Label: KORSUVA- difelikefalin injection, solution


- NDC Code(s): 59353-065-01, 59353-065-12
- Packager: Vifor (International), Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated August 26, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KORSUVA safely and effectively. See full prescribing information for KORSUVA.
KORSUVA® (difelikefalin) injection, for intravenous use
Initial U.S. Approval: 2021INDICATIONS AND USAGE
KORSUVA is a kappa opioid receptor agonist indicated for the treatment of moderate-to-severe pruritus associated with chronic kidney disease (CKD-aP) in adults undergoing hemodialysis (HD). (1)
Limitation of Use
Korsuva has not been studied in patients on peritoneal dialysis and is not recommended for use in this population. (1)
DOSAGE AND ADMINISTRATION
- Recommended dosage is 0.5 mcg/kg. (2.1)
- Administer by intravenous bolus injection into the venous line of the dialysis circuit at the end of each HD treatment. (2.1)
- Do not mix or dilute KORSUVA prior to administration. (2.2)
- Administer within 4 hours of syringe preparation. (2.3)
- See full prescribing information for additional recommendations on preparation and administration of KORSUVA. (2.2, 2.3)
DOSAGE FORMS AND STRENGTHS
Injection: 65 mcg /1.3 mL (50 mcg/mL) of difelikefalin (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Dizziness, Somnolence, Mental Status Changes, and Gait Disturbances: Dizziness, somnolence, mental status changes, and gait disturbances, including falls, have occurred. Centrally-acting depressant medications, sedating antihistamines, and opioid analgesics should be used with caution during treatment with KORSUVA. (5.1)
- Risk of Driving and Operating Machinery: May impair mental or physical abilities. Advise patients not to drive or operate dangerous machinery until the effect of KORSUVA on a patient's ability to drive or operate machinery is known. (5.2)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥2% and ≥1% higher than placebo) were diarrhea, dizziness, nausea, gait disturbances, including falls, hyperkalemia, headache, somnolence, and mental status change. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Vifor (International) Inc. at 1-844-835-8277 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Severe Hepatic Impairment: Not recommended in patients with severe hepatic impairment. (8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
2.2 Preparation Instructions
2.3 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Dizziness, Somnolence, Mental Status Changes, and Gait Disturbances
5.2 Risk of Driving and Operating Machinery
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
- The recommended dosage of KORSUVA is 0.5 mcg/kg administered by intravenous bolus injection into the venous line of the dialysis circuit at the end of each HD treatment [see Dosage and Administration (2.3)].
- If a regularly scheduled HD treatment is missed, resume KORSUVA at the end of the next HD treatment.
2.2 Preparation Instructions
- Do not mix or dilute KORSUVA prior to administration.
- Inspect KORSUVA for particulate matter and discoloration prior to administration. The solution should be clear and colorless. Do not use KORSUVA vials if particulate matter or discoloration is observed.
- KORSUVA is supplied in a single-dose vial. Discard any unused product.
- Injection volume to be administered is determined by patient's target dry body weight in kilograms (one patient may use less than the full contents of the vial or use more than one vial). See Table 1.
Table 1. KORSUVA Injection Volumes Based on Target Dry Body Weight Target Dry Body Weight Range (kg) Injection Volume (mL)* - *
- Total Injection Volume (mL) = Patient Target Dry Body Weight (kg) x 0.01, rounded to the nearest tenth (0.1 mL). For patient target dry body weight outside of the ranges in Table 1, use this formula.
36 – 44 0.4 45 – 54 0.5 55 – 64 0.6 65 – 74 0.7 75 – 84 0.8 85 – 94 0.9 95 – 104 1 105 – 114 1.1 115 – 124 1.2 125 – 134 1.3 135 – 144 1.4 145 – 154 1.5 155 – 164 1.6 165 – 174 1.7 175 – 184 1.8 185 – 194 1.9 195 – 204 2 2.3 Administration Instructions
- KORSUVA is removed by the dialyzer membrane and must be administered after blood is no longer circulating through the dialyzer.
- Administer KORSUVA by intravenous bolus injection into the venous line of the dialysis circuit at the end of each HD session.
- The dose may be given either during or after rinse back of the dialysis circuit.
- If the dose is given after rinse back, administer KORSUVA into the venous line followed by at least 10 mL of normal saline flush.
- If the dose is given during rinse back, no additional normal saline is needed to flush the line.
