Label: LUMIZYME- alglucosidase alfa injection, powder, for solution

- NDC Code(s): 58468-0160-1, 58468-0160-2
- Packager: Genzyme Corporation
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated April 4, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LUMIZYME safely and effectively. See full prescribing information for LUMIZYME.
LUMIZYME® (alglucosidase alfa), for injection, for intravenous use
Initial U.S. Approval: 2010WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS, IMMUNE-MEDIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE
See full prescribing information for complete boxed warning.
Hypersensitivity Reactions Including Anaphylaxis
Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available. If a severe reaction occurs, discontinue LUMIZYME immediately and initiate appropriate medical treatment. (5.1)
Immune-Mediated Reactions
Immune-mediated reactions presenting as proteinuria, nephrotic syndrome, and necrotizing skin lesions have occurred in some patients following LUMIZYME treatment. Monitor patients for the development of systemic immune-mediated reactions involving skin and other organs while receiving LUMIZYME. (5.3)
Risk of Acute Cardiorespiratory Failure
Infantile-onset Pompe disease patients with compromised cardiac or respiratory function may be at risk of serious acute exacerbation of their cardiac or respiratory compromise due to fluid overload and require additional monitoring. (5.4)
RECENT MAJOR CHANGES
Boxed Warnings 3/2024 Dosage and Administration (2.1, 2.2) 3/2024 Warnings and Precautions (5.1, 5.2) 3/2024 INDICATIONS AND USAGE
LUMIZYME® is a hydrolytic lysosomal glycogen-specific enzyme indicated for patients with Pompe disease (GAA deficiency). (1)
DOSAGE AND ADMINISTRATION
- 20 mg per kg body weight administered every 2 weeks as an intravenous infusion. (2)
DOSAGE FORMS AND STRENGTHS
- For injection: 50 mg of LUMIZYME as lyophilized powder in a single-dose vial for reconstitution. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Infusion Associated Reactions (IARs): If an IAR occurs, decreasing the infusion rate, temporarily stopping the infusion, and/or administration of antihistamines and/or antipyretics may ameliorate the symptoms. (5.2)
- Risk of Cardiac Arrhythmia and Sudden Cardiac Death during General Anesthesia for Central Venous Catheter Placement: Caution should be used when administering general anesthesia for the placement of a central venous catheter intended for LUMIZYME infusion. (5.5)
- Risk of Antibody Development: Patients with infantile-onset Pompe disease should have a cross-reactive immunologic material (CRIM) assessment early in their disease course and be managed by a clinical specialist knowledgeable in immune tolerance induction in Pompe disease to optimize treatment. (5.6)
ADVERSE REACTIONS
The most frequently reported adverse reactions (≥5%) in clinical trials were hypersensitivity reactions and included: anaphylaxis, rash, pyrexia, flushing/feeling hot, urticaria, headache, hyperhidrosis, nausea, cough, decreased oxygen saturation, tachycardia, tachypnea, chest discomfort, dizziness, muscle twitching, agitation, cyanosis, erythema, hypertension/increased blood pressure, pallor, rigors, tremor, vomiting, fatigue, and myalgia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme at 1-800-633-1610, option 1 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS, IMMUNE-MEDIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommendations prior to LUMIZYME Treatment
2.2 Recommended Dosage and Administration
2.3 Reconstitution and Dilution Instructions
2.4 Storage Instructions for the Reconstituted and Diluted Product
2.5 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
5.2 Infusion Associated Reactions (IARs)
5.3 Immune-Mediated Reactions
5.4 Risk of Acute Cardiorespiratory Failure
5.5 Risk of Cardiac Arrhythmia and Sudden Cardiac Death during General Anesthesia for Central Venous Catheter Placement
5.6 Risk of Antibody Development
5.7 Monitoring: Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Interference with other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Clinical Trials in Infantile-Onset Pompe Disease
14.2 Clinical Trials in Late-Onset Pompe Disease
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS, IMMUNE-MEDIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE
Hypersensitivity Reactions Including Anaphylaxis
Patients treated with LUMIZYME have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during LUMIZYME administration. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue LUMIZYME immediately and initiate appropriate medical treatment [see Warnings and Precautions (5.1)].
Immune-Mediated Reactions
Immune-mediated reactions presenting as proteinuria, nephrotic syndrome, and necrotizing skin lesions have occurred in some patients following LUMIZYME treatment. Monitor patients for the development of systemic immune-mediated reactions involving skin and other organs while receiving LUMIZYME [see Warnings and Precautions (5.3)].
Risk of Acute Cardiorespiratory Failure
Infantile-onset Pompe disease patients with compromised cardiac or respiratory function may be at risk of serious acute exacerbation of their cardiac or respiratory compromise due to fluid overload and require additional monitoring [see Warnings and Precautions (5.4)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommendations prior to LUMIZYME Treatment
- Prior to LUMIZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids [see Warnings and Precautions (5.1, 5.2)].
- LUMIZYME must be reconstituted and diluted prior to use [see Dosage and Administration (2.5)].
- Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during LUMIZYME administration [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage and Administration
-
The recommended dosage of LUMIZYME is 20 mg/kg body weight administered every 2 weeks as an intravenous infusion. The initial infusion rate should be no more than 1 mg/kg/hr [see Dosage and Administration (2.5)].
2.3 Reconstitution and Dilution Instructions
Reconstitute and dilute LUMIZYME in the following manner.
Use aseptic technique during preparation. Do not use filter needles during preparation.
Reconstitute the Lyophilized Powder
- Determine the number of LUMIZYME vials to be reconstituted based on the actual body weight in kg and the recommended dose of 20 mg/kg. Round the number of vials up to the next whole number.
- Remove the required number of LUMIZYME vials from the refrigerator and allow the vials to sit for approximately 30 minutes at reach room temperature 20°C to 25°C (68°F to 77°F) prior to reconstitution.
- Reconstitute each vial by slowly injecting 10.3 mL of Sterile Water for Injection, down the inside wall of each vial. Avoid adding the Sterile Water for Injection to the vial forcefully or directly onto the lyophilized powder to minimize foaming.
- Gently tilt and roll each vial. Do not invert, swirl, or shake the vial. Each vial will yield a concentration of 5 mg/mL of LUMIZYME. The total extractable dose per vial is 50 mg per 10 mL.
