Label: OTEZLA- apremilast kit
OTEZLA- apremilast tablet, film coated
















-
Contains inactivated NDC Code(s)
NDC Code(s): 59572-630-27, 59572-630-99, 59572-631-06, 59572-631-28, view more59572-632-55 - Packager: Celgene Corporation
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated February 5, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OTEZLA safely and effectively. See full prescribing information for OTEZLA.
OTEZLA® (apremilast) tablets, for oral use
Initial U.S. approval: 2014RECENT MAJOR CHANGES
INDICATIONS AND USAGE
OTEZLA, an inhibitor of phosphodiesterase 4 (PDE4), is indicated for the treatment of:
DOSAGE AND ADMINISTRATION
- To reduce risk of gastrointestinal symptoms, titrate to recommended dose of 30 mg twice daily according to the following schedule (2.1)
- Day 1: 10 mg in morning
- Day 2: 10 mg in morning and 10 mg in evening
- Day 3: 10 mg in morning and 20 mg in evening
- Day 4: 20 mg in morning and 20 mg in evening
- Day 5: 20 mg in morning and 30 mg in evening
- Day 6 and thereafter: 30 mg twice daily
DOSAGE FORMS AND STRENGTHS
Tablets: 10 mg, 20 mg, 30 mg (3)
CONTRAINDICATIONS
Known hypersensitivity to apremilast or any excipients in formulation (4)
WARNINGS AND PRECAUTIONS
- Diarrhea, Nausea, and Vomiting: Consider OTEZLA dose reduction or suspension if patients develop severe diarrhea, nausea, or vomiting (5.1)
- Depression: Advise patients, their caregivers, and families to be alert for the emergence or worsening of depression, suicidal thoughts or other mood changes and if such changes occur to contact their healthcare provider. Carefully weigh risks and benefits of treatment with OTEZLA in patients with a history of depression and/or suicidal thoughts or behavior (5.2)
- Weight Decrease: Monitor weight regularly. If unexplained or clinically significant weight loss occurs, evaluate weight loss and consider discontinuation of OTEZLA (5.3)
- Drug Interactions: Use with strong cytochrome P450 enzyme inducers (e.g., rifampin, phenobarbital, carbamazepine, phenytoin) is not recommended because loss of efficacy may occur (5.4, 7.1)
ADVERSE REACTIONS
- Psoriatic Arthritis: The most common adverse reactions (≥5%) are diarrhea, nausea, and headache (6.1)
- Psoriasis: The most common adverse reactions (≥5%) are diarrhea, nausea, upper respiratory tract infection, and headache, including tension headache (6.1)
- Behçet's Disease: The most common adverse reactions (≥10%) are diarrhea, nausea, headache, and upper respiratory tract infection (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Celgene Corporation at 1-888-423-5436 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
USE IN SPECIFIC POPULATIONS
Severe Renal Impairment: Increased systemic exposure of OTEZLA has been observed, reduction in dose to 30 mg once daily is recommended (2.2, 8.6)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2019
- To reduce risk of gastrointestinal symptoms, titrate to recommended dose of 30 mg twice daily according to the following schedule (2.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Psoriatic Arthritis
1.2 Psoriasis
1.3 Oral Ulcers Associated with Behçet's Disease
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Psoriatic Arthritis, Psoriasis, and Behçet's Disease
2.2 Dosage Adjustment in Patients with Severe Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea, Nausea, and Vomiting
5.2 Depression
5.3 Weight Decrease
5.4 Drug Interactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Strong CYP450 Inducers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Psoriatic Arthritis
14.2 Psoriasis
14.3 Oral Ulcers Associated with Behçet's Disease
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Psoriatic Arthritis, Psoriasis, and Behçet's Disease
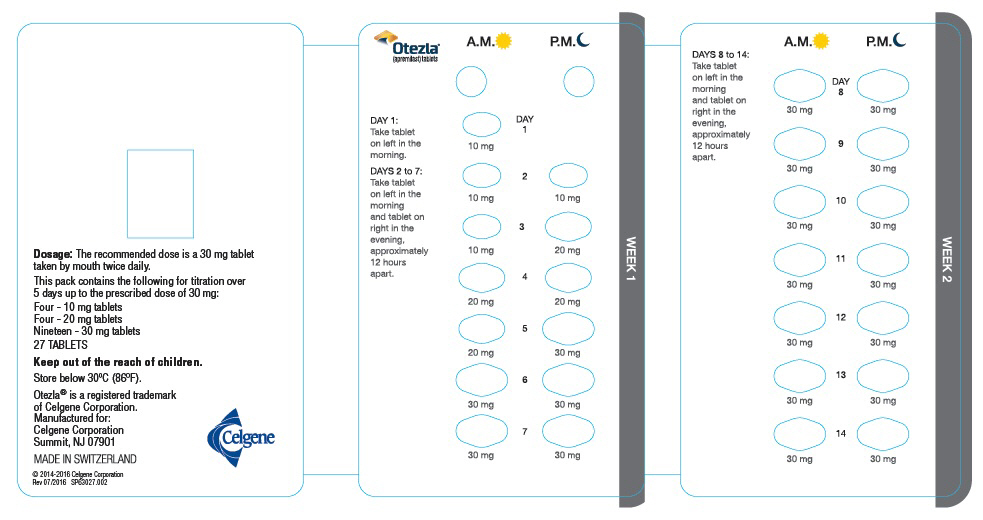
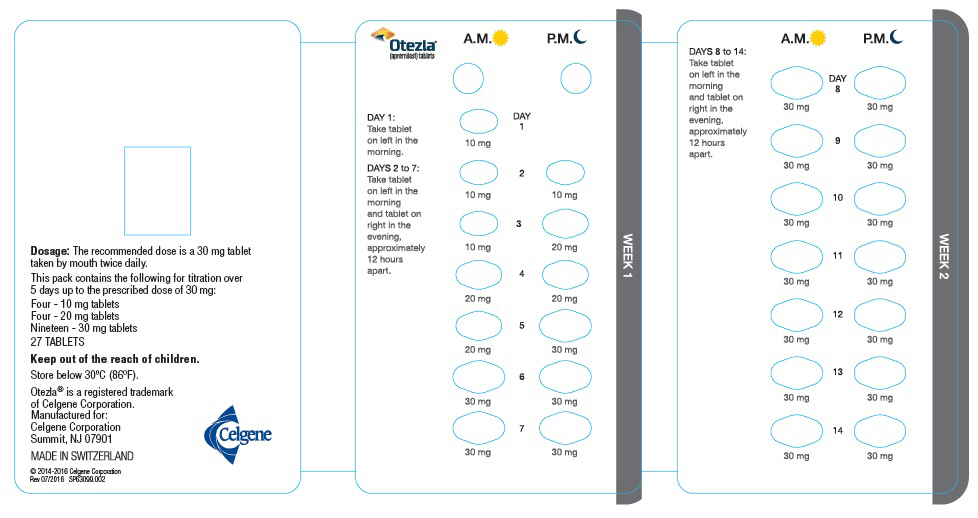
The recommended initial dosage titration of OTEZLA from Day 1 to Day 5 is shown in Table 1. Following the 5-day titration, the recommended maintenance dosage is 30 mg twice daily taken orally starting on Day 6. This titration is intended to reduce the gastrointestinal symptoms associated with initial therapy.
OTEZLA can be administered without regard to meals. Do not crush, split, or chew the tablets.
Table 1: Dosage Titration Schedule Day 1 Day 2 Day 3 Day 4 Day 5 Day 6
& thereafterAM AM PM AM PM AM PM AM PM AM PM 10 mg 10 mg 10 mg 10 mg 20 mg 20 mg 20 mg 20 mg 30 mg 30 mg 30 mg 2.2 Dosage Adjustment in Patients with Severe Renal Impairment
OTEZLA dosage should be reduced to 30 mg once daily in patients with severe renal impairment (creatinine clearance (CLcr) of less than 30 mL per minute estimated by the Cockcroft–Gault equation) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. For initial dosage titration in this group, it is recommended that OTEZLA be titrated using only the AM schedule listed in Table 1 and the PM doses be skipped.