- The dose must be administered within 4 hours of the syringe preparation. Discard any unused product.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Dizziness, Somnolence, Mental Status Changes, and Gait Disturbances
Dizziness, somnolence, mental status changes, and gait disturbances, including falls, have occurred in patients taking KORSUVA and may subside over time with continued treatment [see Adverse Reactions (6.1)]. In Trial 1 and Trial 2, 17.0% of patients randomized to receive KORSUVA reported at least one of these adverse reactions, compared to 12.0% of patients who received placebo. The incidence of somnolence was higher in KORSUVA-treated subjects 65 years of age and older (7.0%) than in KORSUVA-treated subjects less than 65 years of age (2.8%). Concomitant use of centrally-acting depressant medications, sedating antihistamines and opioid analgesics may increase the likelihood of these adverse reactions and should be used with caution during treatment with KORSUVA.
5.2 Risk of Driving and Operating Machinery
Dizziness, somnolence, and mental status changes have occurred in patients taking KORSUVA. KORSUVA may impair the mental or physical abilities needed to perform potentially hazardous activities such as driving a car and operating machinery. Advise patients not to drive or operate dangerous machinery until the effect of KORSUVA on a patient's ability to drive or operate machinery is known.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Dizziness, Somnolence, Mental Status Changes, and Gait Disturbances [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 1306 subjects undergoing HD who had moderate-to-severe pruritus were treated with KORSUVA in placebo-controlled and uncontrolled Phase 3 clinical trials. Of these, 711 were treated for at least 6 months and 400 were treated for at least one year.
Two placebo-controlled Phase 3 trials (Trial 1 and Trial 2), in subjects undergoing HD who had moderate-to-severe pruritus were pooled to evaluate the safety of KORSUVA in comparison to placebo up to 12 weeks. In total, 848 subjects were evaluated (424 in KORSUVA group and 424 in placebo group). The mean age of the subjects was 59 years (range 22 to 88 years), and 59% of the subjects were male. Of the total subjects, 61% were White, 29% were Black or African American, and 5% were Asian.
Table 2 summarizes the adverse reactions that occurred at a rate of ≥2% in the KORSUVA group and ≥1% higher than that of the placebo group during the 12-week placebo-controlled period of Trials 1 and 2. The percentage of subjects who discontinued treatment due to any adverse reaction was 2.6% for subjects taking KORSUVA and 0.7% for subjects taking placebo. The most common adverse reactions (≥0.5% of subjects) leading to discontinuation were dizziness (0.9% for KORSUVA and 0.2% for placebo), mental status change (0.7% and 0.2%, respectively), nausea (0.5% and 0%, respectively), and headache (0.5% and 0%, respectively). The percentage of subjects who developed serious adverse reactions was 4.5% in the KORSUVA group and 2.8% in the placebo group.
Table 2: Adverse Reactions in ≥ 2% of KORSUVA-Treated Subjects with Moderate-to-Severe CKD-aP Undergoing HD and ≥ 1% Higher Than Placebo in Trials 1 and 2 Adverse Reactions Placebo
(N=424)
n (%)KORSUVA
(N=424)
n (%)Diarrhea 24 (5.7) 38 (9.0) Dizziness 16 (3.8) 29 (6.8) Nausea 19 (4.5) 28 (6.6) Gait Disturbances* 23 (5.4) 28 (6.6) Hyperkalemia 15 (3.5) 20 (4.7) Headache 11 (2.6) 19 (4.5) Somnolence 10 (2.4) 18 (4.2) Mental Status Change† 6 (1.4) 14 (3.3) Description of Selected Adverse Reactions
Gait Disturbances, including Falls
Gait disturbances, including falls, were reported in 6.6% of subjects receiving KORSUVA compared to 5.4% of subjects who received placebo. Falls were reported as serious adverse reactions in < 1% of subjects receiving KORSUVA and placebo, with one subject discontinuing KORSUVA due to gait disturbance.
Dizziness
Dizziness was reported in 6.8% of subjects randomized to KORSUVA compared to 3.8% of subjects who received placebo. Dizziness occurred within the first 3 weeks of treatment and was generally transient. Dizziness was serious in 0.2% of KORSUVA-treated subjects compared to 0% of subjects who received placebo and led to discontinuation in 0.9% of KORSUVA-treated subjects compared to 0.2% of subjects who received placebo.
