Label: GAZYVA- obinutuzumab injection, solution, concentrate

- NDC Code(s): 50242-070-01, 50242-070-86
- Packager: Genentech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated August 23, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use GAZYVA safely and effectively. See full prescribing information for GAZYVA.
GAZYVA® (obinutuzumab) injection, for intravenous use
Initial U.S. Approval: 2013WARNING: HEPATITIS B VIRUS REACTIVATION and PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
See full prescribing information for complete boxed warning.
RECENT MAJOR CHANGES
Dosage and Administration, Recommended Dosage for Follicular Lymphoma (2.3) 2/2022 Dosage Modifications for Adverse Reactions (2.5) 2/2022 Dosage and Administration, Preparation and Administration (2.6) 2/2022 Warnings and Precautions, Thrombocytopenia (5.8) and Disseminated Intravascular Coagulation (5.9) 7/2022 INDICATIONS AND USAGE
GAZYVA is a CD20-directed cytolytic antibody indicated:
- in combination with chlorambucil, for the treatment of patients with previously untreated chronic lymphocytic leukemia. (1, 14)
- in combination with bendamustine followed by GAZYVA monotherapy, for the treatment of patients with follicular lymphoma who relapsed after, or are refractory to, a rituximab-containing regimen. (1, 14)
- in combination with chemotherapy followed by GAZYVA monotherapy in patients achieving at least a partial remission, for the treatment of adult patients with previously untreated stage II bulky, III or IV follicular lymphoma. (1, 14)
DOSAGE AND ADMINISTRATION
- Premedicate for infusion-related reactions and tumor lysis syndrome. (2.1, 5.3, 5.4)
- Administer only as intravenous infusion. Do not administer as an intravenous push or bolus. (2.1)
- The recommended dosage for chronic lymphocytic leukemia is 100 mg on day 1 and 900 mg on day 2 of Cycle 1, 1,000 mg on day 8 and 15 of Cycle 1, and 1,000 mg on day 1 of Cycles 2–6. (2.2)
- The recommended dosage for follicular lymphoma is 1,000 mg on day 1, 8 and 15 of Cycle 1, 1,000 mg on day 1 of Cycles 2-6 or Cycles 2-8, and then 1,000 mg every 2 months for up to 2 years. (2.3)
DOSAGE FORMS AND STRENGTHS
Injection: 1,000 mg/40 mL (25 mg/mL) single-dose vial. (3)
CONTRAINDICATIONS
GAZYVA is contraindicated in patients with known hypersensitivity reactions (e.g., anaphylaxis) to obinutuzumab or any of the excipients, including serum sickness with prior obinutuzumab use. (4)
WARNINGS AND PRECAUTIONS
- Infusion-Related Reactions: Premedicate patients with glucocorticoid, acetaminophen, and anti-histamine. Monitor patients closely during infusions. Interrupt, reduce rate, or discontinue for infusion-related reactions based on severity. (2.1, 5.3)
- Hypersensitivity Reactions Including Serum Sickness: Discontinue GAZYVA permanently. (5.4)
- Tumor Lysis Syndrome: Premedicate with anti-hyperuricemics and adequate hydration, especially for patients with high tumor burden, high circulating lymphocyte count or renal impairment. Correct electrolyte abnormalities, provide supportive care, and monitor renal function and fluid balance. (5.5)
- Infections: Do not administer GAZYVA to patients with an active infection. Patients with a history of recurring or chronic infections may be at increased risk of infection. (5.6)
- Neutropenia: In patients with Grade 3 to 4 neutropenia, monitor laboratory tests until resolution and for infection. Consider dose delays and infection prophylaxis, as appropriate. (5.7)
- Thrombocytopenia: Monitor for decreased platelet counts and bleeding. Transfusion may be necessary. (5.8)
- Disseminated Intravascular Coagulation: Evaluate cause and monitor for bleeding, thrombosis, and need for supportive care. (5.9)
- Immunization: Avoid administration of live virus vaccines during GAZYVA treatment and until B-cell recovery. (5.10)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and use effective contraception. (5.11)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥ 20% and ≥ 2% greater in the GAZYVA treated arm) were:
- Previously untreated CLL: infusion-related reactions and neutropenia. (6)
- Relapsed or refractory NHL: infusion-related reactions, fatigue, neutropenia, cough, upper respiratory tract infections, and musculoskeletal pain. (6)
- Previously untreated NHL: infusion-related reactions, neutropenia, upper respiratory tract infections, cough, constipation, and diarrhea. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HEPATITIS B VIRUS REACTIVATION and PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
1 INDICATIONS AND USAGE
1.1 Chronic Lymphocytic Leukemia (CLL)
1.2 Follicular Lymphoma (FL)
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
2.2 Recommended Dosage for Chronic Lymphocytic Leukemia
2.3 Recommended Dosage for Follicular Lymphoma
2.4 Recommended Premedication and Prophylactic Medications
2.5 Dosage Modifications for Adverse Reactions
2.6 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatitis B Virus Reactivation
5.2 Progressive Multifocal Leukoencephalopathy
5.3 Infusion-Related Reactions
5.4 Hypersensitivity Reactions Including Serum Sickness
5.5 Tumor Lysis Syndrome
5.6 Infections
5.7 Neutropenia
5.8 Thrombocytopenia
5.9 Disseminated Intravascular Coagulation (DIC)
5.10 Immunization
5.11 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Chronic Lymphocytic Leukemia
14.2 Follicular Lymphoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HEPATITIS B VIRUS REACTIVATION and PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
- Hepatitis B Virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients receiving CD20-directed cytolytic antibodies, including GAZYVA. Screen all patients for HBV infection before treatment initiation. Monitor HBV-positive patients during and after treatment with GAZYVA. Discontinue GAZYVA and concomitant medications in the event of HBV reactivation [see Warnings and Precautions (5.1)].
- Progressive Multifocal Leukoencephalopathy (PML) including fatal PML, can occur in patients receiving GAZYVA [see Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
1.1 Chronic Lymphocytic Leukemia (CLL)
GAZYVA, in combination with chlorambucil, is indicated for the treatment of patients with previously untreated chronic lymphocytic leukemia.
1.2 Follicular Lymphoma (FL)
GAZYVA, in combination with bendamustine followed by GAZYVA monotherapy, is indicated for the treatment of patients with follicular lymphoma who relapsed after, or are refractory to, a rituximab-containing regimen.
GAZYVA, in combination with chemotherapy followed by GAZYVA monotherapy in patients achieving at least a partial remission, is indicated for the treatment of adult patients with previously untreated stage II bulky, III or IV follicular lymphoma.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
- Premedicate before each infusion [see Dosage and Administration (2.4)].
- Provide prophylactic hydration and anti-hyperuricemics to patients at high risk of tumor lysis syndrome [see Dosage and Administration (2.4) and Warnings and Precautions (5.4)].
- Administer only as an intravenous infusion through a dedicated line [see Dosage and Administration (2.6)].
- Do not administer as an intravenous push or bolus.
- Monitor blood counts at regular intervals.
- GAZYVA should only be administered by a healthcare professional with appropriate medical support to manage severe infusion-related reactions that can be fatal if they occur [see Warnings and Precautions (5.3)].
2.2 Recommended Dosage for Chronic Lymphocytic Leukemia
Each dose of GAZYVA is 1,000 mg administered intravenously with the exception of the first infusions in Cycle 1, which are administered on day 1 (100 mg) and day 2 (900 mg) according to Table 1.
Table 1 Dose of GAZYVA to be Administered During Six 28-Day Treatment Cycles for Patients with CLL Day of treatment cycle Dose of GAZYVA Rate of infusion Cycle 1
(loading doses)Day 1 100 mg Administer at 25 mg/hr over 4 hours. Do not increase the infusion rate. Day 2 900 mg If no infusion-related reaction (IRR) occurred during the previous infusion, administer at 50 mg/hr. The rate of the infusion can be escalated in increments of 50 mg/hr every 30 minutes to a maximum rate of 400 mg/hr.
If an IRR occurred during the previous infusion, administer at 25 mg/hr. The rate of infusion can be escalated in increments of up to 50 mg/hr every 30 minutes to a maximum rate of 400 mg/hr.Day 8 1,000 mg If no IRR occurred during the previous infusion and the final infusion rate was 100 mg/hr or faster, infusions can be started at a rate of 100 mg/hr and increased by 100 mg/hr increments every 30 minutes to a maximum of 400 mg/hr.
If an infusion-related reaction occurred during the previous infusion, administer at 50 mg/hr. The rate of infusion can be escalated in increments of 50 mg/hr every 30 minutes to a maximum rate of 400 mg/hr.Day 15 1,000 mg Cycles 2–6 Day 1 1,000 mg If a planned dose of GAZYVA is missed, administer the missed dose as soon as possible and adjust dosing schedule to maintain the time interval between doses. If appropriate, patients who do not complete the Day 1 Cycle 1 dose may proceed to the Day 2 Cycle 1 dose.
2.3 Recommended Dosage for Follicular Lymphoma
Each dose of GAZYVA is 1,000 mg administered intravenously according to Table 2.
For patients with relapsed or refractory FL, administer GAZYVA in combination with bendamustine in six 28-day cycles. Patients who achieve stable disease, complete response, or partial response to the initial 6 cycles should continue on GAZYVA 1,000 mg as monotherapy for up to two years.
For patients with previously untreated FL, administer GAZYVA with one of the following chemotherapy regimens:
- Six 28-day cycles in combination with bendamustine
- Six 21-day cycles in combination with CHOP, followed by 2 additional 21-day cycles of GAZYVA alone
- Eight 21-day cycles in combination with CVP
Patients with previously untreated FL who achieve a complete response or partial response to the initial 6 or 8 cycles should continue on GAZYVA 1,000 mg as monotherapy for up to two years.
GAZYVA should be administered at the standard infusion rate in Cycle 1 (see Table 2). In patients with FL who do not experience a Grade 3 or higher IRR during Cycle 1, GAZYVA may be administered as a shorter, approximately 90-minute infusion from Cycle 2 onwards (see Table 3) with continued premedication.