- Visually inspect the reconstituted solution in the vials for particulate matter and discoloration. Discard if particles are present or the solution is discolored. The reconstituted solution may occasionally contain some LUMIZYME particles (typically less than 10 in a vial) in the form of thin white strands or translucent fibers subsequent to the initial inspection. This may also happen following dilution for infusion. These particles have been shown to contain LUMIZYME and may appear after the initial reconstitution step and increase over time. Studies have shown that these particles are removed via in-line filtration without having a detectable effect on the purity or strength.
Dilute the Reconstituted Solution
- Select and prepare an appropriate size 0.9% Sodium Chloride for Injection infusion bag with quantity sufficient of 0.9% Sodium Chloride for Injection to obtain the recommended total infusion volume per table 1 based on patient weight. and dilute.
- Slowly withdraw the required volume of reconstituted solution from the LUMIZYME vial(s). Avoid foaming in the syringe. Discard any unused reconstituted solution remaining in the vial.
- Remove airspace from the prepared 0.9% Sodium Chloride for Injection infusion bag to minimize particle formation due to the sensitivity of LUMIZYME to air-liquid interfaces.
- Inject the LUMIZYME reconstituted solution slowly and directly into the port of the prepared 0.9% Sodium Chloride for Injection infusion bag. Avoid foaming and introducing air in the infusion bag.
- Gently invert or massage the infusion bag to mix the solution. Do not shake. After dilution, the solution will have a final concentration of 0.5 to 4 mg/mL of LUMIZYME.
2.4 Storage Instructions for the Reconstituted and Diluted Product
- The reconstituted and diluted solution should be administered without delay. Storage of the reconstituted solution at room temperature is not recommended.
- If immediate use is not possible, the reconstituted and diluted solution is stable for up to 24 hours refrigerated at 2°C to 8°C (36°F to 46°F).
- The reconstituted and diluted LUMIZYME solution should be protected from light.
- Do not freeze or shake.
2.5 Administration Instructions
- The total volume of infusion is determined by the patient's body weight and should be administered over approximately 4 hours.
- Administer LUMIZYME using an in-line low protein binding 0.2-micron filter.
- Infusions should be administered in a step-wise manner using an infusion pump. The initial infusion rate should be no more than 1 mg/kg/hr. The infusion rate may be increased by 2 mg/kg/hr every 30 minutes, after patient tolerance to the infusion rate is established, until a maximum rate of 7 mg/kg/hr is reached.
- Vital signs should be obtained at the end of each step. If the patient is stable, LUMIZYME may be administered at the maximum rate of 7 mg/kg/hr until the infusion is completed.
- The infusion rate may be slowed or temporarily stopped in the event of mild to moderate hypersensitivity reactions. In the event of anaphylaxis or severe hypersensitivity reaction, immediately discontinue administration of LUMIZYME and initiate appropriate medical treatment. See Table 1 below for the rate of infusion at each step, expressed as mL/hr based on the recommended infusion volume by patient weight.
- Do not infuse LUMIZYME in the same intravenous line with other products. Discard any unused product.
Table 1: Recommended LUMIZYME Infusion Volumes and Incremental Rate Steps by Patient Weight Patient Weight Range Total infusion volume Step 1
1 mg/kg/hrStep 2
3 mg/kg/hrStep 3
5 mg/kg/hrStep 4
7 mg/kg/hrInfusion Rate in mL/hr 1.25 to 2.5 kg 25 mL 1.25 3.75 6.25 6.6 2.6 to 10 kg 50 mL 3 8 13 18 10.1 to 20 kg 100 mL 5 15 25 35 20.1 to 30 kg 150 mL 8 23 38 53 30.1 to 35 kg 200 mL 10 30 50 70 35.1 to 50 kg 250 mL 13 38 63 88 50.1 to 60 kg 300 mL 15 45 75 105 60.1 to 100 kg 500 mL 25 75 125 175 100.1 to 120 kg 600 mL 30 90 150 210 120.1 to 140 kg 700 mL 35 105 175 245 140.1 to 160 kg 800 mL 40 120 200 280 160.1 to 180 kg 900 mL 45 135 225 315 180.1 to 200 kg 1,000 mL 50 150 250 350 - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
Anaphylaxis and hypersensitivity reactions have been observed in patients during and up to 3 hours after LUMIZYME infusion. Some of the reactions were life-threatening and included anaphylactic shock, cardiac arrest, respiratory arrest, respiratory distress, hypoxia, apnea, dyspnea, bradycardia, tachycardia, bronchospasm, throat tightness, hypotension, angioedema (including tongue or lip swelling, periorbital edema, and face edema), and urticaria. Other accompanying reactions included chest discomfort/pain, wheezing, tachypnea, cyanosis, decreased oxygen saturation, convulsions, pruritus, rash, hyperhidrosis, nausea, dizziness, hypertension/increased blood pressure, flushing/feeling hot, erythema, pyrexia, pallor, peripheral coldness, restlessness, nervousness, headache, back pain, and paresthesia. Some of these reactions were IgE-mediated.
Prior to LUMIZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids. Appropriate medical support, including cardiopulmonary resuscitation equipment, should be readily available when LUMIZYME is administered.
If anaphylaxis or severe hypersensitivity reactions occur, immediately discontinue administration of LUMIZYME, and initiate appropriate medical treatment. Severe reactions are generally managed with infusion interruption, administration of antihistamines, corticosteroids, intravenous fluids, and/or oxygen, when clinically indicated. In some cases of anaphylaxis, epinephrine has been administered.
The risks and benefits of readministering LUMIZYME following an anaphylactic or hypersensitivity reaction should be considered. Some patients have been rechallenged and have continued to receive LUMIZYME under close clinical supervision. Extreme care should be exercised, with appropriate resuscitation measures available, if the decision is made to readminister the product. Patients may be rechallenged using slower infusion rates at a dosage lower than the recommended dosage [see Adverse Reactions (6.2)].
If a mild or moderate hypersensitivity reaction occurs, consider temporarily holding the infusion or slowing the infusion rate.
5.2 Infusion Associated Reactions (IARs)
Infusion associated reactions (IARs) such as pyrexia, chills, flu-like illness, myalgia, arthralgia, pain, fatigue, urticaria, rash, pruritus, erythema, dyspnea, tachycardia, flushing, nausea, headache and syncope have been observed in patients treated with LUMIZYME [see Adverse Reactions (6.3)].
If an IAR occurs, decreasing the infusion rate, temporarily stopping the infusion, and/or administering antihistamines and/or antipyretics may ameliorate the symptoms. Closely monitor patients who have experienced IARs when re-administering LUMIZYME.
