Label: PEMRYDI RTU- pemetrexed disodium injection






- NDC Code(s): 70121-2453-1, 70121-2461-1, 70121-2462-1
- Packager: Amneal Pharmaceuticals LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated June 9, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PEMRYDI RTU safely and effectively. See full prescribing information for PEMRYDI RTU.
PEMRYDI RTU (pemetrexed injection), for intravenous use
Initial U.S. Approval: 2004INDICATIONS AND USAGE
PEMRYDI RTUTM is a folate analog metabolic inhibitor indicated:
- in combination with pembrolizumab and platinum chemotherapy, for the initial treatment of patients with metastatic non-squamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations. (1.1)
- in combination with cisplatin for the initial treatment of patients with locally advanced or metastatic, non-squamous NSCLC. (1.1)
- as a single agent for the maintenance treatment of patients with locally advanced or metastatic, non-squamous NSCLC whose disease has not progressed after four cycles of platinum-based first-line chemotherapy. (1.1)
- as a single agent for the treatment of patients with recurrent, metastatic non-squamous, NSCLC after prior chemotherapy. (1.1)
Limitations of Use: PEMRYDI RTU is not indicated for the treatment of patients with squamous cell, non-small cell lung cancer. (1.1) - initial treatment, in combination with cisplatin, of patients with malignant pleural mesothelioma whose disease is unresectable or who are otherwise not candidates for curative surgery. (1.2)
DOSAGE AND ADMINISTRATION
- The recommended dose of PEMRYDI RTU administered with pembrolizumab and platinum chemotherapy in patients with a creatinine clearance (calculated by Cockcroft-Gault equation) of 45 mL/min or greater is 500 mg/m2 as an intravenous infusion over 10 minutes, administered after pembrolizumab and prior to platinum chemotherapy, on Day 1 of each 21-day cycle. (2.1)
- The recommended dose of PEMRYDI RTU, administered as a single agent or with cisplatin, in patients with creatinine clearance of 45 mL/minute or greater is 500 mg/m2 as an intravenous infusion over 10 minutes on Day 1 of each 21-day cycle. (2.1, 2.2)
- Initiate folic acid 400 mcg to 1,000 mcg orally, once daily, beginning 7 days prior to the first dose of PEMRYDI RTU and continue until 21 days after the last dose of PEMRYDI RTU. (2.4)
- Administer vitamin B12, 1 mg intramuscularly, 1 week prior to the first dose of PEMRYDI RTU and every 3 cycles. (2.4)
- Administer dexamethasone 4 mg orally, twice daily the day before, the day of, and the day after PEMRYDI RTU administration. (2.4)
DOSAGE FORMS AND STRENGTHS
- Injection: 100 mg/10 mL, 500 mg/50 mL, 1,000 mg/100 mL in single dose vials (3)
CONTRAINDICATIONS
History of severe hypersensitivity reaction to pemetrexed. (4)
WARNINGS AND PRECAUTIONS
- Myelosuppression: Can cause severe bone marrow suppression resulting in cytopenia and an increased risk of infection. Do not administer PEMRYDI RTU when the absolute neutrophil count is less than 1,500 cells/mm3 and platelets are less than 100,000 cells/mm3. Initiate supplementation with oral folic acid and intramuscular vitamin B12 to reduce the severity of hematologic and gastrointestinal toxicity of PEMRYDI RTU. (2.4, 5.1)
- Renal Failure: Can cause severe, and sometimes fatal, renal failure. Do not administer when creatinine clearance is less than 45 mL/min. (2.3, 5.2)
- Bullous and Exfoliative Skin Toxicity: Permanently discontinue for severe and life-threatening bullous, blistering or exfoliating skin toxicity. (5.3)
- Interstitial Pneumonitis: Withhold for acute onset of new or progressive unexplained pulmonary symptoms. Permanently discontinue if pneumonitis is confirmed. (5.4)
- Radiation Recall: Can occur in patients who received radiation weeks to years previously; permanently discontinue for signs of radiation recall. (5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.7, 8.1, 8.3)
ADVERSE REACTIONS
- The most common adverse reactions (incidence ≥ 20%) of PEMRYDI RTU, when administered as a single agent are fatigue, nausea, and anorexia. (6.1)
- The most common adverse reactions (incidence ≥ 20%) of PEMRYDI RTU when administered with cisplatin are vomiting, neutropenia, anemia, stomatitis/pharyngitis, thrombocytopenia, and constipation. (6.1)
- The most common adverse reactions (incidence ≥ 20%) of PEMRYDI RTU when administered in combination with pembrolizumab and platinum chemotherapy are fatigue/asthenia, nausea, constipation, diarrhea, decreased appetite, rash, vomiting, cough, dyspnea, and pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amneal Pharmaceuticals at 1-877-835-5472 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1. INDICATIONS AND USAGE
1.1 Non-Squamous Non-Small Cell Lung Cancer (NSCLC)
1.2 Mesothelioma
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Non-Squamous NSCLC
2.2 Recommended Dosage for Mesothelioma
2.3 Renal Impairment
2.4 Premedication and Concomitant Medications to Mitigate Toxicity
2.5 Dosage Modification of Ibuprofen in Patients with Mild to Moderate Renal Impairment Receiving PEMRYDI RTU
2.6 Dosage Modifications for Adverse Reactions
2.7 Preparation for Administration
3. DOSAGE FORMS AND STRENGTHS
4. CONTRAINDICATIONS
5. WARNINGS AND PRECAUTIONS
5.1 Myelosuppression and Increased Risk of Myelosuppression without Vitamin Supplementation
5.2 Renal Failure
5.3 Bullous and Exfoliative Skin Toxicity
5.4 Interstitial Pneumonitis
5.5 Radiation Recall
5.6 Increased Risk of Toxicity with Ibuprofen in Patients with Renal Impairment
5.7 Embryo-Fetal Toxicity
6. ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7. DRUG INTERACTIONS
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
10. OVERDOSAGE
11. DESCRIPTION
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14. CLINICAL STUDIES
14.1 Non-Squamous NSCLC
14.2 Mesothelioma
15. REFERENCES
16. HOW SUPPLIED/STORAGE AND HANDLING
17. PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1. INDICATIONS AND USAGE
1.1 Non-Squamous Non-Small Cell Lung Cancer (NSCLC)
PEMRYDI RTUTM is indicated:
- in combination with pembrolizumab and platinum chemotherapy, for the initial treatment of patients with metastatic non-squamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
- in combination with cisplatin for the initial treatment of patients with locally advanced or metastatic, non-squamous NSCLC.
- as a single agent for the maintenance treatment of patients with locally advanced or metastatic, non-squamous NSCLC whose disease has not progressed after four cycles of platinum-based first-line chemotherapy.
- as a single agent for the treatment of patients with recurrent, metastatic non-squamous, NSCLC after prior chemotherapy.
Limitations of Use: PEMRYDI RTU is not indicated for the treatment of patients with squamous cell, non-small cell lung cancer [see Clinical Studies (14.1)].
-
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Non-Squamous NSCLC
- The recommended dose of PEMRYDI RTU when administered with pembrolizumab and platinum chemotherapy for the initial treatment of metastatic non-squamous NSCLC in patients with a creatinine clearance (calculated by Cockcroft-Gault equation) of 45 mL/min or greater is 500 mg/m2 as an intravenous infusion over 10 minutes administered after pembrolizumab and prior to carboplatin or cisplatin on Day 1 of each 21-day cycle for 4 cycles. Following completion of platinum-based therapy, treatment with PEMRYDI RTU with or without pembrolizumab is administered until disease progression or unacceptable toxicity. Please refer to the full prescribing information for pembrolizumab and for carboplatin or cisplatin.
- The recommended dose of PEMRYDI RTU when administered with cisplatin for initial treatment of locally advanced or metastatic non-squamous NSCLC in patients with a creatinine clearance (calculated by Cockcroft-Gault equation) of 45 mL/min or greater is 500 mg/m2 as an intravenous infusion over 10 minutes administered prior to cisplatin on Day 1 of each 21-day cycle for up to six cycles in the absence of disease progression or unacceptable toxicity.
- The recommended dose of PEMRYDI RTU for maintenance treatment of non-squamous NSCLC in patients with a creatinine clearance (calculated by Cockcroft-Gault equation) of 45 mL/min or greater is 500 mg/m2 as an intravenous infusion over 10 minutes on Day 1 of each 21-day cycle until disease progression or unacceptable toxicity after four cycles of platinum-based first-line chemotherapy.
- The recommended dose of PEMRYDI RTU for treatment of recurrent non-squamous NSCLC in patients with a creatinine clearance (calculated by Cockcroft-Gault equation) of 45 mL/min or greater is 500 mg/m2 as an intravenous infusion over 10 minutes on Day 1 of each 21-day cycle until disease progression or unacceptable toxicity.
2.2 Recommended Dosage for Mesothelioma
- The recommended dose of PEMRYDI RTU when administered with cisplatin in patients with a creatinine clearance (calculated by Cockcroft-Gault equation) of 45 mL/min or greater is 500 mg/m2 as an intravenous infusion over 10 minutes on Day 1 of each 21-day cycle until disease progression or unacceptable toxicity.
2.3 Renal Impairment
- PEMRYDI RTU dosing recommendations are provided for patients with a creatinine clearance (calculated by Cockcroft-Gault equation) of 45 mL/min or greater [see Dosage and Administration (2.1, 2.2)]. There is no recommended dose for patients whose creatinine clearance is less than 45 mL/min [see Use in Specific Populations (8.6)].
2.4 Premedication and Concomitant Medications to Mitigate Toxicity
Vitamin Supplementation
- Initiate folic acid 400 mcg to 1,000 mcg orally once daily, beginning 7 days before the first dose of PEMRYDI RTU and continuing until 21 days after the last dose of PEMRYDI RTU [see Warnings and Precautions (5.1)].
- Administer vitamin B12, 1 mg intramuscularly, 1 week prior to the first dose of PEMRYDI RTU and every 3 cycles thereafter. Subsequent vitamin B12 injections may be given the same day as treatment with PEMRYDI RTU [see Warnings and Precautions (5.1)]. Do not substitute oral vitamin B12 for intramuscular vitamin B12.
Corticosteroids
- Administer dexamethasone 4 mg orally twice daily for three consecutive days, beginning the day before each PEMRYDI RTU administration.
2.5 Dosage Modification of Ibuprofen in Patients with Mild to Moderate Renal Impairment Receiving PEMRYDI RTU
In patients with creatinine clearances between 45 mL/min and 79 mL/min, modify administration of ibuprofen as follows [see Warnings and Precautions (5.6), Drug Interactions (7) and Clinical Pharmacology (12.3)]:
- Avoid administration of ibuprofen for 2 days before, the day of, and 2 days following administration of PEMRYDI RTU.
- Monitor patients more frequently for myelosuppression, renal, and gastrointestinal toxicity, if concomitant administration of ibuprofen cannot be avoided.
2.6 Dosage Modifications for Adverse Reactions
Obtain complete blood count on Days 1, 8, and 15 of each cycle. Assess creatinine clearance prior to each cycle. Do not administer PEMRYDI RTU if the creatinine clearance is less than 45 mL/min.
Delay initiation of the next cycle of PEMRYDI RTU until:
- recovery of non-hematologic toxicity to Grade 0-2,
- absolute neutrophil count (ANC) is 1,500 cells/mm3 or higher, and
- platelet count is 100,000 cells/mm3 or higher.
Upon recovery, modify the dosage of PEMRYDI RTU in the next cycle as specified in Table 1.
For dosing modifications for cisplatin, carboplatin, or pembrolizumab, refer to their prescribing information.
Table 1: Recommended Dosage Modifications for Adverse Reactionsa
Toxicity in Most Recent Treatment Cycle
PEMRYDI RTU Dose Modification for Next Cycle
Myelosuppressive toxicity [see Warnings and Precautions (5.1)]
ANC less than 500/mm3 and platelets greater than or equal to 50,000/mm3
OR
Platelet count less than 50,000/mm3 without bleeding.