Table 2 Dose and Standard Infusion Rate of GAZYVA to be Administered During 6–8 Treatment Cycles, Followed by GAZYVA Monotherapy for Patients with FL Day of treatment cycle Dose of GAZYVA Rate of infusion Cycle 1
(loading doses)Day 1 1,000 mg Administer at 50 mg/hr. The rate of the infusion can be escalated in 50 mg/hr increments every 30 minutes to a maximum of 400 mg/hr. Day 8 1,000 mg If no infusion-related reaction or an infusion-related reaction of Grade 1 occurred during the previous infusion and the final infusion rate was 100 mg/hr or faster, infusions can be started at a rate of 100 mg/hr and increased by 100 mg/hr increments every 30 minutes to a maximum of 400 mg/hr.
If an infusion-related reaction of Grade 2 or higher occurred during the previous infusion, administer at 50 mg/hr. The rate of infusion can be escalated in increments of 50 mg/hr every 30 minutes to a maximum rate of 400 mg/hr.Day 15 1,000 mg Cycles 2–6 or 2–8 Day 1 1,000 mg Monotherapy Every two months for up to two years 1,000 mg Table 3 Dose and Infusion Rate of a GAZYVA 90-Minute Infusion for Patients with FL Day of treatment cycle Dose of GAZYVA Rate of infusion - *
- Consider an approximately 90-minute infusion in patients with FL who do not experience a Grade 3 or higher infusion-related reaction to GAZYVA in Cycle 1 and subsequent cycles.
Cycle 1 Days 1, 8, 15 1,000 mg See Table 2 Cycles 2–6* or 2-8* Day 1 1,000 mg If no Grade 3 or higher IRR occurred during Cycle 1: 100 mg/hr for 30 minutes, then 900 mg/hr for approximately 60 minutes.
If an IRR of Grade 1-2 with ongoing symptoms or a Grade 3 or higher IRR occurred during the previous approximately 90-minute infusion, administer all subsequent GAZYVA infusions at the standard infusion rate (see Table 2).Monotherapy* Every two months for up to two years 1,000 mg If a planned dose of GAZYVA is missed, administer the missed dose as soon as possible. During GAZYVA and chemotherapy treatment, adjust the dosing schedule accordingly to maintain the time interval between chemotherapy cycles. During monotherapy, maintain the original dosing schedule for subsequent doses. Initiate monotherapy approximately two months after the last dose of GAZYVA administered during the induction phase.
2.4 Recommended Premedication and Prophylactic Medications
Infusion-Related Reactions
Premedication to reduce the risk of IRRs is outlined in Table 4 [see Warnings and Precautions (5.3)].
Hypotension may occur during GAZYVA intravenous infusions. Consider withholding antihypertensive treatments for 12 hours prior to and throughout each GAZYVA infusion and for the first hour after administration [see Warnings and Precautions (5.3)].
Table 4 Premedication for GAZYVA Infusion to Reduce Infusion-Related Reactions Day of Treatment Cycle Patients requiring premedication Premedication Administration Premedication applies to both standard and approximately 90-minute infusions. - *
- Hydrocortisone is not recommended as it has not been effective in reducing the rate of IRRs.
- †
- If a glucocorticoid-containing chemotherapy regimen is administered on the same day as GAZYVA, the glucocorticoid can be administered as an oral medication if given at least 1 hour prior to GAZYVA, in which case additional intravenous glucocorticoid as premedication is not required.
Cycle 1:
CLL
Day 1, Day 2
FL
Day 1All patients Intravenous glucocorticoid:
20 mg dexamethasone or 80 mg methylprednisolone*,†Completed at least 1 hour prior to GAZYVA infusion. 650–1,000 mg acetaminophen At least 30 minutes before GAZYVA infusion. anti-histamine (e.g., 50 mg diphenhydramine) All subsequent infusions, CLL or FL All patients 650–1,000 mg acetaminophen At least 30 minutes before GAZYVA infusion. Patients with an IRR (Grade 1-2) with the previous infusion 650–1,000 mg acetaminophen At least 30 minutes before GAZYVA infusion. anti-histamine (e.g., 50 mg diphenhydramine) Patients with a Grade 3 IRR with the previous infusion OR with a lymphocyte count > 25 × 109/L prior to next treatment Intravenous glucocorticoid:
20 mg dexamethasone or 80 mg methylprednisolone*Completed at least 1 hour prior to GAZYVA infusion. 650–1,000 mg acetaminophen At least 30 minutes before GAZYVA infusion. anti-histamine (e.g., 50 mg diphenhydramine) Tumor Lysis Syndrome Prophylaxis
Patients with high tumor burden, high circulating absolute lymphocyte counts (greater than 25 × 109/L) or renal impairment are considered at risk of tumor lysis syndrome and should receive prophylaxis. Premedicate with anti-hyperuricemics (e.g., allopurinol or rasburicase) and ensure adequate hydration prior to start of GAZYVA therapy. Continue prophylaxis prior to each subsequent GAZYVA infusion, as needed [see Warnings and Precautions (5.4)].
Antimicrobial Prophylaxis
Patients with Grade 3 to 4 neutropenia lasting more than one week are strongly recommended to receive antimicrobial prophylaxis until resolution of neutropenia to Grade 1 or 2. Consider antiviral and antifungal prophylaxis for patients with severe and long lasting (> 1 week) neutropenia.
2.5 Dosage Modifications for Adverse Reactions
Infusion-Related Reactions
If a patient experiences an IRR, adjust the infusion as follows [see Warnings and Precautions (5.3)]:
- Grade 4 (life-threatening): Stop infusion immediately and permanently discontinue GAZYVA.
- Grade 3 (severe): Interrupt infusion and manage symptoms.
- For patients who experience Grade 3 IRRs during standard infusion, upon resolution of symptoms, consider restarting GAZYVA infusion at no more than half the previous rate (the rate being used at the time that the IRR reaction occurred), and if patient does not experience any further IRR symptoms, infusion rate escalation may resume at the increments and intervals as appropriate for the treatment cycle dose. Permanently discontinue treatment if patients experience a Grade 3 or higher IRR at rechallenge.
- For patients with FL who experience Grade 3 IRRs during the approximately 90-minute infusion, upon resolution of symptoms, the infusion can be restarted at no more than half the previous rate (the rate being used at the time that the IRR occurred) and not greater than 400 mg/hr. Administer subsequent infusions at the standard rate. Permanently discontinue treatment if patients experience a Grade 3 or higher IRR at rechallenge.
- For CLL patients only, the Day 1 infusion rate may be increased back up to 25 mg/hr after 1 hour but not increased further.
- Grade 1–2 (mild to moderate): Reduce infusion rate or interrupt infusion and manage symptoms. Upon resolution of symptoms, continue or resume GAZYVA infusion, and if patient does not experience any further IRR symptoms, infusion rate escalation may resume at the increments and intervals as appropriate for the treatment cycle dose.
- For CLL patients only, the Day 1 infusion rate may be increased back up to 25 mg/hr after 1 hour but not increased further.
2.6 Preparation and Administration
Preparation
Prepare the solution for infusion, using aseptic technique, as follows:
- Inspect visually for any particulate matter and discoloration prior to administration.
- Use a sterile needle and syringe to prepare GAZYVA.
- Dilute into a 0.9% Sodium Chloride Injection, USP PVC or non-PVC polyolefin infusion bag.
Chronic Lymphocytic Leukemia- Preparation of solution for infusion on day 1 (100 mg) and day 2 (900 mg) of Cycle 1:
- Prepare day 1 (100 mg) and day 2 (900 mg) infusion bags at the same time using one vial (1,000 mg/40 mL) on day 1.
- Withdraw 40 mL of GAZYVA solution from the vial.
- Dilute 4 mL (100 mg) of GAZYVA into a 100 mL 0.9% Sodium Chloride Injection, USP infusion bag for immediate administration.
- Dilute the remaining 36 mL (900 mg) into a 250 mL 0.9% Sodium Chloride Injection, USP infusion bag at the same time for use on day 2 and store at 2°C to 8°C (36°F to 46°F) for up to 24 hours. After allowing the diluted bag to come to room temperature, use immediately.
- Clearly label each infusion bag.
- Preparation of solution for infusion on day 8 and 15 of Cycle 1 and day 1 of Cycles 2–6:
- Withdraw 40 mL of GAZYVA solution from the vial.
- Dilute 40 mL (1,000 mg) into a 250 mL 0.9% Sodium Chloride Injection, USP infusion bag.
- Preparation of solution for infusion:
- Withdraw 40 mL of GAZYVA solution from the vial.
- Dilute 40 mL (1,000 mg) into a 250 mL 0.9% Sodium Chloride Injection, USP infusion bag.
- Preparation of solution for infusion on day 1 (100 mg) and day 2 (900 mg) of Cycle 1:
- Mix diluted solution by gentle inversion. Do not shake or freeze.
- For microbiological stability, immediately use diluted GAZYVA infusion solution. If not used immediately, store in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours prior to use.
The product can be administered at a final concentration of 0.4 mg/mL to 4 mg/mL.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
GAZYVA is contraindicated in patients with known hypersensitivity reactions (e.g., anaphylaxis) to obinutuzumab or to any of the excipients, or serum sickness with prior obinutuzumab use [see Warnings and Precautions (5.4)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatitis B Virus Reactivation
Hepatitis B virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients treated with anti-CD20 antibodies such as GAZYVA. HBV reactivation has been reported in patients who are hepatitis B surface antigen (HBsAg) positive and also in patients who are HBsAg negative but are hepatitis B core antibody (anti-HBc) positive. Reactivation has also occurred in patients who appear to have resolved hepatitis B infection (i.e., HBsAg negative, anti-HBc positive, and hepatitis B surface antibody [anti-HBs] positive).
HBV reactivation is defined as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level or detection of HBsAg in a person who was previously HBsAg negative and anti-HBc positive. Reactivation of HBV replication is often followed by hepatitis, i.e., increase in transaminase levels and, in severe cases, increase in bilirubin levels, liver failure, and death.
