Label: VIDAZA- azacitidine injection, powder, lyophilized, for solution


- NDC Code(s): 59572-102-01
- Packager: Celgene Corporation
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated January 29, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VIDAZA safely and effectively. See full prescribing information for VIDAZA.
VIDAZA (azacitidine for injection), for subcutaneous or intravenous use
Initial U.S. Approval: 2004INDICATIONS AND USAGE
VIDAZA is a nucleoside metabolic inhibitor indicated for the treatment of:
- •
- Adult patients with the following FAB myelodysplastic syndrome (MDS) subtypes: Refractory anemia (RA) or refractory anemia with ringed sideroblasts (RARS) (if accompanied by neutropenia or thrombocytopenia or requiring transfusions), refractory anemia with excess blasts (RAEB), refractory anemia with excess blasts in transformation (RAEB-T), and chronic myelomonocytic leukemia (CMMoL). (1.1)
- •
- Pediatric patients aged 1 month and older with newly diagnosed Juvenile Myelomonocytic Leukemia (JMML). (1.2)
DOSAGE AND ADMINISTRATION
- •
- Do not substitute VIDAZA for oral azacitidine. The indications and dosing regimen for VIDAZA differ from that of oral azacitidine (2.1, 5.1).
- •
- MDS: The recommended starting dosage for the first treatment cycle, for all patients regardless of baseline hematology values, is VIDAZA 75 mg/m2 daily for 7 days to be administered by subcutaneous injection or intravenous infusion. See full prescribing information for schedule for subsequent cycles. Premedicate for nausea and vomiting (2.2).
- •
- JMML: See full prescribing information for recommended dosage and schedule.
- •
- Continue treatment as long as the patient continues to benefit; up to 6 cycles for pediatric patients with JMML (2.3, 2.4).
- •
- Monitor all patients for hematologic response and for renal toxicity; delay or reduce dosage as appropriate (2.3, 2.4, 2.6, 2.7).
DOSAGE FORMS AND STRENGTHS
- •
- Lyophilized powder in 100 mg single-dose vials (3).
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- •
- Risks of Substitution with Other Azacitidine Products: Do not substitute VIDAZA for oral azacitidine (2.1, 5.1).
- •
- Anemia, Neutropenia and Thrombocytopenia: Monitor complete blood counts (CBC) frequently (5.2).
- •
- Hepatotoxicity: Patients with severe preexisting hepatic impairment are at higher risk for toxicity (5.3).
- •
- Renal Toxicity: Monitor patients with renal impairment for toxicity since azacitidine and its metabolites are primarily excreted by the kidneys (5.4).
- •
- Tumor Lysis Syndrome: VIDAZA may cause fatal or serious tumor lysis syndrome, including in patients with MDS. Assess baseline risk and monitor and treat as appropriate (5.5).
- •
- Embryo-Fetal Toxicity: VIDAZA can cause fetal harm. Advise female patients and male patients with female partners of reproductive potential of the potential risk to a fetus and to use effective contraception (5.6).
ADVERSE REACTIONS
Most common adverse reactions (>30%) in adult patients with MDS by subcutaneous route are: nausea, anemia, thrombocytopenia, vomiting, pyrexia, leukopenia, diarrhea, injection site erythema, constipation, neutropenia and ecchymosis. Most common adverse reactions by intravenous route also included petechiae, rigors, weakness and hypokalemia (6.1).
Most common adverse reactions (>30%) by intravenous route in pediatric patients with JMML are pyrexia, rash, upper respiratory tract infection, and anemia (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- •
- Lactation: Advise not to breastfeed (8.2).
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Myelodysplastic Syndromes (MDS)
1.2 Juvenile Myelomonocytic Leukemia (JMML)
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
2.2 First Treatment Cycle for Adults
2.3 Subsequent Treatment Cycles for Adults
2.4 Pediatric Patients with JMML
2.5 Dosage Adjustment Based on Hematology Laboratory Values
2.6 Dosage Adjustment Based on Serum Electrolytes and Renal Toxicity
2.7 Use in Geriatric Patients
2.8 Preparation of VIDAZA
2.9 Instructions for Subcutaneous Administration
2.10 Instructions for Intravenous Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Advanced Malignant Hepatic Tumors
4.2 Hypersensitivity to Azacitidine or Mannitol
5 WARNINGS AND PRECAUTIONS
5.1 Risks of Substitution with Other Azacitidine Products
5.2 Anemia, Neutropenia and Thrombocytopenia
5.3 Hepatotoxicity in Patients with Severe Pre-existing Hepatic Impairment
5.4 Renal Toxicity
5.5 Tumor Lysis Syndrome
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Myelodysplastic Syndromes (MDS)
14.2 Juvenile Myelomonocytic Leukemia (JMML)
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Myelodysplastic Syndromes (MDS)
VIDAZA® is indicated for treatment of adult patients with the following French-American-British (FAB) myelodysplastic syndrome subtypes: refractory anemia (RA) or refractory anemia with ringed sideroblasts (if accompanied by neutropenia or thrombocytopenia or requiring transfusions), refractory anemia with excess blasts (RAEB), refractory anemia with excess blasts in transformation (RAEB-T), and chronic myelomonocytic leukemia (CMMoL).
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
Do not substitute VIDAZA for oral azacitidine. The indications and dosing regimen for VIDAZA differ from that of oral azacitidine [see Warnings and Precautions (5.1)].
2.2 First Treatment Cycle for Adults
The recommended starting dose for the first treatment cycle, for all patients regardless of baseline hematology laboratory values, is 75 mg/m2 subcutaneously or intravenously, daily for 7 days. Premedicate patients for nausea and vomiting.
Obtain complete blood counts, liver chemistries and serum creatinine prior to the first dose.
2.3 Subsequent Treatment Cycles for Adults
Repeat cycles every 4 weeks. The dose may be increased to 100 mg/m2 if no beneficial effect is seen after 2 treatment cycles and if no toxicity other than nausea and vomiting has occurred. It is recommended that patients be treated for a minimum of 4 to 6 cycles. However, complete or partial response may require additional treatment cycles. Treatment may be continued as long as the patient continues to benefit.
Monitor patients for hematologic response and renal toxicities [see Warnings and Precautions (5.4)], and delay or reduce dosage if necessary [see Dosage and Administration (2.6)].
2.4 Pediatric Patients with JMML
Table 1: Dosage and Administration for Pediatric Patients (JMML)
Pediatric Patients with JMML
Recommended Dose
Age 1 month to less than 1 year OR
weighing less than 10 kg
2.5 mg/kg
Age 1 year and older AND weighing 10 kg
or greater
75 mg/m2
The recommended dosage for pediatric patients with JMML is provided in Table 1. VIDAZA is administered as an intravenous infusion daily for 7 days in a 28-day cycle. Patients should be treated for a minimum of 3 cycles and maximum of 6 cycles.
A delay in dose not exceeding 14 days can be considered for non-hematologic toxicities.
Monitor patients for hematologic response and renal toxicities [see Warnings and Precautions (5.4)], and delay or reduce dosage if necessary. Treatment may be continued up to six cycles as long as the patient continues to benefit.
