. Radioactivity in the placenta declined rapidly and was below the limit of quantification by 24 hours after dosing. Concentrations of radioactivity in the amniotic sac remained measurable at the final sampling time (72 hours), peaking at 6 hours post-dose. The amniotic fluid did not contain radioactivity at any time after dose administration.
8.2 Lactation
Risk Summary
There are no data on the presence of XENLETA in human milk, its effects on the breastfed infant, or its effects on milk production. Animal studies indicate that lefamulin was concentrated in the milk of lactating rats (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions, including QT prolongation, a woman should pump and discard human milk for the duration of treatment with XENLETA and for 2 days after the final dose.
Administration of a single intravenous dose of 30 mg/kg radio-labelled lefamulin to lactating rats resulted in maximal mean concentrations of radioactivity in plasma and milk at 0.25-hour post-dose (3.29 and 10.7 mcg equivalents/g, respectively) that were markedly reduced at 24 hours post-dose (0.00663 and 0.0700 mcg equivalents/g, respectively). Milk/plasma ratios increased from 3.27 at 0.25-hour post-dose to 8.33 at 6 hours post-dose. These data indicate that pups would be exposed to lefamulin and its metabolites in maternal milk.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with XENLETA and for 2 days after the final dose. XENLETA may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of XENLETA in patients less than 18 years of age has not yet been established.
8.5 Geriatric Use
Of the 646 patients randomized to XENLETA in Trials 1 and 2, 268 (41.5%) were ≥65 years of age. Early clinical response (ECR) rates in the subgroup of patients ≥65 were similar to ECR rates in subjects <65 years of age and comparable across treatment groups (XENLETA versus moxifloxacin).
The adverse reaction profiles in patients ≥65 years and in patients <65 years of age were similar. The percentage of patients in the XENLETA group who had at least one adverse reaction was 30% in patients ≥65 years and 38% in patients <65 years.
8.6 Hepatic Impairment
XENLETA Injection
Dosage of XENLETA Injection should be reduced by extending the dosing interval for patients with severe hepatic impairment (Child-Pugh Class C). No dosage adjustment of XENLETA Injection is needed for patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment.
XENLETA Tablets:
XENLETA Tablets have not been studied in patients with hepatic impairment. It is not recommended to use XENLETA Tablets for patients with moderate or severe hepatic impairment [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
XENLETA is a semi-synthetic antibacterial agent for oral and intravenous administration.
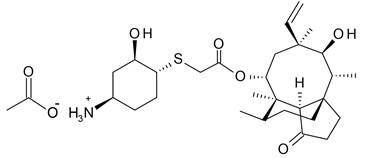
XENLETA, a pleuromutilin derivative, is available as 14-O-{[(1R,2R,4R)-4-amino-2-hydroxy-cyclohexylsulfanyl]-acetyl}-mutilin in the form of an acetic acid salt (acetate). It is a chemical substance with a molecular weight of 567.79 grams per mole. Its empirical formula is C30H49NO7S and its chemical structure is:
XENLETA Tablets for oral administration are available as blue, oval, film-coated tablets containing 671 mg lefamulin acetate equivalent to 600 mg lefamulin. The inactive ingredients are colloidal silicon dioxide, croscarmellose sodium, FD&C Blue No 2 aluminum lake, ferrosoferric oxide, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol (partially hydrolyzed), povidone K30, shellac glaze, talc, and titanium dioxide.
XENLETA Injection supplied as a sterile injection for intravenous use is available as a clear colorless solution in a glass vial containing 168 mg of lefamulin acetate equivalent to 150 mg of lefamulin in 15 mL of 0.9% sodium chloride. This is equivalent to 10 mg/mL lefamulin. The inactive ingredients are sodium chloride and water for injection.