Screen all patients for HBV infection by measuring HBsAg and anti-HBc before initiating treatment with GAZYVA. For patients who show evidence of hepatitis B infection (HBsAg positive [regardless of antibody status] or HBsAg negative but anti-HBc positive), consult healthcare providers with expertise in managing hepatitis B regarding monitoring and consideration for HBV antiviral therapy.
Monitor patients with evidence of current or prior HBV infection for clinical and laboratory signs of hepatitis or HBV reactivation during and for several months following treatment with GAZYVA. HBV reactivation has been reported for other CD20-directed cytolytic antibodies following completion of therapy.
In patients who develop reactivation of HBV while receiving GAZYVA, immediately discontinue GAZYVA and any concomitant chemotherapy and institute appropriate treatment. Resumption of GAZYVA in patients whose HBV reactivation resolves should be discussed with healthcare providers with expertise in managing hepatitis B. Insufficient data exist regarding the safety of resuming GAZYVA in patients who develop HBV reactivation.
5.2 Progressive Multifocal Leukoencephalopathy
John Cunningham (JC) virus infection resulting in progressive multifocal leukoencephalopathy (PML), which can be fatal, occurred in patients treated with GAZYVA. Consider the diagnosis of PML in any patient presenting with new onset or changes to preexisting neurologic manifestations. Evaluation of PML includes, but is not limited to, consultation with a neurologist, brain MRI, and lumbar puncture. Discontinue GAZYVA therapy and consider discontinuation or reduction of any concomitant chemotherapy or immunosuppressive therapy in patients who develop PML.
5.3 Infusion-Related Reactions
GAZYVA can cause severe and life-threatening infusion-related reactions (IRRs). Sixty-five percent of patients with CLL experienced a reaction to the first 1,000 mg of GAZYVA infused. Thirty-seven percent of patients with relapsed or refractory NHL and 60% of patients with previously untreated NHL experienced a reaction on Day 1 of GAZYVA infusion. IRRs have occurred within 24 hours of receiving GAZYVA. IRRs can also occur with subsequent infusions. Symptoms may include hypotension, tachycardia, dyspnea, and respiratory symptoms (e.g., bronchospasm, larynx and throat irritation, wheezing, laryngeal edema). The most frequently reported symptoms include nausea, fatigue, chest discomfort, dyspnea, dizziness, vomiting, diarrhea, rash, hypertension, hypotension, flushing, headache, pyrexia, and chills [see Adverse Reactions (6.1)].
Premedicate patients with acetaminophen, anti-histamine, and a glucocorticoid [see Dosage and Administration (2.4)]. Closely monitor patients during the entire infusion. Reduce infusion rate, interrupt infusion or permanently discontinue GAZYVA for IRRs based on severity [see Dosage and Administration (2.5)]. Institute medical management (e.g., glucocorticoids, epinephrine, bronchodilators, and/or oxygen) for IRRs as needed.
For patients with preexisting cardiac or pulmonary conditions, monitor more frequently throughout the infusion and the post-infusion period since they may be at greater risk of experiencing more severe reactions. Hypotension may occur as part of the IRR to GAZYVA. Consider withholding antihypertensive treatments for 12 hours prior to, during each GAZYVA infusion, and for the first hour after administration until blood pressure is stable. For patients at increased risk of hypertensive crisis, consider the benefits versus the risks of withholding their antihypertensive medication as is suggested here.
5.4 Hypersensitivity Reactions Including Serum Sickness
Hypersensitivity reactions have been reported in patients treated with GAZYVA. Signs of immediate-onset hypersensitivity included dyspnea, bronchospasm, hypotension, urticaria and tachycardia. Late-onset hypersensitivity diagnosed as serum sickness has also been reported, with symptoms that include chest pain, diffuse arthralgia and fever. Hypersensitivity reactions may be difficult to clinically distinguish from IRRs. However, hypersensitivity very rarely occurs with the first infusion and, when observed, often occurs after previous exposure.
If a hypersensitivity reaction is suspected during or after an infusion, stop the infusion and permanently discontinue treatment. GAZYVA is contraindicated in patients with known hypersensitivity reactions to GAZYVA, including serum sickness with prior GAZYVA use [see Contraindications (4)].
5.5 Tumor Lysis Syndrome
Tumor lysis syndrome (TLS), including fatal cases, has been reported in patients receiving GAZYVA. Patients with high tumor burden, high circulating lymphocyte count (> 25 × 109/L) or renal impairment are at greater risk for TLS.
Administer appropriate tumor lysis prophylaxis with anti-hyperuricemics (e.g., allopurinol or rasburicase) and hydration prior to the infusion of GAZYVA for patients at risk for TLS [see Dosage and Administration (2.4)]. During the initial days of GAZYVA treatment, monitor the laboratory parameters of patients considered at risk for TLS. For treatment of TLS, correct electrolyte abnormalities, monitor renal function and fluid balance, and administer supportive care, including dialysis as indicated.
5.6 Infections
Fatal and serious bacterial, fungal, and new or reactivated viral infections can occur during and following GAZYVA therapy. When GAZYVA is administered with chemotherapy followed by GAZYVA monotherapy as in the GALLIUM study, Grade 3 to 5 infections have been reported in up to 8% of patients during combination therapy, up to 13% of patients during monotherapy, and up to 8% of patients after treatment [see Adverse Reactions (6.1)].
In GALLIUM, more Grade 3 to 5 infections were reported in the recipients of GAZYVA and bendamustine (117/410 patients, 29%) as compared to GAZYVA plus CHOP or CVP (43/281 patients, 15%). More fatal infections were reported in patients treated with GAZYVA and bendamustine (3%), as compared to GAZYVA plus CHOP or CVP (< 1%), including during the monotherapy phase and after completion of treatment.
Do not administer GAZYVA to patients with an active infection. Patients with a history of recurring or chronic infections may be at increased risk of infection.
5.7 Neutropenia
Severe and life-threatening neutropenia, including febrile neutropenia, has been reported during treatment with GAZYVA. Monitor patients with Grade 3 to 4 neutropenia frequently with regular laboratory tests until resolution. Anticipate, evaluate, and treat any symptoms or signs of developing infection. Consider dose delays for Grade 3 or 4 neutropenia. Consider administration of granulocyte colony-stimulating factors (GCSF) in patients with Grade 3 or 4 neutropenia.
Neutropenia can also be of late onset (occurring more than 28 days after completion of treatment) and/or prolonged (lasting longer than 28 days). Patients with severe and long lasting (> 1 week) neutropenia are strongly recommended to receive antimicrobial prophylaxis until resolution of neutropenia to Grade 1 or 2. Consider antiviral and antifungal prophylaxis.
5.8 Thrombocytopenia
Severe and life-threatening thrombocytopenia has been reported during treatment with GAZYVA in combination with chemotherapy. Fatal hemorrhagic events have been reported in patients with NHL and CLL treated with GAZYVA in combination with chemotherapy, including during Cycle 1.
Monitor all patients frequently for thrombocytopenia and hemorrhagic events, especially during the first cycle and if clinically indicated, evaluate laboratory coagulation parameters [see Warnings and Precautions (5.9)]. In patients with Grade 3 or 4 thrombocytopenia, monitor platelet counts more frequently until resolution and consider dose delays of GAZYVA and chemotherapy or dose reductions of chemotherapy. Transfusion of blood products (i.e., platelet transfusion) may be necessary. Consider withholding concomitant medications that may increase bleeding risk (platelet inhibitors, anticoagulants), especially during the first cycle.
5.9 Disseminated Intravascular Coagulation (DIC)
Fatal and severe DIC has been reported in patients receiving GAZYVA for treatment of follicular lymphoma and chronic lymphocytic leukemia. The majority of DIC cases have involved changes in platelets and laboratory coagulation parameters following the first infusion, with spontaneous resolution usually occurring by Day 8. In some cases, DIC was associated with IRRs, TLS, or both [see Adverse Reactions (6.1)]. In patients with suspected DIC, evaluate potential causes, and monitor coagulation parameters, platelet counts, and for signs and symptoms of bleeding or thrombosis. Manage according to standard guidelines for DIC. Supportive care, including transfusion of blood products and other medical management, may be necessary.
5.10 Immunization
The safety and efficacy of immunization with live or attenuated viral vaccines during or following GAZYVA therapy have not been studied. Immunization with live virus vaccines is not recommended during treatment and until B-cell recovery.
5.11 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animals, GAZYVA can cause B-cell depletion in infants exposed to obinutuzumab in-utero. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception while receiving GAZYVA and for 6 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hepatitis B virus reactivation [see Warnings and Precautions (5.1)]
- Progressive multifocal leukoencephalopathy [see Warnings and Precautions (5.2)]
- Infusion-related reactions [see Warnings and Precautions (5.3)]
- Hypersensitivity reactions including serum sickness [see Warnings and Precautions (5.4)]
- Tumor lysis syndrome [see Warnings and Precautions (5.5)]
- Infections [see Warnings and Precautions (5.6)]
- Neutropenia [see Warnings and Precautions (5.7)]
- Thrombocytopenia [see Warnings and Precautions (5.8)]
- Disseminated intravascular coagulation [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Chronic Lymphocytic Leukemia
The data below are based on a safety population of 773 previously untreated patients with CLL in the CLL11 study. Patients were treated with chlorambucil alone, GAZYVA in combination with chlorambucil, or rituximab product in combination with chlorambucil. The Stage 1 analysis compared GAZYVA in combination with chlorambucil vs. chlorambucil alone and Stage 2 compared GAZYVA in combination with chlorambucil vs. rituximab product in combination with chlorambucil. Adverse reactions rates and laboratory abnormalities from the Stage 2 phase are presented below and are consistent with the rates in Stage 1. In addition to the adverse reactions observed in Stage 2, in Stage 1, back pain (5% vs. 2%), anemia (12% vs. 10%) and cough (10% vs. 7%) were observed at a higher incidence in the GAZYVA treated patients. The incidence of Grade 3 to 4 back pain (< 1% vs. 0%), cough (0% vs. < 1%) and anemia (5% vs. 4%) was similar in both treatment arms. With regard to laboratory abnormalities, in Stage 1 hyperkalemia (33% vs. 18%), creatinine increased (30% vs. 20%) and alkaline phosphatase increased (18% vs. 11%) were observed at a higher incidence in patients treated with GAZYVA with similar incidences of Grade 3 to 4 abnormalities between the two arms.