2.5 Dosage Adjustment Based on Hematology Laboratory Values
- •
- For adult patients with baseline (start of treatment) WBC greater than or equal to 3 x109/L, ANC greater than or equal to 1.5 x109/L, and platelets greater than or equal to 75 x109/L, adjust the dose as follows, based on nadir counts for any given cycle:
Nadir Counts
% Dose in the Next
CourseANC (x109/L)
Less than 0.5
0.5 –1.5
Greater than 1.5Platelets (x109/L)
Less than 25
25-50
Greater than 50
50%
67%
100%- •
- For adult patients whose baseline counts are WBC less than 3 x109/L, ANC less than 1.5 x109/L, or platelets less than 75 x109/L, base dose adjustments on nadir counts and bone marrow biopsy cellularity at the time of the nadir as noted below, unless there is clear improvement in differentiation (percentage of mature granulocytes is higher and ANC is higher than at onset of that course) at the time of the next cycle, in which case continue the current dose.
WBC or Platelet
Nadir
% decrease in
counts
from baselineBone Marrow
Biopsy Cellularity at Time of Nadir
(%)30-60
15-30
Less than 15
% Dose in the Next Course50 - 75
100
50
33
Greater than 75
75
50
33
If a nadir as defined in the table above has occurred, give the next course 28 days after the start of the preceding course, provided that both the WBC and the platelet counts are greater than 25% above the nadir and rising. If a greater than 25% increase above the nadir is not seen by day 28, reassess counts every 7 days. If a 25% increase is not seen by day 42, reduce the scheduled dose by 50%.
Pediatric Patients with JMML
As hematological toxicity will be difficult to assess and to differentiate from the natural course of the underlying disorder, dose reductions are not recommended due to hematological toxicity within the first 3 cycles. However, if the patient has a neutrophil count of less than 0.5 x109/L at end of Cycle 3 or on Day 1 of Cycles 5 or 6, discontinue the treatment.
2.6 Dosage Adjustment Based on Serum Electrolytes and Renal Toxicity
If unexplained reductions in serum bicarbonate levels to less than 20 mEq/L occur, reduce the dosage by 50% for the next course. Similarly, if unexplained elevations of BUN or serum creatinine occur, delay the next cycle until values return to normal or baseline and reduce the dose by 50% for the next course [see Warnings and Precautions (5.4)].
2.7 Use in Geriatric Patients
Azacitidine and its metabolites are known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, select the dose carefully and monitor renal function [see Warnings and Precautions (5.4) and Use in Specific Populations (8.5)].
2.8 Preparation of VIDAZA
VIDAZA is a hazardous drug. Follow applicable special handling and disposal procedures.
The VIDAZA vial is single-dose and does not contain any preservatives. Discard unused portions of each vial properly [see How Supplied/Storage and Handling (16)]. Do not save any unused portions for later administration.
2.9 Instructions for Subcutaneous Administration
Reconstitute VIDAZA aseptically with 4 mL Sterile Water for Injection, USP to obtain a concentration of 25 mg/mL. Inject the diluent slowly into the vial. Vigorously shake or roll the vial until a uniform suspension is achieved. The suspension will be cloudy. Do not filter the suspension after reconstitution. Doing so could remove the active substance.
Preparation for Immediate Subcutaneous Administration: For doses requiring more than 1 vial, divide the dose equally between the syringes (e.g., dose 150 mg = 6 mL, 2 syringes with 3 mL in each syringe) and inject into two separate sites. Due to retention in the vial and needle, it may not be feasible to withdraw all of the suspension from the vial. The product may be held at room temperature for up to 1 hour, but must be administered within 1 hour after reconstitution.
Preparation for Delayed Subcutaneous Administration: The reconstituted product may be kept in the vial or drawn into a syringe. For doses requiring more than 1 vial, divide the dose equally between the syringes (e.g., dose 150 mg = 6 mL, 2 syringes with 3 mL in each syringe) and inject into two separate sites. Due to retention in the vial and needle, it may not be feasible to withdraw all of the suspension from the vial. The product must be refrigerated immediately. See Table 2 for suspension stability storage timelines based on the temperature of the diluent for delayed subcutaneous administration.
After removal from refrigerated conditions, the suspension may be allowed to equilibrate to room temperature for up to 30 minutes prior to administration.
Subcutaneous Administration
To provide a homogeneous suspension, the contents of the dosing syringe must be re-suspended immediately prior to administration. To re-suspend, vigorously roll the syringe between the palms until a uniform, cloudy suspension is achieved.
VIDAZA suspension is administered subcutaneously. Rotate sites for each injection (thigh, abdomen, or upper arm). New injections should be given at least one inch from an old site and never into areas where the site is tender, bruised, red, or hard.
Table 2 Suspension Stability: Storage timelines based on the temperature of the diluent for suspension stability storage:
Suspension Stability Storage timelines
Diluent
Storage
Temperature/Duration
Room temperature (25°C / 77°F)
Sterile Water for Injection, USP
Store at room
temperature at 25°C
(77°F) for up to 1 hour
or refrigerated at 2°C to
8°C (36°F to 46°F) for
up to 8 hours.
Cold (2°C to 8°C / 36°F to 46°F)
Sterile Water for Injection, USP
Store refrigerated at
2°C to 8°C
(36°F to 46°F) for up to
22 hours.
2.10 Instructions for Intravenous Administration
Parenteral drug product should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use the product if there is evidence of particulate matter or discoloration.
Adult Patients with MDS
Reconstitute the appropriate number of VIDAZA vials to achieve the desired dose. Reconstitute each vial with 10 mL Sterile Water for Injection, USP. Vigorously shake or roll the vial until all solids are dissolved. The resulting solution will contain azacitidine 10 mg/mL. The solution should be clear.
Withdraw the required amount of VIDAZA solution to deliver the desired dose and inject into a 50 -100 mL infusion bag of either 0.9% Sodium Chloride Injection, USP or Lactated Ringer’s Injection, USP.
Pediatric Patients with JMML
For pediatric patients with JMML, withdraw the required amount of VIDAZA solution to deliver the desired dose and inject into an infusion bag (volume up to 100 mL) of either 0.9% Sodium Chloride Injection, USP or Lactated Ringer’s Injection, USP to achieve a final VIDAZA concentration between 0.9 mg/mL and 4 mg/mL.
Intravenous Solution Incompatibility
VIDAZA is incompatible with 5% Dextrose Injection, USP solutions, Hespan, or solutions that contain bicarbonate. These solutions have the potential to increase the rate of degradation of VIDAZA and should therefore be avoided.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Advanced Malignant Hepatic Tumors
VIDAZA is contraindicated in patients with advanced malignant hepatic tumors [see Warnings and Precautions (5.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Risks of Substitution with Other Azacitidine Products
Due to substantial differences in the pharmacokinetic parameters [see Clinical Pharmacology (12.3)], the recommended dose and schedule for VIDAZA are different from those of oral azacitidine products. Treatment of patients using VIDAZA at the recommended dosage of oral azacitidine may result in a fatal adverse reaction. Treatment of patients using oral azacitidine at the doses recommended for VIDAZA may not be effective.
Do not substitute VIDAZA for oral azacitidine [see Dosage and Administration (2.1)].
5.2 Anemia, Neutropenia and Thrombocytopenia
VIDAZA causes anemia, neutropenia and thrombocytopenia in adult patients with MDS and in pediatric patients with JMML. Monitor complete blood counts frequently for response and/or toxicity, at a minimum, prior to each dosing cycle.
In adult patients with MDS, after administration of the recommended dosage for the first cycle, adjust dosage for subsequent cycles based on nadir counts and hematologic response [see Dosage and Administration (2.5)].
In pediatric patients with JMML, dose reductions due to hematological toxicity are not recommended within the first 3 cycles as hematological toxicity will be difficult to assess and to differentiate from the natural course of the underlying disorder [see Dosage and Administration (2.5)].
