Label: LUBIPROSTONE capsule, gelatin coated

- NDC Code(s): 80425-0355-2
- Packager: Advanced Rx Pharmacy of Tennessee, LLC
- This is a repackaged label.
- Source NDC Code(s): 63304-352
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application Authorized Generic
Drug Label Information
Updated August 16, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
Lubiprostone 24mcg Capsule
These highlights do not include all the information needed to use LUBIPROSTONE CAPSULES safely and effectively. See full prescribing information for LUBIPROSTONE CAPSULES.
Lubiprostone capsules, for oral use
Initial U.S. Approval: 2006INDICATIONS AND USAGE
Lubiprostone is a chloride channel activator indicated for the treatment of:
- chronic idiopathic constipation (CIC) in adults. ( 1.1)
- opioid-induced constipation (OIC) in adult patients with chronic, non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent (e.g., weekly) opioid dosage escalation. ( 1.2)
- irritable bowel syndrome with constipation (IBS-C) in women ≥ 18 years old. ( 1.3)
DOSAGE AND ADMINISTRATION
Recommended Dosage ( 2.1)
- CIC and OIC: 24 mcg twice daily.
- IBS-C: 8 mcg twice daily.
- See full prescribing information for dosage adjustment by indication and degree of hepatic impairment.
Administration Instructions ( 2.2)
- Swallow capsules whole and do not break apart or chew,
- Take capsules with food and water,
- Assess periodically the need for continuous therapy.
DOSAGE FORMS AND STRENGTHS
Capsules: 8 mcg and 24 mcg ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Nausea: Patients may experience nausea; concomitant administration of food may reduce this symptom. ( 2.2, 5.1)
- Diarrhea: Avoid use in patients with severe diarrhea. Instruct patients to discontinue lubiprostone and contact their healthcare provider if severe diarrhea occurs during treatment. ( 5.2)
- Syncope and Hypotension: May occur after taking the first dose or with subsequent doses. Generally resolves prior to the next dose, but may recur with repeat dosing. Instruct patients to discontinue lubiprostone and contact their healthcare provider if symptoms occur. ( 5.3)
- Dyspnea: May occur within an hour of first dose. Generally resolves within 3 hours, but may recur with repeat dosing. Instruct patients to contact their healthcare provider if symptoms occur. ( 5.4)
- Bowel Obstruction: Evaluate patients with symptoms suggestive of mechanical gastrointestinal obstruction prior to initiating treatment with lubiprostone. ( 4, 5.5)
ADVERSE REACTIONS
Most common adverse reactions (> 4%) are:
- CIC: nausea, diarrhea, headache, abdominal pain, abdominal distension, and flatulence. ( 6.1)
- OIC: nausea and diarrhea. ( 6.1)
- IBS-C: nausea, diarrhea, and abdominal pain. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Sun Pharmaceutical Industries, Inc. at 1-800-406-9784 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Chronic Idiopathic Constipation in Adults
1.2 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
1.3 Irritable Bowel Syndrome with Constipation
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Nausea
5.2 Diarrhea
5.3 Syncope and Hypotension
5.4 Dyspnea
5.5 Bowel Obstruction
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Methadone
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Chronic Idiopathic Constipation in Adults
14.2 Opioid-Induced Constipation in Adults with Chronic Non-Cancer Pain
14.3 Irritable Bowel Syndrome with Constipation
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Chronic Idiopathic Constipation in Adults
Lubiprostone is indicated for the treatment of chronic idiopathic constipation (CIC) in adults.
1.2 Opioid-Induced Constipation in Adult Patients with Chronic Non-Cancer Pain
Lubiprostone is indicated for the treatment of opioid-induced constipation (OIC) in adult patients with chronic non-cancer pain, including patients with chronic pain related to prior cancer or its treatment who do not require frequent (e.g., weekly) opioid dosage escalation.
Limitations of Use:
Effectiveness of Lubiprostone in the treatment of opioid-induced constipation in patients taking diphenylheptane opioids (e.g., methadone) has not been established. [see Clinical Studies (14.2)]
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended oral dosage of Lubiprostone by indication and adjustments for patients with moderate (Child Pugh Class B) and severe (Child Pugh Class C) hepatic impairment are shown in Table 1.
Table 1. Recommended Dosage Regimen CIC and OIC IBS-C - *
- If the dose is tolerated and an adequate response has not been obtained after an appropriate interval, doses can then be escalated to full dosing with appropriate monitoring of patient response.
Recommended Adult Dosage Regimen 24 mcg twice daily 8 mcg twice daily Dosage Adjustment for Hepatic Impairment [see Use in Specific Populations (8.6)] Moderate Impairment (Child-Pugh Class B):
16 mcg twice daily *Moderate Impairment (Child-Pugh Class B):
No adjustment necessarySevere Impairment (Child-Pugh Class C):
8 mcg twice daily *Severe Impairment (Child-Pugh Class C):
8 mcg once daily * - 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Lubiprostone is contraindicated in patients with known or suspected mechanical gastrointestinal obstruction [see Warnings and Precautions (5.5)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Nausea
Patients taking lubiprostone may experience nausea. Concomitant administration of food with lubiprostone may reduce symptoms of nausea [see Adverse Reactions (6.1)] .
5.2 Diarrhea
Avoid use of lubiprostone in patients with severe diarrhea. Patients should be aware of the possible occurrence of diarrhea during treatment. Instruct patients to discontinue lubiprostone and contact their healthcare provider if severe diarrhea occurs [see Adverse Reactions (6.1)] .