5.3 Immune-Mediated Reactions
Immune-mediated cutaneous reactions have been reported with LUMIZYME including necrotizing skin lesions [see Adverse Reactions (6.3)]. Systemic immune-mediated reactions, including possible type III immune-mediated reactions have been observed with LUMIZYME. These reactions occurred several weeks to 3 years after initiation of LUMIZYME infusions. Skin biopsy in one patient demonstrated deposition of anti-rhGAA antibodies in the lesion. Another patient developed severe inflammatory arthropathy in association with pyrexia and elevated erythrocyte sedimentation rate. Nephrotic syndrome secondary to membranous glomerulonephritis was observed in some Pompe disease patients treated with LUMIZYME who had persistently positive anti-rhGAA IgG antibody titers. In these patients, renal biopsy was consistent with immune complex deposition. Patients improved following treatment interruption. Therefore, patients receiving LUMIZYME should undergo periodic urinalysis [see Adverse Reactions (6.3)].
Patients should be monitored for the development of systemic immune-mediated reactions involving skin and other organs while receiving LUMIZYME. If immune-mediated reactions occur, consider discontinuation of the administration of LUMIZYME, and initiate appropriate medical treatment. The risks and benefits of readministering LUMIZYME following an immune-mediated reaction should be considered. Some patients have been able to be rechallenged and have continued to receive LUMIZYME under close clinical supervision. Immune tolerance induction administered in conjunction with LUMIZYME may also aide tolerability of LUMIZYME under the management of a clinical specialist knowledgeable in immune tolerance induction in pediatric Pompe disease.
5.4 Risk of Acute Cardiorespiratory Failure
Patients with acute underlying respiratory illness or compromised cardiac and/or respiratory function may be at risk of serious exacerbation of their cardiac or respiratory compromise during infusions. Appropriate medical support and monitoring measures should be readily available during LUMIZYME infusion, and some patients may require prolonged observation times that should be individualized based on the needs of the patient. Acute cardiorespiratory failure has been observed in infantile-onset Pompe disease patients with underlying cardiac hypertrophy, possibly associated with fluid overload with intravenous administration of LUMIZYME [see Dosage and Administration (2.2)].
5.5 Risk of Cardiac Arrhythmia and Sudden Cardiac Death during General Anesthesia for Central Venous Catheter Placement
Administration of general anesthesia can be complicated by the presence of severe cardiac and skeletal (including respiratory) muscle weakness. Therefore, caution should be used when administering general anesthesia. Ventricular arrhythmias and bradycardia, resulting in cardiac arrest or death, or requiring cardiac resuscitation or defibrillation have been observed in infantile-onset Pompe disease patients with cardiac hypertrophy during general anesthesia for central venous catheter placement.
5.6 Risk of Antibody Development
Patients with infantile-onset Pompe disease should have a cross-reactive immunologic material (CRIM) assessment early in their disease course and be managed by a clinical specialist knowledgeable in immune tolerance induction in Pompe disease to optimize treatment. Immune tolerance induction administered prior to and in conjunction with initiation of LUMIZYME has been reported to aide tolerability of LUMIZYME in CRIM-negative patients. CRIM status has been shown to be associated with immunogenicity and patients' responses to enzyme replacement therapies. CRIM-negative infants with infantile-onset Pompe disease treated with LUMIZYME have shown poorer clinical response in the presence of high sustained IgG antibody titers and positive inhibitory antibodies compared to CRIM-positive infants [see Adverse Reactions (6.2)].
In clinical studies, the majority of patients developed IgG antibodies to LUMIZYME, typically within 3 months of treatment. There is evidence to suggest that some patients who develop high and sustained IgG antibody titers, including CRIM-negative patients (i.e., patients in whom no endogenous GAA protein was detected by Western blot analysis and/or predicted based on the genotype), may experience reduced clinical LUMIZYME treatment efficacy, such as loss of motor function, ventilator dependence, or death.
5.7 Monitoring: Laboratory Tests
Patients should be monitored for IgG antibody formation every 3 months for 2 years and then annually thereafter. Testing for IgG titers may also be considered if patients develop hypersensitivity reactions, other immune-mediated reactions, or lose clinical response. Patients who experience reduced clinical response may also be tested for inhibitory antibody activity. Patients who experience anaphylactic or hypersensitivity reactions may also be tested for IgE antibodies to LUMIZYME and other mediators of anaphylaxis [see Adverse Reactions (6.2)].
Testing services for antibodies against LUMIZYME are available through Genzyme Corporation. Contact Genzyme Corporation at 1-800-745-4447 for information on testing.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The following serious adverse reactions are described below and elsewhere in the labeling:
- Anaphylaxis and hypersensitivity reactions [see Warnings and Precautions (5.1)]
In clinical trials, the most common adverse reactions (≥5%) following alglucosidase alfa treatment were hypersensitivity reactions, and included anaphylaxis, rash, pyrexia, flushing/feeling hot, urticaria, headache, hyperhidrosis, nausea, cough, decreased oxygen saturation, tachycardia, tachypnea, chest discomfort, dizziness, muscle twitching, agitation, cyanosis, erythema, hypertension/increased blood pressure, pallor, rigors, tremor, vomiting, fatigue, and myalgia.
Clinical Trials in Infantile-Onset and Juvenile-Onset Pompe Disease
Two multicenter, open-label clinical trials were conducted in 39 infantile-onset Pompe disease patients, ages 1 month to 3.5 years old. Approximately half of the patients (54%) were male. Patients were treated with alglucosidase alfa 20 or 40 mg/kg every other week for periods ranging from 1 to 106 weeks (mean: 61 weeks).
The most serious adverse reactions reported with alglucosidase alfa treatment included anaphylaxis and acute cardiorespiratory failure.
The most common adverse reactions requiring intervention in clinical trials were hypersensitivity reactions, occurring in 20 of 39 (51%) patients treated with alglucosidase alfa, and included rash, pyrexia, urticaria, flushing, decreased oxygen saturation, cough, tachypnea, tachycardia, hypertension/increased blood pressure, pallor, rigors, vomiting, cyanosis, agitation, and tremor. These reactions were more likely to occur with higher infusion rates or doses. Some patients who were pretreated with antihistamines, antipyretics and/or corticosteroids still experienced hypersensitivity reactions.
Table 2 summarizes all adverse reactions occurring in ≥5% of patients (2 or more patients) treated with alglucosidase alfa in clinical trials described above.