5.3 Hepatotoxicity in Patients with Severe Pre-existing Hepatic Impairment
Because azacitidine is potentially hepatotoxic in patients with severe pre-existing hepatic impairment, caution is needed in patients with liver disease. Patients with extensive tumor burden due to metastatic disease have been reported to experience progressive hepatic coma and death during azacitidine treatment, especially in such patients with baseline albumin <30 g/L. Azacitidine is contraindicated in patients with advanced malignant hepatic tumors [see Contraindications (4.1)]. Monitor liver chemistries prior to initiation of therapy and with each cycle.
Safety and effectiveness of VIDAZA in patients with MDS or in pediatric patients with JMML and hepatic impairment have not been studied as these patients were excluded from the clinical trials.
5.4 Renal Toxicity
Renal toxicity ranging from elevated serum creatinine to renal failure and death have been reported in patients treated with intravenous azacitidine in combination with other chemotherapeutic agents for non-MDS conditions. In addition, renal tubular acidosis, defined as a fall in serum bicarbonate to <20 mEq/L in association with an alkaline urine and hypokalemia (serum potassium <3 mEq/L) developed in 5 patients with CML treated with azacitidine and etoposide. Monitor serum creatinine and electrolytes prior to initiation of therapy and with each cycle. If unexplained reductions in serum bicarbonate <20 mEq/L or elevations of BUN or serum creatinine occur, reduce or hold the dose [see Dosage and Administration (2.6)].
Patients with renal impairment may be at increased risk for renal toxicity. Also, azacitidine and its metabolites are primarily excreted by the kidney. Therefore, monitor these patients closely for toxicity [see Dosage and Administration (2.6, 2.7)]. Patients with MDS or pediatric patients with JMML and renal impairment were excluded from the clinical studies.
5.5 Tumor Lysis Syndrome
VIDAZA may cause fatal or serious tumor lysis syndrome, including in patients with MDS. Tumor lysis syndrome may occur despite concomitant use of allopurinol. Assess baseline risk and monitor and treat as appropriate.
5.6 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, VIDAZA can cause fetal harm when administered to a pregnant woman. Azacitidine administered to pregnant rats via a single intraperitoneal (IP) dose approximating 8% of the recommended human daily dose caused fetal death and anomalies.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with VIDAZA and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with VIDAZA and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are described in other labeling sections:
- o
- Anemia, Neutropenia and Thrombocytopenia [see Warnings and Precautions (5.2)]
- o
- Hepatotoxicity in Patients with Severe Pre-existing Hepatic Impairment [see Warnings and Precautions (5.3)]
- o
- Renal Toxicity [see Warnings and Precautions (5.4)]
- o
- Tumor Lysis Syndrome [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
MDS
The data described below reflect exposure to VIDAZA in 443 patients with MDS from 4 clinical studies. Study 1 was a supportive-care controlled trial (subcutaneous administration), Studies 2 and 3 were single arm studies (one with subcutaneous administration and one with intravenous administration), and Study 4 was an international randomized trial (subcutaneous administration) [see Clinical Studies (14.1)].
In Studies 1, 2 and 3, a total of 268 patients were exposed to VIDAZA, including 116 exposed for 6 cycles (approximately 6 months) or more and 60 exposed for greater than 12 cycles (approximately one year). VIDAZA was studied primarily in supportive-care controlled and uncontrolled trials (n=150 and n=118, respectively). The population in the subcutaneous studies (n=220) was 23 to 92 years old (mean 66.4 years), 68% male, and 94% white, and had MDS or AML. The population in the intravenous study (n=48) was 35 to 81 years old (mean 63.1 years), 65% male, and 100% white. Most patients received average daily doses between 50 and 100 mg/m2.
In Study 4, a total of 175 patients with higher-risk MDS (primarily RAEB and RAEB-T subtypes) were exposed to VIDAZA. Of these patients, 119 were exposed for 6 or more cycles, and 63 for at least 12 cycles. The mean age of this population was 68.1 years (ranging from 42 to 83 years), 74% were male, and 99% were white. Most patients received daily VIDAZA doses of 75 mg/m2.
Most Commonly Occurring Adverse Reactions (Subcutaneous or Intravenous Route) in Adult Patients with MDS: nausea, anemia, thrombocytopenia, vomiting, pyrexia, leukopenia, diarrhea, injection site erythema, constipation, neutropenia, ecchymosis. The most common adverse reactions by intravenous route also included petechiae, rigors, weakness and hypokalemia.
Adverse Reactions Most Frequently (>2%) Resulting in Clinical Intervention (Subcutaneous or Intravenous Route) in Adult Patients with MDS:
Discontinuation: leukopenia, thrombocytopenia, neutropenia.
Dose Held: leukopenia, neutropenia, thrombocytopenia, pyrexia, pneumonia, febrile neutropenia.
Dose Reduced: leukopenia, neutropenia, thrombocytopenia.
Table 3 presents adverse reactions occurring in at least 5% of patients treated with VIDAZA (subcutaneous) in Studies 1 and 2. It is important to note that duration of exposure was longer for the VIDAZA-treated group than for the observation group: patients received VIDAZA for a mean of 11.4 months while mean time in the observation arm was 6.1 months.
Table 3: Most Frequently Observed Adverse Reactions (≥ 5% in All Subcutaneous VIDAZA Treated Patients; Studies 1 and 2) Number (%) of Patients System Organ Class
Preferred TermaAll VIDAZAb
(N=220)Observationc
(N=92)a Multiple terms of the same preferred terms for a patient are only counted once within each treatment group.
b Includes adverse reactions from all patients exposed to VIDAZA, including patients after crossing over from observations.
c Includes adverse reactions from observation period only; excludes any adverse events after crossover to VIDAZA.Blood and lymphatic system disorders
Anemia
153 (70)
59 (64)
Anemia aggravated
12 (6)
5 (5)
Febrile neutropenia
36 (16)
4 (4)
Leukopenia
106 (48)
27 (29)
Neutropenia
71 (32)
10 (11)
Thrombocytopenia
144 (66)
42 (46)
Gastrointestinal disorders
Abdominal tenderness
26 (12)
1 (1)
Constipation
74 (34)
6 (7)
Diarrhea
80 (36)
13 (14)
Gingival bleeding
21 (10)
4 (4)
Loose stools
12 (6)
0
Mouth hemorrhage
11 (5)
1 (1)
Nausea
155 (71)
16 (17)
Stomatitis
17 (8)
0
Vomiting
119 (54)
5 (5)
General disorders and administration site conditions
Chest pain
36 (16)
5 (5)
Injection site bruising
31 (14)
0
Injection site erythema
77 (35)
0
Injection site granuloma
11 (5)
0
Injection site pain
50 (23)
0
Injection site pigmentation changes
11 (5)
0
Injection site pruritus
15 (7)
0
Injection site reaction
30 (14)
0
Injection site swelling
11 (5)
0
Lethargy
17 (8)
2 (2)
Malaise
24 (11)
1 (1)
Pyrexia
114 (52)
28 (30)
Infections and infestations
Nasopharyngitis
32 (15)
3 (3)
Pneumonia
24 (11)
5 (5)
Upper respiratory tract infection
28 (13)
4 (4)
Injury, poisoning, and procedural complications
Post procedural hemorrhage
13 (6)
1 (1)
Metabolism and nutrition disorders
Anorexia
45 (21)
6 (7)
Musculoskeletal and connective tissue disorders
Arthralgia
49 (22)
3 (3)
Chest wall pain
11 (5)
0
Myalgia
35 (16)
2 (2)
Nervous system disorders
Dizziness
41 (19)
5 (5)
Headache
48 (22)
10 (11)
Psychiatric disorders
Anxiety
29 (13)
3 (3)
Insomnia
24 (11)
4 (4)
Respiratory, thoracic and mediastinal disorders
Dyspnea
64 (29)
11 (12)
Skin and subcutaneous tissue disorders
Dry skin
11 (5)
1 (1)
Ecchymosis
67 (31)
14 (15)
Erythema
37 (17)
4 (4)
Rash
31 (14)
9 (10)
Skin nodule
11 (5)
1 (1)
Urticaria
13 (6)
1 (1)
Vascular disorders
Hematoma
19 (9)
0
Hypotension
15 (7)
2 (2)
Petechiae
52 (24)
8 (9)
Table 4 presents adverse reactions occurring in at least 5% of patients treated with VIDAZA in Study 4. Similar to Studies 1 and 2 described above, duration of exposure to treatment with VIDAZA was longer (mean 12.2 months) compared with best supportive care (mean 7.5 months).