5.3 Syncope and Hypotension
Syncope and hypotension have been reported with lubiprostone in the postmarketing setting and a few of these adverse reactions resulted in hospitalization. Most cases occurred in patients taking 24 mcg twice daily and some occurred within an hour after taking the first dose or subsequent doses of lubiprostone. Some patients had concomitant diarrhea or vomiting prior to developing the adverse reaction. Syncope and hypotension generally resolved following lubiprostone discontinuation or prior to next dose, but recurrence has been reported with subsequent doses. Several cases reported concomitant use of medications known to lower blood pressure, which may increase the risk for the development of syncope or hypotension.
Patients should be aware of the risk of syncope and hypotension during treatment and that other adverse reactions may increase this risk, such as diarrhea or vomiting.
5.4 Dyspnea
In clinical trials, dyspnea was reported by 3%, 1%, and < 1% of the treated CIC, OIC, and IBS-C populations receiving lubiprostone, respectively, compared to 0%, 1%, and < 1% of placebo-treated patients. There have been postmarketing reports of dyspnea when using lubiprostone 24 mcg twice daily. Some patients have discontinued treatment because of dyspnea. These events have usually been described as a sensation of chest tightness and difficulty taking in a breath, and generally have an acute onset within 30 to 60 minutes after taking the first dose. They generally resolve within a few hours after taking the dose, but recurrence has been frequently reported with subsequent doses. Instruct patients to contact their healthcare provider if dyspnea occurs.
5.5 Bowel Obstruction
In patients with symptoms suggestive of mechanical gastrointestinal obstruction, perform a thorough evaluation to confirm the absence of an obstruction prior to initiating therapy with lubiprostone [see Contraindication (4)] .
-
6 ADVERSE REACTIONS
The following adverse reactions are described below and elsewhere in labeling:
- Nausea [see Warnings and Precautions (5.1)]
- Diarrhea [see Warnings and Precautions (5.2)]
- Syncope and Hypotension [see Warnings and Precautions (5.3)]
- Dyspnea [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
During clinical development of lubiprostone for CIC, OIC, and IBS-C, 1648 patients were treated with lubiprostone for 6 months and 710 patients were treated for 1 year (not mutually exclusive).
Chronic Idiopathic Constipation
Adverse reactions in adult dose-finding, efficacy, and long-term clinical studies:The data described below reflect exposure to Lubiprostone 24 mcg twice daily in 1113 patients with CIC over 3- or 4-week, 6-month, and 12-month treatment periods; and from 316 patients receiving placebo over short-term exposure (≤4 weeks). The placebo population (N = 316) had a mean age of 48 (range 21 to 81) years; was 87% female; 81% Caucasian, 10% African American, 7% Hispanic, 1% Asian, and 12% elderly (≥65 years of age). Of those patients treated with lubiprostone 24 mcg twice daily (N=1113), the mean age was 50 (range 19-86) years; 87% were female; 86% Caucasian, 8% African American, 5% Hispanic, 1% Asian, and 17% elderly (≥65 years of age).
The most common adverse reactions (>4%) in CIC were nausea, diarrhea, headache, abdominal pain, abdominal distension, and flatulence.
Table 2 presents data for the adverse reactions that occurred in at least 1% of patients and that occurred more frequently with Lubiprostone than placebo.
Table 2. Adverse Reactions *in Clinical Trials of Adults with CIC System/Adverse Reaction Placebo Lubiprostone 24 mcg Twice Daily N = 316
%N = 1113
%Nausea 3 29 Diarrhea 1 12 Headache 5 11 Abdominal pain 3 8 Abdominal distension 2 6 Flatulence 2 6 Vomiting 0 3 Loose stools 0 3 Edema <1 3 Abdominal discomfort † 1 3 Dizziness 1 3 Chest discomfort/pain 0 2 Dyspnea 0 2 Dyspepsia <1 2 Fatigue 1 2 Dry mouth <1 1 Nausea:Approximately 29% of patients who received lubiprostone experienced nausea; 4% of patients had severe nausea and 9% of patients discontinued treatment due to nausea. The rate of nausea was lower among male (8%) and elderly (19%) patients. No patients in the clinical studies were hospitalized due to nausea.
Diarrhea:Approximately 12% of patients who received lubiprostone experienced diarrhea; 2% of patients had severe diarrhea and 2% of patients discontinued treatment due to diarrhea.
Electrolytes:No serious adverse reactions of electrolyte imbalance were reported in clinical studies, and no clinically significant changes were seen in serum electrolyte levels in patients receiving lubiprostone.
Less common adverse reactions (<1%):fecal incontinence, muscle cramp, defecation urgency, frequent bowel movements, hyperhidrosis, pharyngolaryngeal pain, intestinal functional disorder, anxiety, cold sweat, constipation, cough, dysgeusia, eructation, influenza, joint swelling, myalgia, pain, syncope, tremor, decreased appetite.
Opioid-Induced Constipation
Adverse reactions in adult efficacy and long-term clinical studies:The data described below reflect exposure to lubiprostone 24 mcg twice daily in 860 patients with OIC for up to 12 months and from 632 patients receiving placebo twice daily for up to 12 weeks. The total population (N = 1492) had a mean age of 50 (range 20–89) years; was 63% female; 83% Caucasian, 14% African American, 1% American Indian/Alaska Native, 1% Asian; 5% were of Hispanic ethnicity, and 9% were elderly (≥65 years of age).
