Label: CEFOTAN- cefotetan disodium injection, powder, for solution
CEFOTAN- cefotetan disodium injection, powder, for solution


-
Contains inactivated NDC Code(s)
NDC Code(s): 52565-052-10, 52565-053-10 - Packager: Teligent Pharma, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated November 30, 2018
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
CEFOTAN® (cefotetan for Injection, USP), as cefotetan disodium, is a sterile, semisynthetic, broad-spectrum, beta-lactamase resistant, cephalosporin (cephamycin) antibacterial for parenteral administration. It is the disodium salt of [6R-(6α,7α)]-7-[[[4-(2-amino-1-carboxy-2-oxoethylidene)-1,3-dithietan-2-yl]carbonyl]amino]-7-methoxy-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid. Structural formula:
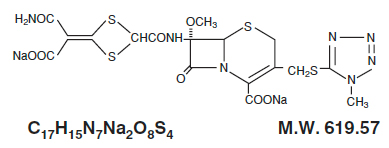
CEFOTAN® (cefotetan for Injection, USP) is supplied in vials containing 80 mg (3.5 mEq) of sodium per gram of cefotetan activity. It is a white to pale yellow powder which is very soluble in water. Reconstituted solutions of CEFOTAN® (cefotetan for Injection, USP) are intended for intravenous and intramuscular administration. The solution varies from colorless to yellow depending on the concentration. The pH of freshly reconstituted solutions is usually between 4.5 to 6.5.
CEFOTAN® (cefotetan for Injection, USP) is available in two vial strengths. Each 1 gram vial contains cefotetan disodium equivalent to 1 gram cefotetan activity. Each 2 gram vial contains cefotetan disodium equivalent to 2 grams cefotetan activity.
-
CLINICAL PHARMACOLOGY
High plasma levels of cefotetan are attained after intravenous and intramuscular administration of single doses to normal volunteers.
PLASMA CONCENTRATIONS AFTER 1 GRAM INTRAVENOUSa OR INTRAMUSCULAR DOSE
Mean Plasma Concentration (mcg/mL)
Time After Injection
Route
15 min
30 min
1 h
2 h
4 h
8 h
12 h
IV
92
158
103
72
42
18
9
IM
34
56
71
68
47
20
9
a 30-minute infusion
PLASMA CONCENTRATIONS AFTER 2 GRAM INTRAVENOUSa OR INTRAMUSCULAR DOSE
Mean Plasma Concentration (mcg/mL)
Time After Injection
Route
5 min
10 min
1 h
3 h
5 h
9 h
12 h
IV
237
223
135
74
48
22
12b
IM
—
20
75
91
69
33
19
a Injected over 3 minutes
b Concentrations estimated from regression line
Repeated administration of CEFOTAN® does not result in accumulation of the drug in normal subjects.
Distribution
Cefotetan is 88% plasma protein bound.
Therapeutic concentrations of cefotetan are achieved in many body tissues and fluids including:
skin
ureter
muscle
bladder
fat
maxillary sinus mucosa
myometrium
tonsil
endometrium
bile
cervix
peritoneal fluid
ovary
umbilical cord serum
kidney
amniotic fluid
Metabolism and Excretion
The plasma elimination half-life of cefotetan is 3 to 4.6 hours after either intravenous or intramuscular administration.
No active metabolites of cefotetan have been detected; however, small amounts (less than 7%) of cefotetan in plasma and urine may be converted to its tautomer, which has antimicrobial activity similar to the parent drug.
In normal patients, from 51% to 81% of an administered dose of CEFOTAN® is excreted unchanged by the kidneys over a 24-hour period, which results in high and prolonged urinary concentrations. Following intravenous doses of 1 gram and 2 grams, urinary concentrations are highest during the first hour and reach concentrations of approximately 1700 and 3500 mcg/mL, respectively.
Specific Populations
Patients with Renal Impairment
In volunteers with reduced renal function, the plasma half-life of cefotetan is prolonged. The mean terminal half-life increases with declining renal function, from approximately 4 hours in volunteers with normal renal function to about 10 hours in those with moderate renal impairment. There is a linear correlation between the systemic clearance of cefotetan and creatinine clearance. When renal function is impaired, a reduced dosing schedule based on creatinine clearance must be used (see DOSAGE AND ADMINISTRATION).
