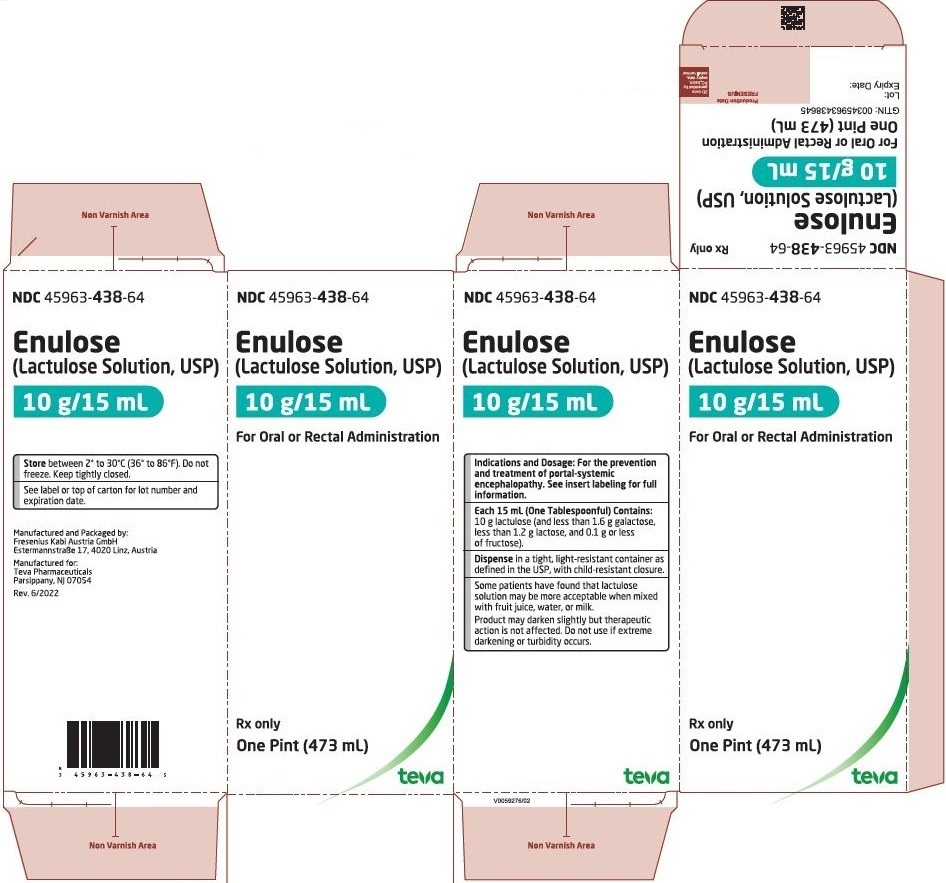
Label: ENULOSE- lactulose solution

- NDC Code(s): 45963-438-64
- Packager: Actavis Pharma, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated June 1, 2022
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Lactulose is a synthetic disaccharide in solution form for oral or rectal administration.
Each 15 mL of lactulose solution, USP contains: 10 g lactulose (and less than 1.6 g galactose, less than 1.2 g lactose, and 0.1 g or less of fructose).
Lactulose is a colonic acidifier for treatment and prevention of portal-systemic encephalopathy.
The chemical name for lactulose is 4-O-β-D-galactopyranosyl-D-fructofuranose. It has the following structural formula:
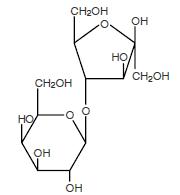
The molecular weight is 342.30. It is freely soluble in water.
-
CLINICAL PHARMACOLOGY
Lactulose causes a decrease in blood ammonia concentration and reduces the degree of portalsystemic encephalopathy. These actions are considered to be results of the following:
Bacterial degradation of lactulose in the colon acidifies the colonic contents.
This acidification of colonic contents results in the retention of ammonia in the colon as the ammonium ion. Since the colonic contents are then more acid than the blood, ammonia can be expected to migrate from the blood into the colon to form the ammonium ion.
The acid colonic contents convert NH3 to the ammonium ion (NH4)+, trapping it and preventing its absorption.
The laxative action of the metabolites of lactulose then expels the trapped ammonium ion from the colon.
Experimental data indicate that lactulose is poorly absorbed. Lactulose given orally to man and experimental animals resulted in only small amounts reaching the blood. Urinary excretion has been determined to be 3% or less and is essentially complete within 24 hours.