The most common adverse reactions (>4%) in OIC were nausea and diarrhea.
Table 3 presents data for the adverse reactions that occurred in at least 1% of patients and that occurred more frequently with study drug than placebo.
Nausea:Approximately 11% of patients who received lubiprostone experienced nausea; 1% of patients had severe nausea and 2% of patients discontinued treatment due to nausea.
Irritable Bowel Syndrome with Constipation
Adverse reactions in adult dose-finding, efficacy, and long-term clinical studies:The data described below reflect exposure to lubiprostone 8 mcg twice daily in 1011 patients with IBS-C for up to 12 months and from 435 patients receiving placebo twice daily for up to 16 weeks. The total population (N = 1267) had a mean age of 47 (range 18–85) years; was 92% female; 78% Caucasian, 13% African American, 9% Hispanic, 0.4% Asian, and 8% elderly (≥65 years of age).
The most common adverse reactions (>4%) in IBS-C were nausea, diarrhea, and abdominal pain.
Table 4 presents data for the adverse reactions that occurred in at least 1% of patients and that occurred more frequently with study drug than placebo.
Nausea:Approximately 8% of patients who received lubiprostone 8 mcg twice daily experienced nausea; 1% of patients had severe nausea and 1% of patients discontinued treatment due to nausea.
Diarrhea:Approximately 7% of patients who received lubiprostone 8 mcg twice daily experienced diarrhea; <1% of patients had severe diarrhea and <1% of patients discontinued treatment due to diarrhea.
Less common adverse reactions (<1%):dyspepsia, loose stools, vomiting, fatigue, dry mouth, edema, increased alanine aminotransferase, increased aspartate aminotransferase, constipation, eructation, gastroesophageal reflux disease, dyspnea, erythema, gastritis, increased weight, palpitations, urinary tract infection, anorexia, anxiety, depression, fecal incontinence, fibromyalgia, hard feces, lethargy, rectal hemorrhage, pollakiuria.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post-approval use of lubiprostone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiovascular:syncope and/or hypotension [see Warnings and Precautions (5.3)] , tachycardia
Gastrointestinal:ischemic colitis
General:asthenia
Immune System:hypersensitivity reactions including rash, swelling, and throat tightness malaise
Muscoskeletal:muscle cramps or muscle spasms.
-
7 DRUG INTERACTIONS
7.1 Methadone
Diphenylheptane opioids (e.g., methadone) have been shown in nonclinical studies to dose-dependently reduce the activation of ClC-2 by lubiprostone in the gastrointestinal tract. There is a possibility of a dose-dependent decrease in the efficacy of lubiprostone in patients using diphenylheptane opioids. No in vivointeraction studies have been conducted.
The effectiveness of lubiprostone in the treatment of OIC in patients taking diphenylhepatane opioids (e.g., methadone) has not been established [see Indications and Usage (1.2)] .
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Following oral administration, concentrations of lubiprostone in plasma are below the level of quantitation; however, one of the metabolites, M3, has measurable systemic concentrations [see Clinical Pharmacology (12.3)] . Limited available data with lubiprostone use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. Animal reproduction studies did not show an increase in structural malformations .Although a dose dependent increase in fetal loss was observed in pregnant guinea pigs that received lubiprostone (doses equivalent to 0.2 to 6 times the maximum recommended human dose (MRHD) based on body surface area (mg/m 2)), these effects were probably secondary to maternal toxicity and occurred after the period of organogenesis (see Data) .
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In developmental toxicity studies, pregnant rats and rabbits received oral lubiprostone during organogenesis at doses up to approximately 338 times (rats) and approximately 34 times (rabbits) the maximum recommended human dose (MRHD) based on body surface area (mg/m 2). Maximal animal doses were 2000 mcg/kg/day (rats) and 100 mcg/kg/day (rabbits). In rats, there were increased incidences of early resorptions and soft tissue malformations ( situs inversus, cleft palate) at the 2000 mcg/kg/day dose; however, these effects were probably secondary to maternal toxicity. A dose-dependent increase in fetal loss occurred when guinea pigs received lubiprostone after the period of organogenesis, on days 40 to 53 of gestation, at daily oral doses of 1, 10, and 25 mcg/kg/day (approximately 0.2, 2 and 6 times the MRHD based on body surface area (mg/m 2)); however, these effects were probably secondary to maternal toxicity. The potential of lubiprostone to cause fetal loss was also examined in pregnant rhesus monkeys. Monkeys received lubiprostone post-organogenesis on gestation days 110 through 130 at daily oral doses of 10 and 30 mcg/kg/day (approximately 3 and 10 times the MRHD based on body surface area (mg/m 2)). Fetal loss was noted in one monkey from the 10-mcg/kg dose group, which is within normal historical rates for this species. There was no drug-related adverse effect seen in monkeys.
8.2 Lactation
Risk Summary
There are no data available on the presence of lubiprostone in human milk or the effect of lubiprostone on milk production. There are limited data available on the effect of lubiprostone on the breastfed infant. Neither lubiprostone nor its active metabolite (M3) were present in the milk of lactating rats. When a drug is not present in animal milk, it is likely that the drug will not be present in human milk. If present, lubiprostone may cause diarrhea in the breastfed infant (see Clinical Considerations) . The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for lubiprostone and any potential adverse effects on the breastfed infant from lubiprostone or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness have not been established in pediatric patients with IBS-C, pediatric functional constipation (PFC), and OIC.
