Label: KADCYLA- ado-trastuzumab emtansine injection, powder, lyophilized, for solution


- NDC Code(s): 50242-087-01, 50242-088-01
- Packager: Genentech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated August 31, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KADCYLA safely and effectively. See full prescribing information for KADCYLA.
KADCYLA® (ado-trastuzumab emtansine) for injection, for intravenous use
Initial U.S. Approval: 2013WARNING: HEPATOTOXICITY, CARDIAC TOXICITY, EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning
- Hepatotoxicity, liver failure and death have occurred in KADCYLA-treated patients. Monitor hepatic function prior to initiation and prior to each dose. Institute dose modifications or permanently discontinue as appropriate. (2.3, 5.1)
- KADCYLA may lead to reductions in left ventricular ejection fraction (LVEF). Assess LVEF prior to initiation. Monitor and withhold dosing or discontinue as appropriate. (2.3, 5.2)
- Embryo-Fetal Toxicity: Exposure to KADCYLA during pregnancy can result in embryo-fetal harm. Advise patients of these risks and the need for effective contraception. (5.3, 8.1, 8.3)
INDICATIONS AND USAGE
KADCYLA is a HER2-targeted antibody and microtubule inhibitor conjugate indicated, as a single agent, for:
- the treatment of patients with HER2-positive, metastatic breast cancer who previously received trastuzumab and a taxane, separately or in combination. Patients should have either:
- received prior therapy for metastatic disease, or
- developed disease recurrence during or within six months of completing adjuvant therapy. (1.1)
- the adjuvant treatment of patients with HER2-positive early breast cancer who have residual invasive disease after neoadjuvant taxane and trastuzumab-based treatment. (1.2)
Select patients for therapy based on an FDA-approved companion diagnostic for KADCYLA [see Dosage and Administration (2.1)]
DOSAGE AND ADMINISTRATION
- Do not substitute KADCYLA for or with trastuzumab.
- HER2 Testing: Perform using FDA-approved tests by laboratories with demonstrated proficiency. (2.1)
- For intravenous infusion only. Do not administer as an intravenous push or bolus. Do not use Dextrose (5%) solution. (2.4)
- The recommended dose of KADCYLA is 3.6 mg/kg given as an intravenous infusion every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity, or a total of 14 cycles for patients with EBC. Do not administer KADCYLA at doses greater than 3.6 mg/kg. (2.2)
- Management of adverse reactions (infusion-related reactions, hepatotoxicity, left ventricular cardiac dysfunction, thrombocytopenia, pulmonary toxicity or peripheral neuropathy) may require temporary interruption, dose reduction, or treatment discontinuation of KADCYLA. (2.3)
DOSAGE FORMS AND STRENGTHS
Lyophilized powder in single-dose vials containing 100 mg per vial or 160 mg per vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Pulmonary Toxicity: Permanently discontinue KADCYLA in patients diagnosed with interstitial lung disease or pneumonitis. For patients with radiation pneumonitis in the adjuvant setting, permanently discontinue KADCYLA for Grade ≥ 3 or for Grade 2 not responding to standard treatment. (2.2, 5.4)
- Infusion-Related Reactions, Hypersensitivity Reactions: Monitor for signs and symptoms during and after infusion. If significant infusion-related reactions or hypersensitivity reactions occur, slow or interrupt the infusion and administer appropriate medical therapies. Permanently discontinue KADCYLA for life threatening infusion-related reaction. (2.1, 2.2, 5.5)
- Hemorrhage: Fatal cases of hemorrhage occurred in clinical trials among patients with no known identified risk factors, as well as among patients with thrombocytopenia and those receiving anti-coagulation and antiplatelet therapy. Use caution with these agents and consider additional monitoring when concomitant use is medically necessary. (5.6)
- Thrombocytopenia: Monitor platelet counts prior to each KADCYLA dose. Institute dose modifications as appropriate. (2.2, 5.7)
- Neurotoxicity: Monitor for signs or symptoms. Withhold dosing temporarily for patients experiencing Grade 3 or 4 peripheral neuropathy. (2.2, 5.8, 13.2)
ADVERSE REACTIONS
Metastatic Breast Cancer
- The most common adverse reactions (≥ 25%) with KADCYLA were fatigue, nausea, musculoskeletal pain, hemorrhage, thrombocytopenia, headache, increased transaminases, constipation and epistaxis. (6.1)
Early Breast Cancer
- The most common adverse reactions (≥ 25%) with KADCYLA were fatigue, nausea, increased transaminases, musculoskeletal pain, hemorrhage, thrombocytopenia, headache, peripheral neuropathy, and arthralgia.
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HEPATOTOXICITY, CARDIAC TOXICITY, EMBRYO-FETAL TOXICITY
1 INDICATIONS AND USAGE
1.1 Metastatic Breast Cancer (MBC)
1.2 Early Breast Cancer (EBC)
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Doses and Schedules
2.3 Dose Modifications
2.4 Preparation for Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Left Ventricular Dysfunction
5.3 Embryo-Fetal Toxicity
5.4 Pulmonary Toxicity
5.5 Infusion-Related Reactions, Hypersensitivity Reactions
5.6 Hemorrhage
5.7 Thrombocytopenia
5.8 Neurotoxicity
5.9 Extravasation
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Post-Marketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Metastatic Breast Cancer
14.2 Early Breast Cancer
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied/Storage
16.2 Special Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HEPATOTOXICITY, CARDIAC TOXICITY, EMBRYO-FETAL TOXICITY
- Hepatotoxicity: Serious hepatotoxicity has been reported, including liver failure and death in patients treated with KADCYLA. Monitor serum transaminases and bilirubin prior to initiation of KADCYLA treatment and prior to each KADCYLA dose. Reduce dose or discontinue KADCYLA as appropriate in cases of increased serum transaminases or total bilirubin. (2.3, 5.1)
- Cardiac Toxicity: KADCYLA administration may lead to reductions in left ventricular ejection fraction (LVEF). Evaluate left ventricular function in all patients prior to and during treatment with KADCYLA. Withhold treatment for clinically significant decrease in left ventricular function. (2.3, 5.2)
- Embryo-Fetal Toxicity: Exposure to KADCYLA during pregnancy can result in embryo-fetal harm. Advise patients of these risks and the need for effective contraception. (5.3, 8.1, 8.3)
-
1 INDICATIONS AND USAGE
1.1 Metastatic Breast Cancer (MBC)
KADCYLA®, as a single agent, is indicated for the treatment of patients with HER2-positive, metastatic breast cancer who previously received trastuzumab and a taxane, separately or in combination. Patients should have either:
- Received prior therapy for metastatic disease, or
- Developed disease recurrence during or within six months of completing adjuvant therapy.
Select patients for therapy based on an FDA-approved companion diagnostic for KADCYLA [see Dosage and Administration (2.1)].
1.2 Early Breast Cancer (EBC)
KADCYLA, as a single agent, is indicated for the adjuvant treatment of patients with HER2-positive early breast cancer who have residual invasive disease after neoadjuvant taxane and trastuzumab -based treatment.
Select patients for therapy based on an FDA-approved companion diagnostic for KADCYLA [see Dosage and Administration (2.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients based on HER2 protein overexpression or HER2 gene amplification in tumor specimens [see Indications and Usage (1), Clinical Studies (14)]. Assessment of HER2 protein overexpression and/or HER2 gene amplification should be performed using FDA-approved tests specific for breast cancers by laboratories with demonstrated proficiency. Information on the FDA-approved tests for the detection of HER2 protein overexpression and HER2 gene amplification is available at: http://www.fda.gov/CompanionDiagnostics.
Improper assay performance, including use of sub-optimally fixed tissue, failure to utilize specified reagents, deviation from specific assay instructions, and failure to include appropriate controls for assay validation, can lead to unreliable results.
2.2 Recommended Doses and Schedules
Do not substitute trastuzumab for or with KADCYLA.
The recommended dose of KADCYLA is 3.6 mg/kg given as an intravenous infusion every 3 weeks (21-day cycle). Do not administer KADCYLA at doses greater than 3.6 mg/kg.
Closely monitor the infusion site for possible subcutaneous infiltration during drug administration [see Warnings and Precautions (5.9)].
First infusion: Administer infusion over 90 minutes. Observe patients during the infusion and for at least 90 minutes following the initial dose for fever, chills, or other infusion-related reactions [see Warnings and Precautions (5.5)].
Subsequent infusions: Administer over 30 minutes if prior infusions were well tolerated. Observe patients during the infusion and for at least 30 minutes after infusion.
2.3 Dose Modifications
Do not re-escalate the KADCYLA dose after a dose reduction is made.
If a planned dose is delayed or missed, administer as soon as possible; do not wait until the next planned cycle. Adjust the schedule of administration to maintain a 3-week interval between doses. Administer the infusion at the dose and rate the patient tolerated in the most recent infusion.
Slow or interrupt the infusion rate of KADCYLA if the patient develops an infusion-related reaction. Permanently discontinue KADCYLA for life-threatening infusion-related reactions [see Warnings and Precautions (5.5)].
Management of increased serum transaminases, hyperbilirubinemia, left ventricular dysfunction, thrombocytopenia, pulmonary toxicity or peripheral neuropathy may require temporary interruption, dose reduction or treatment discontinuation of KADCYLA as per guidelines provided in Tables 1 and 2.