When incubated with extracts of human small intestinal mucosa, lactulose was not hydrolyzed during a 24-hour period and did not inhibit the activity of these extracts on lactose. Lactulose reaches the colon essentially unchanged. There it is metabolized by bacteria with the formation of low molecular weight acids that acidify the colon contents.
-
INDICATIONS AND USAGE
For the prevention and treatment of portal-systemic encephalopathy, including the stages of hepatic pre-coma and coma.
Controlled studies have shown that lactulose solution therapy reduces the blood ammonia levels by 25 to 50%; this is generally paralleled by an improvement in the patients' mental state and by an improvement in EEG patterns. The clinical response has been observed in about 75% of patients, which is at least as satisfactory as that resulting from neomycin therapy. An increase in patients' protein tolerance is also frequently observed with lactulose therapy. In the treatment of chronic portal-systemic encephalopathy, Lactulose has been given for over 2 years in controlled studies.
- CONTRAINDICATIONS
-
WARNINGS
A theoretical hazard may exist for patients being treated with lactulose solution who may be required to undergo electrocautery procedures during proctoscopy or colonoscopy. Accumulation of H2 gas in significant concentration in the presence of an electrical spark may result in an explosive reaction. Although this complication has not been reported with lactulose, patients on lactulose therapy undergoing such procedures should have a thorough bowel cleansing with a non-fermentable solution. Insufflation of CO2 as an additional safeguard may be pursued but is considered to be a redundant measure.
-
PRECAUTIONS
General
Since lactulose solution contains galactose (less than 1.6 g/15 mL) and lactose (less than 1.2 g/15 mL) it should be used with caution in diabetics.
In the overall management of portal-systemic encephalopathy, it should be recognized that there is serious underlying liver disease with complications such as electrolyte disturbance (e.g., hypokalemia) for which other specific therapy may be required.
Infants receiving lactulose may develop hyponatremia and dehydration.
Drug Interactions
There have been conflicting reports about the concomitant use of neomycin and lactulose solution. Theoretically, the elimination of certain colonic bacteria by neomycin and possibly other anti-infective agents may interfere with the desired degradation of lactulose and thus prevent the acidification of colonic contents. Thus the status of the lactulose-treated patient should be closely monitored in the event of concomitant oral anti-infective therapy.
Results of preliminary studies in humans and rats suggest that nonabsorbable antacids given concurrently with lactulose may inhibit the desired lactulose-induced drop in colonic pH. Therefore, a possible lack of desired effect of treatment should be taken into consideration before such drugs are given concomitantly with lactulose.
Other laxatives should not be used, especially during the initial phase of therapy for portal-systemic encephalopathy, because the loose stools resulting from their use may falsely suggest that adequate lactulose dosage has been achieved.
Carcinogenesis, Mutagenesis, Impairment of Fertility
There are no known human data on long-term potential for carcinogenicity, mutagenicity, or impairment of fertility.
There are no known animal data on long-term potential for mutagenicity.
Administration of lactulose solution in the diet of mice for 18 months in concentrations of 3 and 10 percent (v/w) did not produce any evidence of carcinogenicity.
In studies of mice, rats, and rabbits, doses of lactulose solution up to 6 or 12 mL/kg/day produced no deleterious effects in breeding, conception, or parturition.
Pregnancy
Teratogenic Effects
Pregnancy category B. Reproduction studies have been performed in mice, rats, and rabbits at doses up to 2 or 4 times the usual human oral dose and have revealed no evidence of impaired fertility or harm to the fetus due to lactulose. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when lactulose solution is administered to a nursing woman.
Pediatric Use
Very little information on the use of lactulose in pediatric patients has been recorded (see DOSAGE AND ADMINISTRATION).
-
ADVERSE REACTIONS
Precise frequency data are not available.
Lactulose may produce gaseous distention with flatulence or belching and abdominal discomfort such as cramping in about 20% of patients. Excessive dosage can lead to diarrhea with potential complications such as loss of fluids, hypokalemia, and hypernatremia.
Nausea and vomiting have been reported.
To report SUSPECTED ADVERSE EVENTS, contact the FDA at 1-800-FDA-1088 or http://www.fda.gov/ for voluntary reporting of adverse reactions.