Table 4: Most Frequently Observed Adverse Reactions (≥ 5% in the VIDAZA Treated Patients and the Percentage with NCI CTC Grade 3/4 Reactions; Study 4) Number (%) of Patients Any Grade Grade 3/4 System Organ Class
Preferred TermaVIDAZA
(N=175)Best Supportive Care Only
(N=102)VIDAZA
(N=175)Best Supportive Care Only
(N=102)a Multiple reports of the same preferred term from a patient were only counted once within each treatment. Blood and lymphatic system disorders
Anemia
90 (51)
45 (44)
24 (14)
9 (9)
Febrile neutropenia
24 (14)
10 (10)
22 (13)
7 (7)
Leukopenia
32 (18)
2 (2)
26 (15)
1 (1)
Neutropenia
115 (66)
29 (28)
107 (61)
22 (22)
Thrombocytopenia
122 (70)
35 (34)
102 (58)
29 (28)
Gastrointestinal disorders
Abdominal pain
22 (13)
7 (7)
7 (4)
0
Constipation
88 (50)
8 (8)
2 (1)
0
Dyspepsia
10 (6)
2 (2)
0
0
Nausea
84 (48)
12 (12)
3 (2)
0
Vomiting
47 (27)
7 (7)
0
0
General disorders and administration site conditions
Fatigue
42 (24)
12 (12)
6 (3)
2 (2)
Injection site bruising
9 (5)
0
0
0
Injection site erythema
75 (43)
0
0
0
Injection site hematoma
11 (6)
0
0
0
Injection site induration
9 (5)
0
0
0
Injection site pain
33 (19)
0
0
0
Injection site rash
10 (6)
0
0
0
Injection site reaction
51 (29)
0
1 (1)
0
Pyrexia
53 (30)
18 (18)
8 (5)
1 (1)
Infections and infestations
Rhinitis
10 (6)
1 (1)
0
0
Upper respiratory tract infection
16 (9)
4 (4)
3 (2)
0
Urinary tract infection
15 (9)
3 (3)
3 (2)
0
Investigations
Weight decreased
14 (8)
0
1 (1)
0
Metabolism and nutrition disorders
Hypokalemia
11 (6)
3 (3)
3 (2)
3 (3)
Nervous system disorders
Lethargy
13 (7)
2 (2)
0
1 (1)
Psychiatric disorders
Anxiety
9 (5)
1 (1)
0
0
Insomnia
15 (9)
3 (3)
0
0
Renal and urinary disorders
Hematuria
11 (6)
2 (2)
4 (2)
1 (1)
Respiratory, thoracic and mediastinal disorders
Dyspnea
26 (15)
5 (5)
6 (3)
2 (2)
Dyspnea exertional
9 (5)
1 (1)
0
0
Pharyngolaryngeal pain
11 (6)
3 (3)
0
0
Skin and subcutaneous tissue disorders
Erythema
13 (7)
3 (3)
0
0
Petechiae
20 (11)
4 (4)
2 (1)
0
Pruritus
21 (12)
2 (2)
0
0
Rash
18 (10)
1 (1)
0
0
Vascular disorders
Hypertension
15 (9)
4 (4)
2 (1)
2 (2)
In Studies 1, 2 and 4 with subcutaneous administration of VIDAZA, adverse reactions of neutropenia, thrombocytopenia, anemia, nausea, vomiting, diarrhea, constipation, and injection site erythema/reaction tended to increase in incidence with higher doses of VIDAZA. Adverse reactions that tended to be more pronounced during the first 1 to 2 cycles of subcutaneous treatment compared with later cycles included thrombocytopenia, neutropenia, anemia, nausea, vomiting, injection site erythema/pain/bruising/reaction, constipation, petechiae, dizziness, anxiety, hypokalemia, and insomnia. There did not appear to be any adverse reactions that increased in frequency over the course of treatment.
Overall, adverse reactions were qualitatively similar between the intravenous and subcutaneous studies. Adverse reactions that appeared to be specifically associated with the intravenous route of administration included infusion site reactions (e.g. erythema or pain) and catheter site reactions (e.g. infection, erythema, or hemorrhage).
In clinical studies of either subcutaneous or intravenous VIDAZA, the following serious adverse reactions occurring at a rate of <5% (and not described in Tables 2 or 3) were reported:
Blood and lymphatic system disorders: agranulocytosis, bone marrow failure, pancytopenia splenomegaly.
Cardiac disorders: atrial fibrillation, cardiac failure, cardiac failure congestive, cardio-respiratory arrest, congestive cardiomyopathy.
Eye disorders: eye hemorrhage
Gastrointestinal disorders: diverticulitis, gastrointestinal hemorrhage, melena, perirectal abscess.
General disorders and administration site conditions: catheter site hemorrhage, general physical health deterioration, systemic inflammatory response syndrome.
Hepatobiliary disorders: cholecystitis.
Immune system disorders: anaphylactic shock, hypersensitivity.
Infections and infestations: abscess limb, bacterial infection, cellulitis, blastomycosis, injection site infection, Klebsiella sepsis, neutropenic sepsis, pharyngitis streptococcal, pneumonia Klebsiella, sepsis, septic shock, Staphylococcal bacteremia, Staphylococcal infection, toxoplasmosis.
Metabolism and nutrition disorders: dehydration.
Musculoskeletal and connective tissue disorders: bone pain aggravated, muscle weakness, neck pain.
Neoplasms benign, malignant and unspecified: leukemia cutis.
Nervous system disorders: cerebral hemorrhage, convulsions, intracranial hemorrhage.
Renal and urinary disorders: loin pain, renal failure.
Respiratory, thoracic and mediastinal disorders: hemoptysis, lung infiltration, pneumonitis, respiratory distress.
Skin and subcutaneous tissue disorders: pyoderma gangrenosum, rash pruritic, skin induration.
Surgical and medical procedures: cholecystectomy.
Vascular disorders: orthostatic hypotension.
Juvenile Myelomonocytic Leukemia (JMML)
The safety of VIDAZA was evaluated in pediatric patients with JMML (N=18; 0.2-6.9 years old) in Study AZA-JMML-001 [see Clinical Studies (14.2)]. Patients received VIDAZA as intravenous infusion daily for 7 days in a 28-day cycle [see Dosage and Administration (2.4)].
Sixteen patients completed 3 cycles of therapy and 5 of the 16 patients completed 6 cycles. The median treatment duration was 12.3 weeks (range: 4.0 to 30.6 weeks) and the median total number of treatment cycles completed was 3 cycles (range: 1 to 6 cycles).