Patients received three 1,000 mg doses of GAZYVA on the first cycle and a single dose of 1,000 mg once every 28 days for 5 additional cycles in combination with chlorambucil (6 cycles of 28 days each in total). In the last 140 patients enrolled, the first dose of GAZYVA was split between day 1 (100 mg) and day 2 (900 mg) [see Dosage and Administration (2.2)]. In total, 81% of patients received all 6 cycles (of 28 days each) of GAZYVA-based therapy.
Adverse reactions in ≥ 10% of patients in the GAZYVA containing arm were infusion-related reactions, neutropenia, thrombocytopenia, and diarrhea. The most common Grade 3 to 4 adverse reactions (incidence ≥ 10%) in the GAZYVA containing arm were neutropenia, infusion-related reactions, and thrombocytopenia.
Table 5 Adverse Reactions (Incidence ≥ 5% and ≥ 2% Greater in the GAZYVA Arm) in Patients with CLL (Stage 2) Body System
Adverse ReactionsGAZYVA + Chlorambucil
n = 336Rituximab product + Chlorambucil
n = 321All Grades
%Grades 3 to 4
%All Grades
%Grades 3 to 4
%- *
- Adverse reactions reported under "Blood and lymphatic system disorders" reflect those reported by investigator as clinically significant.
Injury, Poisoning and Procedural Complications Infusion-Related Reaction 66 20 38 4 Blood and Lymphatic System Disorders* Neutropenia 38 33 32 28 Thrombocytopenia 14 10 7 3 Gastrointestinal Disorders Diarrhea 10 2 8 < 1 Constipation 8 0 5 0 General Disorders and Administration Site Conditions Pyrexia 9 < 1 7 < 1 Infections and Infestations Nasopharyngitis 6 < 1 3 0 Urinary Tract Infection 5 1 2 < 1 Table 6 Post-Baseline Laboratory Abnormalities (Incidence ≥ 10% and ≥ 2% Greater in the GAZYVA Arm) in Patients with CLL (Stage 2) Laboratory Abnormalities GAZYVA + Chlorambucil
n = 336Rituximab product + Chlorambucil
n = 321All Grades % Grades 3 to 4 % All Grades % Grades 3 to 4 % Hematology Leukopenia 84 35 62 16 Lymphopenia 80 39 50 16 Neutropenia 76 46 69 41 Thrombocytopenia 48 13 40 8 Anemia 39 10 37 10 Chemistry Hypocalcemia 37 3 32 < 1 ALT increased 28 2 21 1 AST increased 27 2 21 < 1 Hyponatremia 26 7 18 2 Hypoalbuminemia 23 < 1 16 < 1 Hypokalemia 14 1 10 < 1 Non-Hodgkin Lymphoma
GADOLIN
The GADOLIN study evaluated safety in 407 patients with relapsed or refractory NHL, including FL (81%), small lymphocytic lymphoma and marginal zone lymphoma (a disease for which GAZYVA is not indicated), who did not respond to or progressed within 6 months of treatment with rituximab product or a rituximab product-containing regimen. In the population of patients with FL, the profile of adverse reactions was consistent with the overall NHL population. Patients received either GAZYVA in combination with bendamustine (204 patients), followed by GAZYVA monotherapy in patients that had not progressed, or bendamustine alone (203 patients).
Patients randomized to the GAZYVA + bendamustine arm received three weekly 1,000 mg doses of GAZYVA in the first cycle and a single dose of 1,000 mg once every 28 days for 5 additional cycles, in combination with bendamustine 90 mg/m2 intravenously on Days 1 and 2 in all 6 cycles. Patients who did not progress on the combination received a single 1,000 mg dose of GAZYVA monotherapy every two months until progression or for a maximum of two years. The control arm received bendamustine 120 mg/m2 on Days 1 and 2 of each cycle for 6 cycles, with a cycle length of 28 days. In the GAZYVA arm, 78% of patients received 6 cycles of bendamustine and 82% received their full 6 cycles of GAZYVA; 72 (46%) of the 158 patients who began GAZYVA monotherapy received all planned doses. In the control arm, 72% of patients received 6 cycles of bendamustine.
Serious adverse reactions occurred in 45% of the GAZYVA arm and 37% of the bendamustine-only arm. Fatal adverse reactions within 90 days of treatment occurred in 3.4% and 2.5%, respectively. During treatment and follow-up combined, fatal adverse reactions occurred in 10% of GAZYVA recipients and in 7.4% of recipients of bendamustine alone, with infection and second primary malignancies being the leading causes.
Dose modification due to adverse reactions occurred in 50% of the GAZYVA arm and 42% of the control arm, and discontinuation of any study drug due to adverse reactions occurred in 20% and 17%, respectively.
Table 7 presents selected adverse reactions in GADOLIN. The most common adverse reactions (incidence ≥ 20%) in GAZYVA recipients included infusion-related reactions, fatigue, neutropenia, cough, upper respiratory tract infections, and musculoskeletal pain.
Table 7 Adverse Reactions (Incidence ≥ 10% and ≥ 2% Greater in the GAZYVA Arm) in Patients with Relapsed or Refractory NHL (GADOLIN) Body System
Adverse Reactions*, †GAZYVA + Bendamustine followed by GAZYVA monotherapy
n = 204Bendamustine
n = 203All Grades
%Grades 3 to 5
%All Grades
%Grades 3 to 5
%- *
- Includes adverse reactions reported throughout study treatment and follow-up.
- †
- Includes grouped terms.
- ‡
- Except where noted, individual events that meet the definition of "infusion-related reaction" are excluded from Table 7 above, as they are included in the grouped term "Infusion-Related Reaction".
- §
- Includes 1 fatal event.
Procedural Complications Infusion-Related Reaction‡ 67 11 63 5 General Disorders Fatigue 40 3 36 3 Pyrexia 19 1 15 1 Blood and Lymphatic System Disorders Neutropenia 37 35§ 29 27 Infections and Infestations Upper Respiratory Tract Infection 36 3 23 1 Respiratory Tract Infection, Unspecified 14 1 8 0 Urinary Tract Infection 13 3 7 0 Respiratory, Thoracic and Mediastinal Disorders Cough 31 <1 21 0 Musculoskeletal and Connective Tissue Disorders Musculoskeletal Pain 28 1 20 0 Arthralgia 12 <1 5 0 Skin and Subcutaneous Tissue Disorders Rash 17 <1 14 <1 Pruritus 11 0 6 0 Infusion-related reactions are defined as any related adverse reaction that occurred during or within 24 hours of infusion.
Fatigue includes fatigue, lethargy, asthenia.
Pyrexia includes pyrexia, hyperthermia, body temperature increased.
Cough includes cough, productive cough, upper-airway cough syndrome.
Neutropenia includes neutropenia, agranulocytosis, granulocytopenia, neutrophil count decreased.
Upper respiratory tract infection includes upper respiratory tract congestion, upper respiratory tract inflammation, upper respiratory fungal infection, rhinovirus infection, and all terms containing: upper respiratory tract infection, laryngitis, nasopharyngitis, pharyngitis, rhinitis, tonsillitis, and sinusitis with the exception of sinobronchitis.
Respiratory tract infection unspecified includes respiratory tract infection, respiratory tract infection viral, influenza, influenza-like illness, sinobronchitis, respiratory syncytial virus infection.
Urinary tract infection includes all terms containing: urinary tract infection, cystitis, pyelonephritis.
Musculoskeletal pain includes non-cardiac chest pain, bone pain, spinal pain, myalgia, back pain, neck pain, musculoskeletal discomfort, pain in extremity, and all terms containing "musculoskeletal pain".
Rash includes drug eruption, skin reaction, all terms containing "rash", urticaria, and selected terms containing "dermatitis".
Pruritus includes pruritus, pruritus generalized.
Other clinically relevant adverse reactions (incidence < 10% and ≥ 2% greater in the GAZYVA arm) included:
- Blood and lymphatic system disorders: febrile neutropenia (6%)
- Infection: sepsis (7%)
During GAZYVA monotherapy (158 patients), adverse reactions in ≥ 10% of patients included upper and lower respiratory tract infections, cough, neutropenia, musculoskeletal pain, fatigue, diarrhea, rash, and urinary tract infection.
Table 8 presents selected new or worsening laboratory abnormalities in the GADOLIN trial.
Table 8 New or Worsening Laboratory Abnormalities (Incidence ≥ 10% and ≥ 2% Greater in the GAZYVA Arm*) in Patients with Relapsed or Refractory NHL (GADOLIN) Laboratory Abnormalities GAZYVA + Bendamustine followed by GAZYVA monotherapy
n = 204Bendamustine
n = 203All Grades % Grades 3 to 4 % All Grades % Grades 3 to 4 % - *
- Two percent difference in either any-grade or Grade 3 to 4 laboratory abnormalities.
Hematology Lymphopenia 97 92 96 84 Leukopenia 84 47 87 34 Neutropenia 76 53 75 42 Chemistry Hypophosphatemia 41 8 38 7 Hypocalcemia 39 3 24 1 ALT/SGPT increased 36 2 31 3 Alkaline phosphatase increased 27 0 23 0 Hyperbilirubinemia 21 2 17 2 Hyperkalemia 20 3 18 0 In the GAZYVA monotherapy phase, new or worsening grade 3 or 4 abnormalities included neutropenia in 25% of patients (Grade 4, 10%) and lymphopenia in 23% (Grade 4, 5%).