Table 2: Adverse Reactions that Occurred in at Least 5% of Infantile-Onset Patients Treated with Alglucosidase Alfa in Clinical Trials Number of Patients
(N=39)
n (%)Adverse Reaction 20 (51) Rash (including rash erythematous, rash macular and maculopapular) 7 (18) Pyrexia 6 (15) Urticaria 5 (13) Flushing 5 (13) Hypertension/Increased Blood Pressure 4 (10) Decreased Oxygen Saturation 3 (8) Cough 3 (8) Tachypnea 3 (8) Tachycardia 3 (8) Erythema 2 (5) Vomiting 2 (5) Rigors 2 (5) Pallor 2 (5) Cyanosis 2 (5) Agitation 2 (5) Tremor 2 (5) An open-label, single-center trial was conducted in 18 treatment-naive infantile-onset Pompe disease patients who were treated exclusively with alglucosidase alfa. Adverse reactions observed in these patients were similar to infantile-onset Pompe disease patients who received alglucosidase alfa in other clinical trials.
Additional hypersensitivity reactions observed in infantile-onset Pompe disease patients treated in other clinical trials and expanded access programs with alglucosidase alfa included livedo reticularis, irritability, retching, increased lacrimation, ventricular extrasystoles, nodal rhythm, rales, respiratory tract irritation, and cold sweat.
Safety was also evaluated in 99 patients (51 male, 48 females) with Pompe disease in an ongoing, open-label, prospective study in patients 12 months of age and older who were previously treated with the 160 L scale of alglucosidase alfa and switched to the 4000 L scale of alglucosidase alfa. Patients were aged 1 to 18 years with a median duration of treatment of 437 days (range 13 to 466 days). No new safety findings were observed following the switch to 4000 L scale of alglucosidase alfa.
Clinical Trials in Late-Onset Pompe Disease
Assessment of adverse reactions in patients with late-onset Pompe disease is based on the exposure of 90 patients (45 male, 45 female), aged 10 to 70 years, to 20 mg/kg alglucosidase alfa or placebo in a randomized, double-blind, placebo-controlled trial. The youngest alglucosidase alfa-treated patient was 16 years of age, and the youngest placebo-treated patient was 10 years of age. All patients were naive to enzyme replacement therapy. Patients were randomized in a 2:1 ratio and received alglucosidase alfa or placebo every other week for 78 weeks (18 months). The study population included 34 males and 26 females (n=60) in the alglucosidase alfa group and 11 males and 19 females (n=30) in the placebo group. Two patients receiving alglucosidase alfa discontinued the trial due to anaphylactic reactions.
Serious adverse reactions reported with alglucosidase alfa included anaphylaxis, which presented as angioedema, throat tightness and chest pain/discomfort. One patient with a history of Wolff-Parkinson-White syndrome experienced a serious adverse reaction of supraventricular tachycardia.
The most common adverse reactions (≥3%; 2 or more patients) observed in alglucosidase alfa-treated patients were hypersensitivity reactions and included anaphylaxis, headache, nausea, urticaria, dizziness, chest discomfort, vomiting, hyperhidrosis, flushing/feeling hot, increased blood pressure, paresthesia, pyrexia, local swelling, diarrhea, pruritus, rash, and throat tightness.
Delayed-onset reactions, defined as adverse reactions occurring 2 to 48 hours after completion of alglucosidase alfa infusion, that were observed in ≥3% more patients in the alglucosidase alfa-treated group compared to patients in the placebo-treated group in the controlled trial, included hyperhidrosis. Additional delayed-onset reactions occurring in alglucosidase alfa-treated patients included fatigue, myalgia, and nausea.
Table 3 summarizes the most common adverse reactions that occurred in at least 3% of alglucosidase alfa-treated patients and with a higher incidence than the placebo-treated patients during the randomized, double-blind, placebo-controlled study described above.
Table 3: Adverse Reactions Occurring in at Least 3% of Alglucosidase Alfa-Treated Late-Onset Patients and with a Higher Incidence than the Placebo-Treated Patients Adverse Reaction Alglucosidase Alfa
n=60
N (%)Placebo
n=30
N (%)Hyperhidrosis 5 (8.3) 0 (0) Urticaria 5 (8.3) 0 (0) Anaphylaxis 4 (6.7) 0 (0) Chest Discomfort 4 (6.7) 1 (3.3) Muscle Twitching 4 (6.7) 1 (3.3) Myalgia 3 (5.0) 1 (3.3) Flushing/Feeling Hot 3 (5.0) 0 (0) Increased Blood Pressure 3 (5.0) 0 (0) Vomiting 3 (5.0) 0 (0) Edema, Peripheral 2 (3.3) 0 (0) Pruritus 2 (3.3) 0 (0) Rash Papular 2 (3.3) 0 (0) Throat Tightness 2 (3.3) 0 (0) In clinical trials, anaphylaxis and hypersensitivity reactions were managed with infusion interruption, decreased infusion rate, administration of antihistamines, corticosteroids, intravenous fluids, and/or oxygen, when clinically indicated. In some cases of anaphylactic reactions, epinephrine was administered. Patients who have experienced anaphylaxis or hypersensitivity reactions should be treated with caution when they are readministered alglucosidase alfa.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other alglucosidase alfa products may be misleading.
In the two clinical trials in infantile-onset patients, the majority of patients (34 of 38; 89%) tested positive for IgG antibodies to alglucosidase alfa. There is evidence to suggest that some patients who develop high sustained titers of anti-alglucosidase alfa antibodies may experience reduced clinical efficacy to alglucosidase alfa treatment [see Warnings and Precautions (5.5)]. Some IgG-positive patients in clinical trials who were retrospectively evaluated for the presence of inhibitory antibodies tested positive for inhibition of enzyme activity and/or uptake in in vitro assays. Furthermore, CRIM-negative infants have shown reduced clinical effect in the presence of high sustained IgG antibody titers with inhibitory activity [see Warnings and Precautions (5.5)]. Alglucosidase alfa-treated patients who experience a decrease in motor function should be tested for the presence of inhibitory antibodies that neutralize enzyme uptake or activity.
Immunogenicity data from clinical trials and published literature in CRIM-negative, infantile-onset Pompe disease patients suggest that the administration of an immune tolerance induction regimen individualized to alglucosidase alfa–naive patients may be effective in preventing or reducing the development of high sustained antibody titer against alglucosidase alfa.