-
3 DOSAGE FORMS AND STRENGTHS
OTEZLA is available as diamond shaped, film coated tablets in the following dosage strengths:
- 10-mg pink tablet engraved with "APR" on one side and "10" on the other side
- 20-mg brown tablet engraved with "APR" on one side and "20" on the other side
- 30-mg beige tablet engraved with "APR" on one side and "30" on the other side.
-
4 CONTRAINDICATIONS
OTEZLA is contraindicated in patients with a known hypersensitivity to apremilast or to any of the excipients in the formulation [see Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea, Nausea, and Vomiting
There have been postmarketing reports of severe diarrhea, nausea, and vomiting associated with the use of OTEZLA. Most events occurred within the first few weeks of treatment. In some cases, patients were hospitalized. Patients 65 years of age or older and patients taking medications that can lead to volume depletion or hypotension may be at a higher risk of complications from severe diarrhea, nausea, or vomiting. Monitor patients who are more susceptible to complications of diarrhea or vomiting. Patients who reduced dosage or discontinued OTEZLA generally improved quickly. Consider OTEZLA dose reduction or suspension if patients develop severe diarrhea, nausea, or vomiting.
5.2 Depression
Treatment with OTEZLA is associated with an increase in adverse reactions of depression. Before using OTEZLA in patients with a history of depression and/or suicidal thoughts or behavior prescribers should carefully weigh the risks and benefits of treatment with OTEZLA in such patients. Patients, their caregivers, and families should be advised of the need to be alert for the emergence or worsening of depression, suicidal thoughts or other mood changes, and if such changes occur to contact their healthcare provider. Prescribers should carefully evaluate the risks and benefits of continuing treatment with OTEZLA if such events occur.
Psoriatic arthritis: During the 0 to 16 week placebo-controlled period of the 3 controlled clinical trials, 1.0% (10/998) of subjects treated with OTEZLA reported depression or depressed mood compared to 0.8% (4/495) treated with placebo. During the clinical trials, 0.3% (4/1441) of subjects treated with OTEZLA discontinued treatment due to depression or depressed mood compared with none in placebo treated subjects (0/495). Depression was reported as serious in 0.2% (3/1441) of subjects exposed to OTEZLA, compared to none in placebo-treated subjects (0/495). Instances of suicidal ideation and behavior have been observed in 0.2% (3/1441) of subjects while receiving OTEZLA, compared to none in placebo treated subjects (0/495). In the clinical trials, 2 subjects who received placebo committed suicide compared to none in OTEZLA-treated subjects.
Psoriasis: During the 0 to 16 week placebo-controlled period of the 3 controlled clinical trials, 1.3% (12/920) of subjects treated with OTEZLA reported depression compared to 0.4% (2/506) treated with placebo. During the clinical trials, 0.1% (1/1308) of subjects treated with OTEZLA discontinued treatment due to depression compared with none in placebo-treated subjects (0/506). Depression was reported as serious in 0.1% (1/1308) of subjects exposed to OTEZLA, compared to none in placebo-treated subjects (0/506). Instances of suicidal behavior have been observed in 0.1% (1/1308) of subjects while receiving OTEZLA, compared to 0.2% (1/506) in placebo-treated subjects. In the clinical trials, one subject treated with OTEZLA attempted suicide while one who received placebo committed suicide.
Behçet's disease: During the placebo-controlled period of the phase 3 study, 1% (1/104) of patients treated with OTEZLA reported depression/depressed mood compared to 1% (1/103) treated with placebo. None of these reports of depression was serious or led to study discontinuation. No instances of suicidal ideation or behavior were reported during the placebo-controlled period of the phase 3 study in patients treated with OTEZLA (0/104) or treated with placebo (0/103).
5.3 Weight Decrease
During the controlled period of the studies in psoriatic arthritis (PsA), weight decrease between 5%-10% of body weight was reported in 10% (49/497) of subjects treated with OTEZLA 30 mg twice daily compared to 3.3% (16/495) treated with placebo.
During the controlled period of the trials in psoriasis, weight decrease between 5%-10% of body weight occurred in 12% (96/784) of subjects treated with OTEZLA compared to 5% (19/382) treated with placebo. Weight decrease of ≥10% of body weight occurred in 2% (16/784) of subjects treated with OTEZLA 30 mg twice daily compared to 1% (3/382) subjects treated with placebo.
During the controlled period of the phase 3 study in Behçet's disease, weight decrease >5% of body weight was reported in 4.9% (5/103) of subjects treated with OTEZLA 30 mg twice daily compared to 3.9% (4/102) patients treated with placebo.
Patients treated with OTEZLA should have their weight monitored regularly. If unexplained or clinically significant weight loss occurs, weight loss should be evaluated, and discontinuation of OTEZLA should be considered [see Adverse Reactions (6.1)].
5.4 Drug Interactions
Co-administration of strong cytochrome P450 enzyme inducer, rifampin, resulted in a reduction of systemic exposure of apremilast, which may result in a loss of efficacy of OTEZLA. Therefore, the use of cytochrome P450 enzyme inducers (e.g., rifampin, phenobarbital, carbamazepine, phenytoin) with OTEZLA is not recommended [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are described elsewhere in the labeling:
- Diarrhea, Nausea, and Vomiting [see Warnings and Precautions (5.1)]
- Depression [see Warnings and Precautions (5.2)]
- Weight Decrease [see Warnings and Precautions (5.3)]
- Drug Interactions [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Psoriatic Arthritis Clinical Trials
OTEZLA was evaluated in 3 multicenter, randomized, double-blind, placebo-controlled trials [Studies PsA-1, PsA-2, and PsA-3] of similar design in adult patients with active psoriatic arthritis [see Clinical Studies (14.1)]. Across the 3 studies, there were 1493 patients randomized equally to placebo, OTEZLA 20 mg twice daily or OTEZLA 30 mg twice daily. Titration was used over the first 5 days [see Dosage and Administration (2.1)]. Placebo patients whose tender and swollen joint counts had not improved by at least 20% were re-randomized 1:1 in a blinded fashion to either OTEZLA 20 mg twice daily or 30 mg twice daily at week 16 while OTEZLA patients remained on their initial treatment. Patients ranged in age from 18 to 83 years, with an overall median age of 51 years.The majority of the most common adverse reactions presented in Table 2 occurred within the first 2 weeks of treatment and tended to resolve over time with continued dosing. Diarrhea, headache, and nausea were the most commonly reported adverse reactions. The most common adverse reactions leading to discontinuation for patients taking OTEZLA were nausea (1.8%), diarrhea (1.8%), and headache (1.2%). The proportion of patients with psoriatic arthritis who discontinued treatment due to any adverse reaction was 4.6% for patients taking OTEZLA 30 mg twice daily and 1.2% for placebo-treated patients.