Table 1 Recommended Dose Reduction Schedule for Adverse Reactions Dose Reduction Schedule Dose Level Starting dose 3.6 mg/kg First dose reduction 3 mg/kg Second dose reduction 2.4 mg/kg Requirement for further dose reduction Discontinue treatment Table 2 Dose Modification Guidelines for KADCYLA ALT = alanine transaminase; AST = aspartate transaminase, CHF = congestive heart failure, DILI = Drug Induced Liver Injury; LVEF = left ventricular ejection fraction, LVSD = left ventricular systolic dysfunction, TBILI = Total Bilirubin, ULN = upper limit of normal - *
- Prior to starting KADCYLA treatment
Dose Modifications for Patients with MBC Adverse reaction Severity Treatment modification Increased Transaminase (AST/ALT) Grade 2
(> 2.5 to ≤ 5× the ULN)Treat at the same dose level. Grade 3
(> 5 to ≤ 20× the ULN)Do not administer KADCYLA until AST/ALT recovers to Grade ≤ 2, and then reduce one dose level Grade 4
(> 20× the ULN)Discontinue KADCYLA Hyperbilirubinemia Grade 2
(> 1.5 to ≤ 3× the ULN)Do not administer KADCYLA until total bilirubin recovers to Grade ≤ 1, and then treat at the same dose level. Grade 3
(> 3 to ≤ 10× the ULN)Do not administer KADCYLA until total bilirubin recovers to Grade ≤ 1 and then reduce one dose level. Grade 4
(> 10× the ULN)Discontinue KADCYLA Drug Induced Liver Injury (DILI) Serum transaminases > 3 × ULN and concomitant total bilirubin > 2 × ULN Permanently discontinue KADCYLA in the absence of another likely cause for the elevation of liver enzymes and bilirubin, e.g. liver metastasis or concomitant medication Nodular Regenerative Hyperplasia (NRH) All Grades Permanently discontinue KADCYLA Thrombocytopenia Grade 3
(25,000 to < 50,000/mm3)Do not administer KADCYLA until platelet count recovers to Grade ≤ 1 (≥ 75,000/mm3), and then treat at the same dose level Grade 4
(< 25,000/mm3)Do not administer KADCYLA until platelet count recovers to Grade ≤ 1 (≥ 75,000/mm3), and then reduce one dose level Left Ventricular Dysfunction Symptomatic CHF Discontinue KADCYLA LVEF < 40% Do not administer KADCYLA
Repeat LVEF assessment within 3 weeks. If LVEF < 40% is confirmed, discontinue KADCYLALVEF 40% to ≤ 45% and decrease is ≥ 10% points from baseline Do not administer KADCYLA
Repeat LVEF assessment within 3 weeks. If the LVEF has not recovered to within 10% points from baseline, discontinue KADCYLALVEF 40% to ≤ 45% and decrease is < 10% points from baseline Continue treatment with KADCYLA.
Repeat LVEF assessment within 3 weeks.LVEF > 45% Continue treatment with KADCYLA. Pulmonary Toxicity Interstitial lung disease (ILD) or pneumonitis Permanently discontinue KADCYLA Peripheral Neuropathy Grade 3-4 Do not administer KADCYLA until resolution Grade ≤ 2 Dose Modification Guidelines for EBC Adverse reaction Severity Treatment modification Increased Alanine Transaminase (ALT) Grade 2-3
(> 3.0 to ≤ 20 × ULN on day of scheduled treatment)Do not administer KADCYLA until ALT recovers to Grade ≤ 1, and then reduce one dose level Grade 4
(> 20 × ULN at any time)Discontinue KADCYLA Increased Aspartate Transaminase (AST) Grade 2
(> 3.0 to ≤ 5 × ULN on day of scheduled treatment)Do not administer KADCYLA until AST recovers to Grade ≤ 1, and then treat at the same dose level Grade 3
(> 5 to ≤ 20 × ULN on day of scheduled treatment)Do not administer KADCYLA until AST recovers to Grade ≤ 1, and then reduce one dose level Grade 4
(> 20 × ULN at any time)Discontinue KADCYLA Hyperbilirubinemia TBILI
> 1.0 to ≤ 2.0 × the ULN on day of scheduled treatmentDo not administer KADCYLA until total bilirubin recovers to ≤ 1.0 × ULN, and then reduce one dose level TBILI
> 2 × ULN at any timeDiscontinue KADCYLA Nodular Regenerative Hyperplasia (NRH) All Grades Permanently discontinue KADCYLA Thrombocytopenia Grade 2-3 on day of scheduled treatment
(25,000 to < 75,000/mm3)Do not administer KADCYLA until platelet count recovers to Grade ≤ 1 (≥ 75,000/mm3), and then treat at the same dose level. If a patient requires 2 delays due to thrombocytopenia, consider reducing dose by one level. Grade 4 at any time
< 25,000/mm3Do not administer KADCYLA until platelet count recovers to Grade ≤ 1 (≥ 75,000/mm3), and then reduce one dose level. Left Ventricular Dysfunction LVEF < 45% Do not administer KADCYLA
Repeat LVEF assessment within 3 weeks. If LVEF < 45% is confirmed, discontinue KADCYLA.LVEF 45% to < 50% and decrease is ≥ 10% points from baseline* Do not administer KADCYLA
Repeat LVEF assessment within 3 weeks. If the LVEF remains < 50% and has not recovered to < 10% points from baseline, discontinue KADCYLA.LVEF 45% to < 50% and decrease is < 10% points from baseline* Continue treatment with KADCYLA.
Repeat LVEF assessment within 3 weeks.LVEF ≥ 50% Continue treatment with KADCYLA. Heart Failure Symptomatic CHF,
Grade 3-4 LVSD or Grade 3-4 heart failure, or
Grade 2 heart failure
accompanied by LVEF < 45%Discontinue KADCYLA Peripheral Neuropathy Grade 3-4 Do not administer KADCYLA until resolution Grade ≤ 2 Pulmonary Toxicity Interstitial lung disease (ILD) or pneumonitis Permanently discontinue KADCYLA Radiotherapy-Related Pneumonitis Grade 2 Discontinue KADCYLA if not resolving with standard treatment Grade 3-4 Discontinue KADCYLA 2.4 Preparation for Administration
In order to prevent medication errors it is important to check the vial labels to ensure that the drug being prepared and administered is KADCYLA (ado-trastuzumab emtansine) and not trastuzumab.
Administration:
- Administer KADCYLA as an intravenous infusion only with a 0.2 or 0.22 micron in-line polyethersulfone (PES) filter. Do not administer as an intravenous push or bolus.
- Do not mix KADCYLA, or administer as an infusion, with other medicinal products.
Reconstitution:
- Use aseptic technique for reconstitution and preparation of dosing solution. Appropriate procedures for the preparation of chemotherapeutic drugs should be used.
- Using a sterile syringe, slowly inject 5 mL of Sterile Water for Injection into the 100 mg KADCYLA vial, or 8 mL of Sterile Water for Injection into the 160 mg KADCYLA vial to yield a solution containing 20 mg/mL. Swirl the vial gently until completely dissolved. Do not shake. Inspect the reconstituted solution for particulates and discoloration.
- The reconstituted solution should be clear to slightly opalescent and free of visible particulates. The color of the reconstituted solution should be colorless to pale brown. Do not use if the reconstituted solution contains visible particulates or is cloudy or discolored.
- The reconstituted lyophilized vials should be used immediately following reconstitution with Sterile Water for Injection. If not used immediately, the reconstituted KADCYLA vials can be stored for up to 24 hours in a refrigerator at 2ºC to 8ºC (36°F to 46°F); discard unused KADCYLA after 24 hours. Do not freeze.
- The reconstituted product contains no preservative and is intended for single-dose only.
Dilution:
Determine the correct dose (mg) of KADCYLA [see Dosage and Administration (2.1)].
- Calculate the volume of the 20 mg/mL reconstituted KADCYLA solution needed.
- Withdraw this amount from the vial and add it to an infusion bag containing 250 mL of 0.9% Sodium Chloride Injection. Do not use Dextrose (5%) solution.
- Gently invert the bag to mix the solution in order to avoid foaming.
- The diluted KADCYLA infusion solution should be used immediately. If not used immediately, the solution may be stored in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours prior to use. This storage time is additional to the time allowed for the reconstituted vials. Do not freeze or shake.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Hepatotoxicity, predominantly in the form of asymptomatic, transient increases in the concentrations of serum transaminases, has been observed in clinical trials with KADCYLA [see Adverse Reactions (6.1)]. Serious hepatotoxicity, including 3 fatal cases, has been observed in clinical trials (n=1624) with KADCYLA as single-agent. All fatal cases occurred in MBC clinical trials with KADCYLA, which included severe drug-induced liver injury and associated hepatic encephalopathy. Some of the patients experiencing hepatotoxicity had comorbidities and/or concomitant medications with known hepatotoxic potential.
Monitor serum transaminases and bilirubin prior to initiation of KADCYLA treatment and prior to each KADCYLA dose. Patients with known active liver disease (such as, hepatitis B virus or hepatitis C virus) were excluded from the EMILIA and KATHERINE studies [see Clinical Studies (14.1)]. Reduce the dose or discontinue KADCYLA as appropriate in cases of increased serum transaminases and/or total bilirubin [see Dosage and Administration (2.2)]. Permanently discontinue KADCYLA treatment in patients with serum transaminases > 3 × ULN and concomitant total bilirubin > 2 × ULN. KADCYLA has not been studied in patients with serum transaminases > 2.5 × ULN or bilirubin > 1.5 × ULN prior to the initiation of treatment.
In clinical trials of KADCYLA, cases of nodular regenerative hyperplasia (NRH) of the liver have been identified from liver biopsies (5 cases out of 1624 treated patients, one of which was fatal). Two of these five cases of NRH were observed in EMILIA and two were observed in KATHERINE [see Adverse Reactions (6.1)]. NRH is a rare liver condition characterized by widespread benign transformation of hepatic parenchyma into small regenerative nodules; NRH may lead to non-cirrhotic portal hypertension. The diagnosis of NRH can be confirmed only by histopathology. NRH should be considered in all patients with clinical symptoms of portal hypertension and/or cirrhosis-like pattern seen on the computed tomography (CT) scan of the liver but with normal transaminases and no other manifestations of cirrhosis. Upon diagnosis of NRH, KADCYLA treatment must be permanently discontinued.
5.2 Left Ventricular Dysfunction
Patients treated with KADCYLA are at increased risk of developing left ventricular dysfunction. A decrease of LVEF to < 40% has been observed in patients treated with KADCYLA. Serious cases of heart failure, with no fatal cases, have been observed in clinical trials with KADCYLA. In EMILIA, left ventricular dysfunction occurred in 1.8% of patients in the KADCYLA-treated group and 3.3% of patients in the lapatinib plus capecitabine-treated group. In KATHERINE, left ventricular dysfunction occurred in 0.4% of patients in the KADCYLA-treated group and 0.6% of patients in the trastuzumab-treated group [see Adverse Reactions (6.1)].