XENLETA Injection must be diluted with the diluent supplied with XENLETA Injection, before administration by intravenous infusion. Each supplied diluent infusion bag contains 250 mL of 10 mM citrate buffered (pH 5) 0.9% sodium chloride. The diluent is a clear, colorless solution. The inactive ingredients are citric acid anhydrous, sodium chloride, trisodium citrate dihydrate, and water for injection. Each 100 mL contains: sodium chloride 900 mg, trisodium citrate dihydrate 200 mg, and citric acid anhydrous 61.5 mg in water for injection. Electrolytes per 1000 mL: sodium 174 mEq; chloride 154 mEq. The osmolality is 280-340 mOsm/kg and the pH is 4.5-5.5.
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
The 24 h free-drug AUC to minimal inhibitory concentration (MIC) ratio has been shown to be the best Pharmacokinetic-Pharmacodynamic (PK-PD) index for the antibacterial activity of lefamulin in animal infection models of Streptococcus pneumoniae and Staphylococcus aureus pneumonia.
Cardiac Electrophysiology
The QTcF interval prolongation risk of XENLETA was evaluated using 2 randomized, double-blind, double-dummy, active controlled (moxifloxacin 400 mg once daily), parallel group, trials (Trials 1 and 2) in adult patients with CABP. A concentration dependent QTc prolongation effect of XENLETA was observed. The mean change from baseline QTcF (90% two-sided upper confidence interval) values around Tmax on day 3 or 4 were 13.6 ms (15.5 ms) for 150 mg injection administered twice daily as infusion and 9.3 ms (10.9 ms) for 600 mg tablet administered twice daily. The mean change from baseline QTcF (90% two-sided upper confidence interval) values around Tmax for the moxifloxacin randomized comparison arm on day 3 or 4 were 16.4 ms (18.3 ms) for 400 mg injection administered once daily as infusion and 11.6 ms (13.2 ms) for 400 mg tablet administered once daily.
12.3 Pharmacokinetics
Following single-dose intravenous administration, the AUC of lefamulin increased approximately dose-proportionally while the Cmax of lefamulin increased less than dose-proportionally over a dose range of 25 mg (0.17 times the approved dose) to 400 mg (2.67 times the approved dose). Following single-dose oral administration, the AUC of lefamulin increased more than dose proportionally over a dose range of 500 mg (0.8 times the approved dose) to 750 mg (1.25 times the approved dose).
Pharmacokinetic (PK) parameters of lefamulin following administration of XENLETA Injection or Tablets to patients with CABP are listed in Table 4.
The mean lefamulin AUC0-24h and Cmax in patients with CABP were 73% and 30% higher, respectively, compared with healthy subjects.
Table 4: Pharmacokinetic (PK) Parameters of Lefamulin Following Single or Multiple Dose (Every 12 Hours) XENLETA Administered as 150 mg (Infused Over 60 Minutes) Intravenously (IV) or 600 mg Orally in Patients with CABP a a Based on population PK modeling (Trial 1 for IV administration and Trial 2 for oral administration)
b Cmax=maximum plasma concentration; Cmin=trough plasma concentration; AUC0–24h=area under the plasma concentration-time curve from time zero to 24 hours
c Dose administered under fasting conditions (1 hour before or 2 hours after a meal)
PK Parameters b Administration Route Arithmetic Mean (% CV) Day 1 Steady State Cmax (mcg/mL) IV 3.50 (11.7) 3.60 (14.6) Oral c 2.24 (36.4) 2.24 (37.1) Cmin (mcg/mL) IV 0.398 (68.1) 0.573 (89.4) Oral c 0.593 (67.3) 0.765 (75.7) AUC0-24h (mcg·h/mL) IV 27.0 (31.8) 28.6 (46.9) Oral c 30.7 (45.0) 32.7 (49.2) Absorption
The mean oral bioavailability of XENLETA Tablets is approximately 25% and peak lefamulin plasma concentration occurred 0.88 to 2 hours after administration to healthy subjects.