Table 2: Adverse Reactions Reported in ≥2% of Patients on OTEZLA 30 mg Twice Daily and ≥1% Than That Observed in Patients on Placebo for up to Day 112 (Week 16) a Of the reported gastrointestinal adverse reactions, 1 subject experienced a serious adverse reaction of nausea and vomiting in OTEZLA 30 mg twice daily; 1 subject treated with OTEZLA 20 mg twice daily experienced a serious adverse reaction of diarrhea; 1 patient treated with OTEZLA 30 mg twice daily experienced a serious adverse reaction of headache. b Of the reported adverse drug reactions none were serious. c n (%) indicates number of patients and percent. Placebo OTEZLA 30 mg BID Preferred Term Day 1 to 5
(N=495)
n (%)cDay 6 to Day 112
(N=490)
n (%)Day 1 to 5
(N=497)
n (%)Day 6 to Day 112
(N=493)
n (%)Diarrheaa 6 (1.2) 8 (1.6) 46 (9.3) 38 (7.7) Nauseaa 7 (1.4) 15 (3.1) 37 (7.4) 44 (8.9) Headachea 9 (1.8) 11 (2.2) 24 (4.8) 29 (5.9) Upper respiratory tract
infectionb3 (0.6) 9 (1.8) 3 (0.6) 19 (3.9) Vomitinga 2 (0.4) 2 (0.4) 4 (0.8) 16 (3.2) Nasopharyngitisb 1 (0.2) 8 (1.6) 1 (0.2) 13 (2.6) Abdominal pain upperb 0 (0.0) 1 (0.2) 3 (0.6) 10 (2.0) Other adverse reactions reported in patients on OTEZLA in clinical studies including extension studies:
Immune system disorders: Hypersensitivity
Investigations: Weight decrease
Gastrointestinal Disorders: Frequent bowel movement, gastroesophageal reflux disease, dyspepsia
Metabolism and Nutrition Disorders: Decreased appetite*
Nervous System Disorders: Migraine
Respiratory, Thoracic, and Mediastinal Disorders: Cough
Skin and Subcutaneous Tissue Disorders: Rash
*1 patient treated with OTEZLA 30 mg twice daily experienced a serious adverse reaction.Psoriasis Clinical Trials
The safety of OTEZLA was assessed in 1426 subjects in 3 randomized, double-blind, placebo-controlled trials in adult subjects with moderate to severe plaque psoriasis who were candidates for phototherapy or systemic therapy. Subjects were randomized to receive OTEZLA 30 mg twice daily or placebo twice daily. Titration was used over the first 5 days [see Dosage and Administration (2.1)]. Subjects ranged in age from 18 to 83 years, with an overall median age of 46 years.Diarrhea, nausea, and upper respiratory tract infection were the most commonly reported adverse reactions. The most common adverse reactions leading to discontinuation for subjects taking OTEZLA were nausea (1.6%), diarrhea (1.0%), and headache (0.8%). The proportion of subjects with psoriasis who discontinued treatment due to any adverse reaction was 6.1% for subjects treated with OTEZLA 30 mg twice daily and 4.1% for placebo-treated subjects.
Table 3: Adverse Reactions Reported in ≥1% of Subjects on OTEZLA and With Greater Frequency Than in Subjects on Placebo; up to Day 112 (Week 16) *Two subjects treated with OTEZLA experienced serious adverse reaction of abdominal pain. Preferred Term Placebo (N=506)
n (%)OTEZLA 30 mg BID (N=920)
n (%)Diarrhea 32 (6) 160 (17) Nausea 35 (7) 155 (17) Upper respiratory tract infection 31 (6) 84 (9) Tension headache 21 (4) 75 (8) Headache 19 (4) 55 (6) Abdominal pain* 11 (2) 39 (4) Vomiting 8 (2) 35 (4) Fatigue 9 (2) 29 (3) Dyspepsia 6 (1) 29 (3) Decreased appetite 5 (1) 26 (3) Insomnia 4 (1) 21 (2) Back pain 4 (1) 20 (2) Migraine 5 (1) 19 (2) Frequent bowel movements 1 (0) 17 (2) Depression 2 (0) 12 (1) Bronchitis 2 (0) 12 (1) Tooth abscess 0 (0) 10 (1) Folliculitis 0 (0) 9 (1) Sinus headache 0 (0) 9 (1) Severe worsening of psoriasis (rebound) occurred in 0.3% (4/1184) subjects following discontinuation of treatment with OTEZLA.
Behçet's Disease Clinical Trials
OTEZLA was evaluated in a Phase 3, multicenter, randomized, placebo-controlled study (BCT-002) in adult patients with Behçet's Disease (BD) with active oral ulcers. A total of 207 patients were randomized to receive OTEZLA 30 mg twice daily or placebo twice daily. Titration was used over the first 5 days [see Dosage and Administration (2.1)]. After Week 12, all patients received treatment with OTEZLA 30 mg twice daily. Patients ranged in age from 19 to 72, with a mean age of 40 years.
Diarrhea, nausea, headache, and upper respiratory tract infection were the most commonly reported adverse reactions. The proportion of patients with BD who discontinued treatment due to any adverse reaction during the placebo-controlled period of the study, was 2.9% for patients treated with OTEZLA 30 mg twice daily and 4.9% for placebo-treated patients.
Table 4: Adverse Reactions Reported in ≥5% of Patients on OTEZLA and with at least 1% Greater Frequency than Patients on Placebo; up to Week 12 Preferred Term Placebo
(N=103)
n (%)OTEZLA 30 mg twice daily
(N=104)
n (%)- *
- There were no serious adverse reactions of diarrhea, nausea or vomiting.
Diarrhea* 21 (20.4) 43 (41.3) Nausea* 11 (10.7) 20 (19.2) Headache 11 (10.7) 15 (14.4) Upper respiratory tract infection 5 (4.9) 12 (11.5) Abdominal pain upper 2 (1.9) 9 (8.7) Vomiting* 2 (1.9) 9 (8.7) Back pain 6 (5.8) 8 (7.7) Viral upper respiratory tract infection 5 (4.9) 7 (6.7) Arthralgia 3 (2.9) 6 (5.8) - 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to OTEZLA during pregnancy. Information about the registry can be obtained by calling 1-877-311-8972 or visiting https://mothertobaby.org/ongoing-study/otezla/.
Risk Summary
Available pharmacovigilance data with OTEZLA use in pregnant women have not established a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes, but these data are extremely limited. Based on findings from animal reproduction studies, OTEZLA may increase the risk for fetal loss. In animal embryo-fetal development studies, the administration of apremilast to pregnant cynomolgus monkeys during organogenesis resulted in dose-related increases in abortion/embryo-fetal death at dose exposures 2.1-times the maximum recommended human therapeutic dose (MRHD) and no adverse effect at an exposure of 1.4-times the MRHD. When administered to pregnant mice, during organogenesis there were no apremilast-induced malformations up to exposures 4.0-times the MRHD (see Data). Advise pregnant women of the potential risk of fetal loss. Consider pregnancy planning and prevention for females of reproductive potential.
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal developmental study, pregnant cynomolgus monkeys were administered apremilast at doses of 20, 50, 200, or 1000 mg/kg/day during the period of organogenesis (gestation Days 20 through 50). There was a dose-related increase in spontaneous abortions, with most abortions occurring during Weeks 3 to 4 of dosing in the first trimester, at doses approximately 2.1-times the MRHD and greater (on an area under the curve [AUC] basis at doses ≥50 mg/kg/day). No abortifacient effects were observed at a dose approximately 1.4-times the MRHD (on an AUC basis at a dose of 20 mg/kg/day). Although, there was no evidence for a teratogenic effect at doses of 20 mg/kg/day and greater when examined at day 100, aborted fetuses were not examined.
In an embryo-fetal development study in mice, apremilast was administered at doses of 250, 500, or 750 mg/kg/day to dams during organogenesis (gestation Day 6 through 15). In a combined fertility and embryo-fetal development study in mice, apremilast was administered at doses of 10, 20, 40, or 80 mg/kg/day starting 15 days before cohabitation and continuing through gestation Day 15. No teratogenic findings attributed to apremilast were observed in either study; however, there was an increase in postimplantation loss at doses corresponding to a systemic exposure of 2.3-times the MRHD and greater (≥20 mg/kg/day). At doses of ≥20 mg/kg/day skeletal variations included incomplete ossification sites of tarsals, skull, sternebra, and vertebrae. No effects were observed at a dose approximately 1.3-times the MRHD (10 mg/kg/day).