Geriatric patients
In pharmacokinetic studies of eight elderly patients (greater than 65 years) with normal renal function and six healthy volunteers (aged 25 to 28 years), mean±SD) Total Body Clearance (1.8±0.1 vs. 1.8±0.3 L/h) and mean±SD Volume of Distribution (10.4±1.2 vs. 10.3±1.6 L) were similar following administration of a one gram intravenous bolus dose.Microbiology
Mechanism of Action
Cefotetan is a bactericidal agent that acts by inhibition of bacterial cell wall synthesis. Cefotetan has activity in the presence of some beta-lactamases, both penicillinases and cephalosporinases, of gram-negative and gram-positive bacteria.
Resistance
Resistance to cefotetan is primarily through hydrolysis by some beta-lactamases, alteration of penicillin –binding proteins (PBPs) and decreased permeability.
Antimicrobial Activity
Cefotetan has been shown to be active against most isolates of the following microorganisms both in vitro and in clinical infections (see INDICATIONS AND USAGE).
Gram-Negative Bacteria
Escherichia coli
Haemophilus influenza
Klebsiella species (including K. pneumoniae)
Morganella morganii
Neisseria gonorrhoeae
Proteus mirabilis
Proteus vulgaris
Providencia rettgeri
Serratia marcescens
Gram-Positive Bacteria
Staphylococcus aureus (methicillin-susceptible isolates only)
Staphylococcus epidermidis (methicillin-susceptible isolates only)
Streptococcus agalactiae
Streptococcus pneumoniae
Streptococcus pyogenes
Streptococcus species
Anaerobes
Prevotella bivia
Prevotella disiens
Bacteroides fragilis
Prevotella melaninogenica
Bacteroides vulgatus
Fusobacterium species
Clostridium species
Peptococcus niger
Peptostreptococcus species
The following in vitro data are available but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for cefotetan against isolates of similar genus or organism group. However, the efficacy of cefotetan in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-Negative Bacteria
Citrobacter species (including C. koseri and C. freundii )
Moraxella catarrhalis
Salmonella species
Serratia species
Shigella species
Yersinia enterocolitica
Anaerobes
Porphyromonas asaccharolytica
Prevotella oralis
Bacteroides splanchnicus
Propionibacterium species
Veillonella species
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
-
INDICATIONS AND USAGE
To reduce the development of drug-resistant bacteria and maintain the effectiveness of CEFOTAN® and other antibacterial drugs, CEFOTAN® should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
Treatment
CEFOTAN® (cefotetan for Injection, USP) is indicated for the therapeutic treatment of the following infections when caused by susceptible strains of the designated organisms:
Urinary Tract Infections caused by E. coli, Klebsiella spp (including K. pneumoniae ), Proteus mirabilis, Proteus vulgaris , Providencia rettgeri , and Morganella morganii ).
Lower Respiratory Tract Infections caused by Streptococcus pneumoniae , Staphylococcus aureus(methicillin-susceptible), Haemophilus influenzae, Klebsiella species (including K. pneumoniae ), E. coli , Proteus mirabilis , and Serratia marcescens.*
Skin and Skin Structure Infections due to Staphylococcus aureus (methicillin-susceptible), Staphylococcus epidermidis (methicillin susceptible), Streptococcus pyogenes, Streptococcus species, Escherichia coli, Klebsiella pneumoniae, Peptococcus niger*, Peptostreptococcus species.
Gynecologic Infections caused by Staphylococcus aureus (methicillin susceptible), Staphylococcus epidermidis (methicillin susceptible, Streptococcus species, Streptococcus agalactiae, E. coli, Proteus mirabilis, Neisseria gonorrhoeae, Bacteroides fragilis, Prevotella melaninogenica Bacteroides vulgatus, Fusobacterium species*, and gram- positive anaerobic cocci (including Peptococcus niger and Peptostreptococcus species).
Cefotetan, like other cephalosporins, has no activity against Chlamydia trachomatis. Therefore, when cephalosporins are used in the treatment of pelvic inflammatory disease, and C. trachomatis is one of the suspected pathogens, appropriate antichlamydial coverage should be added.