Based on limited data from a retrospective observational study, 22% (7 of 32) of patients with HER2-positive metastatic breast cancer (MBC) with a baseline LVEF of 40-49% treated with KADCYLA developed a congestive heart failure (CHF) or a > 10% reduction in LVEF [see Adverse Reactions (6.3)].
Assess LVEF prior to initiation of KADCYLA and at regular intervals (e.g. every three months) during treatment to ensure the LVEF is within the institution's normal limits. KADCYLA has not been studied in an adequately controlled study in patients with LVEF < 50%.
For patients with MBC, if, at routine monitoring, LVEF is < 40%, or is 40% to 45% with a 10% or greater absolute decrease below the pretreatment value, withhold KADCYLA and repeat LVEF assessment within approximately 3 weeks. Permanently discontinue KADCYLA if the LVEF has not improved or has declined further.
For patients with EBC, if, at routine monitoring, LVEF is < 45%, or is 45% to 49% with a 10% or greater absolute decrease below the pretreatment value, withhold KADCYLA and repeat LVEF assessment within approximately 3 weeks. Permanently discontinue KADCYLA if the LVEF has not improved or has declined further [see Dosage and Administration (2.2)].
Patients with a history of symptomatic CHF, serious cardiac arrhythmia, or history of myocardial infarction or unstable angina within 6 months were excluded from the EMILIA and KATHERINE studies [see Clinical Studies (14.1)].
5.3 Embryo-Fetal Toxicity
KADCYLA can cause fetal harm when administered to a pregnant woman. Cases of oligohydramnios, and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities and neonatal death were observed in the post-marketing setting in patients treated with trastuzumab, the antibody component of KADCYLA. DM1, the cytotoxic component of KADCYLA, can cause embryo-fetal toxicity based on its mechanism of action.
Verify the pregnancy status of females of reproductive potential prior to the initiation of KADCYLA. Advise pregnant women and females of reproductive potential that exposure to KADCYLA during pregnancy or within 7 months prior to conception can result in fetal harm. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of KADCYLA [see Use in Specific Populations (8.1, 8.3)].
5.4 Pulmonary Toxicity
Cases of interstitial lung disease (ILD), including pneumonitis, some leading to acute respiratory distress syndrome or fatal outcome have been reported in clinical trials with KADCYLA. Signs and symptoms include dyspnea, cough, fatigue, and pulmonary infiltrates.
In patients with MBC, pneumonitis was reported at an incidence of 0.8% (7 out of 884 treated patients), with one case of Grade 3 pneumonitis. The overall incidence of pneumonitis was 1.2% in EMILIA. In KATHERINE, pneumonitis was reported at an incidence of 1.1% (8 out of 740 patients treated with KADCYLA), with one case of Grade 3 pneumonitis.
Radiation pneumonitis was reported at an incidence of 1.8% (11 out of 623 patients treated with adjuvant radiotherapy and KADCYLA), with 2 cases of Grade 3 radiation pneumonitis [see Adverse Reactions (6.1)].
Permanently discontinue treatment with KADCYLA in patients diagnosed with ILD or pneumonitis. For patients with radiation pneumonitis in the adjuvant setting, KADCYLA should be permanently discontinued for Grade ≥ 3 or for Grade 2 not responding to standard treatment [see Dose Modifications (2.2)].
Patients with dyspnea at rest due to complications of advanced malignancy, co-morbidities, and receiving concurrent pulmonary radiation therapy may be at increased risk of pulmonary toxicity.
5.5 Infusion-Related Reactions, Hypersensitivity Reactions
Treatment with KADCYLA has not been studied in patients who had trastuzumab permanently discontinued due to infusion-related reactions (IRRs) and/or hypersensitivity; treatment with KADCYLA is not recommended for these patients.
Infusion-related reactions, characterized by one or more of the following symptoms − flushing, chills, pyrexia, dyspnea, hypotension, wheezing, bronchospasm, and tachycardia have been reported in clinical trials of KADCYLA. In EMILIA, the overall incidence of IRRs in patients treated with KADCYLA was 1.4%. In KATHERINE, the overall incidence of IRRs in patients treated with KADCYLA was 1.6% [see Adverse Reactions (6.1)]. In most patients, these reactions resolved over the course of several hours to a day after the infusion was terminated. KADCYLA treatment should be interrupted in patients with severe IRR. KADCYLA treatment should be permanently discontinued in the event of a life-threatening IRR [see Dosage and Administration (2.2)]. Patients should be observed closely for IRR reactions, especially during the first infusion.
One case of a serious, allergic/anaphylactic-like reaction has been observed in clinical trials of single-agent KADCYLA. Medications to treat such reactions, as well as emergency equipment, should be available for immediate use.
5.6 Hemorrhage
Cases of hemorrhagic events, including central nervous system, respiratory, and gastrointestinal hemorrhage, have been reported in clinical trials with KADCYLA. Some of these bleeding events resulted in fatal outcomes. In EMILIA, the overall incidence of hemorrhage was 32% in the KADCYLA-treated group and 16% in the lapatinib plus capecitabine-treated group. The incidence of Grade ≥ 3 hemorrhage was 1.8% in the KADCYLA-treated group and 0.8% in the lapatinib plus capecitabine-treated group. In KATHERINE, the overall incidence of hemorrhage was 29% in the KADCYLA-treated group and 10% in the trastuzumab-treated group. The incidence of Grade ≥ 3 hemorrhage was 0.4% in the KADCYLA-treated group, with one fatal case of intracranial hemorrhage, and 0.3% in the trastuzumab-treated group [see Adverse Reactions (6.1)]. Although, in some of the observed cases the patients were also receiving anti-coagulation therapy, antiplatelet therapy, or had thrombocytopenia, in others there were no known additional risk factors. Use caution with these agents and consider additional monitoring when concomitant use is medically necessary.
5.7 Thrombocytopenia
Thrombocytopenia, or decreased platelet count, was reported in clinical trials of KADCYLA (145 of 1624 treated patients with Grade ≥ 3; 494 of 1624 treated patients with any Grade). The majority of these patients had Grade 1 or 2 events (< LLN to ≥ 50,000/mm3) with the nadir occurring by day 8 and generally improving to Grade 0 or 1 (≥ 75,000/mm3) by the next scheduled dose. In clinical trials of KADCYLA, the incidence and severity of thrombocytopenia were higher in Asian patients.
In EMILIA, the overall incidence of thrombocytopenia was 31% in the KADCYLA-treated group and 3.3% in the lapatinib plus capecitabine-treated group [see Adverse Reactions (6.1)]. The incidence of Grade ≥ 3 thrombocytopenia was 15% in the KADCYLA-treated group and 0.4% in the lapatinib plus capecitabine-treated group. In Asian patients, the incidence of Grade ≥ 3 thrombocytopenia was 45% in the KADCYLA-treated group and 1.3% in the lapatinib plus capecitabine-treated group.
In KATHERINE, the overall incidence of thrombocytopenia was 29% in the KADCYLA-treated group and 2.4% in the trastuzumab-treated group [see Adverse Reactions (6.1)]. The incidence of Grade ≥ 3 thrombocytopenia was 6% in the KADCYLA-treated group and 0.3% in the trastuzumab-treated group. In Asian patients, the incidence of Grade ≥ 3 thrombocytopenia was 19% in the KADCYLA-treated group and 0% in the trastuzumab-treated group. The overall incidence of thrombocytopenia in the KADCYLA-treated group for Asian patients was 50%.
Monitor platelet counts prior to initiation of KADCYLA and prior to each KADCYLA dose [see Dosage and Administration (2.2)]. KADCYLA has not been studied in patients with platelet counts < 100,000/mm3 prior to initiation of treatment. In the event of decreased platelet count to Grade ≥ 3 (< 50,000/mm3) do not administer KADCYLA until platelet counts recover to Grade 1 (≥ 75,000/mm3) [see Dosage and Administration (2.2)]. Closely monitor patients with thrombocytopenia (< 100,000/mm3) and patients on anti-coagulant treatment during treatment with KADCYLA.
5.8 Neurotoxicity
Peripheral neuropathy, mainly as Grade 1 and predominantly sensory, was reported in clinical trials of KADCYLA (26 of 1624 treated patients with Grade ≥ 3; 435 of 1624 treated patients with any Grade). In EMILIA, the overall incidence of peripheral neuropathy was 21% in the KADCYLA-treated group and 14% in the lapatinib plus capecitabine-treated group [see Adverse Reactions (6.1)]. The incidence of Grade ≥ 3 peripheral neuropathy was 2.2% in the KADCYLA-treated group and 0.2% in the lapatinib plus capecitabine-treated group. In KATHERINE, the overall incidence of peripheral neuropathy was 32% in the KADCYLA-treated group and 17% in the trastuzumab-treated group. Peripheral neuropathy, including sensory and motor peripheral neuropathy, for KADCYLA treated patients 30% of cases were not resolved at the time of the primary IDFS analysis for KATHERINE. The incidence of Grade ≥ 3 peripheral neuropathy was 1.6% in the KADCYLA-treated group and 0.1% in the trastuzumab-treated group.
KADCYLA should be temporarily discontinued in patients experiencing Grade 3 or 4 peripheral neuropathy until resolution to Grade ≤ 2. Patients should be clinically monitored on an ongoing basis for signs or symptoms of neurotoxicity [see Nonclinical Toxicology (13.2)].
5.9 Extravasation
In KADCYLA clinical studies, reactions secondary to extravasation have been observed. These reactions, observed more frequently within 24 hours of infusion, were usually mild and comprised erythema, tenderness, skin irritation, pain, or swelling at the infusion site. Specific treatment for KADCYLA extravasation is unknown. The infusion site should be closely monitored for possible subcutaneous infiltration during drug administration.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Hepatotoxicity [See Warnings and Precautions (5.1)]
- Left Ventricular Dysfunction [See Warnings and Precautions (5.2)]
- Embryo-Fetal Toxicity [See Warnings and Precautions (5.3)]
- Pulmonary Toxicity [See Warnings and Precautions (5.4)]
- Infusion-Related Reactions, Hypersensitivity Reactions [See Warnings and Precautions (5.5)]
- Hemorrhage [See Warnings and Precautions (5.6)]
- Thrombocytopenia [See Warnings and Precautions (5.7)]
- Neurotoxicity [See Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the WARNINGS AND PRECAUTIONS reflect exposure to KADCYLA as a single agent at 3.6 mg/kg given as an intravenous infusion every 3 weeks (21-day cycle) in 1624 patients including 884 patients with HER2-positive metastatic breast cancer and 740 patients with HER2-positive early breast cancer (KATHERINE trial).