Serious adverse reactions occurred in 67% of patients who received VIDAZA for JMML.
Permanent discontinuation of VIDAZA due to an adverse reaction occurred in a single patient who had abdominal pain and acute respiratory distress syndrome.
The most common (≥30%) adverse reactions were pyrexia, rash, upper respiratory tract infection, and anemia.
Table 5 summarizes the adverse reactions in pediatric JMML.
Table 5: Most Frequently Observed Adverse Reactions (≥10%) in Pediatric Patients with JMML Receiving VIDAZA in Study AZA-JMML-001
Adverse Reaction
All Grades
N=18
%
Grade 3 or 4
N=18
%
General disorders and administration site conditions
Pyrexia
61
0
Edemaa
11
0
Skin and subcutaneous tissue disorders
Rashb
44
0
Pruritusc
22
0
Dermatitis allergic
11
0
Petechiae
11
0
Infections and infestations
Upper respiratory tract infectiond
44
6
Febrile infection
11
6
Fungal infectione
11
6
Pneumoniaf
11
0
Blood and lymphatic system disorders
Anemia
39
33
Thrombocytopenia
28
22
Neutropenia
17
11
Febrile Neutropenia
11
6
Lymphadenopathy
11
6
Gastrointestinal disorders
Vomiting
28
0
Abdominal paing
22
11
Constipation
22
0
Diarrhea
22
6
Hemorrhageh
11
0
Nausea
11
0
Ascites
11
6
Respiratory, thoracic and mediastinal disorders
Cough
22
0
Epistaxis
17
6
Metabolism and nutrition disorders
Hyperuricemia
17
0
Fluid retention
11
0
Musculoskeletal and connective tissue disorders
Musculoskeletal paini
11
0
a Grouped term includes: face edema, generalized edema, edema peripheral.
b Grouped term includes: rash, rash macular, dermatitis bullous, rash maculo-papular, rash papular, rash pruritic.
c Grouped term includes: pruritis, pruritis generalized.
d Grouped term includes: influenza, nasopharyngitis, Parainfluenzae virus infection, respiratory tract infection, rhinitis, upper respiratory tract infection, and viral upper respiratory tract infection
e Grouped term includes: anal candidiasis, Candida infection, oral candidiasis.
f Grouped term includes: lung infection, pneumonia respiratory syncytial viral.
g Grouped term includes: abdominal pain, abdominal pain upper.
h Grouped term includes: mouth hemorrhage, rectal hemorrhage.
i Grouped term includes: back pain, musculoskeletal pain, non-cardiac chest pain, pain in extremity.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of VIDAZA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- ‑
- Interstitial lung disease
- ‑
- Tumor lysis syndrome
- ‑
- Injection site necrosis
- ‑
- Sweet’s syndrome (acute febrile neutrophilic dermatosis)
- ‑
- Necrotizing fasciitis (including fatal cases)
- ‑
- Differentiation syndrome
- ‑
- Pericardial effusion
- ‑
- Pericarditis
- ‑
- Cutaneous vasculitis
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings in animals, VIDAZA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no data on the use of azacitidine in pregnant women. Azacitidine was teratogenic and caused embryo-fetal lethality in animals at doses lower than the recommended human daily dose (see Data). Advise pregnant women of the potential risk to the fetus.The background rate of major birth defects and miscarriage is unknown for the indicated population. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Data
Animal Data
Early embryotoxicity studies in mice revealed a 44% frequency of intrauterine embryonal death (increased resorption) after a single IP (intraperitoneal) injection of 6 mg/m2 (approximately 8% of the recommended human daily dose on a mg/m2 basis) azacitidine on gestation day 10. Developmental abnormalities in the brain have been detected in mice given azacitidine on or before gestation day 15 at doses of ~3-12 mg/m2 (approximately 4%-16% the recommended human daily dose on a mg/m2 basis).In rats, azacitidine was clearly embryotoxic when given IP on gestation days 4-8 (postimplantation) at a dose of 6 mg/m2 (approximately 8% of the recommended human daily dose on a mg/m2 basis), although treatment in the preimplantation period (on gestation days 1-3) had no adverse effect on the embryos. Azacitidine caused multiple fetal abnormalities in rats after a single IP dose of 3 to 12 mg/m2 (approximately 8% the recommended human daily dose on a mg/m2 basis) given on gestation day 9, 10, 11 or 12. In this study azacitidine caused fetal death when administered at 3-12 mg/m2 on gestation days 9 and 10; average live animals per litter was reduced to 9% of control at the highest dose on gestation day 9. Fetal anomalies included: CNS anomalies (exencephaly/encephalocele), limb anomalies (micromelia, club foot, syndactyly, oligodactyly), and others (micrognathia, gastroschisis, edema, and rib abnormalities).
8.2 Lactation
Risk Summary
There is no information regarding the presence of azacitidine in human milk, the effects of VIDAZA on the breastfed infant, or the effects of VIDAZA on milk production. Because many drugs are excreted in human milk and because of the potential for tumorigenicity shown for azacitidine in animal studies [see Nonclinical Toxicology (13.1)] and the potential for serious adverse reactions in nursing infants from VIDAZA, advise patients not to breastfeed during treatment with VIDAZA and for 1 week after the last dose.8.3 Females and Males of Reproductive Potential
Based on its mechanism of action and findings in animals, VIDAZA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating VIDAZA.Contraception
Females
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with VIDAZA and for 6 months after the last dose.Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with VIDAZA and for 3 months after the last dose.Infertility
Based on animal data, azacitidine could have an effect on male or female fertility [see Nonclinical Toxicology (13.1)].8.4 Pediatric Use
Safety and effectiveness of VIDAZA in pediatric patients with MDS have not been established.
The safety and effectiveness of VIDAZA have been established in pediatric patients with newly diagnosed JMML aged 1 month and older and the information on this use is discussed throughout the labeling. The safety and effectiveness of VIDAZA have not been established in pediatric patients younger than 1 month old [see Clinical Studies (14.2)].
8.5 Geriatric Use
Of the total number of patients in Studies 1, 2 and 3, 62% were 65 years and older and 21% were 75 years and older. No overall differences in effectiveness were observed between these patients and younger patients. In addition, there were no relevant differences in the frequency of adverse reactions observed in patients 65 years and older compared to younger patients.
Of the 179 patients randomized to azacitidine in Study 4, 68% were 65 years and older and 21% were 75 years and older. Survival data for patients 65 years and older were consistent with overall survival results. The majority of adverse reactions occurred at similar frequencies in patients <65 years of age and patients 65 years of age and older.
Elderly patients are more likely to have decreased renal function. Monitor renal function in these patients [see Dosage and Administration (2.7) and Warnings and Precautions (5.4)].
-
10 OVERDOSAGE
One case of overdose with VIDAZA was reported during clinical trials. A patient experienced diarrhea, nausea, and vomiting after receiving a single intravenous dose of approximately 290 mg/m2, almost 4 times the recommended starting dose. The events resolved without sequelae, and the correct dose was resumed the following day. In the event of overdosage, the patient should be monitored with appropriate blood counts and should receive supportive treatment, as necessary. There is no known specific antidote for VIDAZA overdosage.