Apremilast distributed across the placenta into the fetal compartment in mice and monkeys.
In a pre- and postnatal study in mice, apremilast was administered to pregnant female mice at doses of 10, 80, or 300 mg/kg/day from Day 6 of gestation through Day 20 of lactation, with weaning on Day 21. Dystocia, reduced viability, and reduced birth weights occurred at doses corresponding to ≥4.0-times the MRHD (on an AUC basis at doses ≥80 mg/kg/day). No adverse effects occurred at a dose 1.3-times the MRHD (10 mg/kg/day). There was no evidence for functional impairment of physical development, behavior, learning ability, immune competence, or fertility in the offspring at doses up to 7.5-times the MRHD (on an AUC basis at a dose of 300 mg/kg/day).
8.2 Lactation
Risk Summary
There are no data on the presence of apremilast in human milk, the effects on the breastfed infant, or the effects on milk production. However, apremilast was detected in the milk of lactating mice. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for OTEZLA and any potential adverse effects on the breastfed infant from OTEZLA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of OTEZLA in pediatric patients less than 18 years of age have not been established.
8.5 Geriatric Use
Of the 1493 subjects who enrolled in Studies PsA-1, PsA-2, and PsA-3 a total of 146 psoriatic arthritis subjects were 65 years of age and older, including 19 subjects 75 years and older. No overall differences were observed in the safety profile of elderly subjects ≥65 years of age and younger adult subjects <65 years of age in the clinical studies.
Of the 1257 subjects who enrolled in two placebo-controlled psoriasis trials (PSOR 1 and PSOR 2), a total of 108 psoriasis subjects were 65 years of age and older, including 9 subjects who were 75 years of age and older. No overall differences were observed in the efficacy and safety in elderly subjects ≥65 years of age and younger adult subjects <65 years of age in the clinical trials.
8.6 Renal Impairment
Apremilast pharmacokinetics were characterized in subjects with mild, moderate, and severe renal impairment as defined by a creatinine clearance of 60-89, 30-59, and less than 30 mL per minute, respectively, by the Cockcroft–Gault equation. While no dose adjustment is needed in patients with mild or moderate renal impairment, the dose of OTEZLA should be reduced to 30 mg once daily in patients with severe renal impairment [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
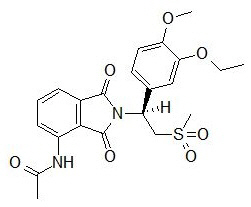
11 DESCRIPTION
The active ingredient in OTEZLA tablets is apremilast. Apremilast is a phosphodiesterase 4 (PDE4) inhibitor. Apremilast is known chemically as N-[2-[(1S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-2,3-dihydro-1,3-dioxo-1H-isoindol-4-yl]acetamide. Its empirical formula is C22H24N2O7S and the molecular weight is 460.5.
The chemical structure is:
OTEZLA tablets are supplied in 10-, 20-, and 30-mg strengths for oral administration. Each tablet contains apremilast as the active ingredient and the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, iron oxide red, iron oxide yellow (20 and 30 mg only) and iron oxide black (30 mg only).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Apremilast is an oral small-molecule inhibitor of phosphodiesterase 4 (PDE4) specific for cyclic adenosine monophosphate (cAMP). PDE4 inhibition results in increased intracellular cAMP levels. The specific mechanism(s) by which apremilast exerts its therapeutic action is not well defined.
12.3 Pharmacokinetics
Absorption
Apremilast when taken orally is absorbed with an absolute bioavailability of ~73%, with peak plasma concentrations (Cmax) occurring at a median time (tmax) of ~2.5 hours. Co-administration with food does not alter the extent of absorption of apremilast.Distribution
Human plasma protein binding of apremilast is approximately 68%. Mean apparent volume of distribution (Vd) is 87 L.Metabolism
Following oral administration in humans, apremilast is a major circulating component (45%) followed by inactive metabolite M12 (39%), a glucuronide conjugate of O-demethylated apremilast. It is extensively metabolized in humans with up to 23 metabolites identified in plasma, urine and feces. Apremilast is metabolized by both cytochrome (CYP) oxidative metabolism with subsequent glucuronidation and non-CYP mediated hydrolysis. In vitro, CYP metabolism of apremilast is primarily mediated by CYP3A4, with minor contributions from CYP1A2 and CYP2A6.Elimination
The plasma clearance of apremilast is about 10 L/hr in healthy subjects, with a terminal elimination half-life of approximately 6-9 hours. Following oral administration of radio-labeled apremilast, about 58% and 39% of the radioactivity is recovered in urine and feces, respectively, with about 3% and 7% of the radioactive dose recovered as apremilast in urine and feces, respectively.Specific Populations
Hepatic Impairment: The pharmacokinetics of apremilast is not affected by moderate or severe hepatic impairment.
Renal Impairment: The pharmacokinetics of apremilast is not affected by mild or moderate renal impairment. In 8 subjects with severe renal impairment administered a single dose of 30 mg apremilast, the AUC and Cmax of apremilast increased by approximately 88% and 42%, respectively [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Age: A single oral dose of 30-mg apremilast was studied in young adults and elderly healthy subjects. The apremilast exposure in elderly subjects (65 to 85 years of age) was about 13% higher in AUC and about 6% higher in Cmax than in young subjects (18 to 55 years of age) [see Use in Specific Populations (8.5)].
Gender: In pharmacokinetic studies in healthy volunteers, the extent of exposure in females was about 31% higher and Cmax was about 8% higher than that in male subjects.
Race and Ethnicity: The pharmacokinetics of apremilast in Chinese and Japanese healthy male subjects is comparable to that in Caucasian healthy male subjects. In addition, apremilast exposure is similar among Hispanic Caucasians, non-Hispanic Caucasians, and African Americans.
Drug Interactions
In vitro data: Apremilast is not an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 and not an inducer of CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP3A4. Apremilast is a substrate, but not an inhibitor of P-glycoprotein (P-gp) and is not a substrate or an inhibitor of organic anion transporter (OAT)1 and OAT3, organic cation transporter (OCT)2, organic anion transporting polypeptide (OATP)1B1 and OATP1B3, or breast cancer resistance protein (BCRP).Drug interaction studies were performed with apremilast and CYP3A4 substrates (oral contraceptive containing ethinyl estradiol and norgestimate), CYP3A and P-gp inhibitor (ketoconazole), CYP450 inducer (rifampin) and frequently co-administered drug in this patient population (methotrexate).
No significant pharmacokinetic interactions were observed when 30-mg oral apremilast was administered with either oral contraceptive, ketoconazole, or methotrexate. Co-administration of the CYP450 inducer rifampin (600 mg once daily for 15 days) with a single oral dose of 30-mg apremilast resulted in reduction of apremilast AUC and Cmax by 72% and 43%, respectively [see Warnings and Precautions (5.3) and Drug Interactions (7.1)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies were conducted in mice and rats with apremilast to evaluate its carcinogenic potential. No evidence of apremilast-induced tumors was observed in mice at oral doses up to 8.8-times the Maximum Recommended Human Dose (MRHD) on an AUC basis (1000 mg/kg/day) or in rats at oral doses up to approximately 0.08- and 1.1-times the MRHD, (20 mg/kg/day in males and 3 mg/kg/day in females, respectively).
Apremilast tested negative in the Ames assay, in vitro chromosome aberration assay of human peripheral blood lymphocytes, and the in vivo mouse micronucleus assay.
In a fertility study of male mice, apremilast at oral doses up to approximately 3-times the MRHD based on AUC (up to 50 mg/kg/day) produced no effects on male fertility. In a fertility study of female mice, apremilast was administered at oral doses of 10, 20, 40, or 80 mg/kg/day. At doses ≥1.8-times the MRHD (≥20 mg/kg/day), estrous cycles were prolonged, due to lengthening of diestrus which resulted in a longer interval until mating. Mice that became pregnant at doses of 20 mg/kg/day and greater also had increased incidences of early postimplantation losses. There was no effect of apremilast approximately 1.0-times the MRHD (10 mg/kg/day).