Intra-abdominal lnfections caused by E. coli, Klebsiella species (including K. pneumoniae), Streptococcus species, Bacteroides fragilis, Prevotella melaninogenica, Bacteroides vulgatus and Clostridium species (other than Clostridium difficile [see WARNINGS])*.
Bone and Joint Infections caused by Staphylococcus aureus (methicillin susceptible)*.
* Efficacy for this organism in this organ system was studied in fewer than ten infections in clinical studies
Specimens for bacteriological examination should be obtained in order to isolate and identify causative organisms and to determine their susceptibilities to cefotetan. Therapy may be instituted before results of susceptibility studies are known; however, once these results become available, the antibiotic treatment should be adjusted accordingly.
In cases of confirmed or suspected gram-positive or gram-negative sepsis or in patients with other serious infections in which the causative organism has not been identified, it is possible to use CEFOTAN® concomitantly with an aminoglycoside. Cefotetan combinations with aminoglycosides have been shown to be synergistic in vitro against many Enterobacteriaceae and also some other gram- negative bacteria. The dosage recommended in the labeling of both antibiotics may be given and depends on the severity of the infection and the patient's condition.
NOTE: Increases in serum creatinine have occurred when CEFOTAN® was given alone. If CEFOTAN® and an aminoglycoside are used concomitantly, renal function should be carefully monitored, because nephrotoxicity may be potentiated.
Prophylaxis
The preoperative administration of CEFOTAN® may reduce the incidence of certain postoperative infections in patients undergoing surgical procedures that are classified as clean contaminated or potentially contaminated (e.g., cesarean section, abdominal or vaginal hysterectomy, transurethral surgery, biliary tract surgery, and gastrointestinal surgery).
If there are signs and symptoms of infection, specimens for culture should be obtained for identification of the causative organism so that appropriate therapeutic measures may be initiated.
- CONTRAINDICATIONS
-
WARNINGS
BEFORE THERAPY WITH CEFOTAN® IS INSTITUTED, CAREFUL INQUIRY SHOULD BE MADE TO DETERMINE WHETHER THE PATIENT HAS HAD PREVIOUS HYPERSENSITIVITY REACTIONS TO CEFOTETAN DISODIUM, CEPHALOSPORINS, PENICILLINS, OR OTHER DRUGS. IF THIS PRODUCT IS TO BE GIVEN TO PENICILLIN-SENSITIVE PATIENTS, CAUTION SHOULD BE EXERCISED BECAUSE CROSS-HYPERSENSITIVITY AMONG BETA-LACTAM ANTIBIOTICS HAS BEEN CLEARLY DOCUMENTED AND MAY OCCUR IN UP TO 10% OF PATIENTS WITH A HISTORY OF PENICILLIN ALLERGY. IF AN ALLERGIC REACTION TO CEFOTAN® OCCURS, DISCONTINUE THE DRUG. SERIOUS ACUTE HYPERSENSITIVITY REACTIONS MAY REQUIRE TREATMENT WITH EPINEPHRINE AND OTHER EMERGENCY MEASURES, INCLUDING OXYGEN, INTRAVENOUS FLUIDS, INTRAVENOUS ANTIHISTAMINES, CORTICOSTEROIDS, PRESSOR AMINES, AND AIRWAY MANAGEMENT, AS CLINICALLY INDICATED.
AN IMMUNE MEDIATED HEMOLYTIC ANEMIA HAS BEEN OBSERVED IN PATIENTS RECEIVING CEPHALOSPORIN CLASS ANTIBIOTICS. SEVERE CASES OF HEMOLYTIC ANEMIA, INCLUDING FATALITIES, HAVE BEEN REPORTED IN ASSOCIATION WITH THE ADMINISTRATION OF CEFOTETAN. SUCH REPORTS ARE UNCOMMON. THERE APPEARS TO BE AN INCREASED RISK OF DEVELOPING HEMOLYTIC ANEMIA ON CEFOTETAN RELATIVE TO OTHER CEPHALOSPORINS OF AT LEAST 3 FOLD. IF A PATIENT DEVELOPS ANEMIA ANYTIME WITHIN 2 TO 3 WEEKS SUBSEQUENT TO THE ADMINISTRATION OF CEFOTETAN, THE DIAGNOSIS OF A CEPHALOSPORIN ASSOCIATED ANEMIA SHOULD BE CONSIDERED AND THE DRUG STOPPED UNTIL THE ETIOLOGY IS DETERMINED WITH CERTAINTY. BLOOD TRANSFUSIONS MAY BE CONSIDERED AS NEEDED (see CONTRAINDICATIONS).