In the randomized, double-blind, placebo-controlled trial in late-onset patients, all alglucosidase alfa-treated patients with available samples (N=59, 100%) developed IgG antibodies to alglucosidase alfa. These patients were all CRIM positive, consistent with late-onset Pompe disease. Most patients who developed IgG antibodies did so within the first 3 months of exposure (median time to seroconversion was 4 weeks). There was no apparent association between mean or peak IgG antibody titers and the occurrence of adverse reactions.
None of the 59 evaluable patients tested positive for inhibition of enzyme activity. Antibody titers for cellular uptake inhibition were present in 18 of 59 (31%) patients by Week 78. All other patients tested negative for inhibition of cellular uptake. Patients who tested positive for uptake inhibition tended to have higher IgG titers than patients who tested negative for uptake inhibition. Among the 32 patients with evaluable pharmacokinetic (PK) samples, 5 patients tested positive for uptake inhibition. The clinical relevance of this in vitro inhibition is not fully understood. The clearance values for 4 of these 5 patients were approximately 1.2-fold to 1.8-fold greater in the presence of inhibitory antibodies (Week 52) as compared to in the absence of inhibitory antibodies (Week 0) [see Clinical Pharmacology (12.3)].
Some patients in the clinical studies or in the postmarketing setting have undergone testing for alglucosidase alfa-specific IgE antibodies. Testing was performed in patients who experienced moderate to severe or recurrent hypersensitivity reactions, for which mast-cell activation was suspected. Some of the patients who tested positive for alglucosidase alfa-specific IgE antibodies experienced anaphylactic reactions [see Boxed Warning and Warnings and Precautions (5.1)].
Some patients who tested positive for alglucosidase alfa-specific IgE antibodies and experienced hypersensitivity reactions were able to be rechallenged with alglucosidase alfa using a slower infusion rate at lower starting doses and have continued to receive treatment under close clinical supervision [see Warnings and Precautions (5.1)]. Since patients who develop IgE antibodies to alglucosidase alfa appear to be at a higher risk for developing anaphylaxis and hypersensitivity reactions, these patients should be monitored more closely during administration of alglucosidase alfa.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of alglucosidase alfa. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In the postmarketing experience with LUMIZYME, serious adverse reactions have been reported, including anaphylaxis [see Boxed Warning and Warnings and Precautions (5.1)]. Acute cardiorespiratory failure, possibly associated with fluid overload, has been reported in infantile-onset Pompe disease patients with pre-existing hypertrophic cardiomyopathy [see Boxed Warning and Warning and Precautions (5.3)].
Infusion associated reactions, including pyrexia, chills, fatigue, urticaria, rash, pruritus, erythema, dyspnea, hypotension, bradycardia, tachycardia, flushing, nausea, headache, and syncope have been reported with alglucosidase alfa.
In addition to the hypersensitivity reactions reported in clinical trials [see Adverse Reactions (6.1)], the following hypersensitivity reactions have been reported in at least 2 patients and included: anaphylactic shock, respiratory failure, respiratory arrest, cardiac arrest, hypoxia, dyspnea, wheezing, convulsions, peripheral coldness, restlessness, nervousness, back pain, stridor, pharyngeal edema, abdominal pain, apnea, muscle spasm, and conjunctivitis. In addition, one case of hyperparathyroidism has been reported.
Systemic and cutaneous immune-mediated reactions, including proteinuria and nephrotic syndrome secondary to membranous glomerulonephritis, and necrotizing skin lesions have been reported in postmarketing safety experience with alglucosidase alfa [see Warnings and Precautions (5.2)].
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Data from postmarketing reports and published case reports with alglucosidase alfa use in pregnant women have not identified a LUMIZYME-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. The continuation of treatment for Pompe disease during pregnancy should be individualized to the pregnant woman. Untreated Pompe disease may result in worsening disease symptoms in pregnant women [see Clinical Considerations].
Reproduction studies performed in mice and rabbits at doses resulting in exposures up to 0.4 or 0.5 times the human steady-state AUC (area under the plasma concentration-time curve), respectively, during the period of organogenesis revealed no evidence of effects on embryo-fetal development. In mice there was an increase in pup mortality during lactation at maternal exposures 0.4 times the human steady-state AUC [see Data].
The background risk of major birth defects and miscarriage in the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Pregnant women and women of reproductive potential should be encouraged to enroll in the Pompe patient registry. The registry will monitor the effect of LUMIZYME on pregnant women and their offspring. For more information, visit www.registrynxt.com or call 1-800-745-4447, extension 15500.
Data
Animal Data
All reproductive studies included pretreatment with diphenhydramine to prevent or minimize hypersensitivity reactions. The effects of alglucosidase alfa were evaluated based on comparison to a control group treated with diphenhydramine alone. Daily intravenous administration of alglucosidase alfa up to 40 mg/kg in mice and rabbits (0.4 and 0.5 times the human steady-state AUC, respectively, at the recommended biweekly dose) during the period of organogenesis had no effects on embryo-fetal development. Administration of 40 mg/kg intravenously every other day in mice (0.4 times the human steady-state AUC at the recommended biweekly dose) during the period of organogenesis through lactation produced an increase in mortality of offspring during the lactation period.
8.2 Lactation
Risk Summary
Available published literature suggests the presence of alglucosidase alfa in human milk. There are no reports of adverse effects of alglucosidase alfa on the breastfed infant. There is no information on the effects of alglucosidase alfa on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for LUMIZYME and any potential adverse effects on the breastfed child from LUMIZYME or from the underlying maternal condition.
Lactating women with Pompe disease treated with LUMIZYME should be encouraged to enroll in the Pompe disease registry [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of alglucosidase alfa have been established in pediatric patients with Pompe disease [see Adverse Reactions (6.2)].
The safety and effectiveness of alglucosidase alfa were assessed in 57 treatment-naive infantile-onset Pompe disease patients, aged 0.2 month to 3.5 years at first infusion, in three separate clinical trials [see Clinical Studies (14.1)].
The safety and effectiveness of alglucosidase alfa were assessed in pediatric patients with late (non-infantile) onset Pompe disease in a randomized, double-blind, placebo-controlled study in 90 patients, including 2 patients 16 years of age or less [see Clinical Studies (14.2)].
Anaphylaxis, hypersensitivity reactions, and acute cardiorespiratory failure have occurred in pediatric patients [see Boxed Warning, Warnings and Precautions (5.1, 5.3)]. Additionally, cardiac arrhythmia and sudden cardiac death have occurred in pediatric patients during general anesthesia for central venous catheter placement [see Warnings and Precautions (5.4)].