-
14 CLINICAL STUDIES
14.1 Psoriatic Arthritis
The safety and efficacy of OTEZLA was evaluated in 3 multicenter, randomized, double-blind, placebo-controlled trials (Studies PsA-1, PsA-2, and PsA-3) of similar design. A total of 1493 adult patients with active PsA (≥3 swollen joints and ≥3 tender joints) despite prior or current treatment with disease-modifying antirheumatic drug (DMARD) therapy were randomized. Patients enrolled in these studies had a diagnosis of PsA for at least 6 months. One qualifying psoriatic skin lesion of at least 2 cm in diameter was required in Study PsA- 3. Previous treatment with a biologic, including TNF-blockers was allowed (up to 10% could be TNF-blocker therapeutic failures). Across the 3 studies, patients were randomly assigned to placebo (n=496), OTEZLA 20 mg (n=500), or OTEZLA 30 mg (n=497) given orally twice daily. Titration was used over the first 5 days [see Dosage and Administration (2.1)]. Patients were allowed to receive stable doses of concomitant methotrexate [MTX (≤25 mg/week)], sulfasalazine [SSZ (≤2 g/day)], leflunomide [LEF (≤20 mg/day)], low dose oral corticosteroids (equivalent to ≤10 mg of prednisone a day), and/or nonsteroidal anti-inflammatory drugs (NSAIDs) during the trial. Treatment assignments were stratified based on small-molecule DMARD use at baseline in Studies PsA-1, PsA-2 and PsA-3. There was an additional stratification of BSA >3% with psoriasis in study PsA-3. The patients who were therapeutic failures of >3 agents for PsA (small molecules or biologics), or >1 biologic TNF blocker were excluded.
The primary endpoint was the percentage of patients achieving American College of Rheumatology (ACR) 20 response at Week 16. Placebo-controlled efficacy data were collected and analyzed through Week 24. Patients whose tender and swollen joint counts had not improved by at least 20% were considered non-responders at Week 16. Placebo non-responders were re-randomized 1:1 in a blinded fashion to either OTEZLA 20 mg twice daily or 30 mg twice daily following the titration schema [see Dosage and Administration (2.1)]. OTEZLA patients remained on their initial treatment. At Week 24, all remaining placebo patients were re-randomized to either 20 mg twice daily or 30 mg twice daily.
Patients with subtypes of PsA were enrolled across the 3 studies, including symmetric polyarthritis (62.0%), asymmetric oligoarthritis (27.0%), distal interphalangeal (DIP) joint arthritis (6.0%), arthritis mutilans (3.0%), and predominant spondylitis (2.1%). The median duration of PsA disease was 5 years. Patients received concomitant therapy with at least one DMARD (65.0%), MTX (55.0%), SSZ (9.0%), LEF (7.0%), low dose oral corticosteroids (14.0%), and NSAIDs (71.0%). Prior treatment with small-molecule DMARDs only was reported in 76.0% of patients and prior treatment with biologic DMARDs was reported in 22.0% of patients, which includes 9.0% who had failed prior biologic DMARD treatment.
Clinical Response in Patients with Psoriatic Arthritis
The percent of patients achieving ACR 20, 50 and 70 responses in Studies PsA-1, PsA-2, and PsA-3 are presented in Table 5 below. OTEZLA ± DMARDs, compared with Placebo ± DMARDs resulted in a greater improvement in signs and symptoms of psoriatic arthritis as demonstrated by the proportion of patients with an ACR 20 response at Week 16.
Table 5: Proportion of Patients With ACR Responses in Studies PsA-1, PsA-2 and PsA-3 a N is number of randomized and treated patients.
b Statistically significantly different from placebo (p<0.05).PsA-1 PsA-2 PsA-3 Na Placebo
±
DMARDs
N=168OTEZLA
30 mg
twice daily ±
DMARDs
N=168Placebo
±
DMARDs
N=159OTEZLA
30 mg
twice daily ±
DMARDs
N=162Placebo
±
DMARDs
N=169OTEZLA
30 mg
twice daily ±
DMARDs
N=167ACR 20
Week 1619% 38% b 19% 32% b 18% 41% b ACR 50
Week 166% 16% 5% 11% 8% 15% ACR 70
Week 161% 4% 1% 1% 2% 4% OTEZLA 30 mg twice daily resulted in improvement for each ACR component, compared to placebo at Week 16 in Study PsA-1 (Table 6). Consistent results were observed in Studies PsA-2 and PsA-3.
Table 6: ACR Components Mean Change from Baseline at Week 16 in Study PsA- 1 Mean changes from baseline are least square means from analyses of covariance.
a Scale 0-78.
b Scale 0-76.
c VAS=Visual Analog Scale; 0=best, 100=worst.
d HAQ-DI=Health Assessment Questionnaire-Disability Index; 0=best, 3=worst; measures the subject's ability to perform the following: dress/groom, arise, eat, walk, reach, grip, maintain hygiene, and maintain daily activity.
e CRP=C-reactive protein; Reference range 0-0.5 mg/dL.
* N reflects randomized patients; actual number of patients evaluable for each endpoint may vary by timepoint.Placebo
(N*=168)OTEZLA 30 mg
twice daily
(N*=168)Number of tender jointsa
Sample Size
Baseline
Mean Change at Week 16166
23
-2164
23
-7Number of swollen jointsb
Sample Size
Baseline
Mean Change at Week 16166
13
-2164
13
-5Patient's assessment of painc
Sample Size
Baseline
Mean Change at Week 16165
61
-6159
58
-14Patient's global assessment of disease
activityc
Sample Size
Baseline
Mean Change at Week 16165
59
-3159
56
-10Physician's global assessment of disease
activityc
Sample Size
Baseline
Mean Change at Week 16158
55
-8159
56
-19HAQ-DId score
Sample Size
Baseline
Mean Change at Week 16165
1.2
-0.09159
1.2
-0.2CRPe
Sample Size
Baseline
Mean Change at Week 16166
1.1
0.1167
0.8
-0.1Treatment with OTEZLA resulted in improvement in dactylitis and enthesitis in patients with pre-existing dactylitis or enthesitis.
Physical Function Response
OTEZLA 30 mg twice daily demonstrated a greater improvement compared to placebo in mean change from baseline for the Health Assessment Questionnaire Disability Index (HAQ-DI) score at Week 16 [-0.244 vs. -0.086, respectively; 95% CI for the difference was (-0.26, -0.06)] in Study PsA-1. The proportions of HAQ-DI responders (≥0.3 improvement from baseline) at Week 16 for the OTEZLA 30 mg twice daily group were 38%, compared to 27%, for the placebo group in Study PsA-1. Consistent results were observed in Studies PsA-2 and PsA-3.
14.2 Psoriasis
Two multicenter, randomized, double-blind, placebo-controlled trials (Studies PSOR-1 and PSOR-2) enrolled a total of 1257 subjects 18 years of age and older with moderate to severe plaque psoriasis [body surface area (BSA) involvement of ≥10%, static Physician Global Assessment (sPGA) of ≥3 (moderate or severe disease), Psoriasis Area and Severity Index (PASI) score ≥12, candidates for phototherapy or systemic therapy]. Subjects were allowed to use low-potency topical corticosteroids on the face, axilla and groin. Subjects with scalp psoriasis were allowed to use coal tar shampoo and/or salicylic acid scalp preparations on scalp lesions.