-
11 DESCRIPTION
VIDAZA (azacitidine for injection) contains azacitidine, which is a nucleoside metabolic inhibitor. Azacitidine is
4-amino-1-β-D-ribofuranosyl-s-triazin-2(1H)-one. The structural formula is as follows:
The empirical formula is C8H12N4O5. The molecular weight is 244. Azacitidine is a white to off-white solid. Azacitidine was found to be insoluble in acetone, ethanol, and methyl ethyl ketone; slightly soluble in ethanol/water (50/50), propylene glycol, and polyethylene glycol; sparingly soluble in water, water saturated octanol, 5% dextrose in water, N-methyl-2-pyrrolidone, normal saline and 5% Tween 80 in water; and soluble in dimethylsulfoxide (DMSO).
The finished product is supplied in a sterile form for reconstitution as a suspension for subcutaneous injection or reconstitution as a solution with further dilution for intravenous infusion. Vials of VIDAZA contain 100 mg of azacitidine and 100 mg mannitol as a sterile lyophilized powder.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
VIDAZA is a pyrimidine nucleoside analog of cytidine. VIDAZA is believed to exert its antineoplastic effects by causing hypomethylation of DNA and direct cytotoxicity on abnormal hematopoietic cells in the bone marrow. The concentration of azacitidine required for maximum inhibition of DNA methylation in vitro does not cause major suppression of DNA synthesis. Hypomethylation may restore normal function to genes that are critical for differentiation and proliferation. The cytotoxic effects of azacitidine cause the death of rapidly dividing cells, including cancer cells that are no longer responsive to normal growth control mechanisms. Non-proliferating cells are relatively insensitive to azacitidine.
12.2 Pharmacodynamics
In pediatric patients with JMML post azacitidine treatment (75 mg/m2 or 2.5 mg/kg), reductions in genome-wide DNA methylation (global DNA methylation scores) were attained in bone marrow granulocytes during the first treatment cycle, confirming the DNA-hypomethylating activity of azacitidine.
12.3 Pharmacokinetics
The pharmacokinetics of azacitidine were studied in 6 adult patients with MDS following a single 75 mg/m2 subcutaneous dose and a single 75 mg/m2 intravenous dose.
Absorption
Azacitidine is rapidly absorbed after subcutaneous administration; the peak plasma azacitidine concentration of 750 ± 403 ng/ml occurred in 0.5 hour after subcutaneous administration.
Distribution
The bioavailability of subcutaneous azacitidine relative to intravenous azacitidine is approximately 89%, based on area under the curve. Mean volume of distribution following intravenous dosing is 76 ± 26 L. Mean apparent subcutaneous clearance is 167 ± 49 L/hour and mean half-life after subcutaneous administration is 41 ± 8 minutes. The AUC and Cmax of subcutaneous administration of azacitidine in 21 patients with cancer were approximately dose proportional within the 25 to 100 mg/m2 dose range. Multiple dosing at the recommended dose-regimen does not result in drug accumulation with intravenous or subcutaneous administration.
Elimination
Metabolism
An in vitro study of azacitidine incubation in human liver fractions indicated that azacitidine is not metabolized by the cytochrome P450 (CYP) enzymes. Azacitidine undergoes spontaneous hydrolysis and deamination mediated by cytidine deaminase.
Excretion
Published studies indicate that urinary excretion is the primary route of elimination of azacitidine and its metabolites. Following intravenous administration of radioactive azacitidine to 5 cancer patients, the cumulative urinary excretion was 85% of the radioactive dose. Fecal excretion accounted for <1% of administered radioactivity over 3 days. Mean excretion of radioactivity in urine following subcutaneous administration of 14C-azacitidine was 50%. The mean elimination half-lives of total radioactivity (azacitidine and its metabolites) were similar after intravenous and subcutaneous administrations, about 4 hours.
Specific Populations
The effects of hepatic impairment, gender, or race/ethnicity on the pharmacokinetics of intravenous and subcutaneous azacitidine have not been studied.
Pediatric Patients
Following intravenous administration of a 75 mg/m2 or 2.5 mg/kg dose in pediatric patients with JMML, population pharmacokinetic analysis determined that azacitidine rapidly reached a geometric mean Cmax of 4510 ng/mL (CV%: 65.6%), and a geometric mean AUC0-24 of 1550 ng∙h/mL (CV%: 56.6%). The geometric mean t1/2 was 0.3 hours (CV%: 59.9%), and the geometric mean clearance was 21.8 L/h (CV%: 102.2%).
Population PK analysis of 6 adults and 34 pediatric patients did not identify any clinically meaningful differences in the pharmacokinetics of intravenous azacitidine according to sex (62.5% male), and tumor type (MDS, JMML, AML). Increased clearance was associated with increased baseline body weight (4.6 to 102 kg). BSA or body weight-based dosing resulted in consistent plasma azacitidine exposure in pediatric patients with JMML across the body weight range (4.6 to 18.5 kg) and age range (0.2 to 6.9 years).
Patients with Renal Impairment
In adult patients with cancer, the pharmacokinetics of azacitidine in 6 patients with normal renal function (CLcr >80 mL/min) and 6 patients with severe renal impairment (CLcr <30 mL/min) were compared following daily subcutaneous dosing (Days 1 through 5) at 75 mg/m2/day. Severe renal impairment increased azacitidine exposure by approximately 70% after single and 41% after multiple subcutaneous administrations. This increase in exposure was not correlated with an increase in adverse events. The exposure was similar to exposure in patients with normal renal function receiving 100 mg/m2.
Drug-Drug Interactions
No formal clinical drug interaction studies with azacitidine have been conducted.
In vitro Studies
Cytochrome P450 (CYP) Enzymes: An in vitro study at azacitidine concentrations up to 100 µM (IV Cmax = 10.6 µM) in human liver microsomes indicated that azacitidine does not cause any inhibition of CYP isoforms CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, or CYP2E1 at clinically achievable concentrations.
In vitro studies with human cultured hepatocytes indicate that azacitidine at concentrations of 1.0 μM to 100 μM does not induce CYP 1A2, 2C19, or 3A4/5.
Transporter Systems: An in vitro study with LLC-PK1 cells expressing P-glycoprotein (P-gp) indicated that azacitidine is not a substrate or inhibitor of P-gp.
Azacitidine does not inhibit, breast cancer resistance protein (BCRP), organic anion transporters (OAT) OAT1 and OAT3, organic anion transporting polypeptides (OATP) OATP1B1 and OATP1B3, or organic cation transporter (OCT) OCT2 at clinically relevant concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The potential carcinogenicity of azacitidine was evaluated in mice and rats. Azacitidine induced tumors of the hematopoietic system in female mice at 2.2 mg/kg (6.6 mg/m2, approximately 8% the recommended human daily dose on a mg/m2 basis) administered IP three times per week for 52 weeks. An increased incidence of tumors in the lymphoreticular system, lung, mammary gland, and skin was seen in mice treated with azacitidine IP at 2.0 mg/kg (6.0 mg/m2, approximately 8% the recommended human daily dose on a mg/m2 basis) once a week for 50 weeks. A tumorigenicity study in rats dosed twice weekly at 15 or 60 mg/m2 (approximately 20%-80% the recommended human daily dose on a mg/m2 basis) revealed an increased incidence of testicular tumors compared with controls.
The mutagenic and clastogenic potential of azacitidine was tested in in vitro bacterial systems Salmonella typhimurium strains TA100 and several strains of trpE8, Escherichia coli strains WP14 Pro, WP3103P, WP3104P, and CC103; in in vitro forward gene mutation assay in mouse lymphoma cells and human lymphoblast cells; and in an in vitro micronucleus assay in mouse L5178Y lymphoma cells and Syrian hamster embryo cells. Azacitidine was mutagenic in bacterial and mammalian cell systems. The clastogenic effect of azacitidine was shown by the induction of micronuclei in L5178Y mouse cells and Syrian hamster embryo cells.