Efficacy was not demonstrated for the treatment of PFC in patients 6 years of age and older in a 12 week, randomized, double-blind, placebo-controlled trial conducted in 606 patients 6 to 17 years with PFC comparing lubiprostone to placebo. The primary efficacy endpoint was an overall response based on spontaneous bowel movement frequency over the duration of the trial; the treatment difference from placebo was not statistically significant. In this age group, adverse reactions to lubiprostone were similar to those reported in adults. In a 36-week, long-term safety extension trial after approximately 9 months of treatment with lubiprostone, a single case of reversible elevation of ALT (17-times upper limit of normal [ULN]), AST (13-times ULN), and GGT (9-times [ULN]) was observed in a child with baseline elevated values (less than or equal to 2.5-times ULN).
Juvenile Animal Toxicity Data
In a 13-week oral toxicity study in juvenile rats, a significant decrease in total bone mineral density was observed in female pups at 0.5 mg/kg/day; in male pups, a significantly lower cortical thickness at the tibial diaphysis was observed at 0.5 mg/kg. The 0.5 mg/kg/day dose is approximately 101 times the maximum recommended adult dose of 48 mcg/day, based on body surface area (mg/m 2).
8.5 Geriatric Use
Chronic Idiopathic Constipation
The efficacy of lubiprostone 24 mcg twice daily in the elderly (at least 65 years of age) subpopulation with CIC was consistent with the efficacy in the overall study population. Of the total number of patients treated in the dose-finding, efficacy, and long-term studies of lubiprostone, 16% were at least 65 years of age, and 4% were at least 75 years of age. Elderly patients taking lubiprostone experienced a lower rate of associated nausea compared to the overall study population taking lubiprostone (19% vs. 29%, respectively).
Opioid-Induced Constipation
The safety profile of lubiprostone in the elderly (at least 65 years of age) subpopulation with OIC (9% were at least 65 years of age and 2% were at least 75 years of age) was consistent with the safety profile in the overall study population. Clinical studies of lubiprostone did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients.
Irritable Bowel Syndrome with Constipation
The safety profile of lubiprostone in the elderly (at least 65 years of age) subpopulation with IBS-C (8% were at least 65 years of age and 2% were at least 75 years of age) was consistent with the safety profile in the overall study population. Clinical studies of lubiprostone did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients.
8.6 Hepatic Impairment
Patients with moderate hepatic impairment (Child-Pugh Class B) and severe hepatic impairment (Child-Pugh Class C) experienced markedly higher systemic exposure of lubiprostone active metabolite M3, when compared to subjects with normal hepatic function [see Clinical Pharmacology (12.3)] . Clinical safety results demonstrated an increased incidence and severity of adverse events in subjects with greater severity of hepatic impairment.
Adjust the dosage of lubiprostone in patients with severe hepatic impairment for all indications. Dosage adjustment is also needed for patients with moderate hepatic impairment treated for CIC, and OIC [see Dosage and Administration (2.1)] . No dosing adjustment is required in patients with mild hepatic impairment (Child-Pugh Class A).
-
10 OVERDOSAGE
There have been six reports of overdosage with lubiprostone during clinical development. Of these six cases, only two subjects reported adverse events: one reported vomiting, diarrhea and stomach ache after taking 168 to 192 mcg of lubiprostone, and another reported diarrhea and a joint injury on the day of overdose after taking 36 mcg of lubiprostone. Adverse reactions that occurred in at least 1% of healthy subjects given a single oral dose of 144 mcg of lubiprostone (6 times the highest recommended dose) in a cardiac repolarization study included nausea (45%), diarrhea (35%), vomiting (27%), dizziness (14%), headache (12%), abdominal pain (8%), flushing/hot flash (8%), retching (8%), dyspnea (4%), pallor (4%), stomach discomfort (4%), anorexia (2%), asthenia (2%), chest discomfort (2%), dry mouth (2%), hyperhidrosis (2%), and syncope (2%).
-
11 DESCRIPTION
Lubiprostone is a chloride channel activator for oral use.
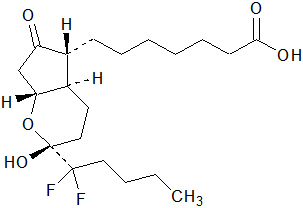
The chemical name for lubiprostone is (–)-7-[(2 R,4a R,5 R,7a R)-2-(1,1-difluoropentyl)-2-hydroxy-6-oxooctahydrocyclopenta[ b]pyran-5-yl]heptanoic acid. The molecular formula of lubiprostone is C 20H 32F 2O 5with a molecular weight of 390.46 and a chemical structure as follows:
Lubiprostone drug substance occurs as white, odorless crystals or crystalline powder, is very soluble in ether and ethanol, and is practically insoluble in hexane and water. Lubiprostone is available as an imprinted, oval, soft gelatin capsule in two strengths. Pink capsules contain 8 mcg of lubiprostone and the following inactive ingredients: ferric oxide, gelatin, medium-chain triglycerides, purified water, sorbitol, and titanium dioxide. Orange capsules contain 24 mcg of lubiprostone and the following inactive ingredients: D&C Yellow #10, FD&C Red #40, gelatin, medium-chain triglycerides, purified water, and sorbitol.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lubiprostone is a locally acting chloride channel activator that enhances a chloride-rich intestinal fluid secretion without altering sodium and potassium concentrations in the serum. Lubiprostone acts by specifically activating ClC-2, which is a normal constituent of the apical membrane of the human intestine, in a protein kinase A–independent fashion.