75% of previous dose
Platelet count less than 50,000/mm3 with bleeding
50% of previous dose
Recurrent Grade 3 or 4 myelosuppression after 2 dose reductions
Discontinue
Non-hematologic toxicity
Any Grade 3 or 4 toxicities EXCEPT mucositis or neurologic toxicity
OR
Diarrhea requiring hospitalization
75% of previous dose
Grade 3 or 4 mucositis
50% of previous dose
Renal toxicity [see Warnings and Precautions (5.2)]
Withhold until creatinine
clearance is 45 mL/min or greater
Grade 3 or 4 neurologic toxicity
Permanently discontinue
Recurrent Grade 3 or 4 non-hematologic toxicity after 2 dose reductions
Permanently discontinue
Severe and life-threatening Skin Toxicity [see Warnings and Precautions (5.3)]
Permanently discontinue
Interstitial Pneumonitis [see Warnings and Precautions (5.4)]
Permanently discontinue
a National Cancer Institute Common Toxicity Criteria for Adverse Events version 2 (NCI CTCAE v2).
2.7 Preparation for Administration
PEMRYDI RTU is a hazardous drug. Follow applicable special handling and disposal procedures.1
- Calculate the dose of PEMRYDI RTU and determine the volume of needed PEMRYDI RTU. Each vial contains an excess of PEMRYDI RTU to facilitate delivery of labeled amount.
- Withdraw the calculated dose of PEMRYDI RTU from the vial(s) and discard the vial(s) with any unused portion.
- Transfer the calculated dose into an empty intravenous bag. Do NOT further dilute PEMRYDI RTU.
- Visually inspect for particulate matter and discoloration prior to administration. Discard if particulate matter or discoloration is observed.
- Immediately administer PEMRYDI RTU undiluted, as an intravenous infusion over 10 minutes using an infusion pump.
- 3. DOSAGE FORMS AND STRENGTHS
-
4. CONTRAINDICATIONS
PEMRYDI RTU is contraindicated in patients with a history of severe hypersensitivity reaction to pemetrexed [see Adverse Reactions (6.1)].
-
5. WARNINGS AND PRECAUTIONS
5.1 Myelosuppression and Increased Risk of Myelosuppression without Vitamin Supplementation
PEMRYDI RTU can cause severe myelosuppression resulting in a requirement for transfusions and which may lead to neutropenic infection. The risk of myelosuppression is increased in patients who do not receive vitamin supplementation. In Study JMCH, incidences of Grade 3-4 neutropenia (38% versus 23%), thrombocytopenia (9% versus 5%), febrile neutropenia (9% versus 0.6%), and neutropenic infection (6% versus 0) were higher in patients who received PEMRYDI RTU plus cisplatin without vitamin supplementation as compared to patients who were fully supplemented with folic acid and vitamin B12 prior to and throughout PEMRYDI RTU plus cisplatin treatment.
Initiate supplementation with oral folic acid and intramuscular vitamin B12 prior to the first dose of PEMRYDI RTU; continue vitamin supplementation during treatment and for 21 days after the last dose of PEMRYDI RTU to reduce the severity of hematologic and gastrointestinal toxicity of PEMRYDI RTU [see Dosage and Administration (2.4)]. Obtain a complete blood count at the beginning of each cycle. Do not administer PEMRYDI RTU until the ANC is at least 1,500 cells/mm3 and platelet count is at least 1,00,000 cells/mm3. Permanently reduce PEMRYDI RTU in patients with an ANC of less than 500 cells/mm3 or platelet count of less than 50,000 cells/mm3 in previous cycles [see Dosage and Administration (2.6)].
In Studies JMDB and JMCH, among patients who received vitamin supplementation, incidence of Grade 3-4 neutropenia was 15% and 23%, the incidence of Grade 3-4 anemia was 6% and 4%, and incidence of Grade 3-4 thrombocytopenia was 4% and 5%, respectively. In Study JMCH, 18% of patients in the PEMRYDI RTU arm required red blood cell transfusions compared to 7% of patients in the cisplatin arm [see Adverse Reactions (6.1)]. In Studies JMEN, PARAMOUNT, and JMEI, where all patients received vitamin supplementation, incidence of Grade 3-4 neutropenia ranged from 3% to 5%, and incidence of Grade 3-4 anemia ranged from 3% to 5%.
5.2 Renal Failure
PEMRYDI RTU can cause severe, and sometimes fatal, renal toxicity. The incidences of renal failure in clinical studies in which patients received PEMRYDI RTU with cisplatin were 2.1% in Study JMDB and 2.2% in Study JMCH. The incidence of renal failure in clinical studies in which patients received PEMRYDI RTU as a single agent ranged from 0.4% to 0.6% (Studies JMEN, PARAMOUNT, and JMEI [see Adverse Reactions (6.1)]. Determine creatinine clearance before each dose and periodically monitor renal function during treatment with PEMRYDI RTU. Withhold PEMRYDI RTU in patients with a creatinine clearance of less than 45 mL/minute [see Dosage and Administration (2.3)].
5.3 Bullous and Exfoliative Skin Toxicity
Serious and sometimes fatal, bullous, blistering and exfoliative skin toxicity, including cases suggestive of Stevens-Johnson Syndrome/Toxic epidermal necrolysis can occur with PEMRYDI RTU. Permanently discontinue PEMRYDI RTU for severe and life-threatening bullous, blistering or exfoliating skin toxicity.
5.4 Interstitial Pneumonitis
Serious interstitial pneumonitis, including fatal cases, can occur with PEMRYDI RTU treatment. Withhold PEMRYDI RTU for acute onset of new or progressive unexplained pulmonary symptoms such as dyspnea, cough, or fever pending diagnostic evaluation. If pneumonitis is confirmed, permanently discontinue PEMRYDI RTU.
5.5 Radiation Recall
Radiation recall can occur with PEMRYDI RTU in patients who have received radiation weeks to years previously. Monitor patients for inflammation or blistering in areas of previous radiation treatment. Permanently discontinue PEMRYDI RTU for signs of radiation recall.
5.6 Increased Risk of Toxicity with Ibuprofen in Patients with Renal Impairment
Exposure to PEMRYDI RTU is increased in patients with mild to moderate renal impairment who take concomitant ibuprofen, increasing the risks of adverse reactions of PEMRYDI RTU. In patients with creatinine clearances between 45 mL/min and 79 mL/min, avoid administration of ibuprofen for 2 days before, the day of, and 2 days following administration of PEMRYDI RTU. If concomitant ibuprofen use cannot be avoided, monitor patients more frequently for PEMRYDI RTU adverse reactions, including myelosuppression, renal, and gastrointestinal toxicity [see Dosage and Administration (2.5), Drug Interactions (7) and Clinical Pharmacology (12.3)].
5.7 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, PEMRYDI RTU can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, intravenous administration of pemetrexed to pregnant mice during the period of organogenesis was teratogenic, resulting in developmental delays and increased malformations at doses lower than the recommended human dose of 500 mg/m2. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
-
6. ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Myelosuppression [see Warnings and Precautions (5.1)]
- Renal failure [see Warnings and Precautions (5.2)]
- Bullous and exfoliative skin toxicity [see Warning and Precautions (5.3)]
- Interstitial pneumonitis [see Warnings and Precautions (5.4)]
- Radiation recall [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
In clinical trials, the most common adverse reactions (incidence ≥ 20%) of PEMRYDI RTU, when administered as a single agent, are fatigue, nausea, and anorexia. The most common adverse reactions (incidence ≥ 20 %) of PEMRYDI RTU, when administered in combination with cisplatin are vomiting, neutropenia, anemia, stomatitis/pharyngitis, thrombocytopenia, and constipation. The most common adverse reactions (incidence ≥ 20%) of PEMRYDI RTU, when administered in combination with pembrolizumab and platinum chemotherapy, are fatigue/asthenia, nausea, constipation, diarrhea, decreased appetite, rash, vomiting, cough, dyspnea, and pyrexia.
Non-Squamous NSCLC
First-line Treatment of Metastatic Non-squamous NSCLC with Pembrolizumab and Platinum Chemotherapy
The safety of PEMRYDI RTU, in combination with pembrolizumab and investigator’s choice of platinum (either carboplatin or cisplatin), was investigated in Study KEYNOTE-189, a multicenter, double-blind, randomized (2:1), active-controlled trial in patients with previously untreated, metastatic non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations. A total of 607 patients received PEMRYDI RTU, pembrolizumab, and platinum every 3 weeks for 4 cycles followed by PEMRYDI RTU and pembrolizumab (n=405), or placebo, PEMRYDI RTU, and platinum every 3 weeks for 4 cycles followed by placebo and PEMRYDI RTU (n=202). Patients with autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible [see Clinical Studies (14.1)].
The median duration of exposure to PEMRYDI RTU was 7.2 months (range: 1 day to 1.7 years). Seventy-two percent of patients received carboplatin. The study population characteristics were: median age of 64 years (range: 34 to 84), 49% age 65 years or older, 59% male, 94% White and 3% Asian, and 18% with history of brain metastases at baseline.
PEMRYDI RTU was discontinued for adverse reactions in 23% of patients in the PEMRYDI RTU, pembrolizumab, and platinum arm. The most common adverse reactions resulting in discontinuation of PEMRYDI RTU in this arm were acute kidney injury (3%) and pneumonitis (2%). Adverse reactions leading to interruption of PEMRYDI RTU occurred in 49% of patients in the PEMRYDI RTU, pembrolizumab, and platinum arm. The most common adverse reactions or laboratory abnormalities leading to interruption of PEMRYDI RTU in this arm (≥ 2%) were neutropenia (12%), anemia (7%), asthenia (4%), pneumonia (4%), thrombocytopenia (4%), increased blood creatinine (3%), diarrhea (3%), and fatigue (3%).
Table 2 summarizes the adverse reactions that occurred in ≥ 20% of patients treated with PEMRYDI RTU, pembrolizumab, and platinum.
Table 2: Adverse Reactions Occurring in ≥ 20% of Patients in KEYNOTE-189
PEMRYDI RTU Pembrolizumab
Platinum Chemotherapy
n=405Placebo
PEMRYDI RTU
Platinum Chemotherapy
n=202Adverse Reaction All Gradesa
(%)Grade 3-4
(%)All Grades
(%)Grade 3-4
(%)Gastrointestinal Disorders Nausea 56 3.5 52 3.5 Constipation 35 1.0 32 0.5 Diarrhea 31 5 21 3.0 Vomiting 24 3.7 23 3.0 General Disorders and Administration Site Conditions Fatigueb 56 12 58 6 Pyrexia 20 0.2 15 0 Metabolism and Nutrition Disorders Decreased appetite 28 1.5 30 0.5 Skin and Subcutaneous Tissue Disorders Rashc 25 2.0 17 2.5 Respiratory, Thoracic and Mediastinal Disorders Cough 21 0 28 0 Dyspnea 21 3.7 26 5 a Graded per NCI CTCAE version 4.03.
b Includes asthenia and fatigue.
c Includes genital rash, rash, rash generalized, rash macular, rash maculo-papular, rash papular, rash pruritic, and rash pustular.Table 3 summarizes the laboratory abnormalities that worsened from baseline in at least 20% of patients treated with PEMRYDI RTU, pembrolizumab, and platinum.
Table 3: Laboratory Abnormalities Worsened from Baseline in ≥ 20% of Patients in KEYNOTE-189
PEMRYDI RTU
Pembrolizumab
Platinum Chemotherapy
Placebo
PEMRYDI RTU
Platinum Chemotherapy
Laboratory Testa
All Gradesb
%
Grades 3-4
%
All Grades
%
Grades 3-4
%
Chemistry
Hyperglycemia
63
9
60
7
Increased ALT
47
3.8
42
2.6
Increased AST
47
2.8
40
1.0
Hypoalbuminemia
39
2.8
39
1.1
Increased creatinine
37
4.2
25
1.0
Hyponatremia
32
7
23
6
Hypophosphatemia
30
10
28
14
Increased alkaline phosphatase
26
1.8
29
2.1
Hypocalcemia
24
2.8
17
0.5
Hyperkalemia
24
2.8
19
3.1
Hypokalemia
21
5
20
5
Hematology
Anemia
85
17
81
18
Lymphopenia
64
22
64
25
Neutropenia
48
20
41
19
Thrombocytopenia
30
12
29
8
a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: PEMRYDI RTU/pembrolizumab/platinum chemotherapy (range: 381 to 401 patients) and placebo/PEMRYDI RTU/platinum chemotherapy (range: 184 to 197 patients).
b Graded per NCI CTCAE version 4.03.