Metastatic Breast Cancer
In clinical trials, KADCYLA has been evaluated as single-agent in 884 patients with HER2-positive metastatic breast cancer. The most common (≥ 25%) adverse reactions were fatigue, nausea, musculoskeletal pain, hemorrhage, thrombocytopenia, headache, increased transaminases, constipation and epistaxis.
The adverse reactions described in Table 3 were identified in patients with HER2-positive metastatic breast cancer treated in the EMILIA trial [see Clinical Studies (14.1)]. Patients were randomized to receive KADCYLA or lapatinib plus capecitabine. The median duration of study treatment was 7.6 months for patients in the KADCYLA-treated group and 5.5 months and 5.3 months for patients treated with lapatinib and capecitabine, respectively.
In the EMILIA trial, 43% of patients experienced Grade ≥ 3 adverse reactions in the KADCYLA-treated group compared with 59% of patients in the lapatinib plus capecitabine-treated group.
Dose adjustments for KADCYLA were permitted [see Dosage and Administration (2.2)]. Thirty-two patients (7%) discontinued KADCYLA due to an adverse reaction, compared with 41 patients (8%) who discontinued lapatinib, and 51 patients (10%) who discontinued capecitabine due to an adverse reaction. The most common adverse reactions leading to KADCYLA discontinuation were thrombocytopenia and increased transaminases. Eighty patients (16%) treated with KADCYLA had adverse reactions leading to dose reductions. The most frequent adverse reactions leading to dose reduction of KADCYLA (in ≥ 1% of patients) included thrombocytopenia, increased transaminases, and peripheral neuropathy. Adverse reactions that led to dose delays occurred in 116 (24%) of KADCYLA treated patients. The most frequent adverse reactions leading to a dose delay of KADCYLA (in ≥ 1% of patients) were neutropenia, thrombocytopenia, leukopenia, fatigue, increased transaminases and pyrexia.
Table 3 reports the adverse reactions that occurred in patients in the KADCYLA-treated group (n=490) of the EMILIA trial. Selected laboratory abnormalities are shown in Table 4. The most common adverse reactions seen with KADCYLA in the randomized trial (frequency > 25%) were nausea, fatigue, musculoskeletal pain, hemorrhage, thrombocytopenia, increased transaminases, headache, and constipation. The most common NCI–CTCAE (version 3) Grade ≥ 3 adverse reactions (frequency > 2%) were thrombocytopenia, increased transaminases, anemia, hypokalemia, peripheral neuropathy and fatigue.
Table 3 Adverse Reactions Occurring in ≥ 10% of Patients on the KADCYLA Treatment Arm in the EMILIA Trial* Adverse Reactions KADCYLA
(3.6 mg/kg)
n=490Lapatinib (1250 mg) + Capecitabine (2000 mg/m2)
n=488All Grades
(%)Grade 3 – 4
(%)All Grades
(%)Grade 3 – 4
(%)SMQ=standardized MedDRA queries - *
- Grouped terms were used for the following Adverse Reactions:
Thrombocytopenia: thrombocytopenia, platelet count decreased
Anemia: anemia, hemoglobin decreased
Abdominal pain: abdominal pain, abdominal pain upper
Stomatitis: stomatitis, mucosal inflammation, oropharyngeal pain
Transaminases Increased: transaminases increased, aspartate aminotransferase increased, alanine aminotransferase increased, gamma-glutamyltransferase increased, liver function test abnormal, hepatic enzyme increased, hepatic function abnormal
Hypokalemia: hypokalemia, blood potassium decreased
Musculoskeletal Pain: muscle spasms, musculoskeletal discomfort, musculoskeletal chest pain, back pain, pain in extremity, bone pain, musculoskeletal pain
Peripheral neuropathy: neuropathy peripheral, peripheral sensory neuropathy, peripheral motor neuropathy, paresthesia
Hemorrhage: Hemorrhage terms (excl laboratory terms) (SMQ, wide), Hemorrhage laboratory terms (SMQ, narrow).
Blood and Lymphatic System Disorders Thrombocytopenia 31 15 3.3 0.4 Anemia 14 4.1 11 2.5 Gastrointestinal Disorders Nausea 40 0.8 45 2.5 Constipation 27 0.4 11 0 Diarrhea 24 1.6 80 21 Vomiting 19 0.8 30 4.5 Abdominal pain 19 0.8 18 1.6 Dry Mouth 17 0 4.9 0.2 Stomatitis 14 0.2 33 2.5 General Disorders and Administration Fatigue 36 2.5 28 3.5 Pyrexia 19 0.2 8 0.4 Asthenia 18 0.4 18 1.6 Investigations Transaminases increased 29 8.0 14 2.5 Metabolism and Nutrition Disorders Hypokalemia 10 2.7 9 4.7 Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain 36 1.8 31 1.4 Arthralgia 19 0.6 8 0 Myalgia 14 0.6 3.7 0 Nervous System Disorders Headache 28 0.8 15 0.8 Peripheral neuropathy 21 2.2 14 0.2 Dizziness 10 0.4 11 0.2 Psychiatric Disorders Insomnia 12 0.4 9 0.2 Respiratory, Thoracic, and Mediastinal Disorders Epistaxis 23 0.2 8 0 Cough 18 0.2 13 0.2 Dyspnea 12 0.8 8 0.4 Skin and Subcutaneous Tissue Disorders Rash 12 0 28 1.8 Vascular Disorders Hemorrhage 32 1.8 16 0.8 The following clinically relevant adverse reactions were reported in < 10% of patients in the KADCYLA-treated group in EMILIA: dyspepsia (9%), urinary tract infection (9%), chills (8%), dysgeusia (8%), neutropenia (7%), peripheral edema (7%), pruritus (6%), hypertension (5%), blood alkaline phosphatase increased (4.7%), vision blurred (4.5%), conjunctivitis (3.9%), dry eye (3.9%), lacrimation increased (3.3%), drug hypersensitivity (2.2%), left ventricular dysfunction (1.8%), infusion-related reaction (1.4%), pneumonitis (1.2%), nodular regenerative hyperplasia (0.4%), portal hypertension (0.4%).
Table 4 Selected Laboratory Abnormalities (EMILIA) Parameter KADCYLA
(3.6 mg/kg)Lapatinib (1250 mg) + Capecitabine (2000 mg/m2) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Chemistry Increased AST 98 7 0.5 65 3 0 Increased ALT 82 5 0.2 54 3 0 Decreased potassium 33 3 0 31 6 0.8 Increased bilirubin 17 0.6 0 57 2 0 Hematology Decreased platelet count 83 14 3 21 0.4 0.6 Decreased hemoglobin 60 4 1 64 3 0.2 Decreased neutrophils 39 3 0.6 38 6 2 Early Breast Cancer
KADCYLA has been evaluated as a single-agent in 740 patients with HER2-positive early breast cancer.
The adverse reactions described in Table 5 were identified in patients with HER2-positive early breast cancer treated in the KATHERINE trial [see Clinical Studies (14.2)]. Patients were randomized to receive KADCYLA or trastuzumab. The median duration of study treatment was 10 months for patients in the KADCYLA-treated group and 10 months for patients treated with trastuzumab.
One hundred and ninety (26%) patients experienced Grade ≥ 3 adverse reactions in the KADCYLA-treated group compared with 111 (15%) patients in the trastuzumab group. One hundred and thirty-three patients (18%) discontinued KADCYLA due to an adverse reaction, compared with 15 patients (2.1%) who discontinued trastuzumab due to an adverse reaction.
The most common adverse reactions leading to KADCYLA discontinuation (in ≥ 1% of patients) were platelet count decreased, blood bilirubin increased, ejection fraction decreased, AST increased, ALT increased, and peripheral neuropathy.
Dose adjustments for KADCYLA were permitted [see Dosage and Administration (2.2)]. One hundred and six patients (14%) treated with KADCYLA had dose reductions. The most frequent adverse reactions leading to dose reduction of KADCYLA (in ≥ 1% of patients) included thrombocytopenia, increased transaminases, blood bilirubin and fatigue. Adverse reactions that led to dose delays occurred in 106 (14%) of KADCYLA treated patients. The most frequent adverse reactions leading to a dose delay of KADCYLA (in ≥ 1% of patients) were neutropenia, thrombocytopenia and AST increased.
Selected laboratory abnormalities are shown in Table 6. The most common adverse reactions seen with KADCYLA in the randomized trial (frequency > 25%) were fatigue, nausea, increased transaminases, musculoskeletal pain, hemorrhage, thrombocytopenia, headache, peripheral neuropathy, and arthralgia. The most common NCI–CTCAE (version 3) Grade ≥ 3 adverse reactions (> 2%) were thrombocytopenia and hypertension.
Table 5 Adverse Reactions Occurring in ≥ 10% of Patients in the KATHERINE Trial* Adverse Reactions KADCYLA
n=740Trastuzumab
n=720All grades
(%)Grade 3 – 4
(%)All grades
(%)Grade 3 – 4
(%)SMQ=standardized MedDRA queries - *
- Grouped terms were used for the following Adverse Reactions:
Thrombocytopenia: thrombocytopenia, platelet count decreased
Anemia: anemia, hemoglobin decreased
Stomatitis: stomatitis, mucosal inflammation, oropharyngeal pain
Abdominal pain: abdominal pain, abdominal pain upper
Urinary Tract Infection: urinary tract infection, cystitis
Transaminases Increased: transaminases increased, aspartate aminotransferase increased, alanine aminotransferase increased, gamma-glutamyltransferase increased, liver function test abnormal, hepatic enzyme increased, hepatic function abnormal
Musculoskeletal Pain: muscle spasms, musculoskeletal discomfort, musculoskeletal chest pain, back pain, pain in extremity, bone pain, musculoskeletal pain
Peripheral neuropathy: neuropathy peripheral, peripheral sensory neuropathy, peripheral motor neuropathy, paresthesia
Hemorrhage: Hemorrhage terms (excl laboratory terms) (SMQ, wide), Hemorrhage laboratory terms (SMQ, narrow) - †
- Included one fatal hemorrhage.