PATIENTS WHO RECEIVE COURSES OF CEFOTETAN FOR TREATMENT OR PROPHYLAXIS OF INFECTIONS SHOULD HAVE PERIODIC MONITORING FOR SIGNS AND SYMPTOMS OF HEMOLYTIC ANEMIA INCLUDING A MEASUREMENT OF HEMATOLOGICAL PARAMETERS WHERE APPROPRIATE.
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including cefotetan, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
In common with many other broad-spectrum antibiotics, CEFOTAN® may be associated with a fall in prothrombin activity and, possibly, subsequent bleeding. Those at increased risk include patients with renal or hepatobiliary impairment or poor nutritional state, the elderly, and patients with cancer. Prothrombin time should be monitored and exogenous vitamin K administered as indicated.
-
PRECAUTIONS
General
Prescribing CEFOTAN® in the absence of proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
As with other broad-spectrum antibiotics, prolonged use of CEFOTAN® may result in overgrowth of nonsusceptible organisms. Careful observation of the patient is essential. If superinfection does occur during therapy, appropriate measures should be taken.
CEFOTAN® should be used with caution in individuals with a history of gastrointestinal disease, particularly colitis.
Information for Patients
Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
Patients should be counseled that antibacterial drugs, including CEFOTAN®, should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When CEFOTAN is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by CEFOTAN® (cefotetan for Injection, USP) or other antibacterial drugs in the future.
As with some other cephalosporins, a disulfiram-like reaction characterized by flushing, sweating, headache, and tachycardia may occur when alcohol (beer, wine, etc.) is ingested within 72 hours after CEFOTAN® (cefotetan for Injection, USP) administration. Patients should be cautioned about the ingestion of alcoholic beverages following the administration of CEFOTAN®.
Drug Interactions
Increases in serum creatinine have occurred when CEFOTAN® was given alone. If CEFOTAN® and an aminoglycoside are used concomitantly, renal function should be carefully monitored, because nephrotoxicity may be potentiated.
Drug/Laboratory Test Interactions
The administration of CEFOTAN® may result in a false positive reaction for glucose in the urine using Clinitest®‡, Benedict's solution, or Fehling's solution. It is recommended that glucose tests based on enzymatic glucose oxidase be used.
As with other cephalosporins, high concentrations of cefotetan may interfere with measurement of serum and urine creatinine levels by Jaffé reaction and produce false increases in the levels of creatinine reported.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Although long-term studies in animals have not been performed to evaluate carcinogenic potential, no mutagenic potential of cefotetan was found in standard laboratory tests.
Cefotetan has adverse affects on the testes of prepubertal rats. Subcutaneous administration of 500 mg/kg/day (approximately 0.8 times the maximum adult human dose on a body surface area basis) on days 6 to 35 of life (thought to be developmentally analogous to late childhood and prepuberty in humans) resulted in reduced testicular weight and seminiferous tubule degeneration in 10 of 10 animals. Affected cells included spermatogonia and spermatocytes; Sertoli and Leydig cells were unaffected. Incidence and severity of lesions were dose-dependant; at 120 mg/kg/day (0.2 times the maximum human dose on a body surface area basis) only 1 of 10 treated animals was affected, and the degree of degeneration was mild.
Similar lesions have been observed in experiments of comparable design with other methylthiotetrazole-containing antibiotics and impaired fertility has been reported, particularly at high dose levels. No testicular effects were observed in 7-week-old rats treated with up to 1000 mg/kg/day subcutaneous for 5 weeks, or in infant dogs (3 weeks old) that received up to 300 mg/kg/day intravenous for 5 weeks (both 1.6 times the maximum recommended human dose on a body surface area basis). The relevance of these findings to humans is unknown.