Effect of Food
The concomitant administration of a single oral dose of 600 mg XENLETA Tablets with a high fat (approximately 50% of total calories from fat), high calorie breakfast (approximately 800-1000 calories) slightly reduced bioavailability. The mean relative reduction for oral XENLETA (fasted vs. fed) was on average 22.9% [90% CI: 12.2; 32.3] for the Cmax and 18.43% [90% CI: 11.7; 24.7] for the AUC0-inf.
Distribution
Mean plasma protein binding of lefamulin ranges from 94.8% at 2.35 mcg/mL to 97.1% at 0.25 mcg/mL in healthy adults.
The mean (min to max) steady state volume of distribution of lefamulin is 86.1 L (34.2 to 153 L) in patients with CABP after administration of XENLETA Injection.
Following a single IV administration of lefamulin 150 mg to healthy subjects, the highest lefamulin epithelial lining fluid (ELF) concentrations were observed at the end of infusion. The mean ELF and plasma AUC0-8 was 3.87 mcg·h/mL and 5.27 mcg·h/mL, respectively. The estimated ratio of ELF AUC to unbound plasma AUC is approximately 15.
Elimination
The mean (min to max) total body clearance of lefamulin is 11.9 L/h (2.94 to 30.0 L/h) in patients with CABP after XENLETA Injection administration.
The mean (min to max) elimination half-life of lefamulin is approximately 8 hours (3 to 20 h) in patients with CABP.
Metabolism
Lefamulin is primarily metabolized by CYP3A4.
Excretion
In healthy adult subjects, the mean % of total radioactivity excreted in feces was 77.3% (4.2% to 9.1% unchanged) and 88.5% (7.8% to 24.8% unchanged), and in urine was 15.5% (9.6% to 14.1% unchanged) and 5.3% (unchanged not determined) following 150 mg IV or 600 mg oral XENLETA, respectively.
Specific Populations
No clinically significant differences in the pharmacokinetics of XENLETA were observed based on age, sex, race, weight, or renal impairment including patients receiving hemodialysis.
Patients with Hepatic Impairment
The disposition of lefamulin was evaluated in non-infected subjects with normal hepatic function and with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment following administration of XENLETA Injection. The half-life of lefamulin is prolonged in subjects with severe hepatic impairment compared to that in subjects with normal hepatic function (17.5 h versus 11.5 h). Protein binding of lefamulin is reduced in subjects with hepatic impairment. Therefore, unbound (biologically active) lefamulin concentrations increased with the degree of hepatic impairment. On average, unbound lefamulin plasma AUC0-inf was increased 3-fold in subjects with severe hepatic impairment compared to that in subjects with normal hepatic function. There is no information to evaluate the effect of hepatic impairment on the disposition of lefamulin following administration of XENLETA Tablets. Thus, XENLETA Tablets are not recommended in patients with moderate or severe hepatic impairment [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
Effect of Other Drugs on the Pharmacokinetics of Lefamulin
Strong CYP3A inducers or P-gp inducers: oral rifampin (strong inducer) reduced the mean lefamulin AUC0-inf and Cmax by 28% and 8%, respectively, when administered concomitantly with XENLETA Injection. Additionally, oral rifampin reduced the mean lefamulin AUC0-inf and Cmax by 72% and 57%, respectively, when administered concomitantly with XENLETA Tablets.
Strong CYP3A inhibitors or P-gp inhibitors: oral ketoconazole (strong inhibitor) increased the mean lefamulin AUC0-inf and Cmax by 31% and 6%, respectively, when administered concomitantly with XENLETA Injection. Additionally, oral ketoconazole (strong inhibitor) increased the lefamulin AUC0-inf and Cmax by 165% and 58%, respectively, when administered concomitantly with XENLETA tablets.
Effect of Lefamulin on the Pharmacokinetics of Other Drugs
CYP3A Substrates: No clinically significant differences in the pharmacokinetics of midazolam were observed when administered concomitantly with XENLETA Injection. Mean AUC0-inf and Cmax of midazolam were increased by approximately 200% and 100%, respectively, when oral midazolam (CYP3A substrate) was administered concomitantly with and at 2 or 4 hours after administration of XENLETA Tablets.
