8.1 Pregnancy
-
Pregnancy Exposure Registry
-
There is a pregnancy exposure registry that monitors pregnancy outcomes in women who are exposed to WAKIX during pregnancy. Patients should be ...
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women who are exposed to WAKIX during pregnancy. Patients should be encouraged to enroll in the WAKIX pregnancy registry if they become pregnant. To enroll or obtain information from the registry, patients can call 1-800-833-7460.
Risk Summary
Available case reports from clinical trials and postmarketing reports with WAKIX use in pregnant women have not determined a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproductive studies, administration of pitolisant during organogenesis caused maternal and embryofetal toxicity in rats and rabbits at doses ≥13 and >4 times the maximum recommended human dose (MRHD) of 35.6 mg based on mg/m2 body surface area, respectively. Oral administration of pitolisant to female rats during pregnancy and lactation adversely affected maternal and fetal health and produced developmental delay at doses ≥13 times the MRHD, based on mg/m2 body surface area and increased the incidence of major malformations at 22 times the MRHD (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Pitolisant was administered orally to pregnant rats during the period of organogenesis at doses of 30, 52, 90 and 110 mg/kg/day, which are approximately 7, 13, 22 and 27 times the MRHD, based on mg/m2 body surface area, respectively. Maternal toxicity occurred at >22 times the MRHD and included convulsions and decreases in body weight and food consumption. At these maternally toxic doses, no adverse effects on embryofetal development were noted and the no observed-adverse-effect-level for embryofetal toxicity is 27 times the MRHD based on mg/m2 body surface area.
Pitolisant was administered intramuscularly to pregnant rabbits during the period of organogenesis at doses of 4, 8, and 16 mg/kg/day, which are approximately 2, 4 and 8 times the MRHD, based on mg/m2 body surface area, respectively. Maternal toxicity occurred at ≥4 times the MRHD and included significant body weight loss and decreased food consumption. Mortality (1 animal) and convulsions (2 animals) occurred at 8 times the MRHD. At the maternally toxic dose (8 times the MRHD), the incidence of pre-implantation loss and abortions increased with a consequent decrease in both the number of implantations and live fetuses. Pitolisant was not teratogenic at doses up to 8 times the MRHD; however, delayed skeletal development (incomplete ossification and supernumerary ribs) was observed. The no-observed-adverse-effect-level for maternal toxicity and embryofetal development is 2 and 4 times the MRHD based on mg/m2 body surface area, respectively.
Pitolisant was administered orally to pregnant rats from gestation day 7 through lactation day 20 post-partum at doses of 30, 52, and 90 mg/kg/day, which are 7, 13 and 22 times the MRHD, based on mg/m2 body surface area, respectively. Maternal toxicity included death, CNS signs including convulsions, and significant decrease in body weight and food consumption at 22 times the MRHD based on mg/m2 body surface area. At the maternally toxic dose (22 times the MRHD), fetal toxicity included stillbirths, postnatal pup mortality (due to lack of milk and/or failure to nurse), and decreased pup length and weight. A single female at the mid dose (13 times the MRHD) also failed to produce milk resulting in pup mortality. At the maternally toxic dose (22 times the MRHD), pitolisant was teratogenic causing major malformations (cleft palate, abnormal limb flexure). F1 toxicity included delay in postnatal development (decrease in body weight and length, delay in incisor eruption, and delay in testes descent), which occurred at ≥13 times the MRHD; however, there was no effect on sexual maturation or reproductive capacity of the F1 generation. The no-observed-adverse-effect-level for developmental toxicity is approximately 7 times the MRHD, based on mg/m2 body surface area.
8.2 Lactation
Risk Summary
The transfer of pitolisant into breastmilk is low based on data from a lactation study. The mean infant dose was 0.009 mg/day, and the relative infant dose was less than 1% of the maternal weight-adjusted dose (see Data). There are no data on the effects of pitolisant on the breastfed infant, or the effect of this drug on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for WAKIX and any potential adverse effects on the breastfed infant from WAKIX or from the underlying maternal condition.
Data
An open-label study in 8 healthy lactating women who were 11 to 96 weeks post-partum evaluated the concentration of pitolisant in breast milk samples collected over 24 hours and serum samples collected over 120 hours after a single dose administration of 35.6 mg of pitolisant. Pitolisant was present in breast milk with a mean Cmax of 47.5 ng/mL, while the mean Cmax of pitolisant in serum was 61.4 ng/mL. Following a single dose of pitolisant 35.6 mg, approximately 50% of the amount of pitolisant measured in breast milk occurred during the first 4 hours post dose. Based on single dose data, the mean infant dosage of pitolisant was calculated to be 0.009 mg/day, which represented a mean of 0.564% of the maternal dose received.
8.3 Females and Males of Reproductive Potential
Contraception
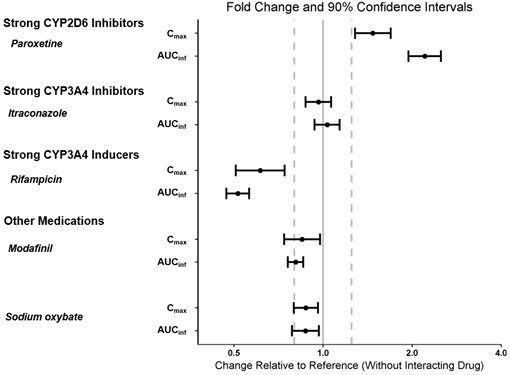
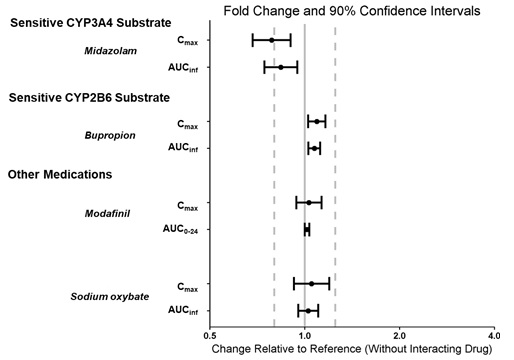
WAKIX may reduce the effectiveness of hormonal contraceptives. Patients using hormonal contraception should be advised to use an alternative non-hormonal contraceptive method during treatment with WAKIX and for at least 21 days after discontinuing treatment [see Drug interactions (7.1), Clinical Pharmacology (12.3)].