Pregnancy
Teratogenic Effects.
Pregnancy Category BThere are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Reproduction studies have been performed in rats and monkeys at doses of up to 2000 and 600 mg/kg/day, or 3 and 2 times the maximum recommended human dose on a body surface area basis. and have revealed no evidence of impaired fertility or harm to the fetus due to cefotetan.
Nursing Mothers
Cefotetan is excreted in human milk in very low concentrations. Caution should be exercised when cefotetan is administered to a nursing woman.
Geriatric Use
Of the 925 subjects who received cefotetan in clinical studies, 492 (53%) were 60 years and older, while 76 (8%) were 80 years and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and the other reported clinical experience has not identified differences in responses between elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function (see DOSAGE AND ADMINISTRATION, Impaired Renal Function).
-
ADVERSE REACTIONS
In clinical studies, the following adverse effects were considered related to CEFOTAN therapy. Those appearing in italics have been reported during postmarketing experience.
Gastrointestinal: symptoms occurred in 1.5% of patients, the most frequent were diarrhea (1 in 80) and nausea (1 in 700); pseudomembranous colitis. Onset of pseudomembranous colitis symptoms may occur during or after antibiotic treatment or surgical prophylaxis (see WARNINGS).
Hematologic: laboratory abnormalities occurred in 1.4% of patients and included eosinophilia (1 in 200), positive direct Coombs test (1 in 250), and thrombocytosis (1 in 300); agranulocytosis, hemolytic anemia, leukopenia, thrombocytopenia, and prolonged prothrombin time with or without bleeding.
Hepatic: enzyme elevations occurred in 1.2% of patients and included a rise in ALT (SGPT) (1 in 150), AST (SGOT) (1 in 300), alkaline phosphatase (1 in 700), and LDH (1 in 700).
Hypersensitivity: reactions were reported in 1.2% of patients and included rash (1 in 150) and itching (1 in 700); anaphylactic reactions and urticaria.
Local: effects were reported in less than 1% of patients and included phlebitis at the site of injection (1 in 300), and discomfort (1 in 500).
Renal: Elevations in BUN and serum creatinine have been reported.
Urogenital: Nephrotoxicity has rarely been reported.
Miscellaneous: Fever
In addition to the adverse reactions listed above which have been observed in patients treated with cefotetan, the following adverse reactions and altered laboratory tests have been reported for cephalosporin-class antibiotics: pruritus, Stevens-Johnson syndrome, erythema multiforme, toxic epidermal necrolysis, vomiting, abdominal pain, colitis, superinfection, vaginitis including vaginal candidiasis, renal dysfunction, toxic nephropathy, hepatic dysfunction including cholestasis, aplastic anemia, hemorrhage, elevated bilirubin, pancytopenia, and neutropenia.
Several cephalosporins have been implicated in triggering seizures, particularly in patients with renal impairment, when the dosage was not reduced (see DOSAGE AND ADMINISTRATION and OVERDOSAGE). If seizures associated with drug therapy occur, the drug should be discontinued. Anticonvulsant therapy can be given if clinically indicated.
To report SUSPECTED ADVERSE REACTIONS, contact Teligent Pharma, Inc. at 1-856-697-1441, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
- OVERDOSAGE
-
DOSAGE AND ADMINISTRATION
Treatment
The usual adult dosage is 1 gram (g) or 2 grams of CEFOTAN® (cefotetan for Injection, USP) administered intravenously or intramuscularly. Proper dosage and route of administration should be determined by the condition of the patient, severity of the infection, and susceptibility of the causative organism.