Initial Treatment in Combination with Cisplatin
The safety of PEMRYDI RTU was evaluated in Study JMDB, a randomized (1:1), open-label, multicenter trial conducted in chemotherapy-naive patients with locally advanced or metastatic NSCLC. Patients received either PEMRYDI RTU 500 mg/m2 intravenously and cisplatin 75 mg/m2 intravenously on Day 1 of each 21-day cycle (n=839) or gemcitabine 1,250 mg/m2 intravenously on Days 1 and 8 and cisplatin 75 mg/m2 intravenously on Day 1 of each 21-day cycle (n=830). All patients were fully supplemented with folic acid and vitamin B12.
Study JMDB excluded patients with an Eastern Cooperative Oncology Group Performance Status (ECOG PS of 2 or greater), uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B12 or corticosteroids were also excluded from the study.
The data described below reflect exposure to PEMRYDI RTU plus cisplatin in 839 patients in Study JMDB. Median age was 61 years (range 26-83 years); 70% of patients were men; 78% were White, 16% were Asian, 2.9% were Hispanic or Latino, 2.1% were Black or African American, and <1% were other ethnicities; 36% had an ECOG PS 0. Patients received a median of 5 cycles of PEMRYDI RTU.
Table 4 provides the frequency and severity of adverse reactions that occurred in ≥ 5% of 839 patients receiving PEMRYDI RTU in combination with cisplatin in Study JMDB. Study JMDB was not designed to demonstrate a statistically significant reduction in adverse reaction rates for PEMRYDI RTU, as compared to the control arm, for any specified adverse reaction listed in Table 4.
Table 4: Adverse Reactions Occurring in ≥ 5% of Fully Vitamin-Supplemented Patients Receiving PEMRYDI RTU in Combination with Cisplatin Chemotherapy in Study JMDB
Adverse Reactiona
PEMRYDI RTU/Cisplatin
(N=839)
Gemcitabine/Cisplatin
(N=830)
All Grades
(%)
Grade 3-4
(%)
All Grades
(%)
Grade 3-4
(%)
All adverse reactions
90
37
91
53
Laboratory
Hematologic
Anemia
33
6
46
10
Neutropenia
29
15
38
27
Thrombocytopenia
10
4
27
13
Renal
Elevated creatinine
10
1
7
1
Clinical Gastrointestinal
Nausea
56
7
53
4
Vomiting
40
6
36
6
Anorexia
27
2
24
1
Constipation
21
1
20
0
Stomatitis/pharyngitis
14
1
12
0
Diarrhea
12
1
13
2
Dyspepsia/heartburn
5
0
6
0
Constitutional symptoms Fatigue 43 7 45 5 Dermatology/Skin
Alopecia
12
0
21
1
Rash/Desquamation
7
0
8
1
Neurology Sensory neuropathy 9 0 12 1 Taste disturbance 8 0 9 0 a NCI CTCAE version 2.0.
The following additional adverse reactions were observed in patients assigned to receive PEMRYDI RTU.
Incidence 1% to < 5%
Body as a Whole — febrile neutropenia, infection, pyrexia
General Disorders — dehydration
Metabolism and Nutrition — increased AST, increased ALT
Renal —renal failure
Eye Disorder — conjunctivitis
Incidence < 1%
Cardiovascular — arrhythmia
General Disorders — chest pain
Metabolism and Nutrition — increased GGT
Neurology — motor neuropathy
Maintenance Treatment Following First-line Non-PEMRYDI RTU Containing Platinum-Based Chemotherapy
In Study JMEN, the safety of PEMRYDI RTU was evaluated in a randomized (2:1), placebo-controlled, multicenter trial conducted in patients with non-progressive locally advanced or metastatic NSCLC following four cycles of a first-line, platinum-based chemotherapy regimen. Patients received either PEMRYDI RTU 500 mg/m2 or matching placebo intravenously every 21 days until disease progression or unacceptable toxicity. Patients in both study arms were fully supplemented with folic acid and vitamin B12.
Study JMEN excluded patients with an ECOG PS of 2 or greater, uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B12 or corticosteroids were also excluded from the study.
The data described below reflect exposure to PEMRYDI RTU in 438 patients in Study JMEN. Median age was 61 years (range 26-83 years), 73% of patients were men; 65% were White, 31% were Asian, 2.9% were Hispanic or Latino, and < 2% were other ethnicities; 39% had an ECOG PS 0. Patients received a median of 5 cycles of PEMRYDI RTU and a relative dose intensity of PEMRYDI RTU of 96%. Approximately half the patients (48%) completed at least six, 21-day cycles and 23% completed ten or more 21-day cycles of PEMRYDI RTU.
Table 5 provides the frequency and severity of adverse reactions reported in ≥ 5% of the 438 PEMRYDI RTU-treated patients in Study JMEN.
Table 5: Adverse Reactions Occurring in ≥ 5% of Patients Receiving PEMRYDI RTU in Study JMEN
Adverse Reactiona
PEMRYDI RTU
(N=438)
Placebo
(N=218)
All Grades
(%)
Grade 3-4
(%)
All Grades
(%)
Grade 3-4
(%)
All adverse reactions
66
16
37
4
Laboratory
Hematologic
Anemia
15
3
6
1
Neutropenia
6
3
0
0
Hepatic
Increased ALT
10
0
4
0
Increased AST
8
0
4
0
Clinical
Constitutional symptoms
Fatigue
25
5
11
1
Gastrointestinal
Nausea
19
1
6
1
Anorexia
19
2
5
0
Vomiting
9
0
1
0
Mucositis/stomatitis
7
1
2
0
Diarrhea
5
1
3
0
Dermatology/Skin
Rash/desquamation
10
0
3
0
Neurology Sensory neuropathy 9 1 4 0 Infection 5 2 2 0 a NCI CTCAE version 3.0.
The requirement for transfusions (9.5% versus 3.2%), primarily red blood cell transfusions, and for erythropoiesis stimulating agents (5.9% versus 1.8%) were higher in the PEMRYDI RTU arm compared to the placebo arm.
The following additional adverse reactions were observed in patients who received PEMRYDI RTU.
Incidence 1% to <5%
Dermatology/Skin — alopecia, pruritus/itching
Gastrointestinal — constipation
General Disorders — edema, fever
Hematologic — thrombocytopenia
Eye Disorder — ocular surface disease (including conjunctivitis), increased lacrimation
Incidence <1%
Cardiovascular — supraventricular arrhythmia
Dermatology/Skin — erythema multiforme
General Disorders — febrile neutropenia, allergic reaction/hypersensitivity
Neurology — motor neuropathy
Renal — renal failure
Maintenance Treatment Following First-line PEMRYDI RTU Plus Platinum Chemotherapy
The safety of PEMRYDI RTU was evaluated in PARAMOUNT, a randomized (2:1), placebo-controlled study conducted in patients with non-squamous NSCLC with non-progressive (stable or responding disease) locally advanced or metastatic NSCLC following four cycles of PEMRYDI RTU in combination with cisplatin as first-line therapy for NSCLC. Patients were randomized to receive PEMRYDI RTU 500 mg/m2 or matching placebo intravenously on Day 1 of each 21-day cycle until disease progression or unacceptable toxicity. Patients in both study arms received folic acid and vitamin B12 supplementation.
PARAMOUNT excluded patients with an ECOG PS of 2 or greater, uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B12 or corticosteroids were also excluded from the study.
The data described below reflect exposure to PEMRYDI RTU in 333 patients in PARAMOUNT. Median age was 61 years (range 32 to 83 years); 58% of patients were men; 94% were White, 4.8% were Asian, and < 1% were Black or African American; 36% had an ECOG PS 0. The median number of maintenance cycles was 4 for PEMRYDI RTU and placebo arms. Dose reductions for adverse reactions occurred in 3.3% of patients in the PEMRYDI RTU arm and 0.6% in the placebo arm. Dose delays for adverse reactions occurred in 22% of patients in the PEMRYDI RTU arm and 16% in the placebo arm.
Table 6 provides the frequency and severity of adverse reactions reported in ≥ 5% of the 333 PEMRYDI RTU-treated patients in PARAMOUNT.
Table 6: Adverse Reactions Occurring in ≥ 5% of Patients Receiving PEMRYDI RTU in PARAMOUNT
Adverse Reactiona
PEMRYDI RTU
(N=333)
Placebo
(N=167)
All Grades
(%)
Grade 3-4
(%)
All Grades
(%)
Grades 3-4
(%)
All adverse reactions
53
17
34
4.8
Laboratory
Hematologic
Anemia
15
4.8
4.8
0.6
Neutropenia
9
3.9
0.6
0
Clinical
Constitutional symptoms
Fatigue
18
4.5
11
0.6
Gastrointestinal
Nausea
12
0.3
2.4
0
Vomiting
6
0
1.8
0
Mucositis/stomatitis
5
0.3
2.4
0
General disorders
Edema
5
0
3.6
0
a NCI CTCAE version 3.0.
The requirement for red blood cell (13% versus 4.8%) and platelet (1.5% versus 0.6%) transfusions, erythropoiesis stimulating agents (12% versus 7%), and granulocyte colony stimulating factors (6% versus 0%) were higher in the PEMRYDI RTU arm compared to the placebo arm.
The following additional Grade 3 or 4 adverse reactions were observed more frequently in the PEMRYDI RTU arm.
Incidence 1% to <5%
Blood/Bone Marrow — thrombocytopenia
General Disorders — febrile neutropenia
Incidence <1%
Cardiovascular — ventricular tachycardia, syncope
General Disorders — pain
Gastrointestinal — gastrointestinal obstruction
Neurologic — depression
Renal — renal failure
Vascular — pulmonary embolism
Treatment of Recurrent Disease After Prior Chemotherapy
The safety of PEMRYDI RTU was evaluated in Study JMEI, a randomized (1:1), open-label, active-controlled trial conducted in patients who had progressed following platinum-based chemotherapy. Patients received PEMRYDI RTU 500 mg/m2 intravenously or docetaxel 75 mg/m2 intravenously on Day 1 of each 21-day cycle. All patients on the PEMRYDI RTU arm received folic acid and vitamin B12 supplementation.
Study JMEI excluded patients with an ECOG PS of 3 or greater, uncontrolled third-space fluid retention, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to discontinue aspirin or other non-steroidal anti-inflammatory drugs or unable to take folic acid, vitamin B12 or corticosteroids were also excluded from the study.
The data described below reflect exposure to PEMRYDI RTU in 265 patients in Study JMEI. Median age was 58 years (range 22 to 87 years); 73% of patients were men; 70% were White, 24% were Asian, 2.6% were Black or African American, 1.8% were Hispanic or Latino, and < 2% were other ethnicities; 19% had an ECOG PS 0.
Table 7 provides the frequency and severity of adverse reactions reported in ≥ 5% of the 265 PEMRYDI RTU-treated patients in Study JMEI. Study JMEI is not designed to demonstrate a statistically significant reduction in adverse reaction rates for PEMRYDI RTU, as compared to the control arm, for any specified adverse reaction listed in the Table 7 below.
Table 7: Adverse Reactions Occurring in ≥ 5% of Fully Supplemented Patients Receiving PEMRYDI RTU in Study JMEI
Adverse Reactiona
PEMRYDI RTU
(N=265)
Docetaxel
(N=276)
All Grades
(%)
Grades 3-4
(%)
All Grade
(%)
Grades 3-4
(%)
Laboratory
Hematologic
Anemia
19
4
22
4
Neutropenia
11
5
45
40
Thrombocytopenia
8
2
1
0
Hepatic
Increased ALT
8
2
1
0
Increased AST
7
1
1
0
Clinical Constitutional symptoms Fatigue 34 5 36 5 Fever 8 0 8 Gastrointestinal
Nausea
31
3
17
2
Anorexia
22
2
24
3
Vomiting
16
2
12
1
Stomatitis/pharyngitis
15
1
17
1
Diarrhea
13
0
24
3
Constipation
6
0
4
0
Dermatology/Skin
Rash/desquamation
14
0
6
0
Pruritus
7
0
2
0
Alopecia
6
1
38
2
a NCI CTCAE version 2.0.