By increasing intestinal fluid secretion, lubiprostone increases motility in the intestine, thereby facilitating the passage of stool and alleviating symptoms associated with chronic idiopathic constipation. Patch clamp cell studies in human cell lines have indicated that the majority of the beneficial biological activity of lubiprostone and its metabolites is observed only on the apical (luminal) portion of the gastrointestinal epithelium.
Lubiprostone, via activation of apical ClC-2 channels in intestinal epithelial cells, bypasses the antisecretory action of opiates that results from suppression of secretomotor neuron excitability.
Activation of ClC-2 by lubiprostone has also been shown to stimulate recovery of mucosal barrier function and reduce intestinal permeability via the restoration of tight junction protein complexes in ex vivostudies of ischemic porcine intestine.
12.2 Pharmacodynamics
Although the pharmacologic effects of lubiprostone in humans have not been fully evaluated, animal studies have shown that oral administration of lubiprostone increases chloride ion transport into the intestinal lumen, enhances fluid secretion into the bowels, and improves fecal transit.
12.3 Pharmacokinetics
Following oral administration, concentrations of lubiprostone in plasma are below the level of quantitation (10 pg/mL). Therefore, standard pharmacokinetic parameters such as area under the curve (AUC), maximum concentration (C max), and half-life (t ½) cannot be reliably calculated. However, the pharmacokinetic parameters of M3 (only measurable active metabolite of lubiprostone) have been characterized.
Absorption
Peak plasma concentrations of M3, after a single oral dose of 24 mcg of lubiprostone, occurred at approximately 1.1 hours. The C maxwas 41.5 pg/mL and the mean AUC 0–twas 57.1 pg∙hr/mL. The AUC 0–tof M3 increases dose proportionally after single 24-mcg and 144-mcg doses of lubiprostone (6-times the maximum recommended 24 mcg dose).
Food Effect
A study was conducted with a single 72-mcg dose of 3H-labeled lubiprostone (3-times the maximum recommended 24 mcg dose) to evaluate the potential of a food effect on lubiprostone absorption, metabolism, and excretion. Pharmacokinetic parameters of total radioactivity demonstrated that C maxdecreased by 55% while AUC 0–∞was unchanged when lubiprostone was administered with a high-fat meal. The clinical relevance of the effect of food on the pharmacokinetics of lubiprostone is not clear. However, lubiprostone was administered with food and water in a majority of clinical trials.
Distribution
In vitroprotein binding studies indicate lubiprostone is approximately 94% bound to human plasma proteins.
Elimination
Metabolism
Lubiprostone is rapidly and extensively metabolized by 15-position reduction, α-chain β-oxidation, and ω-chain ω-oxidation. In vitrostudies using human liver microsomes indicate that cytochrome P450 isoenzymes are not involved in the metabolism of lubiprostone. Further in vitrostudies indicate that M3, a metabolite of lubiprostone, is formed by the reduction of the 15-carbonyl moiety to a hydroxy moiety by microsomal carbonyl reductase. M3 makes up less than 10% of the dose of radiolabeled lubiprostone.
Animal studies have shown that metabolism of lubiprostone rapidly occurs within the stomach and jejunum, most likely in the absence of any systemic absorption.
Excretion
Lubiprostone could not be detected in plasma; however, M3 has a t ½ranging from 0.9 to 1.4 hours. After a single oral dose of 72 mcg of 3H-labeled lubiprostone, 60% of total administered radioactivity was recovered in the urine within 24 hours and 30% of total administered radioactivity was recovered in the feces by 168 hours. Lubiprostone and M3 are only detected in trace amounts in human feces.
Specific Populations
Patients with Renal Impairment
Sixteen subjects, 34 to 47 years old (8 severe renally impaired subjects [creatinine clearance (CrCl) less than 20 mL/min] who required hemodialysis and 8 control subjects with normal renal function [CrCl above 80 mL/min]), received a single oral 24-mcg dose of lubiprostone. Following administration, lubiprostone plasma concentrations were below the limit of quantitation (10 pg/mL). Plasma concentrations of M3 were within the range of exposure from previous clinical experience with lubiprostone.
Patients with Hepatic Impairment
Twenty-five subjects, 38 to 78 years old (9 with severe hepatic impairment [Child-Pugh Class C], 8 with moderate impairment [Child-Pugh Class B], and 8 with normal liver function), received either 12 mcg or 24 mcg of lubiprostone under fasting conditions. Following administration, lubiprostone plasma concentrations were below the limit of quantitation (10 pg/mL) except for two subjects. In moderately and severely impaired subjects, the C maxand AUC 0–tof the active lubiprostone metabolite M3 were increased, as shown in Table 5.