-
OVERDOSAGE
Signs and Symptoms
There have been no reports of accidental overdosage. In the event of overdosage, it is expected that diarrhea and abdominal cramps would be the major symptoms. Medication should be terminated.
Oral LD50: The acute oral LD50 of the drug is 48.8 mL/kg in mice and greater than 30 mL/kg in rats.
Dialysis: Dialysis data are not available for lactulose. Its molecular similarity to sucrose, however, would suggest that it should be dialyzable.
-
DOSAGE AND ADMINISTRATION
Oral
Adult: The usual adult, oral dosage is 2 to 3 tablespoonfuls (30 to 45 ml, containing 20 g to 30 g of lactulose) three or four times daily. The dosage may be adjusted every day or two to produce 2 or 3 soft stools daily.
Hourly doses of 30 to 45 ml of Lactulose may be used to induce the rapid laxation indicated in the initial phase of the therapy of portal-systemic encephalopathy. When the laxative effect has been achieved, the dose of lactulose may then be reduced to the recommended daily dose.
Improvement in the patient's condition may occur within 24 hours but may not begin before 48 hours or even later.
Continuous long-term therapy is indicated to lessen the severity and prevent the recurrence of portal-systemic encephalopathy. The dose of lactulose for this purpose is the same as the recommended daily dose.
Pediatric
Very little information on the use of lactulose in young children and adolescents has been recorded. As with adults, the subjective goal in proper treatment is to produce 2 to 3 soft stools daily. On the basis of information available, the recommended initial daily oral dose in infants is 2.5 to 10 ml in divided doses.
For older children and adolescents, the total daily dose is 40 to 90 ml. If the initial dose causes diarrhea, the dose should be reduced immediately. If diarrhea persists, lactulose should be discontinued.
Rectal
When the adult patient is in the impending coma or coma stage of portal-systemic encephalopathy and the danger of aspiration exists, or when the necessary endoscopic or intubation procedures physically interfere with the administration of the recommended oral doses, lactulose solution may be given as a retention enema via a rectal balloon catheter. Cleansing enemas containing soapsuds or other alkaline agents should not be used.
Three hundred ml of lactulose solution should be mixed with 700 mL of water or physiologic saline and retained for 30 to 60 minutes. Lactulose enema may be repeated every 4 to 6 hours. If the enema is inadvertently evacuated too promptly, it may be repeated immediately.
The goal of treatment is reversal of the coma stage in order that the patient may be able to take oral medication. Reversal of coma may take place within 2 hours of the first enema in some patients. Lactulose, given orally in the recommended doses, should be started before Lactulose by enema is stopped entirely.
-
HOW SUPPLIED
Lactulose Solution USP, 10 g/15 ml, is a natural colored and an unflavored solution supplied in one pint (473 ml) bottles.
Lactulose solution, USP contains lactulose 670 mg/mL (10 g/15 mL).
Store between 36° to 86°F (2° to 30°C). Do not freeze.
Under recommended storage conditions, a normal darkening of color may occur. Such darkening is characteristic of sugar solutions and does not affect therapeutic action. Prolonged exposure to temperatures above 86°F (30°C) or to direct light may cause extreme darkening and turbidity which may be pharmaceutically objectionable. If this condition develops, do not use.
Prolonged exposure to freezing temperatures may cause change to a semi-solid, too viscous to pour. Viscosity will return to normal upon warming to room temperature.
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure.
Manufactured and packaged by:
Fresenius Kabi Austria GmbH
Estermannstraße 17
4020 Linz, AustriaManufactured for:
Teva Pharmaceuticals
Parsippany, NJ 07054
USA
Revised – June 2022
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ENULOSE
lactulose solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:45963-438 Route of Administration ORAL, RECTAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LACTULOSE (UNII: 9U7D5QH5AE) (LACTULOSE - UNII:9U7D5QH5AE) LACTULOSE 10 g in 15 mL Inactive Ingredients Ingredient Name Strength GALACTOSE (UNII: X2RN3Q8DNE) LACTOSE, UNSPECIFIED FORM (UNII: J2B2A4N98G) FRUCTOSE (UNII: 6YSS42VSEV) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:45963-438-64 473 mL in 1 BOTTLE; Type 0: Not a Combination Product 02/28/2011 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090502 02/28/2011 Labeler - Actavis Pharma, Inc. (119723554) Registrant - Fresenius Kabi Austria GmbH (303448575)