The following additional adverse reactions were observed in patients assigned to receive PEMRYDI RTU.
Incidence 1% to <5%
Body as a Whole — abdominal pain, allergic reaction/hypersensitivity, febrile neutropenia, infection
Dermatology/Skin — erythema multiforme
Neurology — motor neuropathy, sensory neuropathy
Incidence <1%
Cardiovascular — supraventricular arrhythmias
Renal — renal failure
Mesothelioma
The safety of PEMRYDI RTU was evaluated in Study JMCH, a randomized (1:1), single-blind study conducted in patients with MPM who had received no prior chemotherapy for MPM. Patients received PEMRYDI RTU 500 mg/m2 intravenously in combination with cisplatin 75 mg/m2 intravenously on Day 1 of each 21-day cycle or cisplatin 75 mg/m2 intravenously on Day 1 of each 21-day cycle administered until disease progression or unacceptable toxicity. Safety was assessed in 226 patients who received at least one dose of PEMRYDI RTU in combination with cisplatin and 222 patients who received at least one dose of cisplatin alone. Among 226 patients who received PEMRYDI RTU in combination with cisplatin, 74% (n=168) received full supplementation with folic acid and vitamin B12 during study therapy, 14% (n=32) were never supplemented, and 12% (n=26) were partially supplemented.
Study JMCH excluded patients with Karnofsky Performance Scale (KPS) of less than 70, inadequate bone marrow reserve and organ function, or a calculated creatinine clearance less than 45 mL/min. Patients unable to stop using aspirin or other non-steroidal anti-inflammatory drugs were also excluded from the study.
The data described below reflect exposure to PEMRYDI RTU in 168 patients that were fully supplemented with folic acid and vitamin B12. Median age was 60 years (range 19 to 85 years); 82% were men; 92% were White, 5% were Hispanic or Latino, 3.0% were Asian, and <1% were other ethnicities; 54% had KPS of 90-100. The median number of treatment cycles administered was 6 in the PEMRYDI RTU/cisplatin fully supplemented group and 2 in the PEMRYDI RTU/cisplatin never supplemented group. Patients receiving PEMRYDI RTU in the fully supplemented group had a relative dose intensity of 93% of the protocol-specified PEMRYDI RTU dose intensity. The most common adverse reaction resulting in dose delay was neutropenia.
Table 8 provides the frequency and severity of adverse reactions ≥ 5% in the subgroup of PEMRYDI RTU-treated patients who were fully vitamin supplemented in Study JMCH. Study JMCH was not designed to demonstrate a statistically significant reduction in adverse reaction rates for PEMRYDI RTU, as compared to the control arm, for any specified adverse reaction listed in the table below.
Table 8: Adverse Reactions Occurring in ≥ 5% of Fully Supplemented Subgroup of Patients Receiving PEMRYDI RTU/Cisplatin in Study JMCHa
Adverse Reactionb
PEMRYDI RTU/cisplatin
(N=168)
Cisplatin
(N=163)
All Grades
(%)
Grade 3-4
(%)
All Grades
(%)
Grade 3-4
(%)
Laboratory
Hematologic
Neutropenia
56
23
13
3
Anemia
26
4
10
0
Thrombocytopenia
23
5
9
0
Renal
Elevated creatinine
11
1
10
1
Decreased creatinine
clearance
16
1
18
2
Clinical
Gastrointestinal Nausea
82
12
77
6
Vomiting
57
11
50
4
Stomatitis/pharyngitis
23
3
6
0
Anorexia
20
1
14
1
Diarrhea
17
4
8
0
Constipation
12
1
7
1
Dyspepsia
5
1
1
0
Constitutional Symptoms
Fatigue
48
10
42
9
Dermatology/Skin Rash 16 1 5 0 Alopecia 11 0 6 0 Neurology Sensory neuropathy 10 0 10 1 Taste disturbance 8 0 6 0 Metabolism and Nutrition Dehydration 7 4 1 1 Eye Disorder Conjunctivitis 5 0 1 0 a In Study JMCH, 226 patients received at least one dose of PEMRYDI RTU in combination with cisplatin and 222 patients received at least one dose of cisplatin. Table 8 provides the ADRs for subgroup of patients treated with PEMRYDI RTU in combination with cisplatin
(168 patients) or cisplatin alone (163 patients) who received full supplementation with folic acid and vitamin B12 during study therapy.
b NCI CTCAE version 2.0.The following additional adverse reactions were observed in patients receiving PEMRYDI RTU plus cisplatin:
Incidence 1% to <5%
Body as a Whole — febrile neutropenia, infection, pyrexia
Dermatology/Skin — urticaria
General Disorders — chest pain
Metabolism and Nutrition — increased AST, increased ALT, increased GGT
Renal — renal failure
Incidence <1%
Cardiovascular — arrhythmia
Neurology — motor neuropathy
Exploratory Subgroup Analyses based on Vitamin Supplementation
Table 9 provides the results of exploratory analyses of the frequency and severity of NCI CTCAE Grade 3 or 4 adverse reactions reported in more PEMRYDI RTU-treated patients who did not receive vitamin supplementation (never supplemented) as compared with those who received vitamin supplementation with daily folic acid and vitamin B12 from the time of enrollment in Study JMCH (fully-supplemented).
Table 9: Exploratory Subgroup Analysis of Selected Grade 3/4 Adverse Reactions Occurring in Patients Receiving PEMRYDI RTU in Combination with Cisplatin with or without Full Vitamin Supplementation in Study JMCHa
Grade 3-4 Adverse Reactions
Fully Supplemented Patients
N=168
(%)
Never Supplemented Patients
N=32
(%)
Neutropenia
23
38
Thrombocytopenia
5
9
Vomiting
11
31
Febrile neutropenia
1
9
Infection with Grade 3/4 neutropenia
0
6
Diarrhea
4
9
a NCI CTCAE version 2.0.
The following adverse reactions occurred more frequently in patients who were fully vitamin supplemented than in patients who were never supplemented:
- hypertension (11% versus 3%),
- chest pain (8% versus 6%),
- thrombosis/embolism (6% versus 3%).
Additional Experience Across Clinical Trials
Sepsis, with or without neutropenia, including fatal cases: 1%
Severe esophagitis, resulting in hospitalization: <1%
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of PEMRYDI RTU. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System — immune-mediated hemolytic anemia
Gastrointestinal — colitis, pancreatitis
General Disorders and Administration Site Conditions — edema
Injury, poisoning, and procedural complications — radiation recall
Respiratory — interstitial pneumonitis
Skin — Serious and fatal bullous skin conditions, Stevens-Johnson syndrome, and toxic epidermal necrolysis
-
7. DRUG INTERACTIONS
Effects of Ibuprofen on Pemetrexed
Ibuprofen increases exposure (AUC) of pemetrexed [see Clinical Pharmacology (12.3)]. In patients with creatinine clearance between 45 mL/min and 79 mL/min:
- Avoid administration of ibuprofen for 2 days before, the day of, and 2 days following administration of PEMRYDI RTU [see Dosage and Administration (2.5)].
- Monitor patients more frequently for myelosuppression, renal, and gastrointestinal toxicity, if concomitant administration of ibuprofen cannot be avoided.
-
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, PEMRYDI RTU can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on PEMRYDI RTU use in pregnant women. In animal reproduction studies, intravenous administration of pemetrexed to pregnant mice during the period of organogenesis was teratogenic, resulting in developmental delays and malformations at doses lower than the recommended human dose of 500 mg/m2 [see Data]. Advise pregnant women of the potential risk to a fetus [see Use in Special Populations (8.3)].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Pemetrexed was teratogenic in mice. Daily dosing of pemetrexed by intravenous injection to pregnant mice during the period of organogenesis increased the incidence of fetal malformations (cleft palate; protruding tongue; enlarged or misshaped kidney; and fused lumbar vertebra) at doses (based on BSA) 0.03 times the human dose of 500 mg/m2. At doses, based on BSA, greater than or equal to 0.0012 times the 500 mg/m2 human dose, pemetrexed administration resulted in dose-dependent increases in developmental delays (incomplete ossification of talus and skull bone; and decreased fetal weight).
8.2 Lactation
Risk Summary
There is no information regarding the presence of pemetrexed or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child from PEMRYDI RTU, advise women not to breastfeed during treatment with PEMRYDI RTU and for one week after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal data, PEMRYDI RTU can cause malformations and developmental delays when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiating Pemetrexed Injection [see Use in Specific Populations (8.1)].
Contraception
Females
Because of the potential for genotoxicity, advise females of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 6 months after the last dose of PEMRYDI RTU.
Males
Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Males
PEMRYDI RTU may impair fertility in males of reproductive potential. It is not known whether these effects on fertility are reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of PEMRYDI RTU in pediatric patients have not been established.
The safety and pharmacokinetics of PEMRYDI RTU were evaluated in two clinical studies conducted in pediatric patients with recurrent solid tumors (NCT00070473 N=32 and NCT00520936 N=72). Patients in both studies received concomitant vitamin B12 and folic acid supplementation and dexamethasone.
No tumor responses were observed. Adverse reactions observed in pediatric patients were similar to those observed in adults.
Single-dose pharmacokinetics of PEMRYDI RTU were evaluated in 22 patients age 4 to 18 years enrolled in NCT00070473 were within range of values in adults.
8.5 Geriatric Use
Of the 3,946 patients enrolled in clinical studies of PEMRYDI RTU, 34% were 65 and over and 4% were 75 and over. No overall differences in effectiveness were observed between these patients and younger patients. The incidences of Grade 3-4 anemia, fatigue, thrombocytopenia, hypertension, and neutropenia were higher in patients 65 years of age and older as compared to younger patients: in at least one of five randomized clinical trials [see Adverse Reactions (6.1) and Clinical Studies (14.1, 14.2)].
8.6 Patients with Renal Impairment
PEMRYDI RTU is primarily excreted by the kidneys. Decreased renal function results in reduced clearance and greater exposure (AUC) to PEMRYDI RTU compared with patients with normal renal function [Warnings and Precautions (5.2, 5.6) and Clinical Pharmacology (12.3)]. No dose is recommended for patients with creatinine clearance less than 45 mL/min [see Dosage and Administration (2.3)].
- 10. OVERDOSAGE
-
11. DESCRIPTION
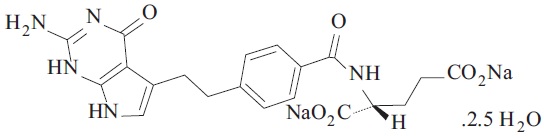
Pemetrexed is a folate analog metabolic inhibitor. The drug substance, Pemetrexed Disodium Hemipentahydrate, has the chemical name disodium N-[4-[2-(2-amino-4,7-dihydro-4-oxo-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl] benzoyl]-L-glutamic acid hemipentahydrate, with a molecular formula of C20H19N5Na2O6•2.5 H2O and a molecular weight of 516.41. The structural formula is as follows:
PEMRYDI RTU is a sterile clear, colorless to pale yellow to green-yellow ready-to-use solution in single-dose vials. Each milliliter of solution contains 10 mg of pemetrexed (equivalent to 12.1 mg pemetrexed disodium hemipentahydrate), 10 mg of mannitol, 9 mg of sodium chloride, 1 mg of L-cysteine hydrochloride, sodium hydroxide and/or hydrochloric acid to adjust pH and water for injection.