Table 5. Pharmacokinetic Parameters of the Metabolite M3 for Subjects with Normal or Impaired Liver Function following Dosing with lubiprostone Liver Function Status Mean (SD) AUC 0–t
(pg∙hr/mL)% Change vs. Normal Mean (SD) C max
(pg/mL)% Change vs. Normal Normal (n=8) 39.6 (18.7) n.a. 37.5 (15.9) n.a. Child-Pugh Class B (n=8) 119 (104) +119 70.9 (43.5) +66 Child-Pugh Class C (n=8) 234 (61.6) +521 114 (59.4) +183 These results demonstrate that there is a correlation between increased exposure of M3 and severity of hepatic impairment. [see Use in Specific Populations (8.6)]
Drug Interaction Studies
Based upon the results of in vitrohuman microsome studies, there is low likelihood of pharmacokinetic drug–drug interactions with lubiprostone. Additionally, in vitrostudies in human liver microsomes demonstrate that lubiprostone does not inhibit cytochrome P450 isoforms 3A4, 2D6, 1A2, 2A6, 2B6, 2C9, 2C19, or 2E1, and in vitrostudies of primary cultures of human hepatocytes show no induction of cytochrome P450 isoforms 1A2, 2B6, 2C9, and 3A4 by lubiprostone. Based on the available information, no protein binding–mediated drug interactions of clinical significance are anticipated.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two 2-year oral (gavage) carcinogenicity studies (one in Crl:B6C3F1 mice and one in Sprague-Dawley rats) were conducted with lubiprostone. In the 2-year carcinogenicity study in mice, lubiprostone doses of 25, 75, 200, and 500 mcg/kg/day (approximately 2, 6, 17, and 42 times the maximum recommended human dose, respectively, based on body surface area (mg/m 2)) were used. In the 2-year rat carcinogenicity study, lubiprostone doses of 20, 100, and 400 mcg/kg/day (approximately 3, 17, and 68 times the maximum recommended human dose, respectively, based on body surface area (mg/m 2)) were used. In the mouse carcinogenicity study, there was no significant increase in any tumor incidences. There was a significant increase in the incidence of interstitial cell adenoma of the testes in male rats at the 400 mcg/kg/day dose. In female rats, treatment with lubiprostone produced hepatocellular adenoma at the 400 mcg/kg/day dose.
Mutagenesis
Lubiprostone was not genotoxic in the in vitroAmes reverse mutation assay, the in vitromouse lymphoma (L5178Y TK +/−) forward mutation assay, the in vitroChinese hamster lung (CHL/IU) chromosomal aberration assay, and the in vivomouse bone marrow micronucleus assay.
Impairment of Fertility
Lubiprostone, at oral doses of up to 1000 mcg/kg/day, had no effect on the fertility and reproductive function of male and female rats. However, the number of implantation sites and live embryos were significantly reduced in rats at the 1000 mcg/kg/day dose as compared to control. The number of dead or resorbed embryos in the 1000 mcg/kg/day group was higher compared to the control group, but was not statistically significant. The 1000 mcg/kg/day dose in rats is approximately 169 times the maximum recommended human dose of 48 mcg/day, based on body surface area (mg/m 2).
-
14 CLINICAL STUDIES
14.1 Chronic Idiopathic Constipation in Adults
Two double-blinded, placebo-controlled studies of identical design were conducted in patients with CIC. CIC was defined as, on average, less than 3 SBMs per week (a SBM is a bowel movement occurring in the absence of laxative use) along with one or more of the following symptoms of constipation for at least 6 months prior to randomization: 1) very hard stools for at least a quarter of all bowel movements; 2) sensation of incomplete evacuation following at least a quarter of all bowel movements; and 3) straining with defecation at least a quarter of the time.
Following a 2-week baseline/washout period, a total of 479 patients (mean age 47 [range 20 to 81] years; 89% female; 81% Caucasian, 10% African American, 7% Hispanic, 2% Asian, 11% at least 65 years of age) were randomized and received lubiprostone 24 mcg twice daily or placebo twice daily for 4 weeks. The primary endpoint of the studies was SBM frequency. The studies demonstrated that patients treated with lubiprostone had a higher frequency of SBMs during Week 1 than the placebo patients. In both studies, results similar to those in Week 1 were also observed in Weeks 2, 3, and 4 of therapy (Table 6).
Table 6. Adult Spontaneous Bowel Movement Frequency Rates *(Efficacy Studies) Trial Study Arm Baseline
Mean ± SD MedianWeek 1
Mean ± SD MedianWeek 2
Mean ± SD MedianWeek 3
Mean ± SD MedianWeek 4
Mean ± SD MedianWeek 1 Change from Baseline
Mean ± SD MedianWeek 4 Change from Baseline
Mean ± SD Median- *
- Frequency rates are calculated as 7 times (number of SBMs) / (number of days observed for that week).
Study 1 Placebo 1.6 ± 1.3
1.53.5 ± 2.3
3.03.2 ± 2.5
3.02.8 ± 2.2
2.02.9 ± 2.4
2.31.9 ± 2.2
1.51.3 ± 2.5
1.0lubiprostone 24 mcg Twice Daily 1.4 ± 0.8
1.55.7 ± 4.4
5.05.1 ± 4.1
4.05.3 ± 4.9
5.05.3 ± 4.7
4.04.3 ± 4.3
3.53.9 ± 4.6
3.0Study 2 Placebo 1.5 ± 0.8
1.54.0 ± 2.7
3.53.6 ± 2.7
3.03.4 ± 2.8
3.03.5 ± 2.9
3.02.5 ± 2.6
1.51.9 ± 2.7
1.5lubiprostone 24 mcg Twice Daily 1.3 ± 0.9
1.55.9 ± 4.0
5.05.0 ± 4.2
4.05.6 ± 4.6
5.05.4 ± 4.8
4.34.6 ± 4.1
3.84.1 ± 4.8
3.0In both studies, lubiprostone demonstrated increases in the percentage of patients who experienced SBMs within the first 24 hours after administration when compared to placebo (57% vs. 37% in Study 1 and 63% vs. 32% in Study 2, respectively). Similarly, the time to first SBM was shorter for patients receiving lubiprostone than for those receiving placebo.