Study PSOR-1 enrolled 844 subjects and Study PSOR-2 enrolled 413 subjects. In both studies, subjects were randomized 2:1 to OTEZLA 30 mg BID or placebo for 16 weeks. Both studies assessed the proportion of subjects who achieved PASI-75 at Week 16 and the proportion of subjects who achieved a sPGA score of clear (0) or almost clear (1) at Week 16. Across both studies, subjects ranged in age from 18 to 83 years, with an overall median age of 46 years. The mean baseline BSA involvement was 25.19% (median 21.0%), the mean baseline PASI score was 19.07 (median 16.80), and the proportion of subjects with sPGA score of 3 (moderate) and 4 (severe) at baseline were 70.0% and 29.8%, respectively. Approximately 30% of all subjects had received prior phototherapy and 54% had received prior conventional systemic and/or biologic therapy for the treatment of psoriasis with 37% receiving prior conventional systemic therapy and 30% receiving prior biologic therapy. Approximately one-third of subjects had not received prior phototherapy, conventional systemic nor biologic therapy. A total of 18% of subjects had a history of psoriatic arthritis.
Clinical Response in Subjects with Plaque Psoriasis
The proportion of subjects who achieved PASI -75 responses, and sPGA score of clear (0) or almost clear (1), are presented in Table 7.
Table 7: Clinical Response at Week 16 in Studies PSOR-1 and PSOR-2 a N is number of randomized and treated patients.
b PASI=Psoriasis Area and Severity Index.
c sPGA=Static Physician Global Assessment.Study PSOR-1 Study PSOR-2 Placebo OTEZLA
30 mg BIDPlacebo OTEZLA
30 mg BIDNa N=282 N=562 N=137 N=274 PASIb-75, n (%) 15 (5.3) 186 (33.1) 8 (5.8) 79 (28.8) sPGAc of Clear or Almost
Clear, n (%)11 (3.9) 122 (21.7) 6 (4.4) 56 (20.4) The median time to loss of PASI-75 response among the subjects re-randomized to placebo at Week 32 during the Randomized Treatment Withdrawal Phase was 5.1 weeks.
14.3 Oral Ulcers Associated with Behçet's Disease
A multicenter, randomized, placebo-controlled trial (BCT-002) enrolled a total of 207 adult patients with BD with active oral ulcers. Patients were previously treated with at least one nonbiologic BD medication and were candidates for systemic therapy. Patients met the International Study Group (ISG) Criteria for BD. Patients had at least 2 oral ulcers at screening and at least 2 oral ulcers at randomization and without currently active major organ involvement. Concomitant treatment for BD was not allowed.
Patients were randomized 1:1 to receive either OTEZLA 30 mg twice daily (n=104) or placebo (n=103) for 12 weeks. After Week 12, all patients received OTEZLA 30 mg twice daily.
Efficacy was assessed based on the number and pain of oral ulcers.
Patients ranged in age from 19 to 72, with a mean age of 40 years. The mean duration of BD was 6.84 years. All subjects had a history of recurrent oral ulcers that were currently active. Subjects had a history of skin lesions (98.6%), genital ulcers (90.3%), musculoskeletal manifestations (72.5%), ocular manifestations (17.4%), central nervous system (9.7%), gastrointestinal (GI) manifestations (9.2%) and vascular involvement (1.4%). The mean baseline oral ulcer counts were 4.2 and 3.9 in the OTEZLA and placebo groups, respectively.
Measures of Oral Ulcers
Improvements in measures of oral ulcers at Week 12 are presented in Table 8.
Table 8: Clinical Response of Oral Ulcers at Week 12 in the BCT-002 Study (ITT* Population) Endpoint Placebo
N=103OTEZLA
30 mg twice daily
N=104Treatment Difference† (95% CI‡) - *
- ITT=intent to treat.
- †
- OTEZLA – Placebo.
- ‡
- CI=confidence interval.
- §
- Mean changes from baseline are least square means from mixed-effects model for repeated measures, adjusting for sex, region, and baseline pain of oral ulcers as measured by the visual analog scale.
- ¶
- VAS=visual analog scale; 0=no pain, 100=worst possible pain.
- #
- Patients for whom data are not available to determine response status are considered non-responders.
- Þ
- Adjusted difference in proportions is the weighted average of the treatment differences across the 4 strata of combined sex and region factors with the Cochran-Mantel-Haenszel weights.
- ß
- Mean daily averages are least squares means from analysis of covariance, after adjusting for sex, region, and baseline number of oral ulcers.
- à
- Based on oral ulcer counts measured at baseline and at Weeks 1, 2, 4, 6, 8, 10, and 12.
Change§ from baseline in the pain of oral ulcers as measured by VAS¶ at Week 12 -18.7 - 42.7 -24.1
(-32.4, -15.7)Proportion# of subjects achieving oral ulcer complete response (oral ulcer-free) at Week 12 22.3% 52.9% 30.6%Þ
(18.1%, 43.1%)Proportion# of subjects achieving oral ulcer complete response (oral ulcer-free) by Week 6, and who remained oral ulcer-free for at least 6 additional weeks during the 12-week Placebo-controlled Treatment Phase 4.9% 29.8% 25.1%Þ
(15.5%, 34.6%)Daily averageß,à number of oral ulcers during the 12-week Placebo-controlled Treatment Phase 2.6 1.5 -1.1
(-1.6, -0.7)Figure 1 displays the mean number of oral ulcers for each treatment group at each visit, while Figure 2 displays the mean oral ulcer pain on a visual analog scale for each treatment group at each visit.
ITT = intent-to-treat; SE = standard error. Figure 1: Mean (± SE) Number of Oral Ulcers by Time Point Through Week 12 (ITT Population) 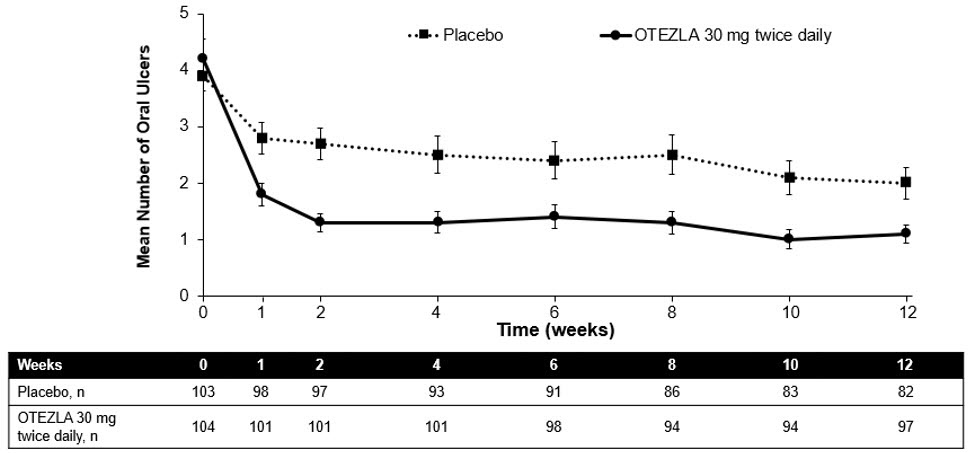
ITT=intent-to-treat; SE=standard error.