Blood and Lymphatic System Disorders Thrombocytopenia 29 6 2.4 0.3 Anemia 10 1.1 9 0.1 Gastrointestinal Disorders Nausea 42 0.5 13 0.3 Constipation 17 0.1 8 0 Stomatitis 15 0.1 8 0.1 Vomiting 15 0.5 5 0.3 Dry Mouth 14 0.1 1.3 0 Diarrhea 12 0.8 13 0.3 Abdominal pain 11 0.4 7 0.3 General Disorders and Administration Fatigue 50 1.1 34 0.1 Pyrexia 10 0 4 0 Infections and Infestations Urinary tract infection 10 0.3 6 0.1 Investigations Transaminases increased 32 1.5 8 0.4 Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain 30 0.7 29 0.7 Arthralgia 26 0.1 21 0 Myalgia 15 0.4 11 0 Nervous System Disorders Headache 28 0 17 0.1 Peripheral neuropathy 28 1.6 14 0.1 Dizziness 10 0.1 8 0.3 Psychiatric Disorders Insomnia 14 0 12 0.1 Respiratory, Thoracic, and Mediastinal Disorders Epistaxis 22 0 3.5 0 Cough 14 0.1 12 0 Vascular Disorders Hemorrhage 29 0.4† 10 0.3 The following clinically relevant adverse reactions were reported in < 10% of patients in the KADCYLA-treated group in KATHERINE: blood alkaline phosphatase increased (8%), dysgeusia (8%), dyspnea (8%), neutropenia (8%), blood bilirubin increased (7%), hypokalemia (7%), pruritus (7%), hypertension (6%), lacrimation increased (6%), chills (5%), dry eye (4.5%), dyspepsia (4.3%), peripheral edema (3.9%),vision blurred (3.9%), conjunctivitis (3.5%), left ventricular dysfunction (3.0%), drug hypersensitivity (2.7%), infusion-related reaction (1.6%), radiation pneumonitis (1.5%), pneumonitis (1.1%), rash (1.1%), asthenia (0.4%), nodular regenerative hyperplasia (0.3%).
Table 6 Selected Laboratory Abnormalities (KATHERINE) Parameter KADCYLA
n=740Trastuzumab
n=720All Grade
(%)Grade 3
(%)Grade 4
(%)All Grade
(%)Grade 3
(%)Grade 4
(%)Chemistry Increased AST 79 0.8 0 21 0.1 0 Increased ALT 55 0.7 0 21 0.1 0 Decreased potassium 26 2 0.5 9 0.7 0.1 Increased bilirubin 12 0 0 4 0.7 0 Hematology Decreased platelet count 51 4 2 13 0.1 0.1 Decreased hemoglobin 31 1 0 29 0.3 0 Decreased neutrophils 24 1 0 19 0.6 0.6 6.2 Immunogenicity
As with all therapeutic proteins, there is the potential for an immune response to KADCYLA. A total of 1243 patients from seven clinical studies were tested at multiple time points for anti-drug antibody (ADA) responses to KADCYLA. Following KADCYLA dosing, 5.1% (63/1243) of patients tested positive for anti-KADCYLA antibodies at one or more post-dose time points. In clinical studies, 6.4% (24/376) of patients tested positive for anti-KADCYLA antibodies. In EMILIA, 5.2% (24/466) of patients tested positive for anti-KADCYLA antibodies, of which 13 were also positive for neutralizing antibodies. In KATHERINE, 3.7% (15/401) of patients tested positive for anti-KADCYLA antibodies, of which 5 were also positive for neutralizing antibodies. Due to the low incidence of ADA, conclusions cannot be made on the impact of anti-KADCYLA antibodies on the pharmacokinetics, safety, and efficacy of KADCYLA. The presence of KADCYLA in patient serum at the time of ADA sampling may interfere with the ability of this assay to detect anti-KADCYLA antibodies. As a result, data may not accurately reflect the true incidence of anti-KADCYLA antibody development.
Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication and the underlying disease. Therefore, comparison of the incidence of antibodies to KADCYLA with the incidence of antibodies to other products may be misleading. Clinical significance of anti-KADCYLA antibodies is not yet known.
6.3 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of KADCYLA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse Reactions from Observational Studies
- CHF and > 10% reduction in LVEF in patients with HER2-positive metastatic breast cancer with a baseline LVEF of 40-49% treated with KADCYLA [see Warnings and Precautions (5.2)].
-
7 DRUG INTERACTIONS
No formal drug-drug interaction studies with KADCYLA have been conducted. In vitro studies indicate that DM1, the cytotoxic component of KADCYLA, is metabolized mainly by CYP3A4 and to a lesser extent by CYP3A5. Concomitant use of strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole) with KADCYLA should be avoided due to the potential for an increase in DM1 exposure and toxicity. Consider an alternate medication with no or minimal potential to inhibit CYP3A4. If concomitant use of strong CYP3A4 inhibitors is unavoidable, consider delaying KADCYLA treatment until the strong CYP3A4 inhibitors have cleared from the circulation (approximately 3 elimination half-lives of the inhibitors) when possible. If a strong CYP3A4 inhibitor is coadministered and KADCYLA treatment cannot be delayed, patients should be closely monitored for adverse reactions.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Pharmacovigilance Program
There is a pregnancy pharmacovigilance program for KADCYLA. If KADCYLA is administered during pregnancy, or if a patient becomes pregnant while receiving KADCYLA or within 7 months following the last dose of KADCYLA, health care providers and patients should immediately report KADCYLA exposure to Genentech at 1-888-835-2555.
Risk Summary
KADCYLA can cause fetal harm when administered to a pregnant woman. There are no available data on the use of KADCYLA in pregnant women. Cases of oligohydramnios and oligohydramnios sequence manifesting as pulmonary hypoplasia, skeletal abnormalities, and neonatal death were observed in the postmarketing setting in patients treated with trastuzumab, the antibody component of KADCYLA [see Data]. Based on its mechanism of action, the DM1 component of KADCYLA can also cause embryo-fetal harm when administered to a pregnant woman [see Data]. Apprise the patient of the potential risks to a fetus. There are clinical considerations if KADCYLA is used in a pregnant woman, or if a patient becomes pregnant within 7 months following the last dose of KADCYLA [see Clinical Considerations].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Human Data
There are no available data on the use of KADCYLA in pregnant women. In the post-marketing setting, cases of oligohydramnios, and of oligohydramnios sequence, manifesting in the fetus as pulmonary hypoplasia, skeletal abnormalities and neonatal death were observed after treatment with trastuzumab during pregnancy. These case reports described oligohydramnios in pregnant women who received trastuzumab either alone or in combination with chemotherapy. In some case reports, amniotic fluid index increased after trastuzumab was stopped. In one case, trastuzumab therapy resumed after amniotic index improved, and oligohydramnios recurred.
Animal Data
There were no reproductive and developmental toxicology studies conducted with ado-trastuzumab emtansine. DM1, the cytotoxic component of KADCYLA, disrupts microtubule function. DM1 is toxic to rapidly dividing cells in animals and is genotoxic, suggesting it has the potential to cause embryotoxicity and teratogenicity. In studies where trastuzumab was administered to pregnant cynomolgus monkeys during the period of organogenesis at doses up to 25 mg/kg given twice weekly (about 7 times the clinical dose), trastuzumab crossed the placental barrier during the early (Gestation Days 20 to 50) and late (Gestation Days 120 to 150) phases of gestation. The resulting concentrations of trastuzumab in fetal serum and amniotic fluid were approximately 33% and 25%, respectively, of those present in the maternal serum but were not associated with adverse developmental effects.
8.2 Lactation
Risk Summary
There is no information regarding the presence of ado-trastuzumab emtansine in human milk, the effects on the breastfed infant, or the effects on milk production. DM1, the cytotoxic component of KADCYLA, may cause serious adverse reactions in breastfed infants based on its mechanism of action [see Data]. Advise women not to breastfeed during treatment and for 7 months following the last dose of KADCYLA.
Data
There were no animal lactation studies conducted with ado-trastuzumab emtansine or the cytotoxic component of KADCYLA (DM1). In lactating cynomolgus monkeys, trastuzumab was present in breast milk at about 0.3% of maternal serum concentrations after pre- (beginning Gestation Day 120) and post-partum (through Post-partum Day 28) doses of 25 mg/kg administered twice weekly (about 7 times the clinical dose of KADCYLA). Infant monkeys with detectable serum levels of trastuzumab did not exhibit any adverse effects on growth or development from birth to 1 month of age.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of KADCYLA.
Contraception
Females
KADCYLA can cause embryo-fetal harm when administered during pregnancy. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of KADCYLA [see Use in Specific Populations (8.1)].
Infertility
Based on results from animal toxicity studies, KADCYLA may impair fertility in females and males of reproductive potential. It is not known if the effects are reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of KADCYLA have not been established in pediatric patients.
8.5 Geriatric Use
Of the 495 patients who were randomized to KADCYLA in EMILIA [see Clinical Studies (14.1)], 65 patients (13%) were ≥ 65 years of age and 11 patients (2%) were ≥ 75 years of age. In patients ≥ 65 years old (n=138 across both treatment arms) the hazard ratios for progression-free survival (PFS) and overall survival (OS) were 1.06 (95% CI: 0.68, 1.66) and 1.05 (95% CI: 0.58, 1.91), respectively. No overall differences in the safety of KADCYLA were observed in patients aged ≥ 65 compared to patients < 65 years of age. EMILIA did not include sufficient numbers of patients aged ≥ 75 years to draw conclusions on the safety or effectiveness of KADCYLA in this age group.
Of the 743 patients who were randomized to KADCYLA in KATHERINE [see Clinical Studies (14.2)], 58 patients (8%) were ≥ 65 years of age and 2 patients (0.3%) were ≥ 75 years of age. No overall differences in the safety or effectiveness of KADCYLA were observed in patients aged ≥ 65 compared to patients < 65 years of age. KATHERINE did not include sufficient numbers of patients aged ≥ 75 years to draw conclusions on the safety or effectiveness of KADCYLA in this age group.