General Guidelines for Dosage of CEFOTAN® (cefotetan for Injection, USP)
Type of Infection
Daily Dose
Frequency and Route
Urinary Tract
1 g to 4 g 500 mg every 12 hours intravenous or intramuscular
1 or 2 g every 24 hours intravenous or intramuscular
1 or 2 g every 12 hours intravenous or intramuscular
Skin & Skin Structure
Mild - Moderatea
Severe
2 g
2 g every 24 hours intravenous
1 g every 12 hours intravenous or intramuscular
4 g
2 g every 12 hours intravenous
Other Sites
2 g to 4 g 1 g or 2 g every 12 hours intravenous or intramuscular Severe
4 grams
2 g every 12 hours intravenous Life-Threatening
6 gramsb
3 g every 12 hours intravenous a Klebsiella pneumoniae skin and skin structure infections should be treated with 1 or 2 grams every 12 hours intravenous or intramuscular.
b Maximum daily dosage should not exceed 6 grams.
If Chlamydia trachomatis is a suspected pathogen in gynecologic infections, appropriate antichlamydial coverage should be added, since cefotetan has no activity against this organism.
Prophylaxis
To prevent postoperative infection in clean contaminated or potentially contaminated surgery in adults, the recommended dosage is 1 or 2 g of CEFOTAN® (cefotetan for Injection, USP) administered once, intravenously, 30 to 60 minutes prior to surgery. In patients undergoing cesarean section, the dose should be administered as soon as the umbilical cord is clamped.
Impaired Renal Function
When renal function is impaired, a reduced dosage schedule must be employed. The following dosage guidelines may be used.
DOSAGE GUIDELINES FOR PATIENTS WITH IMPAIRED RENAL FUNCTION
Creatinine Clearance
mL/min
Dose
Frequency
Greater than 30
Usual Recommended Dosage*
Every 12 hours
10 to 30
Usual Recommended Dosage*
Every 24 hours
Less than 10
Usual Recommended Dosage*
Every 48 hours
* Dose determined by the type and severity of infection, and susceptibility of the causative organism.
Alternatively, the dosing interval may remain constant at 12 hour intervals, but the dose reduced to one-half the usual recommended dose for patients with a creatinine clearance of 10 to 30 mL/min, and one-quarter the usual recommended dose for patients with a creatinine clearance of less than 10 mL/min.
When only serum creatinine levels are available, creatinine clearance may be calculated from the following formula. The serum creatinine level should represent a steady state of renal function.
Males: Weight (kg) x (140 - age)
72 x serum creatinine (mg/100 mL)
Females: 0.85 x value for males
Cefotetan is dialyzable and it is recommended that for patients undergoing intermittent hemodialysis, one-quarter of the usual recommended dose be given every 24 hours on days between dialysis and one-half the usual recommended dose on the day of dialysis.
Preparation of Solution
For Intravenous Use
Reconstitute with Sterile Water for Injection. Shake to dissolve and let stand until clear.
Vial Size
Amount of
Diluent
Added (mL)
Approximate Withdrawable
Volume (mL)
Approximate
Average Concentration (mg/mL)
1 gram
10
10.5
95
2 gram
10 to 20
11 to 21
182 to 95
For Intramuscular Use
Reconstitute with Sterile Water for Injection; Bacteriostatic Water for Injection; Sodium Chloride Injection 0.9%, USP; 0.5% Lidocaine HCl; or 1% Lidocaine HCl. Shake to dissolve and let stand until clear.
Vial Size
Amount of
Diluent
Added (mL)
Approximate Withdrawable
Volume (mL)
Approximate
Average Concentration
(mg/mL)
1 gram
2
2.5
400
2 gram
3
4
500
Intravenous Administration
The intravenous route is preferable for patients with bacteremia, bacterial septicemia, or other severe or life-threatening infections, or for patients who may be poor risks because of lowered resistance resulting from such debilitating conditions as malnutrition, trauma, surgery, diabetes, heart failure, or malignancy, particularly if shock is present or impending.
For intermittent intravenous administration, a solution containing 1 gram or 2 grams of CEFOTAN® (cefotetan for Injection, USP) in Sterile Water for Injection can be injected over a period of three to five minutes. Using an infusion system, the solution may also be given over a longer period of time through the tubing system by which the patient may be receiving other intravenous solutions. Butterfly® or scalp vein-type needles are preferred for this type of infusion. However, during infusion of the solution containing CEFOTAN® (cefotetan for Injection, USP), it is advisable to discontinue temporarily the administration of other solutions at the same site.