8.5 Geriatric Use
The randomized, double-blind, placebo-controlled study of alglucosidase alfa did not include sufficient numbers (n=4) of patients aged 65 years and over to determine whether they respond differently from younger patients [see Clinical Studies (14.1)].
-
11 DESCRIPTION
Alglucosidase alfa is a hydrolytic lysosomal glycogen-specific enzyme encoded by the predominant of nine observed haplotypes of the human acid α-glucosidase (GAA) gene. Alglucosidase alfa is produced by recombinant DNA technology in a Chinese hamster ovary cell line. Alglucosidase alfa degrades glycogen by catalyzing the hydrolysis of α-1,4- and α-1,6- glycosidic linkages of lysosomal glycogen.
Alglucosidase alfa is a glycoprotein with a calculated mass of 99,377 Daltons for the polypeptide chain, and a total mass of approximately 109,000 Daltons, including carbohydrates. Alglucosidase alfa has a specific activity of 3.6 to 5.4 units/mg (one unit is defined as that amount of activity that results in the hydrolysis of 1 micromole of synthetic substrate per minute under specified assay conditions). Alglucosidase alfa is intended for intravenous infusion. It is supplied as a sterile, nonpyrogenic, white to off-white, lyophilized cake or powder for reconstitution with 10.3 mL Sterile Water for Injection, USP. Each 50 mg vial contains 52.5 mg alglucosidase alfa, 210 mg mannitol, 0.5 mg polysorbate 80, 9.9 mg sodium phosphate dibasic heptahydrate, and 31.2 mg sodium phosphate monobasic monohydrate. Following reconstitution as directed, each vial contains 10.5 mL reconstituted solution and a total extractable volume of 10 mL at 5 mg/mL alglucosidase alfa. Alglucosidase alfa does not contain preservatives; each vial is for single dose only.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pompe disease (acid maltase deficiency, glycogen storage disease type II, GSD II, glycogenosis type II) is an inherited disorder of glycogen metabolism caused by the absence or marked deficiency of the lysosomal enzyme GAA.
Alglucosidase alfa provides an exogenous source of GAA. Binding to mannose-6-phosphate receptors on the cell surface has been shown to occur via carbohydrate groups on the GAA molecule, after which it is internalized and transported into lysosomes, where it undergoes proteolytic cleavage that results in increased enzymatic activity. It then exerts enzymatic activity in cleaving glycogen.
12.2 Pharmacodynamics
Clinical pharmacodynamic studies have not been conducted for alglucosidase alfa.
12.3 Pharmacokinetics
The pharmacokinetics of alglucosidase alfa was evaluated in 13 patients with infantile-onset Pompe disease, aged 1 month to 7 months, who received 20 mg/kg (approximately as a 4-hour infusion) or 40 mg/kg (approximately as a 6.5-hour infusion) of alglucosidase alfa every 2 weeks. The measurement of alglucosidase alfa plasma concentration was based on an activity assay using an artificial substrate. Systemic exposure was approximately dose proportional between the 20 and 40 mg/kg doses. Based on the pharmacokinetic blood samples collected for 12 hours after a 4-hour intravenous infusion of 20 mg/kg (n=5), the estimated mean AUC was 811 mcg∙hr/mL with 17% coefficient of variation [CV], Cmax was 162 mcg/mL with 19% CV, clearance was 25 mL/hr/kg with 16% CV, and half-life was 2.3 hours with 17% CV.
The pharmacokinetics of alglucosidase alfa was also evaluated in a separate trial of 14 patients with infantile-onset Pompe disease, aged 6 months to 3.5 years, who received 20 mg/kg of alglucosidase alfa as a 4-hour infusion every 2 weeks. The pharmacokinetic parameters were similar to those observed for the infantile-onset Pompe disease patients aged 1 month to 7 months who received the 20 mg/kg dose.
Nineteen of 21 patients who received treatment with alglucosidase alfa and had pharmacokinetics and antibody titer data available at Week 12 developed antibodies to alglucosidase alfa. Five patients with antibody titers ≥12,800 at Week 12 had an average increase in clearance of 50% (range 5% to 90%) from Week 1 to Week 12. The other 14 patients with antibody titers <12,800 at Week 12 had similar average clearance values at Week 1 and Week 12.
The pharmacokinetics of alglucosidase alfa was evaluated in another trial of 10 adult and 10 pediatric patients with Pompe disease who received a single dose of 20 mg/kg of alglucosidase alfa as a 4-hour infusion. In pediatric patients, aged 7 months to 13.7 years, the estimated mean AUC was 1,110 mcg∙hr/mL with 68% CV and Cmax was 204 mcg/mL with 46% CV. In adult patients, aged 19 to 57 years, the estimated mean AUC was 1,890 mcg∙hr/mL with 51% CV and Cmax was 307 mcg/mL with 47% CV.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate carcinogenic potential or studies to evaluate mutagenic potential have not been performed with alglucosidase alfa.
Intravenous administration of alglucosidase alfa every other day in mice at doses up to 40 mg/kg (0.4 times the human AUC at the recommended biweekly dose) had no effect on fertility and reproductive performance.
-
14 CLINICAL STUDIES
14.1 Clinical Trials in Infantile-Onset Pompe Disease
The safety and efficacy of alglucosidase alfa were assessed in 57 treatment-naive infantile-onset Pompe disease patients, aged 0.2 month to 3.5 years at first infusion, in three separate clinical trials.
Study 1 was an international, multicenter, open-label, clinical trial of 18 infantile-onset Pompe disease patients. This study was conducted between 2003 and 2005. Patients were randomized 1:1 to receive either 20 mg/kg or 40 mg/kg alglucosidase alfa every two weeks, with length of treatment ranging from 52 to 106 weeks. Enrollment was restricted to patients 7 months of age or younger at first infusion with clinical signs of Pompe disease and cardiac hypertrophy, and who did not require ventilatory support at study entry. Fourteen patients were cross reactive immunologic material (CRIM) positive and 4 patients were CRIM negative.
Efficacy was assessed by comparing the proportions of alglucosidase alfa-treated patients who died or needed invasive ventilator support at 18 months of age with the mortality experience of a historical cohort of untreated infantile-onset Pompe disease patients with similar age and disease severity. In the historical cohort, 61 untreated patients with infantile-onset Pompe disease diagnosed by age 6 months, born between 1982 and 2002, were identified by a retrospective review of medical charts. By 18 months of age, 15 of 18 (83%) alglucosidase alfa-treated patients were alive without invasive ventilatory support and 3 (17%) required invasive ventilator support, whereas only one of the 61 (2%) historical control patients was alive. No differences in outcome were observed between patients who received 20 mg/kg versus 40 mg/kg.