Administration of azacitidine to male mice at 9.9 mg/m2 (approximately 9% the recommended human daily dose on a mg/m2 basis) daily for 3 days prior to mating with untreated female mice resulted in decreased fertility and loss of offspring during subsequent embryonic and postnatal development. Treatment of male rats 3 times per week for 11 or 16 weeks at doses of 15-30 mg/m2 (approximately 20%-40%, the recommended human daily dose on a mg/m2 basis) resulted in decreased weight of the testes and epididymides, and decreased sperm counts accompanied by decreased pregnancy rates and increased loss of embryos in mated females. In a related study, male rats treated for 16 weeks at 24 mg/m2 resulted in an increase in abnormal embryos in mated females when examined on day 2 of gestation.
-
14 CLINICAL STUDIES
14.1 Myelodysplastic Syndromes (MDS)
Study 1 was a randomized, open-label, controlled trial carried out in 53 U.S. sites compared the safety and efficacy of subcutaneous VIDAZA plus supportive care with supportive care alone (“observation”) in adult patients with any of the five FAB subtypes of myelodysplastic syndromes (MDS): refractory anemia (RA), RA with ringed sideroblasts (RARS), RA with excess blasts (RAEB), RAEB in transformation (RAEB-T), and chronic myelomonocytic leukemia (CMMoL). RA and RARS patients were included if they met one or more of the following criteria: required packed RBC transfusions; had platelet counts ≤50.0 x 109/L; required platelet transfusions; or were neutropenic (ANC <1.0 x 109/L) with infections requiring treatment with antibiotics. Patients with acute myelogenous leukemia (AML) were not intended to be included. Supportive care allowed in this study included blood transfusion products, antibiotics, antiemetics, analgesics and antipyretics. The use of hematopoietic growth factors was prohibited. Baseline patient and disease characteristics are summarized in Table 6; the 2 groups were similar.
VIDAZA was administered at a subcutaneous dose of 75 mg/m2 daily for 7 days every 4 weeks. The dose was increased to 100 mg/m2 if no beneficial effect was seen after 2 treatment cycles. The dose was decreased and/or delayed based on hematologic response or evidence of renal toxicity. Patients in the observation arm were allowed by protocol to cross over to VIDAZA if they had increases in bone marrow blasts, decreases in hemoglobin, increases in red cell transfusion requirements, or decreases in platelets, or if they required a platelet transfusion or developed a clinical infection requiring treatment with antibiotics. For purposes of assessing efficacy, the primary endpoint was response rate (as defined in Table 7).
Of the 191 patients included in the study, independent review (adjudicated diagnosis) found that 19 had the diagnosis of AML at baseline. These patients were excluded from the primary analysis of response rate, although they were included in an intent-to-treat (ITT) analysis of all patients randomized. Approximately 55% of the patients randomized to observation crossed over to receive VIDAZA treatment.
Table 6. Baseline Demographics and Disease Characteristics
VIDAZA
(N=99)Observation
(N=92)Gender (n%)
Male
72 (72.7)
60 (65.2)
Female
27 (27.3)
32 (34.8)
Race (n%)
White
93 (93.9)
85 (92.4)
Black
1 (1.0)
1 (1.1)
Hispanic
3 (3.0)
5 (5.4)
Asian/Oriental
2 (2.0)
1 (1.1)
Age (years)
N
99
91
Mean ± SD
67.3 ± 10.39
68.0 ± 10.23
Range
31 - 92
35 - 88
Adjudicated MDS diagnosis at
study entry (n%)RA
21 (21.2)
18 (19.6)
RARS
6 (6.1)
5 (5.4)
RAEB
38 (38.4)
39 (42.4)
RAEB-T
16 (16.2)
14 (15.2)
CMMoL
8 (8.1)
7 (7.6)
AML
10 (10.1)
9 (9.8)
Transfusion product used in 3
months before study entry (n%)Any transfusion product
70 (70.7)
59 (64.1)
Blood cells, packed human
66 (66.7)
55 (59.8)
Platelets, human blood
15 (15.2)
12 (13.0)
Hetastarch
0 (0.0)
1 (1.1)
Plasma protein fraction
1 (1.0)
0 (0.0)
Other
2 (2.0)
2 (2.2)
Table 7. Response Criteria
RA
RARS
RAEB
RAEB-T
CMMoL
Complete
Response
(CR),
duration ≥4 weeksMarrow
<5% blasts
Peripheral
BloodNormal CBC if abnormal at baseline
Absence of blasts in the peripheral circulationPartial
Response
(PR),
duration ≥4 weeksMarrow
No marrow requirements
≥50% decrease in blasts
Improvement of marrow dyspoiesis
Peripheral
Blood≥50% restoration in the deficit from normal levels of baseline white cells, hemoglobin and platelets if abnormal at baseline
No blasts in the peripheral circulation
For CMMoL, if WBC is elevated at baseline, a ≥75% reduction in the excess count over the upper limit of normal
The overall response rate (CR + PR) of 15.7% in VIDAZA-treated patients without AML (16.2% for all VIDAZA randomized patients including AML) was statistically significantly higher than the response rate of 0% in the observation group (p<0.0001) (Table 8). The majority of patients who achieved either CR or PR had either 2 or 3 cell line abnormalities at baseline (79%; 11/14) and had elevated bone marrow blasts or were transfusion dependent at baseline. Patients responding to VIDAZA had a decrease in bone marrow blasts percentage, or an increase in platelets, hemoglobin or WBC. Greater than 90% of the responders initially demonstrated these changes by the 5th treatment cycle. All patients who had been transfusion dependent became transfusion independent during PR or CR. The mean and median duration of clinical response of PR or better was estimated as 512 and 330 days, respectively; 75% of the responding patients were still in PR or better at completion of treatment. Response occurred in all MDS subtypes as well as in patients with adjudicated baseline diagnosis of AML.
Table 8. Response Rates
VIDAZA
(N=89)Observation Before Crossover
(N=83)Response
n (%)
n (%)
P value
Overall (CR+PR)
14 (15.7)
0 ( 0.0)
(<0.0001)
Complete (CR)
5 ( 5.6)
0 ( 0.0)
(0.06)
Partial (PR)
9 (10.1)
0 ( 0.0)
--
Patients in the observation group who crossed over to receive VIDAZA treatment (47 patients) had a response rate of 12.8%.
Study 2, a multi-center, open-label, single-arm study of 72 patients with RAEB, RAEB-T, CMMoL, or AML was also carried out. Treatment with subcutaneous VIDAZA resulted in a response rate (CR + PR) of 13.9%, using criteria similar to those described above. The mean and median duration of clinical response of PR or better was estimated as 810 and 430 days, respectively; 80% of the responding patients were still in PR or better at the time of completion of study involvement. In Study 3, another open-label, single-arm study of 48 patients with RAEB, RAEB-T, or AML, treatment with intravenous VIDAZA resulted in a response rate of 18.8%, again using criteria similar to those described above. The mean and median duration of clinical response of PR or better was estimated as 389 and 281 days, respectively; 67% of the responding patients were still in PR or better at the time of completion of treatment. Response occurred in all MDS subtypes as well as in patients with adjudicated baseline diagnosis of AML in both of these studies. VIDAZA dosage regimens in these 2 studies were similar to the regimen used in the controlled study.