Oral ulcer pain was assessed on a 100-mm Visual Analog Scale with 0 = no pain and 100 = worst possible pain. Mean baseline Visual Analog Scale pain scores were 61.2 and 60.8 in the OTEZLA 30 mg twice daily treatment group and placebo treatment group, respectively.Figure 2: Mean (± SE) Oral Ulcer Pain on a Visual Analog Scale by Time Point Through Week 12 (ITT Population) 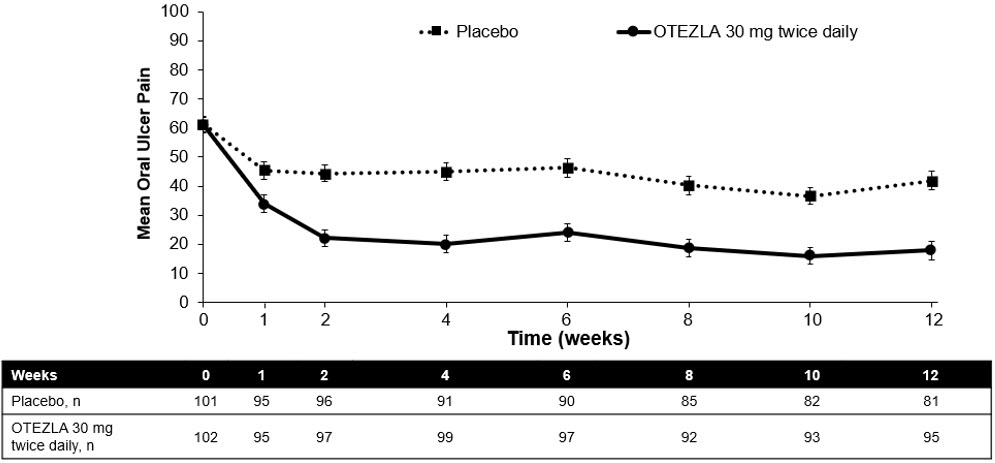
-
16 HOW SUPPLIED/STORAGE AND HANDLING
OTEZLA is available as diamond-shaped, film-coated tablets in the following dosage strengths: 10-mg pink tablet engraved with "APR" on one side and "10" on the other side; 20-mg brown tablet engraved with "APR" on one side and "20" on the other side; 30-mg beige tablet engraved with "APR" on one side and "30" on the other side.
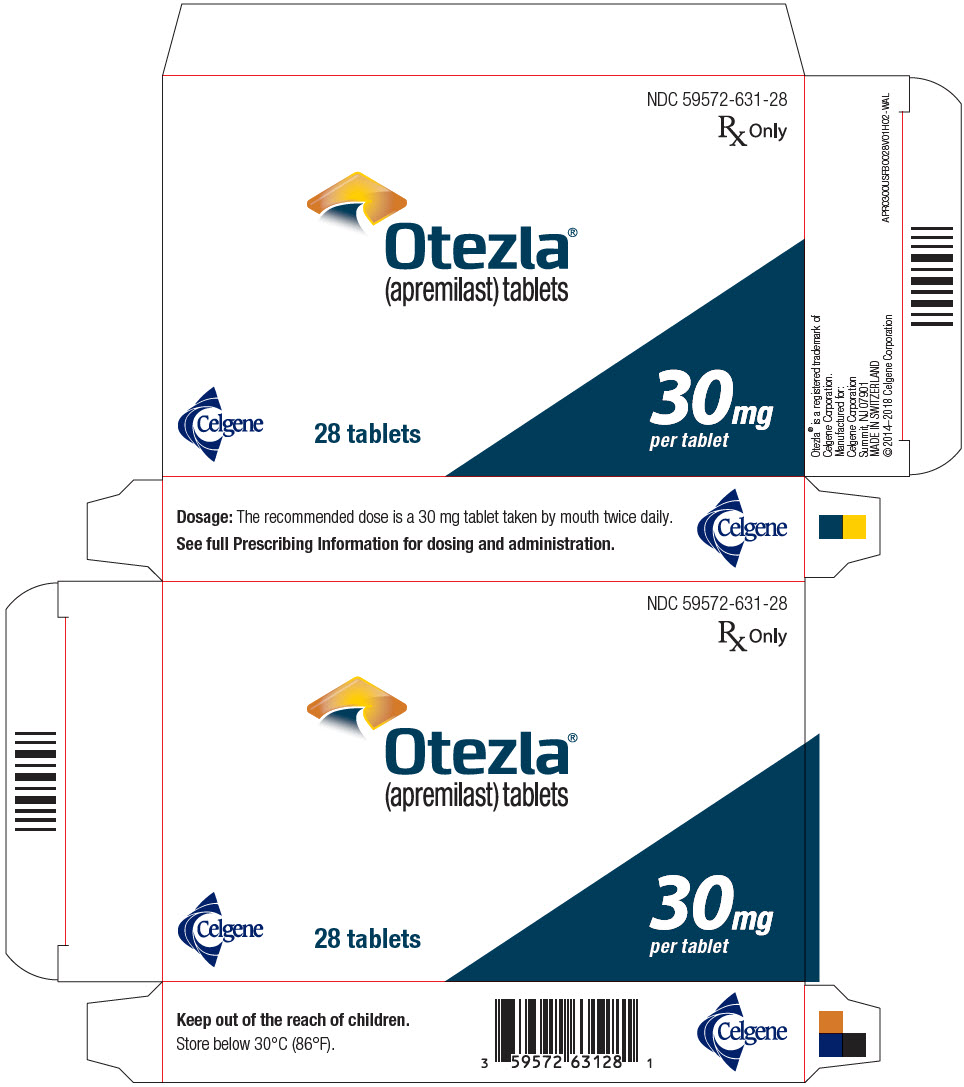
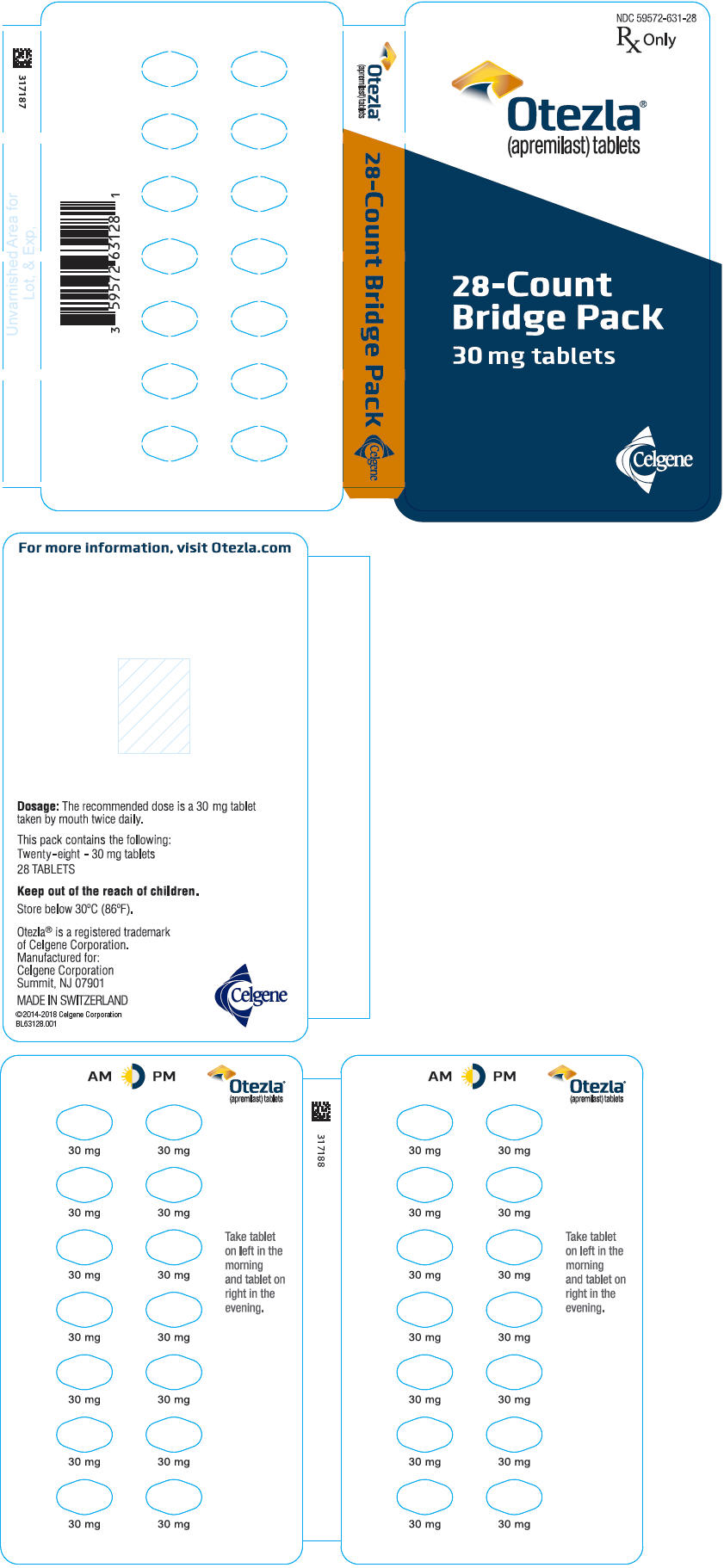
Tablets are supplied in the following strengths and package configurations:
Package configuration
Tablet strength
NDC number
Bottles of 60 30 mg 59572-631-06 Two-week starter pack 13-tablet blister titration pack
containing: (4) 10-mg, (4) 20-mg,
and (5) 30-mg tablets with an
additional (14) 30-mg tablets59572-630-27 28-count carton Two 30-mg blister cards containing
(14) 30-mg tablets59572-631-28 28-day starter pack 13-tablet blister titration pack
containing: (4) 10-mg, (4) 20-mg,
and (5) 30-mg tablets with an
additional (42) 30-mg tablets59572-632-55 Storage and Handling
Store tablets below 30°C (86°F). -
17 PATIENT COUNSELING INFORMATION
-
Diarrhea, Nausea, and Vomiting
Instruct patients to contact their healthcare provider if they experience severe diarrhea, nausea, or vomiting. Prescribers should advise patients of the potential complications of severe diarrhea, nausea, or vomiting. Consider OTEZLA dose reduction or suspension if patients develop severe diarrhea, nausea, or vomiting [see Warnings and Precautions (5.1)]. -
Depression
Before using OTEZLA in patients with a history of depression and/or suicidal thoughts or behavior, prescribers should carefully weigh the risks and benefits of treatment with OTEZLA in such patients. Patients, their caregivers, and families should be advised of the need to be alert for the emergence or worsening of depression, suicidal thoughts or other mood changes, and if such changes occur to contact their healthcare provider. Prescribers should carefully evaluate the risks and benefits of continuing treatment with OTEZLA if such events occur [see Warnings and Precautions (5.2)]. -
Weight Decrease
Patients treated with OTEZLA should have their weight monitored regularly. If unexplained or clinically significant weight loss occurs, weight loss should be evaluated, and discontinuation of OTEZLA should be considered [see Warnings and Precautions (5.3)]. -
Drug Interactions
The use of strong cytochrome P450 enzyme inducers (e.g., rifampin, phenobarbital, carbamazepine, phenytoin) with OTEZLA is not recommended [see Warnings and Precautions (5.4), Drug Interactions (7.1), and Clinical Pharmacology (12.3)]. - Instruct patients to take OTEZLA only as prescribed.
- Advise patients OTEZLA can be taken with or without food.
- Advise patients that the tablets should not be crushed, split, or chewed.
- Advise patients about the side effects associated with OTEZLA [see Adverse Reactions (6.1)].
-
Pregnancy
Inform patients that there is a pregnancy registry for pregnant women who have taken OTEZLA during pregnancy. Advise patients to contact the registry at 1-877-311-8972 to enroll or visit https://mothertobaby.org/ongoing-study/otezla/ [see Use in Specific Populations (8.1)]. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their prescriber of a known or suspected pregnancy.
Manufactured for: Celgene Corporation
Summit, NJ 07901OTEZLA® is a registered trademark of Celgene Corporation.
Pat. http://www.celgene.com/therapies
© 2014-2019 Celgene Corporation. All Rights Reserved.
APRPI.007 07/19
-
Diarrhea, Nausea, and Vomiting
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
PRINCIPAL DISPLAY PANEL - Sample Starter Pack - NDC: 59572-630-98
NDC 59572-630-98
Starter Pack
Rx OnlySAMPLE - NOT FOR SALE
Each pack contains the following for titration over
5 days up to the prescribed dose of 30 mg:
Four - 10 mg tablets
Four - 20 mg tablets
Nineteen - 30 mg tablets
Five starter packs each containing 27 TABLETSCelgene
Otezla®
(apremilast) tablets
- PRINCIPAL DISPLAY PANEL - 27 Tablet Sample Starter Pack Wallet - NDC: 59572-630-97
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL - 30 mg Tablet Blister Pack Carton - NDC: 59572-631-28
- PRINCIPAL DISPLAY PANEL - 30 mg Tablet Blister Pack Carton
- PRINCIPAL DISPLAY PANEL - NDC: 59572-631-99 - Sample 30 mg 28-count Blister Foil
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL - 30 mg Tablet Sample Bridge Pack - NDC: 59572-631-97
- PRINCIPAL DISPLAY PANEL - 30 mg Tablet Sample Bridge Pack Carton - NDC: 59572-631-97
- PRINCIPAL DISPLAY PANEL - 55 Tablet Blister Pack Carton
- PRINCIPAL DISPLAY PANEL - 55 Tablet Blister Pack
-
INGREDIENTS AND APPEARANCE
OTEZLA
apremilast kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59572-630 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59572-630-27 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/25/2014 2 NDC:59572-630-99 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/25/2014 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 4 Part 2 4 Part 3 19 Part 1 of 3 OTEZLA
apremilast tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APREMILAST (UNII: UP7QBP99PN) (APREMILAST - UNII:UP7QBP99PN) APREMILAST 10.0 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color PINK Score no score Shape DIAMOND Size 8mm Flavor Imprint Code APR;10 Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 Part 2 of 3 OTEZLA
apremilast tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APREMILAST (UNII: UP7QBP99PN) (APREMILAST - UNII:UP7QBP99PN) APREMILAST 20.0 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color BROWN Score no score Shape DIAMOND Size 10mm Flavor Imprint Code 20;APR Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 Part 3 of 3 OTEZLA
apremilast tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APREMILAST (UNII: UP7QBP99PN) (APREMILAST - UNII:UP7QBP99PN) APREMILAST 30.0 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color WHITE (Beige) Score no score Shape DIAMOND Size 12mm Flavor Imprint Code 30;APR Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 03/25/2014 OTEZLA
apremilast tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59572-631 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APREMILAST (UNII: UP7QBP99PN) (APREMILAST - UNII:UP7QBP99PN) APREMILAST 30.0 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color WHITE (Beige) Score no score Shape DIAMOND Size 12mm Flavor Imprint Code 30;APR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59572-631-06 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 09/18/2015 2 NDC:59572-631-28 28 in 1 CARTON 08/05/2015 2 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 04/03/2014 OTEZLA
apremilast kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59572-632 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59572-632-55 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 08/05/2015 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 4 Part 2 4 Part 3 47 Part 1 of 3 OTEZLA
apremilast tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APREMILAST (UNII: UP7QBP99PN) (APREMILAST - UNII:UP7QBP99PN) APREMILAST 10.0 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color PINK Score no score Shape DIAMOND Size 8mm Flavor Imprint Code APR;10 Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 Part 2 of 3 OTEZLA
apremilast tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APREMILAST (UNII: UP7QBP99PN) (APREMILAST - UNII:UP7QBP99PN) APREMILAST 20.0 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color BROWN Score no score Shape DIAMOND Size 10mm Flavor Imprint Code 20;APR Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 Part 3 of 3 OTEZLA
apremilast tablet, film coatedProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength APREMILAST (UNII: UP7QBP99PN) (APREMILAST - UNII:UP7QBP99PN) APREMILAST 30.0 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color WHITE (Beige) Score no score Shape DIAMOND Size 12mm Flavor Imprint Code 30;APR Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205437 02/01/2015 Labeler - Celgene Corporation (174201137)