Population pharmacokinetic analysis indicates that age does not have a clinically meaningful effect on the pharmacokinetics of ado-trastuzumab emtansine [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dedicated renal impairment trial for KADCYLA has been conducted. Based on the population pharmacokinetics, as well as analysis of Grade 3 or greater adverse reactions and dose modifications, dose adjustments of KADCYLA are not needed in patients with mild (creatinine clearance [CLcr] 60 to 89 mL/min) or moderate (CLcr 30 to 59 mL/min) renal impairment. No dose adjustment can be recommended for patients with severe renal impairment (CLcr less than 30 mL/min) because of the limited data available [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No adjustment to the starting dose is required for patients with mild or moderate hepatic impairment [see Clinical Pharmacology (12.3)]. KADCYLA was not studied in patients with severe hepatic impairment. Closely monitor patients with hepatic impairment due to known hepatotoxicity observed with KADCYLA [see Warnings and Precautions, Hepatotoxicity (5.1)].
-
10 OVERDOSAGE
There is no known antidote for overdose of KADCYLA. In clinical trials, overdose of KADCYLA has been reported at approximately two times the recommended dose which resulted in Grade 2 thrombocytopenia (resolved 4 days later) and one death. In the fatal case, the patient incorrectly received KADCYLA at 6 mg/kg and died approximately 3 weeks following the overdose; a cause of death and a causal relationship to KADCYLA were not established.
-
11 DESCRIPTION
KADCYLA (ado-trastuzumab emtansine) is a HER2-targeted antibody-drug conjugate (ADC) which contains the humanized anti-HER2 IgG1, trastuzumab, covalently linked to the microtubule inhibitory drug DM1 (a maytansine derivative) via the stable thioether linker MCC (4-[N-maleimidomethyl] cyclohexane-1-carboxylate). Emtansine refers to the MCC-DM1 complex.
The antibody trastuzumab, is a well characterized recombinant monoclonal antibody product produced by mammalian (Chinese hamster ovary) cells, and the small molecule components (DM1 and MCC) are produced by chemical synthesis. Ado-trastuzumab emtansine contains an average of 3.5 DM1 molecules per antibody. Ado-trastuzumab emtansine has the following chemical structure:
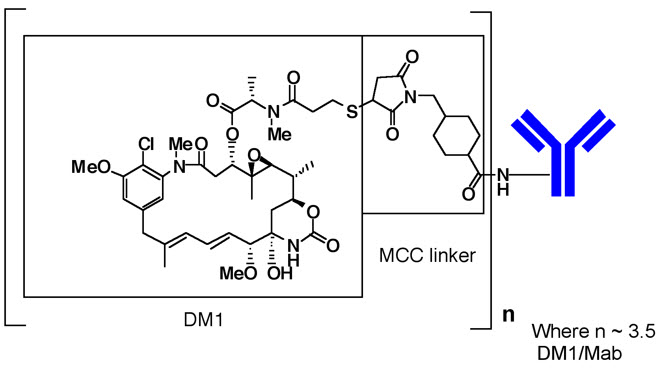
Note: The bracketed structure is DM1 plus MCC which represents the emtansine component. The n is, on average, 3.5 DM1 molecules per trastuzumab (Mab) molecule.
KADCYLA (ado-trastuzumab emtansine) is a sterile, white to off-white preservative free lyophilized powder in single-dose vials. Each vial contains 100 mg or 160 mg ado-trastuzumab emtansine. Following reconstitution, each single-dose vial contains ado-trastuzumab emtansine (20 mg/mL), polysorbate 20 [0.02% (w/v)], sodium succinate (10 mM), and sucrose [6% (w/v)] with a pH of 5.0. The resulting solution containing 20 mg/mL ado-trastuzumab emtansine is administered by intravenous infusion following dilution.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ado-trastuzumab emtansine is a HER2-targeted antibody-drug conjugate. The antibody is the humanized anti-HER2 IgG1, trastuzumab. The small molecule cytotoxin, DM1, is a microtubule inhibitor. Upon binding to sub-domain IV of the HER2 receptor, ado-trastuzumab emtansine undergoes receptor-mediated internalization and subsequent lysosomal degradation, resulting in intracellular release of DM1-containing cytotoxic catabolites. Binding of DM1 to tubulin disrupts microtubule networks in the cell, which results in cell cycle arrest and apoptotic cell death. In addition, in vitro studies have shown that similar to trastuzumab, ado-trastuzumab emtansine inhibits HER2 receptor signaling, mediates antibody-dependent cell-mediated cytotoxicity and inhibits shedding of the HER2 extracellular domain in human breast cancer cells that overexpress HER2.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of multiple doses of KADCYLA (3.6 mg/kg every 3 weeks) on the QTc interval was evaluated in an open label, single arm study in 51 patients with HER2-positive metastatic breast cancer. No large changes in the mean QT interval (i.e., > 20 ms) were detected in the study.
12.3 Pharmacokinetics
The pharmacokinetics of KADCYLA was evaluated in a phase 1 study and in a population pharmacokinetic analysis for the ado-trastuzumab emtansine conjugate (ADC) using pooled data from 5 trials in patients with breast cancer. A linear two-compartment model with first-order elimination from the central compartment adequately describes the ADC concentration-time profile. In addition to ADC, the pharmacokinetics of total antibody (conjugated and unconjugated trastuzumab), DM1 were also determined. The population pharmacokinetic analysis of ADC suggested no difference in KADCYLA exposure based on disease status (adjuvant vs. metastatic setting). The pharmacokinetics of KADCYLA are summarized below.
Distribution
Maximum concentrations (Cmax) of ADC and DM1 were observed close to the end of infusion. In EMILIA, mean (SD) ADC and DM1 Cycle 1 Cmax following KADCYLA administration was 83.4 (16.5) µg/mL and 4.61 (1.61) ng/mL, respectively. In KATHERINE, mean (SD) ADC and DM1 Cycle 1 Cmax following KADCYLA administration was 72.6 (24.3) µg/mL and 4.71 (2.25) ng/mL, respectively.
In vitro, the mean binding of DM1 to human plasma proteins was 93%. In vitro, DM1 was a substrate of P-glycoprotein (P-gp).
Based on population pharmacokinetic analysis, the central volume of distribution of ADC was 3.13 L.
Metabolism
In vitro studies indicate that DM1, the small molecule component of KADCYLA, undergoes metabolism by CYP3A4/5. DM1 did not inhibit or induce major CYP450 enzymes in vitro. In human plasma, ado-trastuzumab emtansine catabolites MCC-DM1, Lys-MCC-DM1, and DM1 were detected at low levels.
Elimination
Based on population pharmacokinetic analysis, following intravenous infusion of KADCYLA, the clearance of the ADC was 0.68 L/day and the elimination half-life (t1/2) was approximately 4 days. No accumulation of KADCYLA was observed after repeated dosing of intravenous infusion every 3 weeks.
Based on population pharmacokinetic analysis (n=671), body weight, sum of longest diameter of target lesions by RECIST, HER2 extracellular domain (ECD) concentrations, AST, albumin, and baseline trastuzumab concentrations were identified as statistically significant covariates for ado-trastuzumab emtansine clearance. However, the magnitude of effect of these covariates on ado-trastuzumab emtansine exposure suggests that, with the exception of body weight, these covariates are unlikely to have a clinically meaningful effect on KADCYLA exposure. Therefore, the body weight based dose of 3.6 mg/kg every 3 weeks without correction for other covariates is considered appropriate.
Effect of Renal Impairment
Based on population pharmacokinetic analysis in 668 patients, including moderate (CLcr 30 - 59 mL/min, n=53) and mild (CLcr 60 - 89 mL/min, n=254) renal impairment, indicate that pharmacokinetics of the ADC is not affected by mild to moderate renal impairment as compared to normal renal function (CLcr ≥ 90 mL/min, n=361). Data from only one patient with severe renal impairment (CLcr < 30 mL/min) is available [see Use in Specific Populations (8.7)].
Effect of Hepatic Impairment
The liver is a primary organ for eliminating DM1 and DM1-containing catabolites. The pharmacokinetics of ado-trastuzumab emtansine and DM1-containing catabolites were evaluated after the administration of 3.6 mg/kg of KADCYLA to metastatic HER2-positive breast cancer patients with normal hepatic function (n=10), mild (Child-Pugh A; n=10) and moderate (Child-Pugh B; n=8) hepatic impairment.
- –
- Plasma concentrations of DM1 and DM1-containing catabolites (Lys-MCC-DM1 and MCC-DM1) were low and comparable between patients with and without hepatic impairment.
- –
- Systemic exposures (AUC) of ado-trastuzumab emtansine at Cycle 1 in patients with mild and moderate hepatic impairment were approximately 38% and 67% lower than that of patients with normal hepatic function, respectively. Ado-trastuzumab emtansine exposure (AUC) at Cycle 3 after repeated dosing in patients with mild or moderate hepatic dysfunction was within the range observed in patients with normal hepatic function.
KADCYLA has not been studied in patients with severe hepatic impairment (Child-Pugh class C).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with ado-trastuzumab emtansine.
DM1 was aneugenic or clastogenic in an in vivo single-dose rat bone marrow micronucleus assay at exposures that were comparable to mean maximum concentrations of DM1 measured in humans administered KADCYLA. DM1 was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
Based on results from animal toxicity studies, KADCYLA may impair fertility in humans. In a single-dose toxicity study of ado-trastuzumab emtansine in rats, degeneration of seminiferous tubules with hemorrhage in the testes associated with increased weights of testes and epididymides at a severely toxic dose level (60 mg/kg; about 4 times the clinical exposure based on AUC) were observed. The same dose in female rats resulted in signs of hemorrhage and necrosis of the corpus luteum in ovaries. In monkeys dosed with ado-trastuzumab emtansine once every three weeks for 12 weeks (four doses), at up to 30 mg/kg (about 7 times the clinical exposure based on AUC), there were decreases in the weights of epididymides, prostate, testes, seminal vesicles and uterus, although the interpretation of these effects is unclear due to the varied sexual maturity of enrolled animals.