Somnolence
Somnolence was reported in 4.2% of subjects randomized to receive KORSUVA compared to 2.4% of subjects who received placebo. Somnolence occurred within the first 3 weeks of treatment and tended to subside with continued dosing. Somnolence was serious in 0.2% of KORSUVA-treated subjects compared to 0% of subjects who received placebo. There were no subjects who discontinued KORSUVA due to an adverse reaction of somnolence.
Mental Status Change
Mental status change (including confusional state) was reported in 3.3% of subjects randomized to receive KORSUVA compared to 1.4% of subjects who received placebo. Most events tended to subside with continued dosing. Mental status change adverse reactions were serious in 1.4% of KORSUVA-treated subjects compared to 0.5% of subjects who received placebo and led to discontinuation in 0.7% of KORSUVA-treated subjects compared to 0.2% of subjects who received placebo.
Hyperkalemia
Hyperkalemia was found in 4.7% of subjects who received KORSUVA compared to 3.5% of subjects who received placebo. The incidence of hyperkalemia was higher in subjects who took concomitant opioids regardless of treatment and was almost doubled in the KORSUVA group (11.7%) compared to the placebo group (6.2%). The clinical relevance of this is unknown.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited human data on use of KORSUVA in pregnant women are not sufficient to evaluate a drug-associated risk for major birth defects or miscarriage. In animal reproduction studies, intravenous injection of difelikefalin to pregnant rats and rabbits during the period of organogenesis at doses 711 and 10 times the maximum recommended human dose (MRHD), respectively, resulted in no adverse effects in either rats or rabbits (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryofetal development study, difelikefalin was administered by intravenous injection to pregnant rats at doses of 0.25, 2.5, and 25 mg/kg/day during the period of organogenesis. Difelikefalin was not associated with embryofetal lethality or fetal malformations. Difelikefalin increased the incidences of skeletal variations (wavy ribs and incompletely ossified ribs) at the dose of 25 mg/kg/day (711 times the MRHD based on AUC comparison).
In an embryofetal development study, difelikefalin was administered by intravenous injection to pregnant rabbits at doses of 0.025, 0.05, and 0.1 mg/kg/day during the period of organogenesis. Maternal toxicity evidenced by decreased maternal body weight gain was noted in all dose groups. Difelikefalin was not associated with embryofetal lethality or fetal malformations at doses up to 0.1 mg/kg/day (10 times the MRHD based on AUC comparison).
In a prenatal and postnatal development study, difelikefalin was administered by intravenous injection to pregnant rats at doses of 0.6, 2.5, and 10 mg/kg/day beginning on gestation day 7 and continuing through lactation day 20. Persisting effects on decreased maternal body weight and/or maternal body weight gain as well as food consumption were noted at doses greater than or equal to 2.5 mg/kg/day (68 times the MRHD based on AUC comparison). No maternal effects were observed at 0.6 mg/kg/day (14 times the MRHD based on AUC comparison). No difelikefalin-related effects on postnatal developmental, neurobehavioral, or reproductive performance of offspring were noted at doses up to 10 mg/kg/day (282 times the MRHD based on AUC comparison).
8.2 Lactation
Risk Summary
There are no data regarding the presence of KORSUVA in human milk or effects on the breastfed infant or on milk production.
Studies in rats showed difelikefalin was transferred into the milk in lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KORSUVA and any potential adverse effects on the breastfed child from KORSUVA or from the underlying maternal condition.
Data
Animal Data
Difelikefalin was administered to lactating rats by intravenous injection at doses of 0.6, 2.5, or 10 mg/kg/day from gestation day 7 through lactation day 14. Difelikefalin was detected in the milk of the lactating rats with the concentration ratio for milk:plasma of 0.04 to 0.05 across the doses. There was no measurable difelikefalin in the plasma of nursing pups.
8.4 Pediatric Use
The safety and effectiveness of KORSUVA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 848 subjects in the placebo-controlled studies who received KORSUVA, 278 subjects (32.8%) were 65 years of age and older and 98 subjects (11.6%) were 75 years of age and older. No overall differences in safety or effectiveness of KORSUVA have been observed between patients 65 years of age and older and younger adult subjects, with the exception of the incidence of somnolence which was higher in KORSUVA-treated subjects 65 years of age and older (7.0%) than in KORSUVA-treated subjects less than 65 years of age (2.8%), and was comparable in both placebo age groups (3.0% and 2.1%, respectively). No differences in plasma concentrations of KORSUVA were observed between subjects 65 years of age and older and younger adult subjects [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
The influence of mild-to-moderate hepatic impairment on the pharmacokinetics of KORSUVA was evaluated in a population pharmacokinetic analysis which concluded that no KORSUVA dosage adjustments are needed in these populations [see Clinical Pharmacology (12.3)]. The influence of severe hepatic impairment on the pharmacokinetics of KORSUVA in subjects undergoing HD has not been evaluated; therefore, use of KORSUVA in this population is not recommended.