Benefit was seen in patients who did not meet the criteria for PR or better, but were considered “improved.” About 24% of VIDAZA-treated patients were considered improved, and about 2/3 of those lost transfusion dependence. In the observation group, only 5/83 patients met criteria for improvement; none lost transfusion dependence. In all 3 studies, about 19% of patients met criteria for improvement with a median duration of 195 days.
Study 4 was an international, multicenter, open-label, randomized trial in patients with MDS with RAEB, RAEB-T or modified CMMoL according to FAB classification and Intermediate-2 and High risk according to IPSS classification. Of the 358 patients enrolled in the study, 179 were randomized to receive azacitidine plus best supportive care (BSC) and 179 were randomized to receive conventional care regimens (CCR) plus BSC (105 to BSC alone, 49 to low dose cytarabine and 25 to chemotherapy with cytarabine and anthracycline). The primary efficacy endpoint was overall survival.
The azacitidine and CCR groups were comparable for baseline parameters. The median age of patients was 69 years (range was 38-88 years), 98% were Caucasian, and 70% were male. At baseline, 95% of the patients were higher risk by FAB classification: RAEB (58%), RAEB-T (34%), and CMMoL (3%). By IPSS classification, 87% were higher risk: Int-2 (41%), High (47%). At baseline, 32% of patients met WHO criteria for AML.
Azacitidine was administered subcutaneously at a dose of 75 mg/m2 daily for 7 consecutive days every 28 days (which constituted one cycle of therapy). Patients continued treatment until disease progression, relapse after response, or unacceptable toxicity. Azacitidine patients were treated for a median of 9 cycles (range 1 to 39), BSC only patients for a median of 7 cycles (range 1 to 26), low dose cytarabine patients for a median of 4.5 cycles (range 1 to 15), and chemotherapy with cytarabine and anthracycline patients for a median of 1 cycle (range 1 to 3, i.e. induction plus 1 or 2 consolidation cycles).
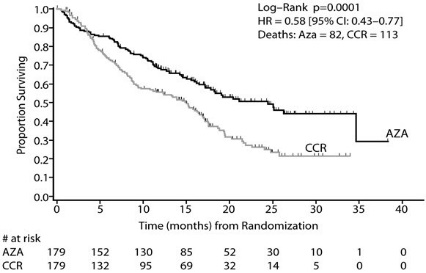
In the Intent-to-Treat analysis, patients treated with azacitidine demonstrated a statistically significant difference in overall survival as compared to patients treated with CCR (median survival of 24.5 months vs. 15.0 months; stratified log-rank p=0.0001). The hazard ratio describing this treatment effect was 0.58 (95% CI: 0.43, 0.77).
Kaplan-Meier Curve of Time to Death from Any Cause: (Intent-to-Treat Population)
Key: AZA = azacitidine; CCR = conventional care regimens; CI = confidence interval; HR = Hazard Ratio
Azacitidine treatment led to a reduced need for red blood cell transfusions (see Table 8). In patients treated with azacitidine who were RBC transfusion dependent at baseline and became transfusion independent, the median duration of RBC transfusion independence was 13.0 months.
Table 9. Effect of Azacitidine on RBC Transfusions in Patients with MDS
Efficacy Parameter
Azacitidine plus BSC
(n= 179)Conventional Care
Regimens
(n= 179)Number and percent of patients who were transfusion dependent at baseline who became transfusion independent on treatment1
50/111 (45.0%)
13/114 (11.4%)
(95% CI: 35.6%, 54.8%)
(95% CI: 6.2%, 18.7%)
Number and percent of patients who were transfusion-independent at baseline who became transfusion-dependent on treatment
10/68 (14.7%)
28/65 (43.1%)
(95% CI: 7.3%, 25.4%)
(95% CI: 30.9%, 56.0%)
1A patient was considered RBC transfusion independent during the treatment period if the patient had no RBC transfusions during any 56 consecutive days or more during the treatment period. Otherwise, the patient was considered transfusion dependent.
14.2 Juvenile Myelomonocytic Leukemia (JMML)
AZA-JMML-001 (NCT02447666) was an international, multicenter, open-label study to evaluate the pharmacokinetics, pharmacodynamics, safety, and activity of VIDAZA prior to hematopoietic stem cell transplantation (HSCT) in a total of 18 pediatric patients (median age of 2.1 years, range 0.2-6.9 years; 61% male; 89% white) with juvenile myelomonocytic leukemia (JMML).
Patients with newly diagnosed JMML were included if they met the following criteria: diagnosis confirmed in peripheral blood and bone marrow, and had one of the following: somatic mutation in PTPN11, KRAS, or NRAS and HbF % > 5 x normal value for age, or clinical diagnosis of neurofibromatosis Type 1 (NF-1). Additionally, patients included had no CNS involvement, isolated extramedullary disease, or germline molecular aberrations in CBL, PTPN11, NRAS, or KRAS.
Eighteen patients with JMML (13 PTPN11, 3 NRAS, 1 KRAS somatic mutations and 1 clinical diagnosis of neurofibromatosis type 1 [NF 1]) were enrolled. Patients were treated with intravenous VIDAZA [see Dosage and Administration (2.4)], daily on Days 1 to 7 of a 28-day cycle for a minimum of 3 cycles and a maximum of 6 cycles, provided the patients did not have disease progression or were ready for HSCT between Cycles 4 and 6. The efficacy of VIDAZA was established based on clinical complete remission (cCR) or clinical partial remission (cPR) according to the International JMML response criteria at 3 months (Cycle 3 Day 28). Responses must have been sustained for at least 4 weeks either in the 4-week period preceding or succeeding Cycle 3 Day 28 (i.e., sustained over the period Cycle 2 Day 28 to Cycle 3 Day 28, or Cycle 3 Day 28 to Cycle 4 Day 28).
There were a total of 9 patients (50%, 95% CI 26-74) with a confirmed clinical response. Of these 9 patients, there were 3 cCR and 6 cPR. The median time to response was 1.2 months (range 1-1.9 months). The proportion of patients with JMML undergoing HSCT was 94% and the median time to HSCT was 4.6 months (range 2.8-19 months).
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
VIDAZA (azacitidine for injection) is supplied as a lyophilized powder in 100 mg single-dose vials packaged in cartons of 1 vial (NDC 59572-102-01).
-
17 PATIENT COUNSELING INFORMATION
Hepatotoxicity in Patients with Severe Pre-Existing Hepatic Impairment
Instruct patients to inform their physician about any underlying liver disease [see Warnings and Precautions (5.3)].Renal Toxicity
Instruct patients to inform their physician about any underlying renal disease [see Warnings and Precautions (5.4)].Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1)].Advise females of reproductive potential to use effective contraception during treatment with VIDAZA and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with VIDAZA and for 3 months after the last dose. Advise patients to report known or suspected pregnancy to their physicians immediately [see Warnings and Precautions (5.6) and Use in Specific Populations (8.3)].
Lactation
Advise patients to avoid breastfeeding while receiving VIDAZA and for 1 week after the last dose [see Use in Specific Populations (8.2)].Infertility
Advise males and females that VIDAZA may impair fertility [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Marketed by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
VIDAZA® is a trademark of Celgene Corporation, a Bristol-Myers Squibb company.
VIDPI.19 - PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
VIDAZA
azacitidine injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59572-102 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AZACITIDINE (UNII: M801H13NRU) (AZACITIDINE - UNII:M801H13NRU) AZACITIDINE 100 mg Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59572-102-01 1 in 1 CARTON; Type 0: Not a Combination Product 07/05/2004 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA050794 07/05/2004 Labeler - Celgene Corporation (174201137)