P-gp substrates: No clinically significant differences in the pharmacokinetics of digoxin (P-gp substrate) were observed when administered concomitantly with XENLETA Tablets.
In Vitro Studies Where Drug Interaction Potential Was Not Further Evaluated Clinically
Lefamulin inhibited CYP2C8 (IC50=37.0 mcg/mL), BCRP (Breast Cancer Resistance Protein) (IC50=21.4 mcg/mL), and MATE1 (IC50=0.15 mcg/mL).
12.4 Microbiology
Mechanism of Action
XENLETA is a systemic pleuromutilin antibacterial. It inhibits bacterial protein synthesis through interactions (hydrogen bonds, hydrophobic interactions, and Van der Waals forces) with the A- and P-sites of the peptidyl transferase center (PTC) in domain V of the 23s rRNA of the 50S subunit. The binding pocket of the bacterial ribosome closes around the mutilin core for an induced fit that prevents correct positioning of tRNA.
XENLETA is bactericidal in vitro against S. pneumoniae, H. influenzae and M. pneumoniae (including macrolide-resistant strains), and bacteriostatic against S. aureus and S. pyogenes at clinically relevant concentrations.
XENLETA is not active against Enterobacteriaceae and Pseudomonas aeruginosa.
Resistance
The resistance frequency to XENLETA due to spontaneous mutations in vitro at 2-8 times the MIC was 2 x 10-9 to <2 x 10-11 for S. aureus, <1 x 10-9 to <3 x 10-10 for S. pneumoniae, and <4 x 10-9 to <2 x 10-10 for S. pyogenes. Resistance development at sub-MIC concentrations required greater than 1 mutational step with no resistant clones detected at ≥4-times MIC.
Resistance mechanisms that affect XENLETA include specific protection or modification of the ribosomal target by ABC-F proteins such as vga (A, B, E), lsa(E), sal(A), Cfr methyl transferase, or by mutations of ribosomal proteins L3 and L4. Cfr methyl transferase has the potential to mediate cross-resistance between lefamulin and phenicols, lincosamides, oxazolidinones, and streptogramin A antibacterials.
Some isolates resistant to β-lactams, glycopeptides, macrolides, mupirocin, quinolones, tetracyclines, and trimethoprim-sulfamethoxazole may be susceptible to XENLETA.
Interaction with Other Antimicrobials
In vitro studies demonstrated no antagonism between XENLETA and other antibacterial drugs (e.g., amikacin, azithromycin, aztreonam, ceftriaxone, levofloxacin, linezolid, meropenem, penicillin, tigecycline, trimethoprim/sulfamethoxazole, and vancomycin).
XENLETA has demonstrated synergy in vitro with doxycycline against S. aureus.
Antimicrobial Activity
XENLETA has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1.1)]:
Gram-positive BacteriaStreptococcus pneumoniae
Staphylococcus aureus (methicillin-susceptible isolates)
Gram-negative BacteriaHaemophilus influenzae
Other BacteriaMycoplasma pneumoniae
Chlamydophila pneumoniae
Legionella pneumophila
At least 90% of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoints for XENLETA against isolates of similar genus or organism group. However, the safety and efficacy of XENLETA in treating clinical infections due to these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-positive BacteriaStaphylococcus aureus (methicillin-resistant [MRSA] isolates)
Streptococcus agalactiae
Streptococcus anginosus
Streptococcus mitis
Streptococcus pyogenes
Streptococcus salivarius
Gram-negative BacteriaHaemophilus parainfluenzae
Moraxella catarrhalis
Susceptibility Test MethodsFor specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies have not been conducted with lefamulin.