-
10 OVERDOSAGE
Single doses of KORSUVA up to 12 times and multiple doses of KORSUVA up to 5 times the recommended dosage of 0.5 mcg/kg were administered in clinical studies in subjects undergoing HD. A dose-dependent increase in adverse reactions, including dizziness, somnolence, mental status changes, paresthesia, fatigue, hypertension, and vomiting, were observed.
In the event of overdosage, provide the appropriate medical attention based on patient's clinical status. Difelikefalin is primarily eliminated by the kidneys with a low plasma protein binding of approximately 23% to 28% in dialysis patients. Hemodialysis for 4 hours using a high-flux dialyzer effectively cleared approximately 70% to 80% of difelikefalin from plasma, and difelikefalin was not detectable in plasma at the end of the second of two dialysis cycles. [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
KORSUVA (difelikefalin) is a kappa opioid receptor agonist. Difelikefalin is a synthetic peptide with a single stereoisomer and is present as an acetate salt. Difelikefalin acetate is a white to off-white powder with a molecular formula of C36H53N7O6∙xAcOH (1.0≤ × ≤2.0) and a molecular weight of 679.4 g/mol (mono- isotopic; free base). It is soluble in water. The chemical name of difelikefalin acetate is 4-amino-1-(D-phenylalanyl-D-phenylalanyl-D-leucyl-D-lysyl)piperidine-4-carboxylic acid, acetate salt.
The chemical structure is:

KORSUVA (difelikefalin) injection is supplied in a single-dose vial containing 65 mcg/1.3 mL (50 mcg/mL) of difelikefalin as a sterile, preservative-free, clear and colorless solution for intravenous injection.
KORSUVA is formulated as an isotonic 40 mM acetate buffer solution with an osmolality of 250 to 350 mOsm and a pH of 4.5.
Each milliliter of KORSUVA injection contains 50 mcg of difelikefalin (equivalent to an average of 58.3 mcg of difelikefalin acetate), 1.3 mg of acetic acid, 2.5 mg of sodium acetate trihydrate, 7.2 mg of sodium chloride (to adjust tonicity), and water for injection.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
KORSUVA is a kappa opioid receptor (KOR) agonist. The relevance of KOR activation to therapeutic effectiveness is not known.
12.2 Pharmacodynamics
Difelikefalin exposure-response relationships and the time course of pharmacodynamic response are unknown.
12.3 Pharmacokinetics
The pharmacokinetics of difelikefalin is dose proportional over a single dosage range from 1 to 3 mcg/kg (2 to 6 times the recommended dosage) and multiple intravenous dosage range from 0.5 to 2.5 mcg/kg (1 to 5 times the recommended dosage) in chronic kidney disease patients undergoing HD. Steady-state was reached after the second administered dosage and the mean accumulation ratio was up to 1.6.
Distribution
The mean volume of distribution of difelikefalin is approximately 238 mL/kg. Difelikefalin binding to human plasma protein in dialysis patients is 23% to 28%.
Elimination
The half-life of difelikefalin in HD subjects prior to dialysis ranged between 23 and 31 hours. Following administration of radiolabeled difelikefalin, >99% of circulating radioactivity was present in plasma as parent. Hemodialysis reduced the difelikefalin plasma concentrations by 70% to 80% and difelikefalin was not detectable in plasma after 2 dialysis cycles.
Specific Populations
No clinically significant differences in the pharmacokinetics of difelikefalin were observed based on age (25 to 80 years of age), sex, race/ethnicity, or mild-to-moderate hepatic impairment. The effect of severe hepatic impairment on the pharmacokinetics of difelikefalin is unknown [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
No clinical studies evaluating the drug interaction potential of difelikefalin were conducted.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Difelikefalin did not inhibit CYP enzymes (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A, or CYP2D6), or induce CYP enzymes (CYP1A2, CYP2B6, or CYP3A) and is not a substrate of CYP450 enzymes (CYP1A2, CYP2C19, CYP2C8, CYP2C9, CYP2D6 or CYP3A).