13.2 Animal Toxicology and/or Pharmacology
In monkeys, treatment with doses of ado-trastuzumab emtansine up to 30 mg/kg (about 7 times the clinical exposure based on AUC) caused dose dependent axonal degeneration in the sciatic nerve with hypertrophy or hyperplasia of the Schwann cells, and axonal degeneration of the dorsal funiculus in the spinal cord. Based on the mechanism of action of the cytotoxic component DM1, there is clinical potential for neurotoxicity [see Warnings and Precautions (5.8)].
-
14 CLINICAL STUDIES
14.1 Metastatic Breast Cancer
The efficacy of KADCYLA was evaluated in a randomized, multicenter, open-label trial (EMILIA) (NCT00829166) of 991 patients with HER2-positive, unresectable locally advanced or metastatic breast cancer. Prior taxane and trastuzumab-based therapy was required before trial enrollment. Patients with only prior adjuvant therapy were required to have disease recurrence during or within six months of completing adjuvant therapy. Breast tumor samples were required to show HER2 overexpression defined as 3+ IHC or FISH amplification ratio ≥ 2.0 determined at a central laboratory. Patients were randomly allocated (1:1) to receive lapatinib plus capecitabine or KADCYLA. Randomization was stratified by world region (United States, Western Europe, other), number of prior chemotherapy regimens for unresectable locally advanced or metastatic disease (0–1, > 1) and visceral versus non-visceral disease as determined by the investigators.
KADCYLA was given intravenously at 3.6 mg/kg on Day 1 of a 21-day cycle. Lapatinib was administered at 1250 mg/day orally once per day of a 21-day cycle and capecitabine was administered at 1000 mg/m2 orally twice daily on Days 1−14 of a 21-day cycle. Patients were treated with KADCYLA or lapatinib plus capecitabine until progression of disease, withdrawal of consent, or unacceptable toxicity. At the time of the primary analysis, median time on study drug was 5.7 months (range: 0–28.4) for KADCYLA, 4.9 months (range: 0–30.8) for lapatinib, and 4.8 months (range: 0–30.4) for capecitabine.
The co-primary efficacy outcomes of the study were progression-free survival (PFS) based on tumor response assessments by an independent review committee (IRC), and overall survival (OS). PFS was defined as the time from the date of randomization to the date of disease progression or death from any cause (whichever occurred earlier). Overall survival was defined as the time from the date of randomization to the date of death from any cause. Additional outcomes included PFS (based on investigator tumor response assessments), objective response rate (ORR), duration of response and time to symptom progression.
Patient demographics and baseline tumor characteristics were balanced between treatment arms. All patients had metastatic disease at study entry. The median age was approximately 53 years (range 24-84 years), 74% were White, 18% were Asian and 5% were Black. All but 5 patients were women. Twenty-seven percent of patients were enrolled in United States, 32% in Europe and 16% in Asia. Tumor prognostic characteristics including hormone receptor status (positive: 55%, negative: 43%), presence of visceral disease (68%) and non-visceral disease only (33%) and the number of metastatic sites (< 3: 61%, ≥ 3: 37%) were similar in the study arms.
The majority of patients (88%) had received prior systemic treatment in the metastatic setting. Twelve percent of patients had prior treatment only in the neoadjuvant or adjuvant setting and had disease relapse within 6 months of treatment. All but one patient received trastuzumab prior to study entry; approximately 85% of patients received prior trastuzumab in the metastatic setting. Over 99% percent of patients had received a taxane, and 61% of patients had received an anthracycline prior to study entry. Overall, patients received a median of 3 systemic agents in the metastatic setting. Among patients with hormone receptor-positive tumors, 44.4% received prior adjuvant hormonal therapy and 44.8% received hormonal therapy for locally advanced/metastatic disease.
The randomized trial demonstrated a statistically significant improvement in IRC-assessed PFS in the KADCYLA-treated group compared with the lapatinib plus capecitabine-treated group [hazard ratio (HR) = 0.65, 95% CI: 0.55, 0.77, p < 0.0001], and an increase in median PFS of 3.2 months (median PFS of 9.6 months in the KADCYLA-treated group vs. 6.4 months in the lapatinib plus capecitabine group). See Table 7 and Figure 1. The results for investigator-assessed PFS were similar to those observed for IRC-assessed PFS.
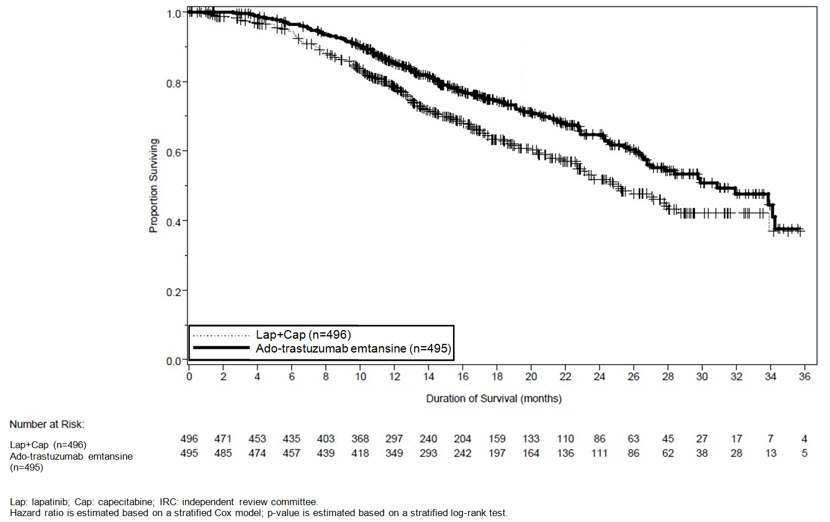
At the time of PFS analysis, 223 patients had died. More deaths occurred in the lapatinib plus capecitabine arm (26%) compared with the KADCYLA arm (19%), however the results of this interim OS analysis did not meet the pre-specified stopping boundary for statistical significance. At the time of the second interim OS analysis, 331 events had occurred. The co-primary endpoint of OS was met; OS was significantly improved in patients receiving KADCYLA (HR = 0.68, 95% CI: 0.55, 0.85, p = 0.0006). This result crossed the pre-specified efficacy stopping boundary (HR = 0.73 or p = 0.0037). The median duration of survival was 30.9 months in the KADCYLA arm vs. 25.1 months in the lapatinib plus capecitabine arm. See Table 7 and Figure 2.
A treatment benefit with KADCYLA in terms of PFS and OS was observed in patient subgroups based on stratification factors, key baseline demographic and disease characteristics, and prior treatments. In the subgroup of patients with hormone receptor-negative disease (n=426), the hazard ratios for PFS and OS were 0.56 (95% CI: 0.44, 0.72) and 0.75 (95% CI: 0.54, 1.03), respectively. In the subgroup of patients with hormone receptor-positive disease (n=545), the hazard ratios for PFS and OS were 0.72 (95% CI: 0.58, 0.91) and 0.62 (95% CI: 0.46, 0.85), respectively. In the subgroup of patients with non-measurable disease (n=205), based on IRC assessments, the hazard ratios for PFS and OS were 0.91 (95% CI: 0.59, 1.42) and 0.96 (95% CI: 0.54, 1.68), respectively; in patients with measurable disease the hazard ratios were 0.62 (95% CI: 0.52, 0.75) and 0.65 (95% CI: 0.51, 0.82), respectively. The PFS and OS hazard ratios in patients who were younger than 65 years old (n=853) were 0.62 (95% CI: 0.52, 0.74) and 0.66 (95% CI: 0.52, 0.83), respectively. In patients ≥ 65 years old (n=138), the hazard ratios for PFS and OS were 1.06 (95% CI: 0.68, 1.66) and 1.05 (95% CI: 0.58, 1.91), respectively.
Table 7 Summary of Efficacy from EMILIA KADCYLA
N=495Lapatinib+Capecitabine
N=496PFS: progression-free survival; OR: objective response - *
- Stratified by world region (United States, Western Europe, other), number of prior chemotherapeutic regimens for locally advanced or metastatic disease (0-1 vs. > 1), and visceral vs. non-visceral disease.
- †
- The second interim analysis for OS was conducted when 331 events were observed and the results are presented in this table.
Progression-Free Survival
(independent review)Number (%) of patients with event 265 (53.5%) 304 (61.3%) Median duration of PFS (months) 9.6 6.4 Hazard Ratio (stratified*) 0.650 95% CI for Hazard Ratio (0.549, 0.771) p-value (Log-Rank test, stratified*) < 0.0001 Overall Survival † Number (%) of patients who died 149 (30.1%) 182 (36.7%) Median duration of survival (months) 30.9 25.1 Hazard Ratio (stratified*) 0.682 95% CI for Hazard Ratio (0.548, 0.849) p-value (Log-Rank test*) 0.0006 Objective Response Rate
(independent review)Patients with measurable disease 397 389 Number of patients with OR (%) 173 (43.6%) 120 (30.8%) Difference (95% CI) 12.7% (6.0, 19.4) Duration of Objective Response
(months)Number of patients with OR 173 120 Median duration (95% CI) 12.6 (8.4, 20.8) 6.5 (5.5, 7.2) Figure 1 Kaplan-Meier Curve of IRC-Assessed Progression-Free Survival for EMILIA
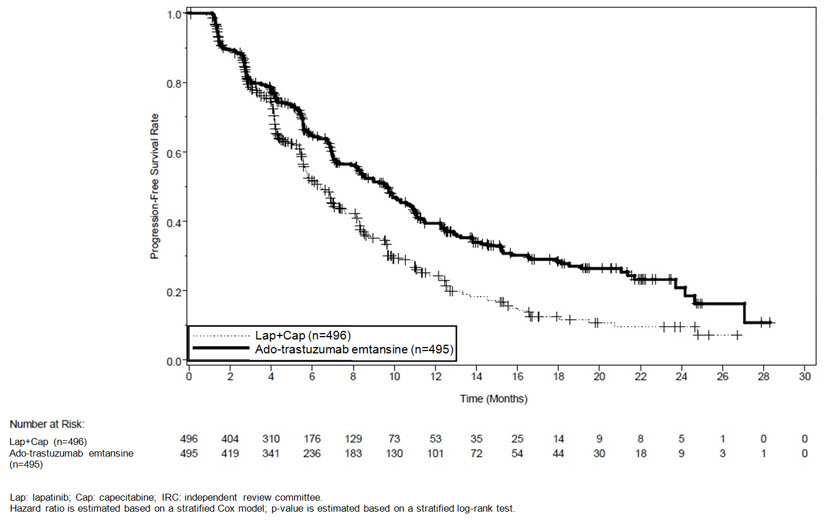
Figure 2 Kaplan-Meier Curve of Overall Survival for EMILIA
14.2 Early Breast Cancer
KATHERINE (NCT01772472) was a randomized, multicenter, open-label trial of 1486 patients with HER2-positive, early breast cancer. Patients were required to have had neoadjuvant taxane and trastuzumab-based therapy with residual invasive tumor in the breast and/or axillary lymph nodes. Patients received radiotherapy and/or hormonal therapy concurrent with study treatment as per local guidelines. Breast tumor samples were required to show HER2 overexpression defined as 3+ IHC or ISH amplification ratio ≥ 2.0 determined at a central laboratory using Ventana's PATHWAY anti-HER2-/neu (4B5) Rabbit Monoclonal Primary Antibody or INFORM HER2 Dual ISH DNA Probe Cocktail assays. Patients were randomized (1:1) to receive KADCYLA or trastuzumab. Randomization was stratified by clinical stage at presentation, hormone receptor status, preoperative HER2-directed therapy (trastuzumab, trastuzumab plus additional HER2-directed agent[s]), and pathological nodal status evaluation after preoperative therapy.