-
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
PEMRYDI RTU is a folate analog metabolic inhibitor that disrupts folate-dependent metabolic processes essential for cell replication. In vitro studies show that pemetrexed inhibits thymidylate synthase (TS), dihydrofolate reductase, and glycinamide ribonucleotide formyltransferase (GARFT), which are folate-dependent enzymes involved in the de novo biosynthesis of thymidine and purine nucleotides. Pemetrexed is taken into cells by membrane carriers such as the reduced folate carrier and membrane folate binding protein transport systems. Once in the cell, pemetrexed is converted to polyglutamate forms by the enzyme folylpolyglutamate synthetase. The polyglutamate forms are retained in cells and are inhibitors of TS and GARFT.
12.2 Pharmacodynamics
Pemetrexed inhibited the in vitro growth of mesothelioma cell lines (MSTO-211H, NCI-H2052) and showed synergistic effects when combined with cisplatin.
Based on population pharmacodynamic analyses, the depth of the absolute neutrophil counts (ANC) nadir correlates with the systemic exposure to pemetrexed and supplementation with folic acid and vitamin B12. There is no cumulative effect of pemetrexed exposure on ANC nadir over multiple treatment cycles.
12.3 Pharmacokinetics
Absorption
The pharmacokinetics of pemetrexed when PEMRYDI RTU was administered as a single agent in doses ranging from 0.2 to 838 mg/m2 infused over a 10-minute period have been evaluated in 426 cancer patients with a variety of solid tumors. Pemetrexed total systemic exposure (AUC) and maximum plasma concentration (Cmax) increased proportionally with increase of dose. The pharmacokinetics of pemetrexed did not change over multiple treatment cycles.
Distribution
Pemetrexed has a steady-state volume of distribution of 16.1 liters. In vitro studies indicated that pemetrexed is 81% bound to plasma proteins.
Elimination
The total systemic clearance of pemetrexed is 91.8 mL/min and the elimination half-life of pemetrexed is 3.5 hours in patients with normal renal function (creatinine clearance of 90 mL/min). As renal function decreases, the clearance of pemetrexed decreases and exposure (AUC) of pemetrexed increases.
Metabolism
Pemetrexed is not metabolized to an appreciable extent.
Excretion
Pemetrexed is primarily eliminated in the urine, with 70% to 90% of the dose recovered unchanged within the first 24 hours following administration. In vitro studies indicated that pemetrexed is a substrate of OAT3 (organic anion transporter 3), a transporter that is involved in the active secretion of pemetrexed.
Specific Populations
Age (26 to 80 years) and sex had no clinically meaningful effect on the systemic exposure of pemetrexed based on population pharmacokinetic analyses.
Racial Groups
The pharmacokinetics of pemetrexed were similar in Whites and Blacks or African Americans. Insufficient data are available for other ethnic groups.
Patients with Hepatic Impairment
Pemetrexed has not been formally studied in patients with hepatic impairment. No effect of elevated AST, ALT, or total bilirubin on the PK of pemetrexed was observed in clinical studies.
Patients with Renal Impairment
Pharmacokinetic analyses of pemetrexed included 127 patients with impaired renal function. Plasma clearance of pemetrexed decreases as renal function decreases, with a resultant increase in systemic exposure. Patients with creatinine clearances of 45, 50, and 80 mL/min had 65%, 54%, and 13% increases, respectively in systemic exposure (AUC) compared to patients with creatinine clearance of 100 mL/min [see Dosage and Administration (2.3) and Warnings and Precautions (5.2)].
Third-Space Fluid
The pemetrexed plasma concentrations in patients with various solid tumors with stable, mild to moderate third-space fluid were comparable to those observed in patients without third space fluid collections. The effect of severe third space fluid on pharmacokinetics is not known.
Drug Interaction Studies
Drugs Inhibiting OAT3 Transporter
Ibuprofen, an OAT3 inhibitor, administered at 400 mg four times a day decreased the clearance of pemetrexed and increased its exposure (AUC) by approximately 20% in patients with normal renal function (creatinine clearance > 80 mL/min).
In Vitro Studies
Pemetrexed is a substrate for OAT3. Ibuprofen, an OAT3 inhibitor inhibited the uptake of pemetrexed in OAT3-expressing cell cultures with an average [Iu]/IC50 ratio of 0.38. In vitro data predict that at clinically relevant concentrations, other NSAIDs (naproxen, diclofenac, celecoxib) would not inhibit the uptake of pemetrexed by OAT3 and would not increase the AUC of pemetrexed to a clinically significant extent [see Drug Interactions (7)].
Pemetrexed is a substrate for OAT4. In vitro, ibuprofen and other NSAIDs (naproxen, diclofenac, celecoxib) are not inhibitors of OAT4 at clinically relevant concentrations.
Aspirin
Aspirin, administered in low to moderate doses (325 mg every 6 hours), does not affect the pharmacokinetics of pemetrexed.
Cisplatin
Cisplatin does not affect the pharmacokinetics of pemetrexed and the pharmacokinetics of total platinum are unaltered by pemetrexed.
Vitamins
Neither folic acid nor vitamin B12 affect the pharmacokinetics of pemetrexed.
Drugs Metabolized by Cytochrome P450 Enzymes
In vitro studies suggest that pemetrexed does not inhibit the clearance of drugs metabolized by CYP3A, CYP2D6, CYP2C9, and CYP1A2.
-
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity studies have been conducted with pemetrexed. Pemetrexed was clastogenic in an in vivo micronucleus assay in mouse bone marrow but was not mutagenic in multiple in vitro tests (Ames assay, Chinese Hamster Ovary cell assay).
Pemetrexed administered intraperitoneally at doses of ≥ 0.1 mg/kg/day to male mice (approximately 0.0006 times the recommended human dose based on BSA) resulted in reduced fertility, hypospermia, and testicular atrophy.
-
14. CLINICAL STUDIES
14.1 Non-Squamous NSCLC
Initial Treatment in Combination with Pembrolizumab and Platinum
The efficacy of PEMRYDI RTU in combination with pembrolizumab and platinum chemotherapy was investigated in Study KEYNOTE-189 (NCT02578680), a randomized, multicenter, double-blind, active-controlled trial conducted in patients with metastatic non-squamous NSCLC, regardless of PD-L1 tumor expression status, who had not previously received systemic therapy for metastatic disease and in whom there were no EGFR or ALK genomic tumor aberrations. Patients with autoimmune disease that required systemic therapy within 2 years of treatment; a medical condition that required immunosuppression; or who had received more than 30 Gy of thoracic radiation within the prior 26 weeks were ineligible. Randomization was stratified by smoking status (never versus former/current), choice of platinum (cisplatin versus carboplatin), and tumor PD-L1 status (TPS < 1% [negative] versus TPS ≥ 1%). Patients were randomized (2:1) to one of the following treatment arms:
- PEMRYDI RTU 500 mg/m2, pembrolizumab 200 mg, and investigator's choice of cisplatin 75 mg/m2 or carboplatin AUC 5 mg/mL/min intravenously on Day 1 of each 21-day cycle for 4 cycles followed by PEMRYDI RTU 500 mg/m2 and pembrolizumab 200 mg intravenously every 3 weeks. PEMRYDI RTU was administered after pembrolizumab and prior to platinum chemotherapy on Day 1.
- Placebo, PEMRYDI RTU 500 mg/m2, and investigator's choice of cisplatin 75 mg/m2 or carboplatin AUC 5 mg/mL/min intravenously on Day 1 of each 21-day cycle for 4 cycles followed by placebo and PEMRYDI RTU 500 mg/m2 intravenously every 3 weeks.
Treatment with PEMRYDI RTU continued until RECIST v1.1 (modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ)-defined progression of disease as determined by the investigator or unacceptable toxicity.
Patients randomized to placebo, PEMRYDI RTU, and platinum chemotherapy were offered pembrolizumab as a single agent at the time of disease progression.
Assessment of tumor status was performed at Week 6, Week 12, and then every 9 weeks thereafter. The main efficacy outcome measures were OS and PFS as assessed by BICR RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of five target lesions per organ. Additional efficacy outcome measures were ORR and duration of response, as assessed by the BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ.
A total of 616 patients were randomized: 410 patients to the PEMRYDI RTU, pembrolizumab, and platinum chemotherapy arm and 206 to the placebo, PEMRYDI RTU, and platinum chemotherapy arm. The study population characteristics were: median age of 64 years (range: 34 to 84); 49% age 65 or older; 59% male; 94% White and 3% Asian; 56% ECOG performance status of 1; and 18% with history of brain metastases. Thirty-one percent had tumor PD-L1 expression TPS < 1%. Seventy-two percent received carboplatin and 12% were never smokers. A total of 85 patients in the placebo, PEMRYDI RTU, and chemotherapy arm received an anti-PD-1/PD-L1 monoclonal antibody at the time of disease progression.
The trial demonstrated a statistically significant improvement in OS and PFS for patients randomized to PEMRYDI RTU in combination with pembrolizumab and platinum chemotherapy compared with placebo, PEMRYDI RTU, and platinum chemotherapy (see Table 10 and Figure 1).
Table 10: Efficacy Results of KEYNOTE-189
Endpoint
PEMRYDI RTU
Pembrolizumab
Platinum Chemotherapy
n=410Placebo
PEMRYDI RTU
Platinum Chemotherapy
n=206
OS
Number (%) of patients with event
127 (31%)
108 (52%)
Median in months (95% CI)
NR
(NR, NR)
11.3
(8.7, 15.1)
Hazard ratioa (95% CI)
0.49 (0.38, 0.64)
p-valueb
< 0.0001
PFS
Number of patients with event (%)
245 (60%)
166 (81%)
Median in months (95% CI)
8.8 (7.6, 9.2)
4.9 (4.7, 5.5)
Hazard ratioa (95% CI)
0.52 (0.43, 0.64)
p-valueb
< 0.0001
ORR
Overall response ratec (95% CI)
48% (43, 53)
19% (14, 25)
Complete response
0.5%
0.5%
Partial response
47%
18%
p-valued
< 0.0001
Duration of Response
Median in months (range)
11.2 (1.1+, 18.0+)
7.8 (2.1+, 16.4+)
a Based on the stratified Cox proportional hazard model.
b Based on stratified log-rank test.
c Response: Best objective response as confirmed complete response or partial response.
d Based on Miettinen and Nurminen method stratified by PD-L1 status, platinum chemotherapy and smoking status.
NR = not reached
At the protocol specified final OS analysis, the median in the PEMRYDI RTU in combination with pembrolizumab and platinum chemotherapy arm was 22.0 months (95% CI: 19.5, 24.5) compared to 10.6 months (95% CI: 8.7, 13.6) in the placebo with PEMRYDI RTU and platinum chemotherapy arm, with an HR of 0.56 (95% CI: 0.46, 0.69).
Figure 1: Kaplan-Meier Curve for Overall Survival in KEYNOTE-189*
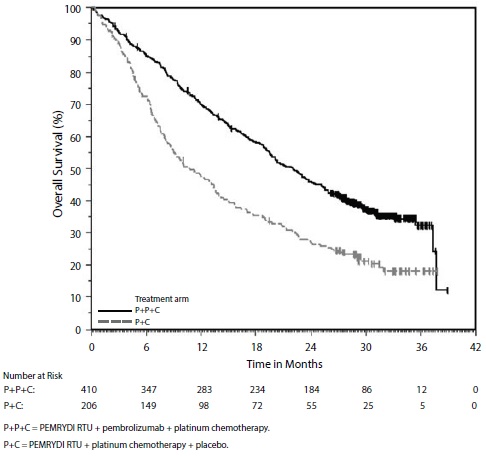
*Based on the protocol-specified final OS analysis
Initial Treatment in Combination with Cisplatin
The efficacy of PEMRYDI RTU was evaluated in Study JMDB (NCT00087711), a multi-center, randomized (1:1), open-label study conducted in 1,725 chemotherapy-naive patients with Stage IIIb/IV NSCLC. Patients were randomized to receive PEMRYDI RTU with cisplatin or gemcitabine with cisplatin. Randomization was stratified by Eastern Cooperative Oncology Group Performance Status (ECOG PS 0 versus 1), gender, disease stage, basis for pathological diagnosis (histopathological/cytopathological), history of brain metastases, and investigative center. PEMRYDI RTU was administered intravenously over 10 minutes at a dose of 500 mg/m2 on Day 1 of each 21-day cycle. Cisplatin was administered intravenously at a dose of 75 mg/m2 approximately 30 minutes after PEMRYDI RTU administration on Day 1 of each cycle, gemcitabine was administered at a dose of 1,250 mg/m2 on Day 1 and Day 8, and cisplatin was administered intravenously at a dose of 75 mg/m2 approximately 30 minutes after administration of gemcitabine, on Day 1 of each 21-day cycle. Treatment was administered up to a total of 6 cycles; patients in both arms received folic acid, vitamin B12, and dexamethasone [see Dosage and Administration (2.4)]. The primary efficacy outcome measure was overall survival.