GALLIUM
A randomized, open-label multicenter trial (GALLIUM) evaluated the safety of GAZYVA as compared to rituximab product in 1385 patients with previously untreated follicular lymphoma (86%) or marginal zone lymphoma (14%). Patients received chemotherapy (bendamustine, CHOP, or CVP) combined with either GAZYVA (691 patients) or rituximab product (694 patients), followed in responding patients by GAZYVA or rituximab product monotherapy every two months until disease progression or for a maximum of two years. The study excluded patients having an absolute neutrophil count (ANC) < 1500 / µL, platelets < 75,000 / µL, CLcr < 40 mL/min and, unless attributable to lymphoma, hepatic transaminases > 2.5 × upper limit of normal.
The median age was 60 (range: 23-88), 47% were male, 82% were white, and 97% had an ECOG performance status of 0 or 1. The chemotherapy was bendamustine in 59%, CHOP in 31% and CVP in 10% of patients. Following combination therapy, 624 patients (90%) in the GAZYVA arm and 612 patients (88%) in the rituximab product arm received monotherapy.
Serious adverse reactions occurred in 50% of patients on the GAZYVA arm and 43% of patients on the rituximab product arm. Fatal adverse reactions were reported during treatment in 3% in the GAZYVA arm and 2% in the rituximab product arm, most often from infections in the GAZYVA arm. During treatment and follow-up combined, fatal adverse reactions were reported in 5% of the GAZYVA arm and 4% of the rituximab product arm, with infections and second malignancies being leading causes. In the GAZYVA arm, fatal infections occurred in 2% of patients compared to < 1% in the rituximab product arm.
During combination therapy, 93% of patients received all treatment cycles in the GAZYVA arm, and 92% received all treatment cycles in the rituximab product arm. Of the responding patients who began monotherapy with GAZYVA or rituximab product, 76% and 73%, respectively, completed the full course. Dose modification due to adverse reactions occurred in 74% of the GAZYVA arm and 63% of the rituximab product arm throughout study treatment, and discontinuation of any study drug due to adverse reactions occurred in 18% and 15%, respectively.
Throughout treatment and follow-up, the most common adverse reactions (incidence ≥ 20%) observed at least 2% more in the GAZYVA arm included infusion-related reactions, neutropenia, upper respiratory tract infections, constipation and diarrhea (Table 9). Neutropenia, infusion-related reactions, febrile neutropenia and thrombocytopenia were the most common Grade 3 to 5 adverse reactions (incidence ≥ 5%) observed more frequently in the GAZYVA arm.
Table 9 Adverse Reactions (Incidence ≥ 10% and ≥ 2% Greater in the GAZYVA Arm) in Patients with Previously Untreated NHL (GALLIUM) Body System
Adverse Reactions *, †GAZYVA + chemotherapy followed by GAZYVA monotherapy
n = 691Rituximab product + chemotherapy followed by rituximab product monotherapy
n = 694All Grades
%Grades 3 to 5
%All Grades
%Grades 3 to 5
%- *
- Includes adverse reactions reported throughout study treatment and follow-up.
- †
- Includes grouped terms.
- ‡
- Except where noted, individual events that meet the definition of "infusion-related reaction" are excluded from Table 9 above, as they are already included in the grouped term "Infusion-Related Reaction". The most common individual terms within the grouped term "Infusion-Related Reaction" in decreasing order of frequency are nausea, chills, pyrexia and vomiting.
- §
- Includes adverse reactions reported as infusion-related reactions.
Injury, Poisoning and Procedural Complications Infusion-Related Reaction ‡ 72 12 60 8 Blood and Lymphatic System Disorders Neutropenia § 53 49 47 41 Thrombocytopenia § 14 7 8 3 Infections and Infestations Upper Respiratory Tract Infection 50 3 43 1 Herpesvirus Infection 18 3 14 1 Pneumonia 14 7 12 6 Respiratory, Thoracic and Mediastinal Disorders Cough 35 < 1 28 < 1 Gastrointestinal Disorders Constipation 32 < 1 29 < 1 Diarrhea 30 3 26 2 Nervous System Disorders Headache 18 < 1 15 < 1 Musculoskeletal and Connective Tissue Disorders Arthralgia 16 0 14 < 1 Psychiatric Disorders Insomnia 15 < 1 12 < 1 Metabolism and Nutrition Disorders Decreased Appetite 14 < 1 12 < 1 Skin and Subcutaneous Tissue Disorders Pruritus 11 < 1 9 0 Infusion-related reactions are defined as any related adverse reaction that occurred during or within 24 hours of infusion.
Neutropenia includes neutropenia, agranulocytosis, granulocytopenia, and neutrophil count decreased.
Febrile neutropenia includes febrile neutropenia, neutropenic infection, neutropenic sepsis, and febrile bone marrow aplasia.
Thrombocytopenia includes thrombocytopenia and platelet count decreased.
Upper respiratory tract infection includes upper respiratory tract congestion, upper respiratory tract inflammation, upper respiratory tract infection, rhinovirus infection, and all terms containing: laryngitis, nasopharyngitis, pharyngitis, rhinitis, tonsillitis, and sinusitis with the exception of sinobronchitis.
Herpesvirus infection includes all terms containing "herpes" or "varicella."
Pneumonia includes all terms containing "pneumonia," bacterial, pneumonia haemophilus, pneumonia pneumococcal, pneumonia fungal, pneumocystis jirovecii infection, lung infection, and lung infiltration.
Diarrhea includes diarrhea, defecation urgency, frequent bowel movement, and all terms containing "gastroenteritis".
Headache includes all terms containing "headache" and migraine.
Insomnia includes all terms containing "insomnia" and sleep disorder.
Pruritus includes pruritus and pruritus generalized.
During the monotherapy period, the common adverse reactions (incidence ≥ 10%) observed at least 2% more with GAZYVA were upper respiratory tract infection (40%), cough (23%), musculoskeletal pain (20%), neutropenia (19%), and herpesvirus infection (13%).
Table 10 summarizes treatment-emergent laboratory abnormalities during treatment and follow-up. The Grade 3 to 4 abnormalities reported at least 2% more in the GAZYVA arm were lymphopenia, leukopenia, neutropenia, thrombocytopenia, and hyperuricemia. Patients in the GAZYVA arm, as compared to the rituximab product arm, had higher incidences of Grade 4 neutropenia (38% vs. 30%, respectively), Grade 4 lymphopenia (33% vs. 22%), and Grade 4 leukopenia (17% vs. 12%).
Table 10 New or Worsening Laboratory Abnormalities (Incidence ≥ 10% and ≥ 2% Greater in the GAZYVA Arm) in Patients with Previously Untreated NHL (GALLIUM) Laboratory Abnormalities * GAZYVA+ chemotherapy followed by GAZYVA monotherapy
n = 691Rituximab product + chemotherapy followed by rituximab product monotherapy
n = 694All Grades % Grades 3 to 4 % All Grades % Grades 3 to 4 % - *
- Includes lab abnormalities, reported throughout treatment and follow-up, that were new or worsening, or worsening from baseline unknown.
Hematology Lymphopenia 97 83 95 67 Leukopenia 92 49 89 39 Neutropenia 84 59 76 50 Thrombocytopenia 68 11 50 4 Chemistry ALT increased 50 3 43 2 AST increased 44 1 41 1 Hypophosphatemia 36 5 33 5 Hypoalbuminemia 33 1 25 1 Hypoproteinemia 32 0 30 0 Hypocalcemia 32 1 26 1 Hyperuricemia 28 28 22 22 Hyponatremia 26 4 20 3 Hyperkalemia 23 1 17 1 Hypernatremia 16 < 1 13 0 In the monotherapy phase, new-onset Grade 3 or 4 neutropenia was reported in 21% of patients in the GAZYVA arm (Grade 4, 10%) and 17% of patients in the rituximab product arm (Grade 4, 9%).
GAZELLE
GAZELLE (NCT03817853) is a single-arm study designed to characterize the safety of GAZYVA administered as a shortened-duration infusion (approximately 90 minutes) in patients with previously untreated FL. All patients received GAZYVA at the standard infusion rate with premedication during Cycle 1. If no Grade 3 or higher IRR occurred with any infusion during Cycle 1, GAZYVA was administered over approximately 90 minutes in Cycle 2 and subsequent cycles. The primary safety measure was the proportion of patients who experienced Grade 3 or higher IRRs with the 90-minute infusion at Cycle 2. GAZYVA was administered in combination with CHOP, CVP or bendamustine for 6 to 8 cycles (induction), followed by monotherapy for up to 2 years.
Of the 113 patients treated with GAZYVA, 99 (88%) received the 90-minute infusion starting in Cycle 2. In total, 97% of patients who received GAZYVA at either a standard or shorter infusion duration received premedication in Cycle 2. IRRs were observed in 63% of patients throughout induction (including IRRs observed after standard-duration infusion). In cycle 1, 58% of patients developed IRRs with the standard infusion rate (Grade ≥3 IRR, 5%). Of the patients who received the 90-minute infusion, 10% had IRRs of any grade in Cycle 2, with 8% and 2% of patients having a Grade 1 IRR or Grade 2 IRR, respectively. Following Cycle 2, one (1%) patient experienced a Grade 3 IRR, which occurred after the 90-minute infusion at Cycle 5.
Infusion-Related Reactions
Chronic Lymphocytic Leukemia
The incidence of IRRs in the CLL11 study was 65% with the first infusion of GAZYVA. The incidence of Grade 3 or 4 IRRs was 20% with 7% of patients discontinuing therapy. The incidence of reactions with subsequent infusions was 3% with the second 1,000 mg and < 1% thereafter. No Grade 3 or 4 IRRs were reported beyond the first 1,000 mg infused.
Of the first 53 patients receiving GAZYVA in CLL11, 47 (89%) experienced an IRR. After this experience, study protocol modifications were made to require pre-medication with a corticosteroid, antihistamine, and acetaminophen. The first dose was also divided into two infusions (100 mg on day 1 and 900 mg on day 2). For the 140 patients for whom these mitigation measures were implemented, 74 patients (53%) experienced a reaction with the first 1,000 mg (64 patients on day 1, 3 patients on day 2, and 7 patients on both days) and < 3% thereafter [see Dosage and Administration (2.2)].