Other outcome measures in this study included unblinded assessments of motor function by the Alberta Infant Motor Scale (AIMS), a measure of infant motor performance that assesses motor maturation of the infant through age 18 months. Although gains in motor function were noted in 13 patients, the motor function was substantially delayed compared to normal infants of comparable age in the majority of patients. Two of 9 patients who had initially demonstrated gains in motor function after 12 months of alglucosidase alfa treatment regressed despite continued treatment.
Changes from baseline to Month 12 in left ventricular mass index (LVMI), a measure of pharmacodynamic effect, were evaluated by echocardiography. Fifteen patients who underwent both baseline and Month 12 echocardiograms demonstrated decreases from baseline in LVMI (mean decrease 118 g/m2, range 45 to 193 g/m2). However, the magnitude of the decrease in LVMI did not correlate with the clinical outcome measure of ventilator-free survival.
Study 2 was an international, multicenter, non-randomized, open-label clinical trial that enrolled 21 infantile-onset patients aged 3 months to 3.5 years at first infusion. Eighteen patients were CRIM positive and 3 patients were CRIM negative. All patients received 20 mg/kg alglucosidase alfa every other week for up to 104 weeks. Five of 21 patients were receiving invasive ventilatory support at the time of first infusion.
The primary outcome measure was the proportion of patients alive at the conclusion of treatment. At the 52–week interim analysis, 16 of 21 patients were alive. Sixteen patients were free of invasive ventilatory support at the time of first infusion; of these, 4 died, 2 required invasive ventilatory support, and 10 were free of invasive ventilatory support after 52 weeks of treatment. For the 5 patients who were receiving invasive ventilatory support at baseline, 1 died, and 4 remained on invasive ventilatory support at Week 52.
Study 3 was an open-label, single-center trial in 18 infantile-onset Pompe disease patients who had a confirmed diagnosis of Pompe disease as identified through a newborn screening program. All patients were CRIM positive. Patients were treated with alglucosidase alfa prior to 6 months of age (0.2 to 5.8 months at first infusion). Sixteen patients reached 18 months of age at the time of analysis, and all (100%) were alive without invasive ventilator support.
14.2 Clinical Trials in Late-Onset Pompe Disease
The safety and efficacy of alglucosidase alfa were assessed in 90 patients with late-onset Pompe disease, aged 10 to 70 years, in a randomized, double-blind, placebo-controlled trial. The youngest alglucosidase alfa-treated patient was 16 years of age, and the youngest placebo-treated patient was 10 years of age. All patients were naive to enzyme replacement therapy. Patients were allocated in a 2:1 ratio and received 20 mg/kg alglucosidase alfa (n=60) or placebo (n=30) every other week for 78 weeks (18 months). The study population included 34 males and 26 females (n=60) in the alglucosidase alfa group and 11 males and 19 females (n=30) in the placebo group. At baseline, all patients were ambulatory (some required assistive walking devices), did not require invasive ventilator support or non-invasive ventilation while awake and sitting upright, and had a forced vital capacity (FVC) between 30 and 79% of predicted in the sitting position. Patients who could not walk 40 meters in 6 minutes or were unable to perform appropriate pulmonary and muscle function testing were excluded from the study.
A total of 81 of 90 patients completed the trial. Of the 9 patients who discontinued, 5 were in the alglucosidase alfa group and 4 were in the placebo group. Three patients discontinued the study due to an adverse event; two patients were in the alglucosidase alfa treatment group and one patient was in placebo group.
At study entry, the mean % predicted FVC in the sitting position among all patients was about 55%. After 78 weeks, the mean % predicted FVC increased to 56.2% for alglucosidase alfa-treated patients and decreased to 52.8% for placebo-treated patients indicating an alglucosidase alfa treatment effect of 3.4% (95% confidence interval: [1.3% to 5.5%]; p=0.004). Stabilization of % predicted FVC in the alglucosidase alfa-treated patients was observed (see Figure 1).
Figure 1: Mean FVC Upright (% Predicted) Over Time
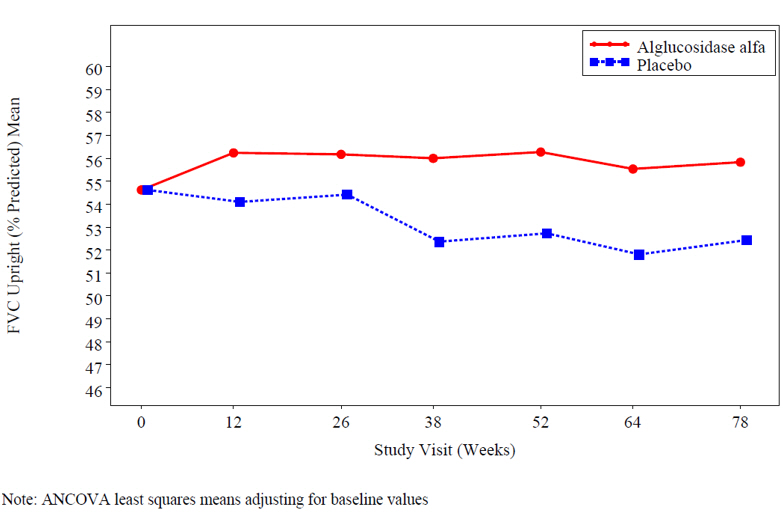
At study entry, the mean 6 minute walk test (6MWT) among all patients was about 330 meters. After 78 weeks, the mean 6MWT increased by 25 meters for alglucosidase alfa-treated patients and decreased by 3 meters for placebo-treated patients indicating an alglucosidase alfa treatment effect of 28 meters (95% confidence interval: [-1 to 52 meters]; p=0.06) (see Figure 2).