A total of 1,725 patients were enrolled with 862 patients randomized to PEMRYDI RTU in combination with cisplatin and 863 patients to gemcitabine in combination with cisplatin. The median age was 61 years (range 26-83 years), 70% were male, 78% were White, 17% were Asian, 2.9% were Hispanic or Latino, and 2.1% were Black or African American, and < 1% were other ethnicities. Among patients for whom ECOG PS (n=1,722) and smoking history (n=1,516) were collected, 65% had an ECOG PS of 1, 36% had an ECOG PS of 0, and 84% were smokers. For tumor characteristics, 73% had non-squamous NSCLC and 27% had squamous NSCLC; 76% had Stage IV disease. Among 1,252 patients with non-squamous NSCLC histology, 68% had a diagnosis of adenocarcinoma, 12% had large cell histology and 20% had other histologic subtypes.
Efficacy results in Study JMDB are presented in Table 11 and Figure 2.
Table 11: Efficacy Results in Study JMDB
Efficacy Parameter
PEMRYDI RTU plus Cisplatin
(N=862)
Gemcitabine plus Cisplatin
(N=863)
Overall Survival
Median (months)
(95% CI)
10.3
(9.8-11.2)
10.3
(9.6-10.9)
Hazard ratio (HR)a,b
(95% CI)
0.94
(0.84-1.05)
Progression-Free Survival
Median (months)
(95% CI)
4.8
(4.6-5.3)
5.1
(4.6-5.5)
Hazard ratio (HR)a,b
(95% CI)
1.04
(0.94-1.15)
Overall Response Rate
(95% CI)
27.1%
(24.2-30.1)
24.7%
(21.8-27.6)
a Unadjusted for multiple comparisons.
b Adjusted for gender, stage, basis of diagnosis, and performance status.
Figure 2: Kaplan-Meier Curves for Overall Survival in Study JMDB
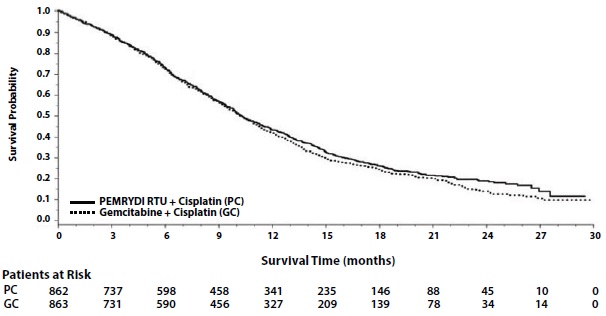
In pre-specified analyses assessing the impact of NSCLC histology on overall survival, clinically relevant differences in survival according to histology were observed. These subgroup analyses are shown in Table 12 and Figures 3 and 4. This difference in treatment effect for PEMRYDI RTU based on histology demonstrating a lack of efficacy in squamous cell histology was also observed in Studies JMEN and JMEI.
Table 12: Overall Survival in NSCLC Histologic Subgroups in Study JMDB
Histologic Subgroups
PEMRYDI RTU plus Cisplatin
(N=862)
Gemcitabine plus Cisplatin
(N=863)
Non-squamous NSCLC (N=1,252)
Median (months)
(95% CI)
11.0
(10.1-12.5)
10.1
(9.3-10.9)
HRa,b
(95% CI)
0.84
(0.74-0.96)
Adenocarcinoma (N=847)
Median (months)
(95% CI)
12.6
(10.7-13.6)
10.9
(10.2-11.9)
HRa,b
(95% CI)
0.84
(0.71-0.99)
Large Cell (N=153)
Median (months)
(95% CI)
10.4
(8.6-14.1)
6.7
(5.5-9.0)
HRa,b
(95% CI)
0.67
(0.48-0.96)
Non-squamous, not otherwise specified (N=252)
Median (months)
(95% CI)
8.6
(6.8-10.2)
9.2
(8.1-10.6)
HRa,b
(95% CI)
1.08
(0.81-1.45)
Squamous Cell (N=473)
Median (months)
(95% CI)
9.4
(8.4-10.2)
10.8
(9.5-12.1)
HRa,b
(95% CI)
1.23
(1.00-1.51)
a Unadjusted for multiple comparisons.
b Adjusted for ECOG PS, gender, disease stage, and basis for pathological diagnosis (histopathological/cytopathological).
Figure 3: Kaplan-Meier Curves for Overall Survival in Non-squamous NSCLC in Study JMDB
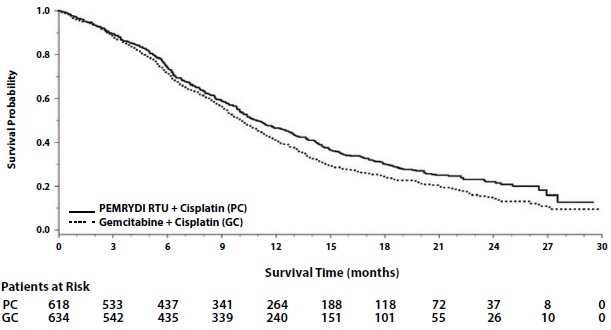
Figure 4: Kaplan-Meier Curves for Overall Survival in Squamous NSCLC in Study JMDB
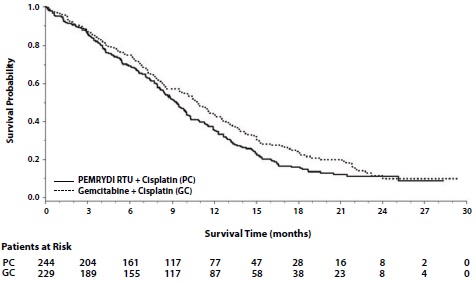
Maintenance Treatment Following First-line Non-PEMRYDI RTU Containing Platinum-Based Chemotherapy
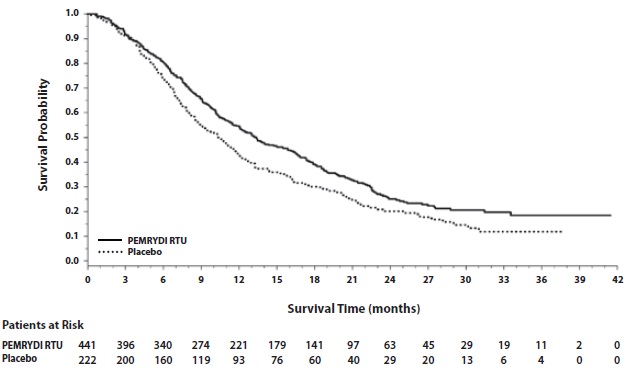
The efficacy of PEMRYDI RTU as maintenance therapy following first-line platinum-based chemotherapy was evaluated in Study JMEN (NCT00102804), a multicenter, randomized (2:1), double-blind, placebo-controlled study conducted in 663 patients with Stage IIIb/IV NSCLC who did not progress after four cycles of platinum-based chemotherapy. Patients were randomized to receive PEMRYDI RTU 500 mg/m2 intravenously every 21 days or placebo until disease progression or intolerable toxicity. Patients in both study arms received folic acid, vitamin B12, and dexamethasone [see Dosage and Administration (2.4)]. Randomization was carried out using a minimization approach [Pocock and Simon (1,975)] using the following factors: gender, ECOG PS (0 versus 1), response to prior chemotherapy (complete or partial response versus stable disease), history of brain metastases (yes versus no), non-platinum component of induction therapy (docetaxel versus gemcitabine versus paclitaxel), and disease stage (IIIb versus IV). The major efficacy outcome measures were progression-free survival based on assessment by independent review and overall survival; both were measured from the date of randomization in Study JMEN.
A total of 663 patients were enrolled with 441 patients randomized to PEMRYDI RTU and 222 patients randomized to placebo. The median age was 61 years (range 26-83 years); 73% were male; 65% were White, 32% were Asian, 2.9% were Hispanic or Latino, and < 2% were other ethnicities; 60% had an ECOG PS of 1; and 73% were current or former smokers. Median time from initiation of platinum-based chemotherapy to randomization was 3.3 months (range 1.6 to 5.1 months) and 49% of the population achieved a partial or complete response to first-line, platinum-based chemotherapy. With regard to tumor characteristics, 81% had Stage IV disease, 73% had non-squamous NSCLC and 27% had squamous NSCLC. Among the 481 patients with non-squamous NSCLC, 68% had adenocarcinoma, 4% had large cell, and 28% had other histologies.
Efficacy results are presented in Table 13 and Figure 5.
Table 13: Efficacy Results in Study JMEN
Efficacy Parameter
PEMRYDI RTU
Placebo
Overall survival
N=441
N=222
Median (months)
(95% CI)
13.4
(11.9-15.9)
10.6
(8.7-12.0)
Hazard ratioa
(95% CI)
0.79
(0.65-0.95)
p-value
p=0.012
Progression-free survival per independent review
N=387
N=194
Median (months)
(95% CI)
4.0
(3.1-4.4)
2.0
(1.5-2.8)
Hazard ratioa
(95% CI)
0.60
(0.49-0.73)
p-value
p < 0.00001
a Hazard ratios are adjusted for multiplicity but not for stratification variables.
Figure 5: Kaplan-Meier Curves for Overall Survival in Study JMEN
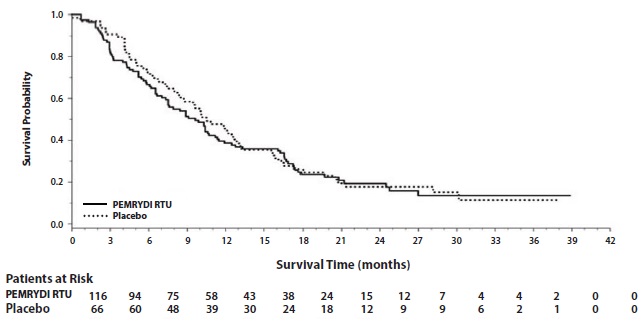
The results of pre-specified subgroup analyses by NSCLC histology are presented in Table 14 and Figures 6 and 7.
Table 14: Efficacy Results in Study JMEN by Histologic Subgroup
Efficacy Parameter
Overall Survival
Progression-Free Survival
Per Independent Review
PEMRYDI RTU
(N=441)
Placebo
(N=222)
PEMRYDI RTU
(N=387)
Placebo
(N=194)
Non-squamous NSCLC (n=481)
Median (months)
15.5
10.3
4.4
1.8
HRa(95% CI)
0.70
(0.56-0.88)
0.47
(0.37-0.60)
Adenocarcinoma (n=328)
Median (months)
16.8
11.5
4.6
2.7
HRa
(95% CI)
0.73
(0.56-0.96)
0.51
(0.38-0.68)
Large cell carcinoma (n=20)
Median (months)
8.4
7.9
4.5
1.5
HRa
(95% CI)
0.98
(0.36-2.65)
0.40
(0.12-1.29)
Otherb (n=133)
Median (months)
11.3
7.7
4.1
1.6
HRa
(95% CI)
0.61
(0.40-0.94)
0.44
(0.28-0.68)
Squamous cell NSCLC (n=182)
Median (months)
9.9
10.8
2.4
2.5
HRa
(95% CI)
1.07
(0.77-1.50)
1.03
(0.71-1.49)
a Hazard ratios are not adjusted for multiplicity
b Primary diagnosis of NSCLC not specified as adenocarcinoma, large cell carcinoma, or squamous cell carcinoma.