Non-Hodgkin Lymphoma
Overall, 67% of patients in the GADOLIN study experienced an IRR (all grades) during treatment with GAZYVA in combination with bendamustine. The incidence of Grade 3 to 4 IRRs in GADOLIN was 11%. In Cycle 1, the incidence of IRRs (all grades) was 53% in patients receiving GAZYVA in combination with bendamustine of which 34 (9%) were Grade 3 to 4 in severity. In patients receiving GAZYVA in combination with bendamustine, the incidence of IRRs was highest on Day 1 (37%), and gradually decreased on Days 2, 8 and 15 (23%, 6% and 4%, respectively).
During Cycle 2, the incidence of IRRs was 24% in patients receiving GAZYVA in combination with bendamustine and decreased with subsequent cycles.
During GAZYVA monotherapy in GADOLIN, IRRs (all grades) were observed in 8% of patients. One Grade 3 and no Grade 4 IRRs were reported during GAZYVA monotherapy.
Overall, 2% of patients in GADOLIN experienced an IRR leading to discontinuation of GAZYVA.
In GALLIUM, 72% of patients in the GAZYVA treated arm experienced an IRR (all grades). The incidence of Grade 3 to 4 IRRs for these patients was 12%. In Cycle 1, the incidence of IRRs (all grades) was 62% in the GAZYVA treated arm with Grade 3 to 4 IRRs reported in 10%. The incidence of IRRs (all grades) was highest on Day 1 (60%) and decreased on Days 8 and 15 (9% and 6%, respectively).
During Cycle 2, the incidence of IRRs (all grades) in the GAZYVA treated arm was 13% and decreased with subsequent cycles.
During GAZYVA monotherapy treatment in GALLIUM, IRRs (all grades) were observed in 9% of patients.
Overall, 1% of patients in GALLIUM experienced an IRR leading to discontinuation of GAZYVA.
In GAZELLE, 10% of patients with FL experienced IRRs of any grade at Cycle 2 when GAZYVA was administered over approximately 90 minutes.
Neutropenia
Chronic Lymphocytic Leukemia
The incidence of neutropenia reported as an adverse reaction in CLL11 was 38% in the GAZYVA treated arm and 32% in the rituximab product treated arm, with the incidence of serious adverse reactions being 1% and < 1%, respectively (Table 5). Cases of late-onset neutropenia (occurring 28 days after completion of treatment or later) were 16% in the GAZYVA treated arm and 12% in the rituximab product treated arm.
Non-Hodgkin Lymphoma
The incidence of neutropenia in GADOLIN was higher in the GAZYVA plus bendamustine arm (37%) compared to the arm treated with bendamustine alone (30%). Cases of prolonged neutropenia (3%) and late onset neutropenia (8%) were also reported in the GAZYVA plus bendamustine arm. The incidence of neutropenia was higher during treatment with GAZYVA in combination with bendamustine (30%) compared to the GAZYVA monotherapy treatment phase (13%).
The incidence of neutropenia in GALLIUM was higher in the GAZYVA treated arm (53%) compared to the rituximab product treated arm (47%). Cases of prolonged neutropenia (1%) and late onset neutropenia (4%) were also reported in the GAZYVA treated arm. The incidence of neutropenia was higher during treatment with GAZYVA in combination with chemotherapy (45%) compared to the GAZYVA monotherapy treatment phase (20%).
Infection
Chronic Lymphocytic Leukemia
The incidence of infections was similar between GAZYVA and rituximab product treated arms. Thirty-eight percent of patients in the GAZYVA treated arm and 37% in the rituximab product treated arm experienced an infection, with Grade 3 to 4 rates being 11% and 13%, respectively. Fatal events were reported in 1% of patients in both arms.
Non-Hodgkin Lymphoma
The incidence of infection in GADOLIN was 68% in the GAZYVA plus bendamustine arm and 59% in the bendamustine arm, with Grade 3 to 4 events reported in 20% and 16%, respectively. Fatal events were reported in 3% of patients in the GAZYVA plus bendamustine arm and 3% in the bendamustine arm.
The incidence of infections in GALLIUM was 82% in the GAZYVA treated arm and 73% in the rituximab product treated arm, with Grade 3 to 4 events reported in 21% and 17%, respectively. In the GAZYVA arm, fatal infections occurred in 2% of patients compared to <1% in the rituximab product arm.
The incidence of Grade 3 to 4 infections in the GAZYVA and rituximab product treated arms was lower in patients receiving GCSF prophylaxis (14%; 16%) compared with patients not receiving GCSF prophylaxis (24%; 18%). The incidence of fatal infections in patients receiving GCSF prophylaxis in the GAZYVA and rituximab product treated arms was 2% and 0%, respectively, and was 2% and < 1% in patients not receiving GCSF prophylaxis.
Thrombocytopenia
Chronic Lymphocytic Leukemia
The overall incidence of thrombocytopenia reported as an adverse reaction was higher in the GAZYVA treated arm (14%) compared to the rituximab product treated arm (7%), with the incidence of Grade 3 to 4 events being 10% and 3%, respectively (Table 5). The difference in incidences between the treatment arms is driven by events occurring during the first cycle. The incidence of thrombocytopenia (all grades) in the first cycle was 11% in the GAZYVA and 3% in the rituximab product treated arms, with Grade 3 to 4 rates being 8% and 2%, respectively. Four percent of patients in the GAZYVA treated arm experienced acute thrombocytopenia (occurring within 24 hours after the GAZYVA infusion).
The overall incidence of hemorrhagic events and the number of fatal hemorrhagic events were similar between the treatment arms, with 3 in the rituximab product and 4 in the GAZYVA treated arms. However, all fatal hemorrhagic events in patients treated with GAZYVA occurred in Cycle 1.
Non-Hodgkin Lymphoma
The incidence of thrombocytopenia in GADOLIN was lower in the GAZYVA plus bendamustine arm (15%) compared to the arm treated with bendamustine alone (25%). The incidence of hemorrhagic events in GAZYVA plus bendamustine treated patients compared to bendamustine alone was 12% and 11%, respectively. Grade 3 to 4 hemorrhagic events were similar in both treatment arms (4% in the GAZYVA plus bendamustine arm and 2% in the bendamustine arm).
In GALLIUM, thrombocytopenia was reported as an adverse reaction in 14% of the GAZYVA treated arm and 8% of the rituximab product treated arm, with the incidence of Grade 3 to 4 events being 7% and 3%, respectively. The difference in incidences between the treatment arms is driven by events occurring during the first cycle. The incidence of thrombocytopenia (all grades) in the first cycle was 9% in the GAZYVA and 3% in the rituximab product treated arms, with Grade 3 to 4 rates being 5% and 1%, respectively. In GALLIUM, both treatment arms had a 12% overall incidence of hemorrhagic events and a < 1% incidence of fatal hemorrhagic events.
Disseminated Intravascular Coagulation
In GALLIUM, DIC was reported as an adverse reaction in 0.3% of the GAZYVA treated patients. All events occurred within 1-2 days after the first infusion.
Tumor Lysis Syndrome
The incidence of Grade 3 or 4 tumor lysis syndrome in GAZYVA treated patients was 2% in CLL11, 0.5% in GADOLIN and 0.9% in GALLIUM.
Musculoskeletal Disorders
Chronic Lymphocytic Leukemia
Adverse reactions related to musculoskeletal disorders (all events from the body system), including pain, have been reported in the GAZYVA treated arm with higher incidence than in the rituximab product treated arm (18% vs. 15%).
Non-Hodgkin Lymphoma
In GADOLIN, adverse reactions related to musculoskeletal disorders (all events from the body system), including pain, have been reported in the GAZYVA plus bendamustine treated arm with higher incidence than in the bendamustine alone arm (44% vs. 30%).
In GALLIUM, musculoskeletal disorders were reported in 54% of patients in the GAZYVA treated arm and 49% of patients in the rituximab product treated arm.
Liver Enzyme Elevations
Hepatic enzyme elevations have occurred in CLL patients who received GAZYVA in clinical trials and had normal baseline hepatic enzyme levels (AST, ALT and ALP). The events occurred most frequently within 24–48 hours of the first infusion. In some patients, elevations in liver enzymes were observed concurrently with IRRs or tumor lysis syndrome. In the CLL11 study, there was no clinically meaningful difference in overall hepatotoxicity adverse reactions between all arms (4% of patients in the GAZYVA treated arm). Medications commonly used to prevent IRRs (e.g., acetaminophen) may also be implicated in these events. Monitor liver function tests during treatment, especially during the first cycle. Consider treatment interruption or discontinuation for hepatotoxicity.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Seven percent (18/271) of patients with CLL tested positive for anti-GAZYVA antibodies at one or more time points in CLL11. No patients developed anti-GAZYVA antibodies during or following GAZYVA treatment in GADOLIN, while 1 patient (1/564, 0.2%) developed anti-GAZYVA antibodies in GALLIUM. Neutralizing activity of anti-GAZYVA antibodies has not been assessed.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of GAZYVA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Immune/Autoimmune Events: Serum sickness
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, GAZYVA can cause fetal B-cell depletion [see Clinical Pharmacology (12.1)]. There are no data with GAZYVA use in pregnant women to inform a drug-associated risk. Monoclonal antibodies are transferred across the placenta. In animal reproduction studies, weekly intravenous administration of obinutuzumab to pregnant cynomolgus monkeys from day 20 of pregnancy until parturition which includes the period of organogenesis at doses with exposures up to 2.4 times the exposure at the clinical dose of 1,000 mg monthly produced opportunistic infections and immune complex mediated hypersensitivity reactions. No embryo-toxic or teratogenic effects were observed in the monkeys (see Data). Advise pregnant women of the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, the estimated background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
GAZYVA is likely to cause fetal B-cell depletion (see Data). Avoid administering live vaccines to neonates and infants exposed to GAZYVA in utero until B-cell recovery occurs [see Warnings and Precautions (5.11) and Clinical Pharmacology (12.2)].
