The safety and efficacy of umeclidinium 62.5 mcg were evaluated in 3 dose-ranging trials, 2 placebo-controlled clinical trials (one 12-week trial and one 24-week trial), and a 12-month long‑term ...
The safety and efficacy of umeclidinium 62.5 mcg were evaluated in 3 dose-ranging trials, 2 placebo-controlled clinical trials (one 12-week trial and one 24-week trial), and a 12-month long‑term safety trial. The efficacy of INCRUSE ELLIPTA is based primarily on the dose‑ranging trials in 624 subjects and the 2 placebo-controlled confirmatory trials in 1,738 subjects with COPD, including chronic bronchitis and/or emphysema. [See Clinical Studies (14.1, 14.2).]
The safety and efficacy of INCRUSE ELLIPTA in combination with an ICS/LABA were also evaluated in four 12-week clinical trials. The efficacy of INCRUSE ELLIPTA in combination with an ICS/LABA is based on 1,637 subjects with COPD. [See Clinical Studies (14.3).]
Evidence of efficacy for INCRUSE ELLIPTA on COPD exacerbations was established by the efficacy of the umeclidinium component as part of a fixed-dose combination with an ICS/LABA, as assessed in a 12-month trial in 10,355 subjects. [See Clinical Studies (14.3).]
14.1 Dose-Ranging Trials
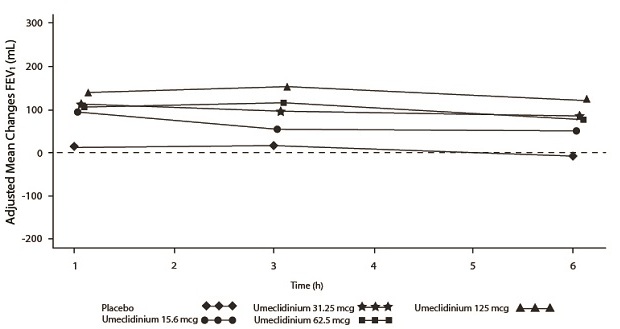
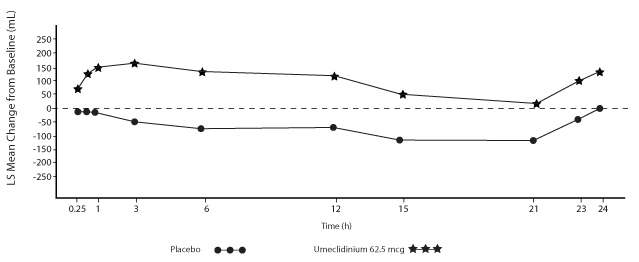
Dose selection for umeclidinium in COPD was supported by a 7-day, randomized, double-blind, placebo-controlled, crossover trial evaluating 4 doses of umeclidinium (15.6 to 125 mcg) or placebo dosed once daily in the morning in 163 subjects with COPD. A dose ordering was observed, with the 62.5- and 125-mcg doses demonstrating larger improvements in FEV1 over 24 hours compared with the lower doses of 15.6 and 31.25 mcg (Figure 2).
The differences in trough FEV1 from baseline after 7 days for placebo and the 15.6-, 31.25-, 62.5-, and 125-mcg doses were -74 mL (95% CI: -118, -31), 38 mL (95% CI: -6, 83), 27 mL (95% CI: -18, 72), 49 mL (95% CI: 6, 93), and 109 mL (95% CI: 65, 152), respectively. Two additional dose-ranging trials in subjects with COPD demonstrated minimal additional benefit at doses above 125 mcg. The dose-ranging results supported the evaluation of 2 doses of umeclidinium, 62.5 and 125 mcg, in the confirmatory COPD trials to further assess dose response.
Evaluations of dosing interval by comparing once- and twice-daily dosing supported selection of a once-daily dosing interval for further evaluation in the confirmatory COPD trials.
Figure 2. Adjusted Mean Change from Baseline in Postdose Serial FEV1 (mL) on Days 1 and 7
Day 1
Day 7
14.2 Maintenance Treatment: Confirmatory Trials
Lung Function
The clinical development program for INCRUSE ELLIPTA included 2 randomized, double‑blind, placebo-controlled, parallel-group trials in subjects with COPD designed to evaluate the efficacy of INCRUSE ELLIPTA on lung function. Trial 1 (NCT01313650) was a 24-week placebo‑controlled trial, and Trial 2 (NCT01387230) was a 12-week placebo‑controlled trial. These trials treated subjects that had a clinical diagnosis of COPD, were 40 years of age or older, had a history of smoking ≥10 pack-years, had a post-albuterol FEV1 ≤70% of predicted normal values, had a ratio of FEV1/FVC of <0.7, and had a Modified Medical Research Council (mMRC) score ≥2. Subjects in Trial 1 had a mean age of 63 years and an average smoking history of 46 pack-years, with 50% identified as current smokers. At screening, the mean postbronchodilator percent predicted FEV1 was 47% (range: 13% to 74%), the mean postbronchodilator FEV1/FVC ratio was 0.47 (range: 0.20 to 0.74), and the mean percent reversibility was 15% (range: -35% to 109%). The majority of subjects (72%) reported not having a COPD exacerbation in the prior 12 months. Baseline demographics and lung function for subjects in Trial 2 were similar to those in Trial 1.