Figure 6: Kaplan-Meier Curves for Overall Survival in Non-squamous NSCLC in Study JMEN
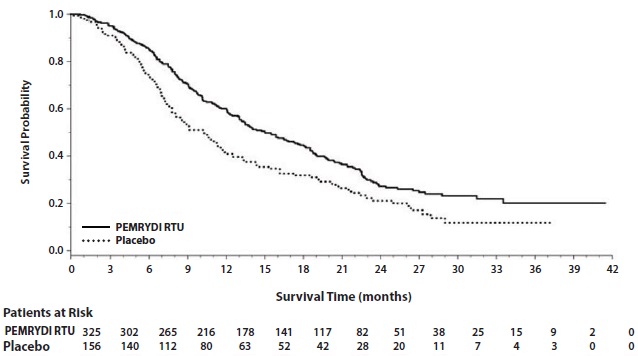
Figure 7: Kaplan-Meier Curves for Overall Survival in Squamous NSCLC in Study JMEN
Maintenance Treatment Following First-line PEMRYDI RTU Plus Platinum Chemotherapy
The efficacy of PEMRYDI RTU as maintenance therapy following first-line platinum-based chemotherapy was also evaluated in PARAMOUNT (NCT00789373), a multi-center, randomized (2:1), double-blind, placebo-controlled study conducted in patients with Stage IIIb/IV non-squamous NSCLC who had completed four cycles of PEMRYDI RTU in combination with cisplatin and achieved a complete response (CR) or partial response (PR) or stable disease (SD). Patients were required to have an ECOG PS of 0 or 1. Patients were randomized to receive PEMRYDI RTU 500 mg/m2 intravenously every 21 days or placebo until disease progression. Randomization was stratified by response to PEMRYDI RTU in combination with cisplatin induction therapy (CR or PR versus SD), disease stage (IIIb versus IV), and ECOG PS (0 versus 1). Patients in both arms received folic acid, vitamin B12, and dexamethasone. The main efficacy outcome measure was investigator-assessed progression-free survival (PFS) and an additional efficacy outcome measure was overall survival (OS); PFS and OS were measured from the time of randomization.
A total of 539 patients were enrolled with 359 patients randomized to PEMRYDI RTU and 180 patients randomized to placebo. The median age was 61 years (range 32 to 83 years); 58% were male; 95% were White, 4.5% were Asian, and < 1% were Black or African American; 67% had an ECOG PS of 1; 78% were current or former smokers; and 43% of the population achieved a partial or complete response to first-line, platinum-based chemotherapy. With regard to tumor characteristics, 91% had Stage IV disease, 87% had adenocarcinoma, 7% had large cell, and 6% had other histologies.
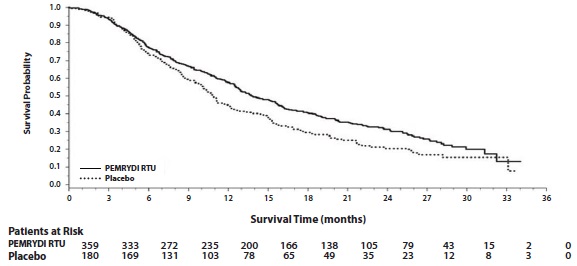
Efficacy results for PARAMOUNT are presented in Table 15 and Figure 8.
Table 15: Efficacy Results in PARAMOUNT
Efficacy Parameter
PEMRYDI RTU
(N=359)
Placebo
(N=180)
Overall survival
Median (months)
(95% CI)
13.9
(12.8-16.0)
11.0
(10.0-12.5)
Hazard ratio (HR)a
(95% CI)
0.78
(0.64-0.96)
p-value
p=0.02
Progression-free survivalb
Median (months)
(95% CI)
4.1
(3.2-4.6)
2.8
(2.6-3.1)
Hazard ratio (HR) a
(95% CI)
0.62
(0.49-0.79)
p-value
p < 0.0001
a Hazard ratios are adjusted for multiplicity but not for stratification variables.
b Based on investigator's assessment.
Figure 8: Kaplan-Meier Curves for Overall Survival in PARAMOUNT
Treatment of Recurrent Disease After Prior Chemotherapy
The efficacy of PEMRYDI RTU was evaluated in Study JMEI (NCT00004881), a multicenter, randomized (1:1), open-label study conducted in patients with Stage III or IV NSCLC that had recurred or progressed following one prior chemotherapy regimen for advanced disease. Patients were randomized to receive PEMRYDI RTU 500 mg/m2 intravenously or docetaxel 75 mg/m2 as a 1-hour intravenous infusion once every 21 days. Patients randomized to PEMRYDI RTU also received folic acid and vitamin B12. The study was designed to show that overall survival with PEMRYDI RTU was non-inferior to docetaxel, as the major efficacy outcome measure, and that overall survival was superior for patients randomized to PEMRYDI RTU compared to docetaxel, as a secondary outcome measure.
A total of 571 patients were enrolled with 283 patients randomized to PEMRYDI RTU and 288 patients randomized to docetaxel. The median age was 58 years (range 22 to 87 years); 72% were male; 71% were White, 24% were Asian, 2.8% were Black or African American, 1.8% were Hispanic or Latino, and < 2% were other ethnicities; 88% had an ECOG PS of 0 or 1. With regard to tumor characteristics, 75% had Stage IV disease; 53% had adenocarcinoma, 30% had squamous histology; 8% large cell; and 9% had other histologic subtypes of NSCLC.
The efficacy results in the overall population and in subgroup analyses based on histologic subtype are provided in Tables 16 and 17, respectively. Study JMEI did not show an improvement in overall survival in the intent-to-treat population. In subgroup analyses, there was no evidence of a treatment effect on survival in patients with squamous NSCLC; the absence of a treatment effect in patients with NSCLC of squamous histology was also observed Studies JMDB and JMEN [see Clinical Studies (14.1)].
Table 16: Efficacy Results in Study JMEI
Efficacy Parameter
PEMRYDI RTU
(N=283)
Docetaxel
(N=288)
Overall survival
Median (months)
(95% CI)
8.3
(7.0-9.4)
7.9
(6.3-9.2)
Hazard ratioa
(95% CI)
0.99
(0.82-1.20)
Progression-free survival
Median (months)
(95% CI)
2.9
(2.4-3.1)
2.9
(2.7-3.4)
Hazard ratioa
(95% CI)
0.97
(0.82-1.16)
Overall response rate
(95% CI)
8.5%
(5.2-11.7)
8.3%
(5.1-11.5)
a Hazard ratios are not adjusted for multiplicity or for stratification variables.
Table 17: Exploratory Efficacy Analyses by Histologic Subgroup in Study JMEI
Histologic Subgroups
PEMRYDI RTU
(N=283)
Docetaxel
(N=288)
Non-squamous NSCLC (N=399)
Median (months)
(95% CI)
9.3
(7.8-9.7)
8.0
(6.3-9.3)
HRa
(95% CI)
0.89
(0.71-1.13)
Adenocarcinoma (N=301)
Median (months)
(95% CI)
9.0
(7.6-9.6)
9.2
(7.5-11.3)
HRa
(95% CI)
1.09
(0.83-1.44)
Large Cell (N=47)
Median (months)
(95% CI)
12.8
(5.8-14.0)
4.5
(2.3-9.1)
HRa
(95% CI)
0.38
(0.18-0.78)
Otherb (N=51)
Median (months)
(95% CI)
9.4
(6.0-10.1)
7.9
(4.0-8.9)
HRa
(95% CI)
0.62
(0.32-1.23)
Squamous NSCLC (N=172)
Median (months)
(95% CI)
6.2
(4.9-8.0)
7.4
(5.6-9.5)
HRa
(95% CI)
1.32
(0.93-1.86)
a Hazard ratio unadjusted for multiple comparisons.
b Primary diagnosis of NSCLC not specified as adenocarcinoma, large cell carcinoma, or squamous cell carcinoma.
14.2 Mesothelioma
The efficacy of PEMRYDI RTU was evaluated in Study JMCH (NCT00005636), a multicenter, randomized (1:1), single-blind study conducted in patients with MPM who had received no prior chemotherapy. Patients were randomized (n=456) to receive PEMRYDI RTU 500 mg/m2 intravenously over 10 minutes followed 30 minutes later by cisplatin 75 mg/m2 intravenously over two hours on Day 1 of each 21-day cycle or to receive cisplatin 75 mg/m2 intravenously over 2 hours on Day 1 of each 21-day cycle; treatment continued until disease progression or intolerable toxicity. The study was modified after randomization and treatment of 117 patients to require that all patients receive folic acid 350 mcg to 1,000 mcg daily beginning 1 to 3 weeks prior to the first dose of PEMRYDI RTU and continuing until 1 to 3 weeks after the last dose, vitamin B12 1,000 mcg intramuscularly 1 to 3 weeks prior to first dose of PEMRYDI RTU and every 9 weeks thereafter, and dexamethasone 4 mg orally, twice daily, for 3 days starting the day prior to each PEMRYDI RTU dose. Randomization was stratified by multiple baseline variables including KPS, histologic subtype (epithelial, mixed, sarcomatoid, other), and gender. The major efficacy outcome measure was overall survival and additional efficacy outcome measures were time to disease progression, overall response rate, and response duration.
A total of 448 patients received at least one dose of protocol-specified therapy; 226 patients were randomized to and received at least one dose of PEMRYDI RTU plus cisplatin, and 222 patients were randomized to and received cisplatin. Among the 226 patients who received cisplatin with PEMRYDI RTU, 74% received full supplementation with folic acid and vitamin B12 during study therapy, 14% were never supplemented, and 12% were partially supplemented. Across the study population, the median age was 61 years (range: 20 to 86 years); 81% were male; 92% were White, 5% were Hispanic or Latino, 3.1% were Asian, and < 1% were other ethnicities; and 54% had a baseline KPS score of 90-100% and 46% had a KPS score of 70-80%. With regard to tumor characteristics, 46% had Stage IV disease, 31% Stage III, 15% Stage II, and 7% Stage I disease at baseline; the histologic subtype of mesothelioma was epithelial in 68% of patients, mixed in 16%, sarcomatoid in 10% and other histologic subtypes in 6%. The baseline demographics and tumor characteristics of the subgroup of fully supplemented patients was similar to the overall study population.
The efficacy results from Study JMCH are summarized in Table 18 and Figure 9.
Table 18: Efficacy Results in Study JMCH
Efficacy Parameter
All Randomized and
Treated Patients
(N=448)
Fully Supplemented
Patients
(N=331)
PEMRYDI RTU /Cisplatin
(N=226)
Cisplatin
(N=222)
PEMRYDI RTU /Cisplatin
(N=168)
Cisplatin
(N=163)
Median overall survival (months)
12.1
9.3
13.3
10.0
(95% CI)
(10.0-14.4)
(7.8-10.7)
(11.4-14.9)
(8.4-11.9)
Hazard ratioa
0.77
0.75
Log rank p-value
0.020
NAb
a Hazard ratios are not adjusted for stratification variables.
b Not a pre-specified analysis.
Figure 9: Kaplan-Meier Curves for Overall Survival in Study JMCH
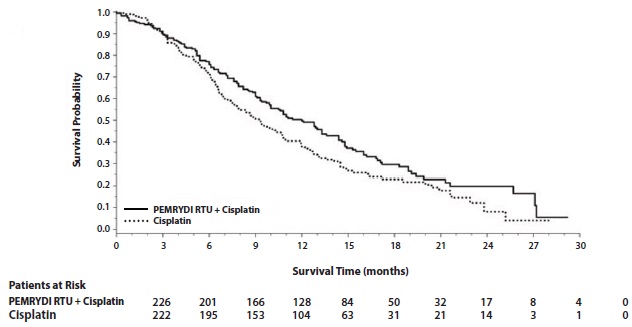
Based upon prospectively defined criteria (modified Southwest Oncology Group methodology) the objective tumor response rate for PEMRYDI RTU plus cisplatin was greater than the objective tumor response rate for cisplatin alone. There was also improvement in lung function (forced vital capacity) in the PEMRYDI RTU plus cisplatin arm compared to the control arm.
- 15. REFERENCES
-
16. HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
PEMRYDI RTU (pemetrexed injection) is supplied as a sterile clear, colorless to pale yellow to green-yellow ready-to-use solution packaged in a USP type-I glass vial with rubber stopper and aluminium flip-off cap.