Lefamulin did not elicit genotoxic potential in an in vivo rat bone marrow micronucleus assay for clastogenicity or in the in vitro Mouse Lymphoma Ly5178Y TK+/- mutation assay. The main human metabolite of lefamulin (2R-hydroxy lefamulin) also did not elicit genotoxic potential in the in vitro Mouse Lymphoma Ly5178Y TK+/- mutation assay.
In rats, there were no effects on male fertility that were considered to be related to lefamulin. Reproductive indices including mating behavior and fertility were not changed in any group in either gender at the highest dose tested (75 mg/kg/day, approximately 0.7 times the mean exposure of CABP patients treated IV, based on AUC0-24h); that dose was the NOAEL for fertility in male rats. In females, abnormal estrous cycling and increased post-implantation loss were observed at the high dose, making the NOAEL for fertility and early embryonic development in female rats the next highest dose, 50 mg/kg/day (approximately 0.5 times the mean exposure of CABP patients treated IV).
13.2 Animal Toxicology and/or Pharmacology
Following IV administration of lefamulin to rats for 4 or 13 weeks, anemia (all doses), increased coagulation times, and lower organ weights and histopathological changes in spleen (decreased peri-arteriolar lymphoid sheath, decreased size of the marginal zone) and thymus (cortical atrophy) were seen in rats at exposures greater than approximately 0.7 times exposure in CABP patients after IV administration in the 4-week study and greater than approximately 0.3 times exposure in CABP patients in the 13-week study.
In cynomolgus monkeys administered IV lefamulin, anemia and pancreatic microvesicular vacuolization of acinar cells were noted at exposures greater than approximately 1.6 times exposure in CABP patients in a 4-week study. In a 13-week study, pancreatic microvesicular vacuolization of acinar cells and minimal alveolar macrophage infiltrates in the lung were observed at all doses, and anemia was noted at exposures greater than approximately 1.0 times clinical exposure.
Lefamulin was evaluated in 4-week oral toxicology studies in rats and cynomolgus monkeys. Findings included partially reversible degenerative changes in the stomach and evidence of lymphoid depletion and hematopoietic cell depletion in rats at exposures greater than approximately 0.6 times exposure following oral administration to CABP patients. Findings in cynomolgus monkeys included myocardial vacuolation and fibrosis at exposures equal to or greater than 0.3 times that in CABP patients.
Evidence of dose-dependent regenerative anemia in both species may indicate that XENLETA was potentially hemolytic at a concentration that is approximately ten times higher than the concentration of the infusion solution which will be used clinically. This effect was not apparent from an in vitro evaluation of blood compatibility using human blood at a concentration of 0.6 mg/mL.
-
14 CLINICAL STUDIES
14.1 Community-Acquired Bacterial Pneumonia
A total of 1289 adults with CABP were randomized in two multicenter, multinational, double-blind, double-dummy, non-inferiority trials (Trial 1 NCT #02559310 and Trial 2 NCT #02813694). Trial 1 compared 5 to 10 days of XENLETA to 7 to 10 days of moxifloxacin ± linezolid. Trial 2 compared 5 days of XENLETA to 7 days of moxifloxacin.
In Trial 1, 276 patients were randomized to XENLETA (150 mg by intravenous [IV] infusion over 60 minutes every 12 hours, with the option to switch to 600 mg orally every 12 hours after at least 3 days of IV treatment) and 275 patients were randomized to moxifloxacin (400 mg IV every 24 hours, with the option to switch to 400 mg orally every 24 hours after at least 3 days of IV treatment). If methicillin-resistant Staphylococcus aureus (MRSA) was suspected at screening, patients randomized to moxifloxacin were to receive adjunctive linezolid (600 mg IV every 12 hours, with the option to switch to 600 mg orally every 12 hours after at least 3 days of IV treatment), and patients randomized to XENLETA were to receive linezolid placebo. Patients were predominantly male (60%) and white (87%). Approximately 72% of patients were PORT Risk Class III and 28% were PORT Risk Class IV or V. Median age was 62 (range 19-91) years, approximately 18% of patients were 75 years or older, and median body mass index (BMI) was 25.8 (range 11-58.4) kg/m2. Approximately 53% of patients had creatinine clearance (CrCl) <90 mL/min. Common comorbid conditions included hypertension (41%), asthma/chronic obstructive pulmonary disease (COPD) (17%), and diabetes mellitus (13%).