Trial 1 evaluated umeclidinium 62.5 mcg and placebo. The primary endpoint was change from baseline in trough (predose) FEV1 at Day 169 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 168) compared with placebo. INCRUSE ELLIPTA 62.5 mcg demonstrated a larger increase in mean change from baseline in trough (predose) FEV1 relative to placebo (Table 2). Similar results were obtained from Trial 2.
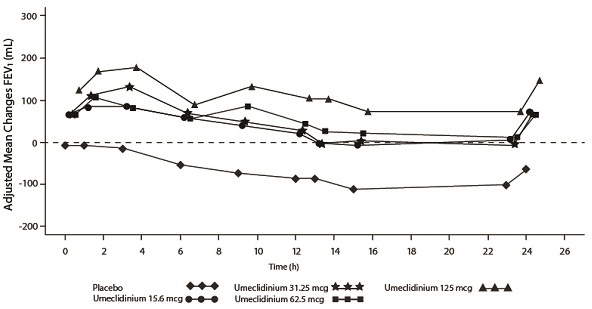
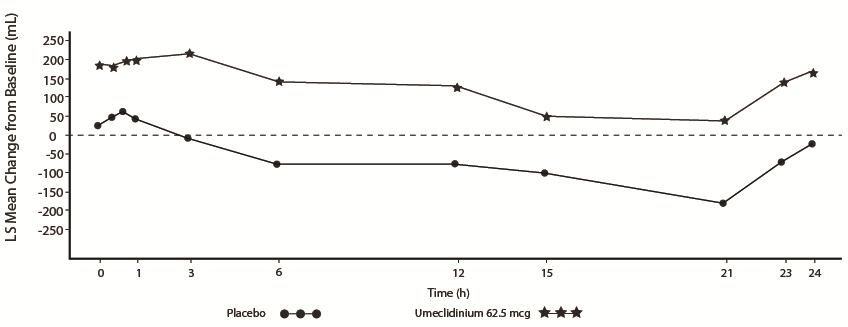
Serial spirometric evaluations throughout the 24-hour dosing interval were performed in a subset of subjects (n = 54, umeclidinium 62.5 mcg; n = 36, placebo) at Days 1, 84, and 168 in Trial 1, and for all patients at Days 1 and 84 in Trial 2. Results from Trial 1 at Day 1 and Day 168 are shown in Figure 3.
Figure 3. Least Squares (LS) Mean Change from Baseline in FEV1 (mL) over Time (0-24 h) on Days 1 and 168 (Trial 1 Subset Population)
Day 1
Day 168
In Trial 1, the mean peak FEV1 (over the first 6 hours relative to baseline) at Day 1 and at Day 168 for the group receiving umeclidinium 62.5 mcg compared with placebo was 126 and 130 mL, respectively.
Health-Related Quality of Life
Health-related quality of life was measured using St. George’s Respiratory Questionnaire (SGRQ). Umeclidinium demonstrated an improvement in mean SGRQ total score compared with placebo treatment at Day 168: -4.69 (95% CI: -7.07,-2.31). The proportion of patients with a clinically meaningful decrease (defined as a decrease of at least 4 units from baseline) at Week 24 was greater for INCRUSE ELLIPTA 62.5 mcg (42%; 172/410) compared with placebo (31%; 86/274).
14.3 Maintenance Treatment: Combination with an ICS/LABA Trials
Lung Function
The efficacy of INCRUSE ELLIPTA in combination with an ICS/LABA was evaluated in 4 randomized, double-blind, parallel-group trials in subjects with COPD. These trials, all of similar study design, were of 12-weeks’ treatment duration. Subjects were randomized to INCRUSE ELLIPTA 62.5 mcg + ICS/LABA or placebo + ICS/LABA. Entry criteria for subjects enrolled in these trials were similar to those described above in Section 14.2. The primary endpoint for these trials was change from baseline in trough (predose) FEV1 at Day 85 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 84). Baseline FEV1 was measured while subjects were on background ICS/LABA.
Combination with Fluticasone Furoate + Vilanterol
Trial 4 (NCT01957163) and Trial 5 (NCT02119286) randomized subjects to INCRUSE ELLIPTA 62.5 mcg + FF/VI 100 mcg/25 mcg administered once daily or placebo + FF/VI 100 mcg/25 mcg administered once daily. Trial population demographics and results for Trials 4 and 5 were similar; therefore, only Trial 4 results are presented below.