It is available as follows:
100 mg/10 mL (10 mg/mL):
1 Single-dose Vial in a Carton: NDC 70121-2453-1
500 mg/50 mL (10 mg/mL):
1 Single-dose Vial in a Carton: NDC 70121-2461-1
1,000 mg/100 mL (10 mg/mL):
1 Single-dose Vial in a Carton: NDC 70121-2462-1
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
PEMRYDI RTU is a hazardous drug. Follow applicable special handling and disposal procedures.1
-
17. PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Premedication and Concomitant Medication: Instruct patients to take folic acid as directed and to keep appointments for vitamin B12 injections to reduce the risk of treatment-related toxicity. Instruct patients of the requirement to take corticosteroids to reduce the risks of treatment-related toxicity [see Dosage and Administration (2.4) and Warnings and Precautions (5.1)].
Myelosuppression: Inform patients of the risk of low blood cell counts and instruct them to immediately contact their physician for signs of infection, fever, bleeding, or symptoms of anemia [see Warnings and Precautions (5.1)].
Renal Failure: Inform patients of the risks of renal failure, which may be exacerbated in patients with dehydration arising from severe vomiting or diarrhea. Instruct patients to immediately contact their healthcare provider for a decrease in urine output [see Warnings and Precautions (5.2)].
Bullous and Exfoliative Skin Disorders: Inform patients of the risks of severe and exfoliative skin disorders. Instruct patients to immediately contact their healthcare provider for development of bullous lesions or exfoliation in the skin or mucous membranes [see Warnings and Precautions (5.3)].
Interstitial Pneumonitis: Inform patients of the risks of pneumonitis. Instruct patients to immediately contact their healthcare provider for development of dyspnea or persistent cough [see Warnings and Precautions (5.4)].
Radiation Recall: Inform patients who have received prior radiation of the risks of radiation recall. Instruct patients to immediately contact their healthcare provider for development of inflammation or blisters in an area that was previously irradiated [see Warnings and Precautions (5.5)].
Increased Risk of Toxicity with Ibuprofen in Patients with Renal Impairment: Advise patients with mild to moderate renal impairment of the risks associated with concomitant ibuprofen use and instruct them to avoid use of all ibuprofen containing products for 2 days before, the day of, and 2 days following administration of PEMRYDI RTU [see Dosage and Administration (2.5), Warnings and Precautions (5.6), and Drug Interactions (7)].
Embryo-Fetal Toxicity: Advise females of reproductive potential and males with female partners of reproductive potential of the potential risk to a fetus [see Warnings and Precautions (5.7) and Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 6 months after the last dose. Advise females to inform their prescriber of a known or suspected pregnancy. Advise males with female partners of reproductive potential to use effective contraception during treatment with PEMRYDI RTU and for 3 months after the last dose [see Warnings and Precautions (5.7) and Use in Specific Populations (8.3)].
Lactation: Advise women not to breastfeed during treatment with PEMRYDI RTU and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
Zydus Lifesciences Limited,
Ahmedabad, India.
Distributed by:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 06-2023-00
-
PATIENT INFORMATION
PEMRYDI RTUTM (PEM-rih-DEE. AR. TEE. YU.)
(pemetrexed injection)
For Intravenous Use
What is PEMRYDI RTU?
PEMRYDI RTU is a prescription medicine used to treat:
- a kind of lung cancer called non-squamous non-small cell lung cancer (NSCLC). PEMRYDI RTU is used:
- as the first treatment in combination with pembrolizumab and platinum chemotherapy when your lung cancer with no abnormal EGFR or ALK gene has spread (advanced NSCLC).
- as the first treatment in combination with cisplatin when your lung cancer has spread (advanced NSCLC).
- alone as maintenance treatment after you have received 4 cycles of chemotherapy that contains platinum for first treatment of your advanced NSCLC and your cancer has not progressed.
- alone when your lung cancer has returned or spread after prior chemotherapy.
PEMRYDI RTU is not for use for the treatment of people with squamous cell non-small cell lung cancer.
- a kind of cancer called malignant pleural mesothelioma. This cancer affects the lining of the lungs and chest wall. PEMRYDI RTU is used in combination with cisplatin as the first treatment for malignant pleural mesothelioma that cannot be removed by surgery or you are not able to have surgery.
PEMRYDI RTU has not been shown to be safe and effective in children.
Do not take PEMRYDI RTU if you have had a severe allergic reaction to any medicine that contains pemetrexed.
Before taking PEMRYDI RTU, tell your healthcare provider about all of your medical conditions, including if you:
- have kidney problems.
- have had radiation therapy.
- are pregnant or plan to become pregnant. PEMRYDI RTU can harm your unborn baby.
Females who are able to become pregnant:
Your healthcare provider will check to see if you are pregnant before you start treatment with PEMRYDI RTU.
You should use effective birth control (contraception) during treatment with PEMRYDI RTU and for 6 months after the last dose. Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with PEMRYDI RTU.
Males with female partners who are able to become pregnant should use effective birth control (contraception) during treatment with PEMRYDI RTU and for 3 months after the last dose.
- are breastfeeding or plan to breastfeed. It is not known if PEMRYDI RTU passes into breast milk. Do not breastfeed during treatment with PEMRYDI RTU and for 1 week after the last dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Tell your healthcare provider if you have kidney problems and take a medicine that contains ibuprofen. You should avoid taking ibuprofen for 2 days before, the day of, and 2 days after receiving treatment with PEMRYDI RTU.
How is PEMRYDI RTU given?
-
It is very important to take folic acid and vitamin B12 during your treatment with PEMRYDI RTU to lower your risk of harmful side effects.
- Take folic acid exactly as prescribed by your healthcare provider 1 time a day, beginning 7 days (1 week) before your first dose of PEMRYDI RTU and continue taking folic acid until 21 days (3 weeks) after your last dose of PEMRYDI RTU.
- Your healthcare provider will give you vitamin B12 injections during treatment with PEMRYDI RTU. You will get your first vitamin B12 injection 7 days (1 week) before your first dose of PEMRYDI RTU, and then every 3 cycles.
- Your healthcare provider will prescribe a medicine called corticosteroid for you to take 2 times a day for 3 days, beginning the day before each treatment with PEMRYDI RTU.
- PEMRYDI RTU is given to you by intravenous (IV) infusion into your vein. The infusion is given over 10 minutes.
- PEMRYDI RTU is usually given once every 21 days (3 weeks).
What are the possible side effects of PEMRYDI RTU?
PEMRYDI RTU can cause serious side effects, including:
- Low blood cell counts. Low blood cell counts can be severe, including low white blood cell counts (neutropenia), low platelet counts (thrombocytopenia), and low red blood cell counts (anemia). Your healthcare provider will do blood tests to check your blood cell counts regularly during your treatment with PEMRYDI RTU. Tell your healthcare provider right away if you have any signs of infection, fever, bleeding, or severe tiredness during your treatment with PEMRYDI RTU.
- Kidney problems, including kidney failure. PEMRYDI RTU can cause severe kidney problems that can lead to death. Severe vomiting or diarrhea can lead to loss of fluids (dehydration) which may cause kidney problems to become worse. Tell your healthcare provider right away if you have a decrease in amount of urine.
- Severe skin reactions. Severe skin reactions that may lead to death can happen with PEMRYDI RTU. Tell your healthcare provider right away if you develop blisters, skin sores, skin peeling, or painful sores, or ulcers in your mouth, nose, throat or genital area.
- Lung problems (pneumonitis). PEMRYDI RTU can cause serious lung problems that can lead to death. Tell your healthcare provider right away if you get any new or worsening symptoms of shortness of breath, cough, or fever.
- Radiation recall. Radiation recall is a skin reaction that can happen in people who have received radiation treatment in the past and are treated with PEMRYDI RTU. Tell your healthcare provider if you get swelling, blistering, or a rash that looks like a sunburn in an area that was previously treated with radiation.
● tiredness
● nausea
● loss of appetite
The most common side effects of PEMRYDI RTU when given with cisplatin are:
● vomiting
● low white blood cell counts (neutropenia)
● swelling or sores in your mouth or sore throat
● low platelet counts (thrombocytopenia)
● constipation
● low red blood cell counts (anemia)
The most common side effects of PEMRYDI RTU when given with pembrolizumab and platinum chemotherapy are:
● tiredness and weakness
● nausea
● constipation
● diarrhea
● loss of appetite
● rash
● vomiting
● cough
● shortness of breath
● fever
PEMRYDI RTU may cause fertility problems in males. This may affect your ability to father a child. It is not known if these effects are reversible. Talk to your healthcare provider if this is a concern for you.
Your healthcare provider will do blood tests to check for side effects during treatment with PEMRYDI RTU. Your healthcare provider may change your dose of PEMRYDI RTU, delay treatment, or stop treatment if you have certain side effects.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the side effects of PEMRYDI RTU. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of PEMRYDI RTU.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about PEMRYDI RTU that is written for health professionals.
What are the ingredients in PEMRYDI RTU?
Active ingredient: pemetrexed.
Inactive ingredients: mannitol, sodium chloride, L-cysteine hydrochloride, sodium hydroxide and/or hydrochloric acid to adjust pH, and water for injection.
Manufactured by:
Zydus Lifesciences Limited,
Ahmedabad, India.
Distributed by:Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 06-2023-00
For more information, call 1-877-835-5472.This Patient Information has been approved by the U.S. Food and Drug Administration.
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
NDC 70121-2453-1
PEMRYDI RTU (pemetrexed injection)
100 mg/10 mL (10 mg/mL)
For Intravenous infusion only
Rx only
Amneal Pharmaceuticals LLC


NDC 70121-2461-1
PEMRYDI RTU (pemetrexed injection)
500 mg/50 mL (10 mg/mL)
For Intravenous infusion only
Rx only
Amneal Pharmaceuticals LLC
NDC 70121-2462-1
PEMRYDI RTU (pemetrexed injection)
1,000 mg/100 mL (10 mg/mL)
For Intravenous infusion only
Rx only
Amneal Pharmaceuticals LLC


-
INGREDIENTS AND APPEARANCE
PEMRYDI RTU
pemetrexed disodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70121-2453 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEMETREXED DISODIUM HEMIPENTAHYDRATE (UNII: F4GSH45R4C) (PEMETREXED - UNII:04Q9AIZ7NO) PEMETREXED DISODIUM HEMIPENTAHYDRATE 100 mg in 10 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SODIUM CHLORIDE (UNII: 451W47IQ8X) CYSTEINE HYDROCHLORIDE (UNII: ZT934N0X4W) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70121-2453-1 1 in 1 CARTON 06/06/2023 1 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215179 06/06/2023 PEMRYDI RTU
pemetrexed disodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70121-2461 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEMETREXED DISODIUM HEMIPENTAHYDRATE (UNII: F4GSH45R4C) (PEMETREXED - UNII:04Q9AIZ7NO) PEMETREXED DISODIUM HEMIPENTAHYDRATE 500 mg in 50 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SODIUM CHLORIDE (UNII: 451W47IQ8X) CYSTEINE HYDROCHLORIDE (UNII: ZT934N0X4W) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70121-2461-1 1 in 1 CARTON 06/06/2023 1 50 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215179 06/06/2023 PEMRYDI RTU
pemetrexed disodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70121-2462 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEMETREXED DISODIUM HEMIPENTAHYDRATE (UNII: F4GSH45R4C) (PEMETREXED - UNII:04Q9AIZ7NO) PEMETREXED DISODIUM HEMIPENTAHYDRATE 1000 mg in 100 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SODIUM CHLORIDE (UNII: 451W47IQ8X) CYSTEINE HYDROCHLORIDE (UNII: ZT934N0X4W) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70121-2462-1 1 in 1 CARTON 06/06/2023 1 100 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215179 06/06/2023 Labeler - Amneal Pharmaceuticals LLC (827748190) Establishment Name Address ID/FEI Business Operations Zydus Lifesciences Limited 650348852 analysis(70121-2453, 70121-2461, 70121-2462) , manufacture(70121-2453, 70121-2461, 70121-2462)