NOTE: Solutions of cefotetan must not be admixed with solutions containing aminoglycosides. If CEFOTAN® and aminoglycosides are to be administered to the same patient, they must be administered separately and not as a mixed injection.
Intramuscular Administration
As with all intramuscular preparations, CEFOTAN® (cefotetan for Injection, USP) should be injected well within the body of a relatively large muscle such as the upper outer quadrant of the buttock (i.e., gluteus maximus); aspiration is necessary to avoid inadvertent injection into a blood vessel.
Compatibility and Stability
Frozen samples should be thawed at room temperature before use. After the periods mentioned below, any unused solutions or frozen material should be discarded. DO NOT REFREEZE.
NOTE: Solutions of CEFOTAN® (cefotetan for Injection, USP) must not be admixed with solutions containing aminoglycosides. If CEFOTAN® (cefotetan for Injection, USP) and aminoglycosides are to be administered to the same patient, they must be administered separately and not as a mixed injection. DO NOT ADD SUPPLEMENTARY MEDICATION.
CEFOTAN® (cefotetan for Injection, USP) reconstituted as described above (see DOSAGE AND ADMINISTRATION,Preparation of Solution) maintains satisfactory potency for 24 hours at room temperature (25ºC/77ºF), for 96 hours under refrigeration (5ºC/41ºF), and for at least 1 week in the frozen state (-20ºC/-4ºF). After reconstitution and subsequent storage in disposable glass or plastic syringes, CEFOTAN® (cefotetan for Injection, USP) is stable for 24 hours at room temperature and 96 hours under refrigeration.
NOTE: Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
-
HOW SUPPLIED
CEFOTAN® (cefotetan for Injection, USP) is a dry, white to pale yellow powder supplied in vials containing cefotetan disodium equivalent to 1 g and 2 g cefotetan activity for intravenous and intramuscular administration. The vials should be stored at 20⁰C to 25⁰ C (68⁰ to 77⁰ F) [see USP Controlled Room Temperature] and should be protected from light.
The following packages are available:
NDC No. Strength
52565-052-10 1 gram 10 mL vial, packaged in a tray of 10 52565-053-10 2 grams 20 mL vial, packaged in a tray of 10 Vial stoppers do not contain natural rubber latex.
‡Clinitest® is a registered trademark of Ames Division, Miles Laboratories, Inc.
CEFOTAN® is a registered trademark of Teligent Pharma, Inc.
Manufactured for:
Teligent Pharma, Inc.
Buena, NJ 08310
Made in Italy
Revised: November 2018
- PACKAGE LABEL. CARTON PRINCIPAL DISPLAY PANEL, 1 Gram Carton NDC 52565-052-10Cefotan® (Cefotetan For Injection) Equivalent to 1 gram Cefotetan per vial 10 x 10mL Vials Protect from Light Must be reconstituted. For intravenous infusion or intramuscular injection.
- PACKAGE LABEL. CARTON PRINCIPAL DISPLAY PANEL, 2 Gram Carton NDC 52565-053-10 Cefotan® (Cefotetan For Injection) Equivalent to 2 grams Cefotetan per vial10 - 20 mL Vials Protect from LightKEEP TOP CLOSEDMust be reconstituted. For intravenous infusion or intramuscular injection.
-
INGREDIENTS AND APPEARANCE
CEFOTAN
cefotetan disodium injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52565-052 Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Cefotetan Disodium (UNII: 0GXP746VXB) (Cefotetan - UNII:48SPP0PA9Q) Cefotetan 1 g in 10 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52565-052-10 10 in 1 TRAY 10/01/2015 1 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA050588 10/01/2015 CEFOTAN
cefotetan disodium injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:52565-053 Route of Administration INTRAVENOUS, INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Cefotetan Disodium (UNII: 0GXP746VXB) (Cefotetan - UNII:48SPP0PA9Q) Cefotetan 2 g in 20 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:52565-053-10 10 in 1 TRAY 10/01/2015 1 20 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA050588 10/01/2015 Labeler - Teligent Pharma, Inc. (011036910) Registrant - Teligent Pharma, Inc. (011036910) Establishment Name Address ID/FEI Business Operations ACSDobfar S.p.A 439962157 api manufacture(52565-052, 52565-053)