KADCYLA was given intravenously at 3.6 mg/kg on Day 1 of a 21-day cycle. Trastuzumab was given intravenously at 6 mg/kg on Day 1 of a 21-day cycle. Patients were treated with KADCYLA or trastuzumab for a total of 14 cycles unless there was recurrence of disease, withdrawal of consent, or unacceptable toxicity. At the time of the major efficacy outcome analysis, median treatment duration was 10 months for both KADCYLA- and trastuzumab-treated patients. Patients who discontinued KADCYLA for reasons other than disease recurrence could complete the remainder of the planned HER2-directed therapy with trastuzumab if appropriate based on toxicity considerations and investigator discretion.
The major efficacy outcome of the study was invasive disease-free survival (IDFS). IDFS was defined as the time from the date of randomization to first occurrence of ipsilateral invasive breast tumor recurrence, ipsilateral local or regional invasive breast cancer recurrence, distant recurrence, contralateral invasive breast cancer, or death from any cause. Additional efficacy outcomes included IDFS including second primary non-breast cancer, disease free survival (DFS), and overall survival (OS).
Patient demographics and baseline tumor characteristics were generally balanced between treatment arms. The median age was approximately 49 years (range 23-80 years), 73% were White, 9% were Asian, 6% were American Indian or Alaska Native and 3% were Black or African American. Most patients (99.7%) were women. Enrollment by region was as follows: 23% in North America, 54% in Europe and 23% throughout the rest of the world. Tumor prognostic characteristics including hormone receptor status (positive: 72%, negative: 28%), clinical stage at presentation (inoperable: 25%, operable: 75%) and pathological nodal status after preoperative therapy (node positive: 46%, node negative or not evaluated: 54%) were similar across study arms.
The majority of patients (77%) had received an anthracycline-containing neoadjuvant chemotherapy regimen. Twenty percent of patients received another HER2-targeted agent in addition to trastuzumab as a component of neoadjuvant therapy; 94% of these patients received pertuzumab.
After a median follow-up of 40 months, a statistically significant improvement in IDFS was observed in patients who received KADCYLA compared with trastuzumab. The OS data were not mature at the time of the IDFS analysis (98 deaths [6.6%] occurred in 1486 patients). The efficacy results from KATHERINE are summarized in Table 8 and Figure 3.
Consistent results were observed with KADCYLA in terms of IDFS across subgroups based on stratification factors, key baseline demographic and disease characteristics, and prior treatments.
Table 8 Efficacy Results from KATHERINE KADCYLA
N=743Trastuzumab
N=743HR: Hazard Ratio; CI: Confidence Intervals, Invasive Disease-Free Survival (IDFS)*,† Number (%) of patients with event 91 (12.2%) 165 (22.2%) HR [95% CI]‡ 0.50 [0.39, 0.64] p-value (Log-Rank test, unstratified) < 0.0001 3-year event-free rate§, % [95% CI] 88.3 [85.8, 90.7] 77.0 [73.8, 80.7] IDFS including second primary non-breast cancer Number (%) of patients with event 95 (12.8%) 167 (22.5%) HR [95% CI]‡ 0.51 [0.40, 0.66] 3-year event-free rate§, % [95% CI] 87.7 [85.2, 90.2] 76.9 [73.7, 80.1] Disease-Free Survival (DFS) Number (%) of patients with event 98 (13.2%) 167 (22.5%) HR [95% CI]‡ 0.53 [0.41, 0.68] 3-year event-free rate§, % [95% CI] 87.4 [84.9, 89.9] 76.9 [73.7, 80.1] Figure 3 Kaplan-Meier Curve of Invasive Disease-Free Survival in KATHERINE
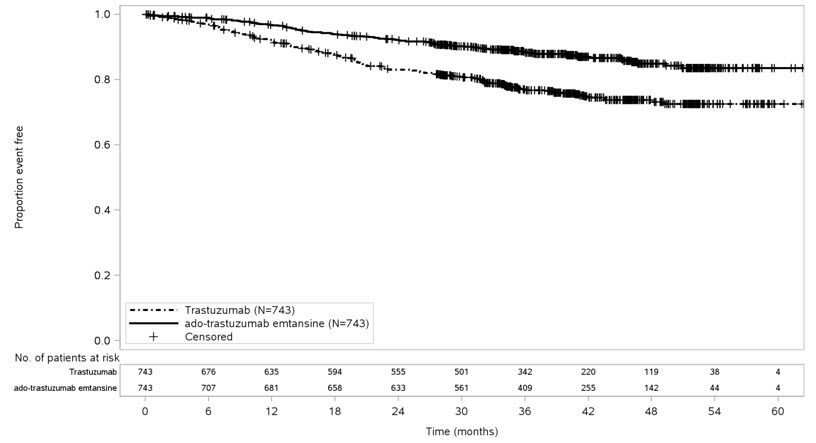
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied/Storage
KADCYLA (ado-trastuzumab emtansine) is supplied as:
Carton Contents NDC One 100 mg vial, single-dose vial NDC 50242-088-01 One 160 mg vial, single-dose vial NDC 50242-087-01 Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) until time of reconstitution. Do not freeze or shake.
-
17 PATIENT COUNSELING INFORMATION
Hepatotoxicity
- Inform patients of the possibility of severe liver injury and advise patients to immediately seek medical attention if they experience symptoms of acute hepatitis such as nausea, vomiting, abdominal pain (especially RUQ abdominal pain), jaundice, dark urine, generalized pruritus, anorexia, etc. [see Warnings and Precautions (5.1)].
Left Ventricular Dysfunction
- Advise patients to contact a health care professional immediately for any of the following: new onset or worsening shortness of breath, cough, swelling of the ankles/legs, palpitations, weight gain of more than 5 pounds in 24 hours, dizziness or loss of consciousness [see Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential that KADCYLA exposure during pregnancy or within 7 months prior to conception can result in fetal harm. Advise female patients to contact their healthcare provider with a known or suspected pregnancy [see Use in Specific Populations (8.1, 8.3)].
- Advise women who are exposed to KADCYLA during pregnancy or who become pregnant within 7 months following the last dose of KADCYLA that there is a pregnancy pharmacovigilance program that monitors pregnancy outcomes. Encourage these patients to report their pregnancy to Genentech [see Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of KADCYLA [see Use in Specific Populations (8.1, 8.3)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 4 months following the last dose of KADCYLA [see Use in Specific Populations (8.3)].
Lactation
- Advise women not to breastfeed during treatment and for 7 months after the last dose of KADCYLA [see Use in Specific Populations (8.2)].
-
SPL UNCLASSIFIED SECTION
KADCYLA® [ado-trastuzumab emtansine]
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
U.S. License No: 1048KADCYLA is a trademark of Genentech, Inc.
©2022 Genentech, Inc.Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
- PRINCIPAL DISPLAY PANEL - 100 mg Vial Carton
- PRINCIPAL DISPLAY PANEL - 160 mg Vial Carton
-
INGREDIENTS AND APPEARANCE
KADCYLA
ado-trastuzumab emtansine injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-088 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TRASTUZUMAB EMTANSINE (UNII: SE2KH7T06F) (ADO-TRASTUZUMAB EMTANSINE - UNII:SE2KH7T06F) TRASTUZUMAB EMTANSINE 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 60 mg in 1 mL SUCCINIC ACID (UNII: AB6MNQ6J6L) 1.18 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) 0.45 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.2 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-088-01 1 in 1 CARTON 02/22/2013 1 5 mL in 1 VIAL, SINGLE-USE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125427 02/22/2013 KADCYLA
ado-trastuzumab emtansine injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-087 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TRASTUZUMAB EMTANSINE (UNII: SE2KH7T06F) (ADO-TRASTUZUMAB EMTANSINE - UNII:SE2KH7T06F) TRASTUZUMAB EMTANSINE 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 60 mg in 1 mL SUCCINIC ACID (UNII: AB6MNQ6J6L) 1.18 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) 0.45 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.2 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-087-01 1 in 1 CARTON 02/22/2013 1 8 mL in 1 VIAL, SINGLE-USE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125427 02/22/2013 Labeler - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 833220176 PACK(50242-087, 50242-088) , LABEL(50242-087, 50242-088) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd 485244961 MANUFACTURE(50242-087, 50242-088) , ANALYSIS(50242-087, 50242-088) , PACK(50242-087, 50242-088) Establishment Name Address ID/FEI Business Operations Roche Diagnostics GmbH 315028860 ANALYSIS(50242-087, 50242-088) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 004074162 ANALYSIS(50242-087, 50242-088) , API MANUFACTURE(50242-087, 50242-088) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 146373191 ANALYSIS(50242-087, 50242-088) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 080129000 ANALYSIS(50242-087, 50242-088) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche AG 482242971 API MANUFACTURE(50242-087, 50242-088) , ANALYSIS(50242-087, 50242-088)