In Trial 2, 370 patients were randomized to XENLETA (600 mg orally every 12 hours for 5 days) and 368 patients were randomized to moxifloxacin (400 mg orally every 24 hours for 7 days). Patients were predominantly male (52%) and white (74%). Approximately 50% of patients were PORT Risk Class II and 49% were PORT Risk Class III or IV. Median age was 59 (range 19-97) years, approximately 16% of patients were 75 years or older, and median BMI was 26.0 (range 13-63.9) kg/m2. Approximately 50% of patients had CrCl <90 mL/min. Common comorbid conditions included hypertension (36%), asthma/COPD (16%), and diabetes mellitus (13%).
In both trials, efficacy was determined by Early Clinical Response (ECR) at 72 to 120 hours after the first dose in the Intent-to-treat (ITT) Analysis Set, which comprised all randomized patients. Patients entered the trials with at least three of four symptoms consistent with CABP (cough, sputum production, chest pain, and/or dyspnea). Response was defined as survival with improvement of at least two symptoms, no worsening of any symptom, and no receipt of non-study antibacterial treatment for CABP. Table 5 summarizes ECR rates in the two trials.
Table 5: Early Clinical Response Rates in Trial 1 and Trial 2 (ITT Analysis Set) *Trial 1 compared XENLETA to moxifloxacin ± linezolid.
**95% confidence interval for the treatment difference.
Study XENLETA
n/N (%)Moxifloxacin
n/N (%)*Treatment Difference
(95% CI)**Trial 1 241/276 (87.3) 248/275 (90.2) -2.9 (-8.5, 2.8) Trial 2 336/370 (90.8) 334/368 (90.8) 0.1 (-4.4, 4.5) Clinical response was also assessed by the Investigator at the Test of Cure (TOC) Visit 5 to 10 days after the last dose of study drug. Response was defined as survival with improvement of signs and symptoms based on the Investigator’s assessment and no receipt of non-study antibacterial treatment for CABP. Table 6 summarizes Investigator-assessed Clinical Response (IACR) rates at TOC in the ITT Analysis Set, which comprised all randomized patients.
Table 6: Investigator-assessed Clinical Response Rates at TOC in Trial 1 and Trial 2 (ITT Analysis Set) *Trial 1 compared XENLETA to moxifloxacin ± linezolid.
**95% confidence interval for the treatment difference.
Study XENLETA
n/N (%)Moxifloxacin
n/N (%)*Treatment Difference
(95% CI)**Trial 1 223/276 (80.8) 230/275 (83.6) -2.8 (-9.6, 3.9) Trial 2 322/370 (87.0) 328/368 (89.1) -2.1 (-7.0, 2.8) Table 7 summarizes IACR rates at TOC by the most common baseline pathogens across both trials in the microITT Analysis Set, which comprised all randomized patients with at least 1 baseline pathogen.
Table 7: Investigator-assessed Clinical Response Rates at TOC by Baseline Pathogen in Trial 1 and Trial 2 (microITT Analysis Set) *Trial 1 compared XENLETA to moxifloxacin ± linezolid.