Uridine diphosphate (UDP)-glucuronosyl transferase (UGT) Enzymes: Difelikefalin is not an inhibitor of UGT1A3, UGT1A9, or UGT2B7.
Transporter Systems: Difelikefalin did not inhibit BCRP, Pgp, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, or, MATE2-K human transporters and is not a substrate of OAT1, OAT2, OAT3, OATP1A2, OCT2, OCT3, LAT1, PEPT1, PEPT2, ASBT, BSEP, MRP2, OATP1B1, OATP1B3, OATP2B1, OCT1, OCTN1, OCTN2, Pgp, BCRP, OSTα/β, MATE1, or, MATE2-K.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year carcinogenicity study in rats, difelikefalin was not carcinogenic when administered via subcutaneous injection at doses up to 1.0 mg/kg/day (597 times the MRHD based on AUC comparison). Difelikefalin was not carcinogenic in a 6-month carcinogenicity study in transgenic rasH2 mice at subcutaneous doses up to 30 mg/kg/day.
Difelikefalin was negative for genotoxicity in a bacterial reverse mutation assay, an in vitro mammalian chromosomal aberration assay, and an in vivo mouse micronucleus assay.
Difelikefalin administered via intravenous injection caused a significant decrease in the number of estrous cycles per 14 days (i.e., prolonged diestrus) in female rats at doses greater than or equal to 2.5 mg/kg/day (56 times the MRHD based on AUC comparison). Difelikefalin had no effects on mating index, fertility index, or any ovarian or uterine parameters in female rats at doses up to 25 mg/kg/day (635 times the MRHD based on AUC comparison). Difelikefalin did not impair male fertility at doses up to 25 mg/kg/day (971 times the MRHD based on AUC comparison).
-
14 CLINICAL STUDIES
The efficacy of KORSUVA was evaluated in two randomized, multicenter, double-blind, placebo-controlled trials (Trial 1 [NCT03422653] and Trial 2 [NCT03636269]) that enrolled a total of 851 subjects 18 years of age and older undergoing HD who had moderate-to-severe pruritus. In both trials, subjects received intravenous bolus injections of KORSUVA 0.5 mcg per kilogram of dry body weight into the venous line of the hemodialysis circuit at the end of each hemodialysis session or placebo three times per week for 12 weeks. In both trials, a 7-day run-in period prior to randomization was used to confirm that each subject had moderate-to-severe pruritus and to establish a baseline itch intensity, as measured by the patient-reported daily 24-hour Worst Itching Intensity Numerical Rating Scale (WI-NRS) scores (0 "no itch" to 10 "worst itch imaginable").
The mean (SD) baseline WI-NRS score was 7.1 (1.5) in Trial 1 and 7.2 (1.4) in Trial 2. At baseline in Trial 1, 61% of subjects were male, 49% were White, 42% were Black or African American, the mean age was 57 years (range 22 to 88 years), and 40% of subjects were using prior anti-pruritic medications (including sedating antihistamines) and continued the use throughout the trial. At baseline in Trial 2, 58% of subjects were male, 70% were White, 19% were Black or African American, the mean age was 60 years (range 23 to 90 years), and 36% of subjects were using prior anti-pruritic medications (including sedating antihistamines) and continued the use throughout the trial.
In each trial, efficacy was assessed based on the proportion of subjects achieving a 4-point or greater improvement (reduction) from baseline in the weekly mean of the daily 24-hour WI-NRS score at Week 12.
The results of the KORSUVA trials (Trials 1 and 2) are presented in Table 3 and Figure 1.