Signs and symptoms related to constipation, including abdominal bloating, abdominal discomfort, stool consistency, and straining, as well as constipation severity ratings, were also improved with lubiprostone versus placebo. The results were consistent in subpopulation analyses for gender, race, and elderly patients at least 65 years of age.
During a 7-week randomized withdrawal study, patients who received lubiprostone during a 4-week treatment period were then randomized to receive either placebo or to continue treatment with lubiprostone. Inlubiprostone-treated patients randomized to placebo, SBM frequency rates returned toward baseline within 1 week and did not result in worsening compared to baseline. Patients who continued on lubiprostone maintained their response to therapy over the additional 3 weeks of treatment.
14.2 Opioid-Induced Constipation in Adults with Chronic Non-Cancer Pain
The efficacy of lubiprostone in the treatment of OIC in patients receiving opioid therapy for chronic, non-cancer-related pain was assessed in three randomized, double-blinded, placebo-controlled studies. In Study 1, the median age was 52 years (range 20 to 82) and 63% were female. In Study 2, the median age was 50 years (range 21 to 77) and 64% were female. In Study 3, the median age was 50 years (range 21 to 89) and 60% were female. Patients had been receiving stable opioid therapy for at least 30 days prior to screening, which was to continue throughout the 12-week treatment period. At baseline, mean oral morphine equivalent daily doses (MEDDs) were 99 mg and 130 mg for placebo-treated and lubiprostone-treated patients, respectively, in Study 1. Baseline mean MEDDs were 237 mg and 265 mg for placebo-treated and lubiprostone-treated patients, respectively, in Study 2. In Study 3, baseline mean MEDDs were 330 mg and 373 mg for placebo-treated and lubiprostone-treated patients, respectively. The Brief Pain Inventory-Short Form (BPI-SF) questionnaire was administered to patients at baseline and monthly during the treatment period to assess pain control. Patients had documented opioid-induced constipation at baseline, defined as having less than 3 spontaneous bowel movements (SBMs) per week, with at least 25% of SBMs associated with one or more of the following conditions: (1) hard to very hard stool consistency; (2) moderate to very severe straining; and/or (3) having a sensation of incomplete evacuation. Laxative use was discontinued at the beginning of the screening period and throughout the study. With the exception of the 48-hour period prior to first dose and for at least 72 hours (Study 1) or 1 week (Study 2 and Study 3) following first dose, use of rescue medication was allowed in cases where no bowel movement had occurred in a 3-day period. Median weekly SBM frequencies at baseline were 1.5 for placebo patients and 1.0 for lubiprostone patients in Study 1 and, for both Study 2 and Study 3, median weekly SBM frequencies at baseline were 1.5 for both treatment groups.
In Study 1, patients receiving non-diphenylheptane (e.g., non-methadone) opioids (n = 431) were randomized to receive placebo (n = 217) or lubiprostone 24 mcg twice daily (n = 214) for 12 weeks. The primary efficacy analysis was a comparison of the proportion of "overall responders" in each treatment arm. A patient was considered an "overall responder" if ≥1 SBM improvement over baseline were reported for all treatment weeks for which data were available and≥3 SBMs/week were reported for at least 9 of 12 treatment weeks. The proportion of patients in Study 1 qualifying as an "overall responder" was 27.1% in the group receiving lubiprostone 24 mcg twice daily compared to 18.9% of patients receiving placebo twice daily (treatment difference = 8.2%; p-value = 0.03). Examination of gender and race subgroups did not identify differences in response to lubiprostone among these subgroups. There were too few elderly patients (≥ 65 years of age) to adequately assess differences in effects in that population.
In Study 2, patients receiving opioids (N = 418) were randomized to receive placebo (n = 208) or lubiprostone 24 mcg twice daily (n = 210) for 12 weeks. Study 2 did not exclude patients receiving diphenylheptane opioids (e.g., methadone). The primary efficacy endpoint was the mean change from baseline in SBM frequency at Week 8; 3.3 vs. 2.4 for lubiprostone and placebo-treated patients, respectively; treatment difference = 0.9; p-value = 0.004. The proportion of patients in Study 2 qualifying as an "overall responder," as prespecified in Study 1, was 24% in the group receiving lubiprostone compared to 15% of patients receiving placebo. In the subgroup of patients in Study 2 taking diphenylheptane opioids (baseline mean [median] MEDDs of 691 [403] mg and 672 [450] mg for placebo and lubiprostone patients, respectively), the proportion of patients qualifying as an "overall responder" was 20.5% (8/39) in the group receiving lubiprostone compared to 6.3% (2/32) of patients receiving placebo. Examination of gender and race subgroups did not identify differences in response to lubiprostone among these subgroups. There were too few elderly patients (≥ 65 years of age) to adequately assess differences in effects in that population.
In Study 3, patients receiving opioids (N = 451) were randomized to placebo (n = 216) or lubiprostone 24 mcg twice daily (n = 235) for 12 weeks. Study 3 did not exclude patients receiving diphenylheptane opioids (e.g., methadone). The primary efficacy endpoint was the change from baseline in SBM frequency at Week 8. The study did not demonstrate a statistically significant improvement in SBM frequency rates at Week 8 (mean change from baseline of 2.7 vs. 2.5 for lubiprostone and placebo-treated patients, respectively; treatment difference = 0.2; p-value = 0.76). The proportion of patients in Study 3 qualifying as an "overall responder," as prespecified in Study 1, was 15% in the patients receiving lubiprostone compared to 13% of patients receiving placebo. In the subgroup of patients in Study 3 taking diphenylheptane opioids (baseline mean [median] MEDDs of 730 [518] mg and 992 [480] mg for placebo and lubiprostone patients, respectively), the proportion of patients qualifying as an "overall responder" was 2% (1/47) in the group receiving lubiprostone compared to 12% (5/41) of patients receiving placebo.