Pathogen XENLETA
n/N (%)Moxifloxacin
n/N (%)*Streptococcus pneumoniae 184/216 (85.2) 193/223 (86.5) Methicillin-susceptible Staphylococcus aureus (MSSA) 14/16 (87.5) 5/5 (100.0) Haemophilus influenzae 95/107 (88.8) 88/105 (83.8) Mycoplasma pneumoniae 35/39 (89.7) 33/34 (97.1) Legionella pneumophila 27/34 (79.4) 26/31 (83.9) Chlamydophila pneumoniae 20/27 (74.1) 23/31 (74.2) -
16 HOW SUPPLIED/STORAGE AND HANDLING
XENLETA is supplied in the following strengths and package configurations:
XENLETA Injection
How Supplied
XENLETA Injection is a clear, colorless, sterile, nonpyrogenic solution for intravenous administration containing 150 mg of lefamulin in 15 mL 0.9% sodium chloride in a single-dose vial intended for dilution in 250 mL of 10 mM citrate buffered (pH 5) 0.9% sodium chloride. The drug product is provided in a clear type I glass 15 mL vial with a gray rubber stopper, aluminum seal and flip off cap. The diluent is provided in infusion bags containing 250 mL of sterile, nonpyrogenic 10 mM citrate buffered (pH 5) 0.9% sodium chloride solution. The vial stopper and infusion bag are not made with natural rubber latex.
They are supplied as follows:
150 mg single dose lefamulin vials (NDC 72000-120-06); packed in cartons of 6.
250 mL citrate buffer diluent bags (NDC 72000-030-06); packed in cartons of 6.
Storage and Handling
XENLETA Injection should be stored at 2°C to 8°C (36°F to 46°F). Store in a refrigerator. Do not freeze. The diluent bags should be stored in barrier overwrap at 2°C to 25°C (36°F to 77°F) until ready to use. [see Dosage and Administration (2.5)].
XENLETA Tablets
How Supplied
XENLETA Tablets are available as blue, oval, film-coated tablets containing 600 mg lefamulin. The tablets are printed with ‘LEF 600’ in black on one side.
They are supplied as follows:
Child-resistant blister cards of 10 tablets (NDC 72000-110-10).
HDPE bottles of 30 tablets with Child-resistant Closure (NDC 72000-110-30).
Storage and Handling
XENLETA Tablets should be stored at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Diarrhea
Advise patients that diarrhea is a common problem caused by antibacterial drugs, including XENLETA, which usually ends when the antibacterial drug is discontinued. Sometimes after starting treatment with an antibacterial drug, patients can develop watery stools (with or without stomach cramps and fever) which may be a sign of a more serious intestinal infection, even as late as 2 or more months after having taken the last dose of the antibacterial drug. If this occurs, instruct patients to contact their healthcare provider as soon as possible [see Warnings and Precautions (5.3), and Adverse Reactions (6.1)].
Nausea and Vomiting
Advise patients that nausea and vomiting are common adverse reactions to XENLETA [see Adverse Reactions (6.1)].
Drug Interactions
Advise patients of the potential interaction other medications can have with XENLETA or the effect XENLETA may have on other medications, as these interactions may result in decreased effectiveness or increased toxicities of either XENLETA or the other medications. Patients should alert their physician if they are currently taking any medication(s) (including herbal or nutritional supplements) or are prescribed new medication(s) during treatment with XENLETA [see Drug Interactions (7)].
Allergic Reactions
Advise patients that allergic reactions, including serious allergic reactions, could occur with XENLETA and that serious allergic reactions require immediate treatment. Ask the patient about any previous hypersensitivity reactions to XENLETA, or other pleuromutilin class antibacterial drugs [see Contraindications (4)].
Administration with Food
Advise patients that XENLETA should be taken at least 1 hour before a meal or 2 hours after a meal and should be swallowed whole with water (6 to 8 ounces). XENLETA should not be crushed or divided [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus, and to inform their healthcare provider of a known or suspected pregnancy. Advise patients to avoid becoming pregnant while receiving this drug [see Warnings and Precautions (5.2) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with XENLETA and for 2 days after the final dose [see Warnings and Precautions (5.2) and Use in Specific Populations (8.3)].
Inform patients that Nabriva Therapeutics has a surveillance program for pregnant women who have inadvertently taken XENLETA during pregnancy. Advise patients to call 1-855-5NABRIVA to enroll [see Use in Specific Populations (8.1)].