Table 3. Efficacy Results of Subjects with Moderate-to-Severe CKD-aP Undergoing HD at Week 12 (Trials 1 & 2) Trial 1 Trial 2 KORSUVA
0.5 mcg/kg
3 times weekly
N=189Placebo
N=189KORSUVA
0.5 mcg/kg
3 times weekly
N=237Placebo
N=236Percentage of subjects with ≥4-point improvement from baseline in WI-NRS score 40% 21% 37% 26% Difference from Placebo (95% CI) 19% (9%, 28%) 12% (3%, 20%) Figure 1: Percentage of Subjects with Moderate-to-Severe CKD-aP Undergoing HD with a ≥4-point Improvement from Baseline on the WI-NRS in Trial 1 and Trial 2
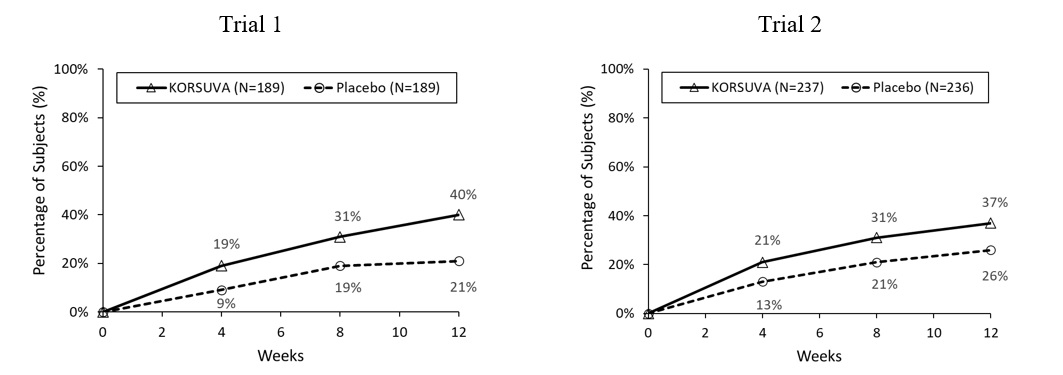
Itch reduction was seen by Week 4 and sustained through Week 12.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
KORSUVA (difelikefalin) injection is supplied as a sterile, clear and colorless solution in 1.3 mL single-dose, glass vials:
NDC 59353-065-01: 65 mcg/1.3 mL (50 mcg/mL) single-dose vial
NDC 59353-065-12: Carton containing 12 vials
Storage and Handling
Store vials at 20°C to 25°C (68°F to 77°F), excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Do not freeze.
KORSUVA injection must be administered within 4 hours of syringe preparation; prepared syringes can be stored at ambient temperature 20°C to 25°C (68°F to 77°F) until dosing [see Dosage and Administration (2.2, 2.3)] .
KORSUVA injection is supplied in a single-dose vial. Any unused drug remaining after injection must be discarded.
-
17 PATIENT COUNSELING INFORMATION
Dizziness, Somnolence, Mental Status Changes, and Gait Disturbances
Inform patients that dizziness, somnolence, mental status changes, and gait disturbances, including falls, can occur during treatment with KORSUVA [see Adverse Reactions (6.1)]. Somnolence is more likely to happen to patients who are age 65 years or older.
Inform patients that concomitant treatment with centrally-acting depressants, sedating antihistamines and opioid analgesics may increase the likelihood of these adverse reactions and these medications should be used with caution during treatment with KORSUVA [see Warnings and Precautions (5.1)].
Driving or Operating Machinery
Inform patients that KORSUVA may impair the ability to perform potentially hazardous activities such as driving a car or operating heavy machinery. Advise patients not to drive or operate machinery until they know how they will react to KORSUVA [see Warnings and Precautions (5.2)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 1.3 mL Vial Label
NDC 59353-065-01
Store at 20°C to 25°C (68°F to 77°F)
Rx onlyKORSUVA™
(difelikefalin) Injection65 mcg/1.3 mL
(50 mcg/mL)For Intravenous Use Only
1.3 mL Single-dose Vial. Discard unused portion
Mfd. for Vifor (International) Inc., Rechenstrasse 37,
9014 St. Gallen, Switzerland. Made in France.3102942-01 48307/16/25

-
PRINCIPAL DISPLAY PANEL - 1.3 mL Tray Carton
NDC 59353-065-12
Rx onlyKORSUVA™
(difelikefalin) Injection65 mcg/1.3 mL
(50 mcg/mL)For Intravenous Use after Hemodialysis
Single-dose vial. Discard unused portion.
Store at 20°C to 25°C (68°F to 77°F), excursions
permitted to 15°C to 30°C (59°F to 86°F) [see USP
Controlled Room Temperature]. Do not freeze.
Sterile, preservative-free solution.12 x 1.3 mL
Single-dose Vials
-
INGREDIENTS AND APPEARANCE
KORSUVA
difelikefalin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59353-065 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DIFELIKEFALIN ACETATE (UNII: 0P70AR5BYB) (DIFELIKEFALIN - UNII:NA1U919MRO) DIFELIKEFALIN 50 ug in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59353-065-12 12 in 1 TRAY 11/01/2021 1 NDC:59353-065-01 1.3 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214916 11/01/2021 Labeler - Vifor (International), Inc. (482603065)