Data
Animal Data
In a pre- and post-natal development study, pregnant cynomolgus monkeys received weekly intravenous doses of 25 or 50 mg/kg obinutuzumab from day 20 of pregnancy until parturition, which includes the period of organogenesis. The high dose results in an exposure (AUC) that is 2.4 times the exposure in patients with CLL at the recommended label dose. There were no embryo-toxic or teratogenic effects in animals. Secondary opportunistic infections, immune complex mediated hypersensitivity reactions, or a combination of both were observed in exposed dams. When first measured on day 28 postpartum, obinutuzumab was detected in offspring at levels in the range of maternal serum levels on the same day, and B-cells were completely depleted. The B-cell counts returned to normal levels, and immunologic function was restored within 6 months after birth.
Obinutuzumab was measured in the milk of lactating cynomolgus monkeys on day 28 postpartum after weekly intravenous administration from day 20 of pregnancy until parturition. Concentrations in milk were approximately 0.04% and 0.13% of concentrations in maternal serum in the 25 and 50 mg/kg groups, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of GAZYVA in human milk, the effects on the breastfed child, or the effects on milk production. However, low levels of obinutuzumab were present in the milk of lactating cynomolgus monkeys [see Use in Specific Populations (8.1)]. Human IgG is known to be present in human milk. Because of the potential of serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with GAZYVA and for 6 months after the last dose.
8.3 Females and Males of Reproductive Potential
GAZYVA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of GAZYVA in pediatric patients have not been established.
8.5 Geriatric Use
Chronic Lymphocytic Leukemia
Of 336 patients with previously untreated CLL who received GAZYVA in combination with chlorambucil, 81% were 65 years and older, while 46% were 75 and older. Of the patients 75 years and older, 46% experienced serious adverse reactions and 7% experienced adverse reactions leading to death. Of the patients younger than 75, 33% experienced a serious adverse reaction and 2% an adverse reaction leading to death. No significant differences in efficacy were observed between younger and older patients [see Clinical Studies (14.1)].
Non-Hodgkin Lymphoma
Of 204 patients in GADOLIN with relapsed or refractory NHL treated with GAZYVA plus bendamustine, 44% were 65 and over, while 14% were 75 and over. In patients 65 and over, 55% of patients experienced serious adverse reactions and 28% experienced adverse reactions leading to treatment withdrawal while in patients under 65, 37% and 14% experienced serious adverse reactions and adverse reactions leading to treatment withdrawal, respectively. No clinically meaningful differences in efficacy were observed between these patients and younger patients in GADOLIN.
Of the 691 patients in GALLIUM treated with GAZYVA plus chemotherapy as first-line therapy, 33% were 65 and over, while 7% were 75 and over. Of patients 65 and over, 63% experienced serious adverse reactions and 26% experienced adverse reactions leading to treatment withdrawal, while in patients under 65, 43% experienced serious adverse reactions and 13% had an adverse reaction leading to treatment withdrawal. No clinically meaningful differences in efficacy were observed between these patients and younger patients in GALLIUM.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Obinutuzumab is a humanized anti-CD20 monoclonal antibody of the IgG1 subclass. It recognizes a specific epitope of the CD20 molecule found on B cells. The molecular mass of the antibody is approximately 150 kDa.
GAZYVA (obinutuzumab) injection is produced by mammalian cell (CHO) suspension culture. GAZYVA was engineered for reduced fucose content as compared to a typical IgG1 produced in CHO cells. GAZYVA is a sterile, clear, colorless to slightly brown, preservative-free liquid concentrate for intravenous use. GAZYVA is supplied at a concentration of 25 mg/mL in 1,000 mg single-dose vials. The product is formulated in 20 mM L-histidine/L-histidine hydrochloride, 240 mM trehalose, 0.02% poloxamer 188. The pH is 6.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Obinutuzumab is a monoclonal antibody that targets the CD20 antigen expressed on the surface of pre-B and mature B lymphocytes. Upon binding to CD20, obinutuzumab mediates B-cell lysis through (1) engagement of immune effector cells, (2) by directly activating intracellular death signaling pathways (direct cell death), and/or (3) activation of the complement cascade. The immune effector cell mechanisms include antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis.
As an antibody with reduced fucose content, obinutuzumab induces greater ADCC activity than rituximab in vitro using human cancer cell lines. Obinutuzumab also demonstrated an increased ability to induce direct cell death when compared to rituximab. Obinutuzumab binds to FcγRIII using purified proteins with a higher affinity than rituximab. Obinutuzumab and rituximab bind with similar affinity to overlapping epitopes on CD20.
12.2 Pharmacodynamics
In patients with CLL, GAZYVA caused CD19 B-cell depletion (defined as CD19 B cell counts < 0.07 × 109/L). Initial CD19 B cell recovery was observed in some patients approximately 9 months after the last GAZYVA dose. At 18 months of follow-up, some patients remain B cell depleted.
Although the depletion of B cells in the peripheral blood is a measurable pharmacodynamic effect, it is not directly correlated with the depletion of B-cells in solid organs or in malignant deposits. B cell depletion has not been shown to be directly correlated to clinical response.
12.3 Pharmacokinetics
The pharmacokinetic parameters of obinutuzumab after 100 mg on day 1 and 900 mg on day 2 of Cycle 1, 1,000 mg on day 8 and 15 of Cycle 1, and 1,000 mg on day 1 of Cycles 2–6 for CLL and after 1,000 mg on day 1, 8 and 15 of Cycle 1, 1,000 mg on day 1 of Cycles 2-6 or Cycles 2-8, and then 1,000 mg every 2 months for up to 2 years for NHL are provided in Table 11. The dosing regimen is within the linear pharmacokinetic behavior of obinutuzumab.
Table 11 Obinutuzumab Measures of Exposure PK Measure CLL* Relapsed or refractory FL* First line FL in combination with chemotherapy GAZYVA + Bendamustine* GAZYVA + CHOP or CVP† Results are presented as geometric mean (% Coefficient of Variation). Cmax, µg/mL 466.3 (35) 553.5 (32) 513.4 (28) 676.4 (30) Ctrough, µg/mL 192.5 (78) 295 (56) 255 (46) 395 (44) AUC, µg/mL*day 8701 (51) 11362 (41) 10088 (35) 10723 (37) Distribution and Elimination
The elimination of obinutuzumab is comprised of a linear clearance pathway and a time-dependent non-linear clearance pathway. As GAZYVA treatment progresses, the impact of the time-dependent pathway diminishes in a manner suggesting target-mediated drug disposition (TMDD) and saturation of the TMDD at the end of the treatment cycle at the proposed clinical dose regimen. The pharmacokinetic properties of obinutuzumab in patients with CLL and NHL are provided in Table 12.
Specific Populations
Age (median [range]: 63 [22, 89] years) and baseline creatinine clearance (CLcr) (median [range] 84 [22, > 120] mL/min) did not affect the pharmacokinetics of GAZYVA. In patients with CLcr ≤ 30 mL/min, the pharmacokinetics of GAZYVA was unaffected. GAZYVA has not been studied in patients with hepatic impairment.
The volume of distribution and steady-state clearance increased with body weight; however, the expected change in exposure does not warrant a dose modification.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or genotoxicity studies have been conducted with obinutuzumab.
No specific studies have been conducted to evaluate potential effects on fertility; however, no adverse effects on male or female reproductive organs were observed in the 26-week repeat-dose toxicity study in cynomolgus monkeys.
-
14 CLINICAL STUDIES
14.1 Chronic Lymphocytic Leukemia
The efficacy of GAZYVA was evaluated in a three-arm, open-label, active-controlled, randomized, multicenter trial (CLL11; NCT01010061) in 781 patients with previously untreated CD20+ CLL requiring treatment who had coexisting medical conditions or reduced renal function as measured by creatinine clearance (CLcr) < 70 mL/min. Patients with CLcr < 30 mL/min, active infections, positive hepatitis B (HBsAg or anti-HBc positive; patients positive for anti-HBc could be included if hepatitis B viral DNA was not detectable) and hepatitis C serology, or immunization with live virus vaccine within 28 days prior to randomization were excluded from the trial. Patients were treated with chlorambucil control (Arm 1), GAZYVA in combination with chlorambucil (Arm 2), or rituximab product in combination with chlorambucil (Arm 3). The safety and efficacy of GAZYVA was evaluated in a Stage 1 comparison of Arm 1 vs. Arm 2 in 356 patients and a Stage 2 comparison of Arm 2 vs. Arm 3 in 663 patients.
The majority of patients received 1,000 mg of GAZYVA on days 1, 8 and 15 of the first cycle, followed by treatment on the first day of 5 subsequent cycles (total of 6 cycles, 28 days each). The first dose of GAZYVA was divided between day 1 (100 mg) and day 2 (900 mg) [see Dosage and Administration (2.2)], which was implemented in 140 patients. Chlorambucil was given orally at 0.5 mg/kg on day 1 and day 15 of all treatment cycles (1 to 6).
In CLL11, the median age was 73 years, 62% were male, and 95% were White. Sixty-five percent had a CLcr < 70 mL/min and 76% had multiple coexisting medical conditions. Twenty-two percent of patients were Binet stage A, 42% were stage B, and 36% were stage C. The median estimated CLcr was 62 mL/min. Eighty-one percent of patients treated with GAZYVA in combination with chlorambucil received all 6 cycles compared to 89% of patients in the rituximab product treated arm and 67% in the chlorambucil alone arm.
In the Stage 1 analysis of CLL11, the median progression-free survival (PFS) in the GAZYVA in combination with chlorambucil arm was 27.2 months and 11.2 months in the chlorambucil alone arm (median observation time 22.8 months) as assessed by independent review and is consistent with investigator-assessed PFS. The median overall survival (OS) was not yet reached with a total of 46 deaths: 22 (9%) in the GAZYVA in combination with chlorambucil arm and 24 (20%) in the chlorambucil arm. The hazard ratio for OS was 0.41 (95% CI: 0.23-0.74).