Subjects in Trial 4 across all treatment arms had a mean age of 64 years and an average smoking history of 50 pack-years, with 42% identified as current smokers. At screening, the mean postbronchodilator percent predicted FEV1 was 45% (range: 13% to 76%), the mean postbronchodilator FEV1/FVC ratio was 0.48 (range: 0.22 to 0.70), and the mean percent reversibility was 14% (range: -20% to 71%). The majority of subjects (85%) reported not having a COPD exacerbation in the prior 12 months.
The primary endpoint was change from baseline in trough (predose) FEV1 at Day 85 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 84) compared with placebo (INCRUSE ELLIPTA + FF/VI vs. placebo + FF/VI). INCRUSE ELLIPTA + FF/VI demonstrated a larger mean change from baseline in trough (predose) FEV1 relative to placebo + FF/VI (Table 3).
Combination with Fluticasone Propionate + Salmeterol
Trial 6 (NCT01772134) and Trial 7 (NCT01772147) randomized subjects to INCRUSE ELLIPTA 62.5 mcg + FP/SAL 250 mcg/50 mcg or placebo + FP/SAL 250 mcg/50 mcg. The treatments with INCRUSE ELLIPTA and placebo were administered once daily, while the FP/SAL treatment was administered twice daily. Trial population demographics and results for Trials 6 and 7 were similar; therefore, only Trial 6 results are presented below.
Subjects in Trial 6 across all treatment arms had a mean age of 63 years and an average smoking history of 50 pack-years, with 54% identified as current smokers. At screening, the mean postbronchodilator percent predicted FEV1 was 47% (range: 12% to 70%), the mean postbronchodilator FEV1/FVC ratio was 0.47 (range: 0.22 to 0.69), and the mean percent reversibility was 16% (range: -36% to 79%). The majority of subjects (79%) reported not having a COPD exacerbation in the prior 12 months.
The primary endpoint was change from baseline in trough (predose) FEV1 at Day 85 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 84) compared with placebo (INCRUSE ELLIPTA + FP/SAL vs. placebo + FP/SAL). INCRUSE ELLIPTA + FP/SAL demonstrated a larger mean change from baseline in trough (predose) FEV1 relative to placebo + FP/SAL (Table 4).
Exacerbations
In Trial 8 (NCT02164513), a total of 10,355 subjects with COPD with a history of 1 or more moderate or severe exacerbations in the prior 12 months were randomized (2:2:1) to receive fluticasone furoate/umeclidinium/vilanterol 100 mcg/62.5 mcg/25 mcg (n = 4,151), fluticasone furoate/vilanterol 100 mcg/25 mcg (n = 4,133), or umeclidinium/vilanterol 62.5 mcg/25 mcg (n = 2,070) administered once daily in a 12-month trial. The population demographics across all treatments were: mean age of 65 years, 77% white, 66% male, and an average smoking history of 46.6 pack-years, with 35% identified as current smokers. At trial entry, the most common COPD medications were ICS + anticholinergic + LABA (34%), ICS + LABA (26%), anticholinergic + LABA (8%), and anticholinergic (7%). The mean postbronchodilator percent predicted FEV1 was 46% (standard deviation: 15%), the mean postbronchodilator FEV1/FVC ratio was 0.47 (standard deviation: 0.12), and the mean percent reversibility was 10% (range: -59% to 125%).
The primary endpoint was annual rate of on-treatment moderate and severe exacerbations in subjects treated with fluticasone furoate/umeclidinium/vilanterol compared with the fixed-dose combinations of fluticasone furoate/vilanterol and umeclidinium/vilanterol. Exacerbations were defined as worsening of 2 or more major symptoms (dyspnea, sputum volume, and sputum purulence) or worsening of any 1 major symptom together with any 1 of the following minor symptoms: sore throat, colds (nasal discharge and/or nasal congestion), fever without other cause, and increased cough or wheeze for at least 2 consecutive days. Exacerbations were considered to be of moderate severity if treatment with systemic corticosteroids and/or antibiotics was required and were considered to be severe if resulted in hospitalization or death.
Evidence of efficacy for INCRUSE ELLIPTA on COPD exacerbations was established by the efficacy of the umeclidinium component of fluticasone furoate/umeclidinium/vilanterol in Trial 8. Treatment with fluticasone furoate/umeclidinium/vilanterol statistically significantly reduced the on-treatment annual rate of moderate/severe exacerbations by 15% compared with fluticasone furoate/vilanterol (Table 5). A reduction in risk of on-treatment moderate/severe exacerbation (as measured by time to first) was also observed for the same comparison.
Close