Figure 2: Mean Six Minute Walk Test Total Distance Walked Over Time
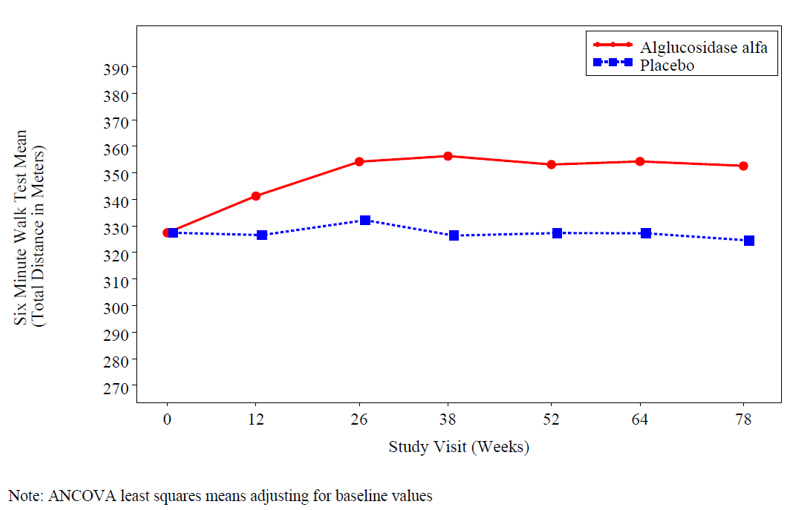
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions Including Anaphylaxis, Infusion-Associated Reactions (IARs) and Immune-Mediated Reactions
Advise the patient and caregiver that reactions related to the infusion may occur during and after LUMIZYME treatment, including life-threatening hypersensitivity reactions, including anaphylaxis, IARs, and immune-mediated reactions.
Advise the patient and caregiver that delayed-onset of hypersensitivity reactions may occur.
Inform the patients and/or caregiver of the signs and symptoms of hypersensitivity reactions including anaphylaxis, IARs, and immune-mediated reactions and have them seek immediate medical care should these signs and symptoms occur [see Warning and Precautions (5.1, 5.2, 5.3)].
Risk of Acute Cardiorespiratory Failure
Advise the patient and caregiver that patients with underlying respiratory illness or compromised cardiac or respiratory function may be at risk of acute cardiorespiratory failure. Patients with compromised cardiac or respiratory function may require close observation during and after alglucosidase alfa administration.
Pompe Registry
Inform the patient and caregiver that the Pompe Registry has been established in order to better understand the variability and progression of Pompe disease, and to continue to monitor and evaluate long-term treatment effects of alglucosidase alfa. The Pompe Registry will also monitor the effect of alglucosidase alfa on pregnant women and their offspring [see Use in Specific Populations (8.1)]. Patients and their caregivers should be encouraged to participate in the Pompe Registry and advised that their participation is voluntary and may involve long-term follow-up. For more information regarding the registry program, visit www.registrynxt.com or call 1-800-745-4447, extension 15500.
- SPL UNCLASSIFIED SECTION
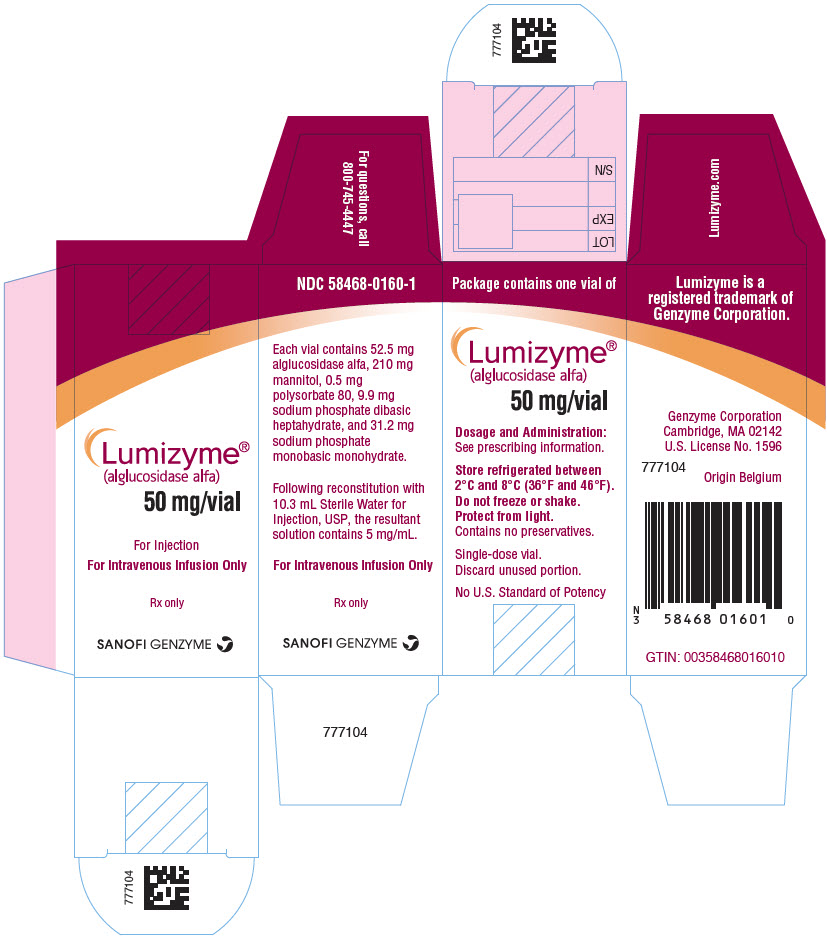
- PRINCIPAL DISPLAY PANEL - 50 mg Vial Carton
-
INGREDIENTS AND APPEARANCE
LUMIZYME
alglucosidase alfa injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:58468-0160 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALGLUCOSIDASE ALFA (UNII: DTI67O9503) (ALGLUCOSIDASE ALFA - UNII:DTI67O9503) ALGLUCOSIDASE ALFA 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 20 mg in 1 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 2.97 mg in 1 mL SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) 0.94 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.05 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:58468-0160-1 1 in 1 CARTON 05/24/2010 1 10.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:58468-0160-2 10 in 1 CARTON 05/24/2010 08/31/2023 2 10.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125291 05/24/2010 Labeler - Genzyme Corporation (025322157) Establishment Name Address ID/FEI Business Operations Genzyme Flanders 372153895 ANALYSIS(58468-0160) , MANUFACTURE(58468-0160) , API MANUFACTURE(58468-0160) Establishment Name Address ID/FEI Business Operations Genzyme Ireland Limited 985127419 ANALYSIS(58468-0160) , MANUFACTURE(58468-0160) , PACK(58468-0160) , LABEL(58468-0160) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 050424395 LABEL(58468-0160) , PACK(58468-0160) Establishment Name Address ID/FEI Business Operations Eurofins Biopharma Product Testing Ireland Limited 238239933 ANALYSIS(58468-0160)