8.4 Pediatric Use
The safety and effectiveness of WAKIX have been established for the treatment of excessive daytime sleepiness in pediatric patients 6 years of age and older with narcolepsy. Use of WAKIX in this age group is supported by one adequate and well-controlled study in 110 pediatric patients with narcolepsy ages 6 to less than 18 years of age [see Clinical Studies (14.1)].
The safety and effectiveness of WAKIX have not been established for treatment of excessive daytime sleepiness in pediatric patients less than 6 years of age with narcolepsy.
The safety and effectiveness of WAKIX have not been established for treatment of cataplexy in pediatric patients with narcolepsy.
Juvenile Animal Toxicity Data
In a juvenile animal study, male and female rats were administered pitolisant at 9, 21, or 48 mg/kg/day by oral gavage from postnatal day (PND) 7 to PND 70. Mortality occurred at the highest dose of 48 mg/kg/day; however, death was primarily related to aspiration/inhalation of food material. No adverse effects on growth and development up to the high dose were observed; however, plasma exposures at this dose were lower than those predicted to occur in pediatric patients at the maximum recommended human dose (MRHD) of 35.6 mg due to low oral bioavailability in juvenile rats.
In a second juvenile animal study, male and female rats were administered pitolisant at 15 or 30 mg/kg/day or 30 mg/kg/twice daily (60 mg/kg/day) by intraperitoneal injection from PND 7 to PND 70. Mortality and convulsions were observed at the top two doses of 30 and 60 mg/kg/day. Similar findings of convulsions and mortality were also observed in studies in adult rats at comparable doses. The no observed adverse effect level (NOAEL) is 15 mg/kg/day in juvenile animals administered pitolisant by intraperitoneal injection, which corresponds to plasma exposures that are approximately 4 times and 1 times the predicted pediatric exposures at the MRHD of 35.6 mg, based on Cmax and AUC, respectively.
8.5 Geriatric Use
Limited pharmacokinetic data are available in healthy elderly subjects. A pharmacokinetic study that compared 12 elderly subjects (age 68 to 82 years) to 12 healthy adults (age 18 to 45 years) did not reveal any significant differences in drug exposure [see Clinical Pharmacology (12.3)].
Of the total number of patients with narcolepsy in clinical studies of WAKIX, 14 patients (5%) were ≥65 years old. No overall differences in safety or effectiveness were observed between these patients and younger patients in these clinical trials, but greater sensitivity of some older individuals cannot be ruled out. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
8.6 Hepatic Impairment
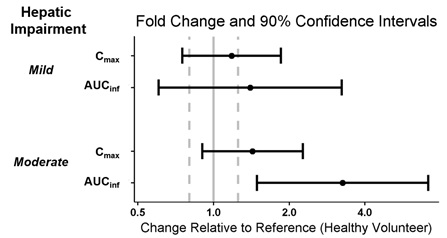
WAKIX is contraindicated in patients with severe hepatic impairment (Child-Pugh C) as it has not been studied in this population. WAKIX is extensively metabolized by the liver and there is a significant increase in WAKIX exposure in patients with moderate hepatic impairment [see Contraindications (4), Clinical Pharmacology (12.3)].
Monitor patients with moderate hepatic impairment (Child-Pugh B) and adjust the dosage of WAKIX [see Dosage and Administration (2.3)].
Monitor patients with mild hepatic impairment (Child-Pugh A). No dosage adjustment of WAKIX is recommended in patients with mild hepatic impairment.
8.7 Renal Impairment
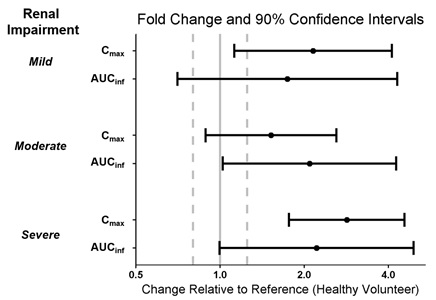
The pharmacokinetics of WAKIX in patients with end-stage renal disease (ESRD) (eGFR of <15 mL/minute/1.73 m2) is unknown [see Clinical Pharmacology (12.3)]. Therefore, WAKIX is not recommended in patients with ESRD [see Dosage and Administration (2.4), Warnings and Precautions (5.1)].
Dosage adjustment of WAKIX is recommended in patients with eGFR <60 mL/minute/1.73 m2[see Dosage and Administration (2.4)].
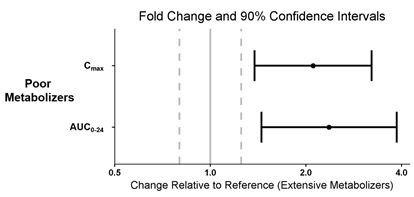
8.8 CYP2D6 Poor Metabolizers
Dosage reduction is recommended in patients known to be poor CYP2D6 metabolizers because these patients have higher pitolisant concentrations than normal CYP2D6 metabolizers [see Dosage and Administration (2.6), Clinical Pharmacology (12.3, 12.5)].
Close