Lactation
Advise lactating women to pump and discard human milk for the duration of treatment with XENLETA and for 2 days after the final dose [see Use in Specific Populations (8.2)].
Antibacterial Resistance
Patients should be counseled that antibacterial drugs including XENLETA should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When XENLETA is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of treatment, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by XENLETA or other antibacterial drugs in the future [see Warnings and Precautions (5.4)].
Distributed by:
Nabriva Therapeutics US, Inc.
Fort Washington, PA 19034
Nabriva Therapeutics is a trademark of Nabriva Therapeutics US, Inc.
XENLETA is a trademark of Nabriva Therapeutics US, Inc.
For patent information: www.nabriva.com/patents
Copyright © 2019-2021 Nabriva Therapeutics US, Inc., an affiliate of Nabriva Therapeutics Ireland DAC. All rights reserved.
- PRINCIPAL DISPLAY PANEL - NDC: 72000-120-01 - Injection Vial Label
- PRINCIPAL DISPLAY PANEL - NDC: 72000-120-06 - Injection Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 72000-030-01 - Diluent Bag Label
- PRINCIPAL DISPLAY PANEL - NDC: 72000-030-06 - Diluent Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 72000-110-30 - Tablet 30-count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 72000-110-10 - 10 Count Blister Pack Label
-
INGREDIENTS AND APPEARANCE
XENLETA
lefamulin acetate injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72000-120 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LEFAMULIN ACETATE (UNII: HDN0B924X4) (LEFAMULIN - UNII:21904A5386) LEFAMULIN 150 mg in 15 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72000-120-06 6 in 1 CARTON 09/09/2019 1 NDC:72000-120-01 15 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211673 09/09/2019 CITRIC BUFFERED NORMAL SALINE
anhydrous citric acid injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72000-030 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) (ANHYDROUS CITRIC ACID - UNII:XF417D3PSL) ANHYDROUS CITRIC ACID 166 mg in 270 mL Inactive Ingredients Ingredient Name Strength TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) SODIUM CHLORIDE (UNII: 451W47IQ8X) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72000-030-06 6 in 1 CARTON 09/09/2019 1 NDC:72000-030-01 270 mL in 1 BAG; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211673 09/09/2019 XENLETA
lefamulin acetate tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72000-110 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LEFAMULIN ACETATE (UNII: HDN0B924X4) (LEFAMULIN - UNII:21904A5386) LEFAMULIN 600 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) POVIDONE K30 (UNII: U725QWY32X) WATER (UNII: 059QF0KO0R) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE (UNII: 029TFK992N) TALC (UNII: 7SEV7J4R1U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color BLUE Score no score Shape OVAL Size 20mm Flavor Imprint Code LEF;600 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72000-110-10 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 09/09/2019 2 NDC:72000-110-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/09/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211672 09/09/2019 Labeler - Nabriva Therapeutics US, Inc. (072863844) Registrant - Nabriva Therapeutics Ireland DAC (985699902) Establishment Name Address ID/FEI Business Operations Hovione Limited 985003840 API MANUFACTURE(72000-120, 72000-110) Establishment Name Address ID/FEI Business Operations Charles River Endotoxin Microbial Detection Europe 264731577 ANALYSIS(72000-120, 72000-110) Establishment Name Address ID/FEI Business Operations HOVIONE FARMACIENCIA, S.A. 449818434 ANALYSIS(72000-120, 72000-110) Establishment Name Address ID/FEI Business Operations Patheon Italia S.p.A 338336589 MANUFACTURE(72000-120) Establishment Name Address ID/FEI Business Operations Almac Pharma Services Limited 233170864 MANUFACTURE(72000-110) Establishment Name Address ID/FEI Business Operations Sharp Packaging Services, LLC 143696495 PACK(72000-120, 72000-030, 72000-110)