14.3 Irritable Bowel Syndrome with Constipation
Two double-blinded, placebo-controlled studies of similar design were conducted in adult patients with IBS-C. IBS was defined as abdominal pain or discomfort occurring over at least 6 months with two or more of the following: 1) relieved with defecation; 2) onset associated with a change in stool frequency; and 3) onset associated with a change in stool form. Patients were sub-typed as having IBS-C if they also experienced two of three of the following: 1) <3 spontaneous bowel movements (SBMs) per week, 2) >25% hard stools, and 3) >25% SBMs associated with straining.
Following a 4-week baseline/washout period, a total of 1154 patients (mean age 47 [range 18 to 85] years; 92% female; 77% Caucasian, 13% African American, 9% Hispanic, 0.4% Asian; 8% at least 65 years of age) were randomized and received lubiprostone 8 mcg twice daily (16 mcg/day) or placebo twice daily for 12 weeks. The primary efficacy endpoint was assessed weekly utilizing the patient's response to a global symptom relief question based on a 7-point, balanced scale ("significantly worse" to "significantly relieved"): "How would you rate your relief of IBS symptoms (abdominal discomfort/pain, bowel habits, and other IBS symptoms) over the past week compared to how you felt before you entered the study?"
The primary efficacy analysis was a comparison of the proportion of "overall responders" in each arm. A patient was considered an "overall responder" if the criteria for being designated a "monthly responder" were met in at least 2 of the 3 months on study. A "monthly responder" was defined as a patient who had reported "significantly relieved" for at least 2 weeks of the month or at least "moderately relieved" in all 4 weeks of that month. During each monthly evaluation period, patients reporting "moderately worse" or "significantly worse" relief, an increase in rescue medication use, or those who discontinued due to lack of efficacy, were deemed non-responders.
The percentage of patients in Study 1 qualifying as an "overall responder" was 14% in the group receiving lubiprostone 8 mcg twice daily compared to 8% of patients receiving placebo twice daily. In Study 2, 12% of patients in the lubiprostone 8 mcg group were "overall responders" versus 6% of patients in the placebo group. In both studies, the treatment differences between the placebo and lubiprostone groups were statistically significant.
Results in men:The two randomized, placebo-controlled, double-blinded studies comprised 97 (8%) male patients, which is insufficient to determine whether men with IBS-C respond differently to lubiprostone from women.
During a 4-week randomized withdrawal period following Study 1, patients who received lubiprostone during the 12-week treatment period were re-randomized to receive either placebo or to continue treatment with lubiprostone. In lubiprostone-treated patients who were "overall responders" during Study 1 and who were re-randomized to placebo, SBM frequency rates did not result in worsening compared to baseline.
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Administration Instructions
- Instruct patients to take lubiprostone orally with food and water to reduce the occurrence of nausea [see Warnings and Precautions (5.1)] .
- Swallow capsules whole and do not break apart or chew.
- Physicians and patients should periodically assess the need for continued therapy.
Diarrhea
Inform patients that they may experience diarrhea during treatment with lubiprostone. Instruct patients to discontinue lubiprostone and contact their healthcare provider if severe diarrhea occurs [see Warnings and Precautions (5.2)].
Syncope and Hypotension
Inform patients that they may experience syncope and hypotension after taking the first dose or subsequent doses of lubiprostone. Syncope and hypotension generally resolve prior to the next dose, but may recur with repeat dosing. Instruct patients to discontinue Lubiprostone and to contact their healthcare provider if these reactions occur [see Warnings and Precautions (5.3)] . Inform patients that other adverse reactions may increase the risk of syncope and hypotension, such as diarrhea or vomiting.
Dyspnea
Inform patients that they may experience dyspnea within an hour of the first dose. Dyspnea generally resolves within 3 hours, but may recur with repeat dosing. Instruct patients to inform their healthcare provider if dyspnea occurs [see Warnings and Precautions (5.4)] .
Lactation
Advise lactating women to monitor their human milk-fed infants for diarrhea while taking lubiprostone [see Use in Specific Populations (8.2)] .
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 24 mcg Capsule Bottle Label
-
INGREDIENTS AND APPEARANCE
LUBIPROSTONE
lubiprostone capsule, gelatin coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:80425-0355(NDC:63304-352) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LUBIPROSTONE (UNII: 7662KG2R6K) (LUBIPROSTONE - UNII:7662KG2R6K) LUBIPROSTONE 24 ug Inactive Ingredients Ingredient Name Strength MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SORBITOL (UNII: 506T60A25R) FD&C RED NO. 40 (UNII: WZB9127XOA) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) WATER (UNII: 059QF0KO0R) Product Characteristics Color orange Score no score Shape OVAL (oval, soft gelatin capsule) Size 9mm Flavor Imprint Code SPI Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:80425-0355-2 60 in 1 BOTTLE; Type 0: Not a Combination Product 08/16/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA021908 08/16/2023 Labeler - Advanced Rx Pharmacy of Tennessee, LLC (117023142) Establishment Name Address ID/FEI Business Operations Advanced Rx Pharmacy of Tennessee, LLC 117023142 repack(80425-0355)