In the Stage 2 analysis of CLL11, the median PFS was 26.7 months in the GAZYVA arm and 14.9 months in the rituximab product arm with a median observation time of 18.7 months (HR: 0.42, 95% CI: 0.33-0.54, p-value < 0.0001). These results were assessed by independent review and are consistent with investigator-assessed PFS. Minimal residual disease (MRD) was evaluated using allele-specific oligonucleotide polymerase chain reaction (ASO-PCR). The cutoff for a negative status was one CLL cell per 104 leukocytes in the sample (i.e., an MRD value of < 10-4 was considered negative). Among patients who achieved complete response (CR) and complete response with incomplete marrow recovery (CRi; 94 patients in the GAZYVA arm and 34 patients in the rituximab product arm), 18 patients (19%) had negative MRD in the bone marrow in the GAZYVA arm compared to 2 patients (6%) in the rituximab product arm. Out of the patients who achieved CR and CRi, 39 patients (41%) in the GAZYVA arm, and 4 patients (12%) in the rituximab product arm were MRD negative in peripheral blood samples collected at least 3 months after the end of treatment.
Efficacy results are shown in Table 13 and Figures 1 and 2.
Table 13 Efficacy Results from CLL11 Endpoint Stage 1 of CLL11 Stage 2 of CLL11 GAZYVA + Chlorambucil* Chlorambucil GAZYVA + Chlorambucil* Rituximab product + Chlorambucil n = 238 n = 118 n = 333 n = 330 Median Progression-Free Survival† 27.2 months 11.2 months 26.7 months 14.9 months (HR 0.19 [0.14; 0.27], p-value < 0.0001 stratified log-rank test) (HR 0.42 [0.33; 0.54], p-value < 0.0001 stratified log-rank test) Overall Response Rate‡ 78.2% 33.1% 79.6% 66.3% Complete Response 28.2% 0 26.1% 8.8% Complete Response with Incomplete Marrow Recovery 2.5% 1.7% 2.1% 1.5% Partial Response 45.0% 30.5% 48.6% 54.1% Nodular Partial Response 2.5% 0.8% 2.7% 1.8% Median Duration of Response 22.4 months 4.7 months 19.6 months 9.7 months Overall Survival HR 0.41 [0.23; 0.74] Not Yet Mature Figure 1
Kaplan-Meier Curve of Overall Survival in Patients with CLL in CLL11 (Stage 1)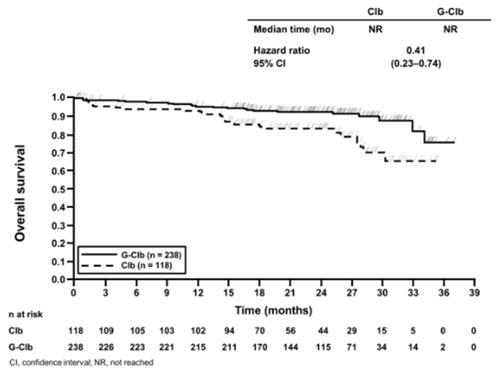
Figure 2
Kaplan-Meier Curve of Progression-Free Survival in Patients with CLL in CLL11 (Stage 2)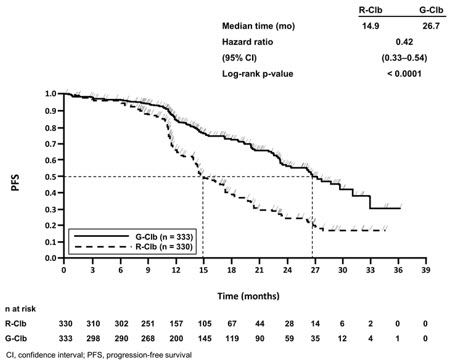
14.2 Follicular Lymphoma
GADOLIN
The efficacy of GAZYVA was evaluated in GADOLIN (NCT01059630), an open-label, multicenter, randomized study that included 335 patients with follicular lymphoma (FL) who had no response to or have progressed during or within 6 months of rituximab product or a rituximab product-containing regimen. These patients were randomized to receive either bendamustine alone (n = 171) or GAZYVA in combination with bendamustine (n = 164) for 6 cycles, each of 28 days duration. Patients in the GAZYVA plus bendamustine arm who did not have disease progression [patients with a complete response (CR), partial response (PR) or stable disease (SD)] at the end of the 6 cycles continued receiving GAZYVA monotherapy for 2 years. Patients were stratified according to the type of refractoriness to rituximab product (refractory to rituximab product monotherapy versus rituximab product in combination with chemotherapy), the number of prior therapies (≤ 2 versus > 2), and geographic region.
GAZYVA was given by intravenous infusion as a flat dose of 1,000 mg on Days 1, 8 and 15 of Cycle 1, on Day 1 of Cycles 2–6, and then every 2 months until disease progression for up to 2 years. Bendamustine was given intravenously on Days 1 and 2 for all treatment cycles (1–6) at 90 mg/m2/day when given in combination with GAZYVA or 120 mg/m2/day when given alone.
The primary analysis included 321 FL patients, including 166 patients randomized to bendamustine alone and 155 patients randomized to GAZYVA in combination with bendamustine. In the primary analysis, patients had a median age of 63 years, 88% were White and 56% were male. Thirty-four percent had bulky disease (> 6 cm), 15% had at least one B-symptom at baseline and 95% had an ECOG performance status of 0–1 at baseline. The median time since initial diagnosis was 3 years and the median number of prior therapies was 2 (range 1 to 10). Forty-six percent of patients received 1 prior therapy and 33% of patients received 2 prior therapies. Twenty percent of patients were refractory to prior rituximab product monotherapy, 37% of patients were refractory to prior rituximab product plus chemotherapy induction treatment, and 41% of patients were refractory to rituximab product maintenance treatment received following rituximab product plus chemotherapy induction. Seventy-nine percent of patients were refractory to both rituximab product and an alkylating agent during any prior regimen (double refractory).
The major efficacy outcome measure was PFS as determined by an independent review committee (IRC). At the time of the primary analysis, median observation time was 21.1 months. The median PFS in the bendamustine arm was 13.8 months. Median PFS was not reached in the GAZYVA plus bendamustine arm (PFS HR = 0.48, 95% CI: 0.34-0.68; stratified log-rank test p-value < 0.0001). The investigator assessed PFS result was consistent with the IRC-assessed PFS. The median investigator-assessed PFS in the bendamustine arm was 13.7 months and the median in the GAZYVA containing arm was 29.2 months (PFS HR = 0.48, 95% CI: 0.35-0.67; stratified log-rank test p-value < 0.0001).
Efficacy results are summarized in Table 14. The Kaplan-Meier curve for IRC-PFS is shown in Figure 3.
Table 14 Primary Analysis Efficacy Results from GADOLIN*, † Endpoint GADOLIN GAZYVA + Bendamustine followed by GAZYVA monotherapy
n = 155Bendamustine
n = 166Median Progression-Free Survival (months) Not Reached 13.8 (HR = 0.48 [0.34; 0.68], p-value < 0.0001 by stratified log-rank test) Best Overall Response‡ 78.7% 74.7% Complete Response 15.5% 18.7% Partial Response 63.2% 56.0% Median duration of response (months) Not Reached 11.6 Figure 3
Kaplan-Meier Curve of IRC-Assessed Progression-Free Survival in Patients with FL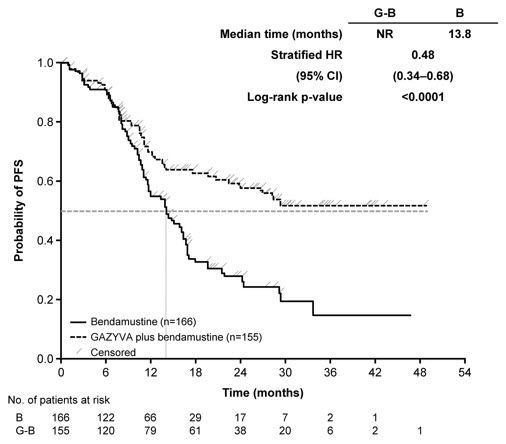
The final analysis included a total of 335 patients with 171 randomized to bendamustine alone and 164 to GAZYVA in combination with bendamustine. With an overall median observation time of 52.2 months (range: 0-100.9 months), there were 66 deaths (40.2%) in the GAZYVA arm and 85 deaths (51.3%) in the bendamustine-alone arm (OS HR = 0.71, 95% CI: 0.51, 0.98). The Kaplan-Meier curve for OS is presented in Figure 4.
Figure 4
Kaplan-Meier Curve of Overall Survival in Patients with FL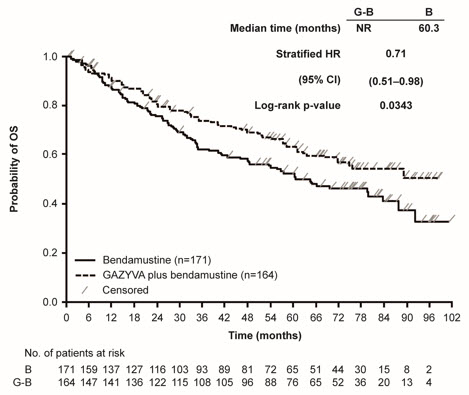
GALLIUM
The efficacy of GAZYVA was evaluated in GALLIUM (NCT01332968), a multicenter, open-label, randomized study that included 1202 patients with previously untreated, stage II bulky, III or IV FL. Patients were randomized 1:1 to receive either GAZYVA (n = 601) or rituximab product (n = 601) in combination with chemotherapy (CHOP, CVP, or bendamustine) for 6–8 cycles. Patients were stratified by chemotherapy (selected by each site; all patients at that site received the chosen chemotherapy regimen), FLIPI (Follicular Lymphoma International Prognostic Index) risk group and geographic region. Patients with at least PR to combination therapy received monotherapy with GAZYVA (1,000 mg) or rituximab product every two months until disease progression or for a maximum of two years. The study excluded patients with follicular lymphoma grade 3b or transformed disease; patients having an ANC < 1500 / µL, platelets < 75,000 / µL, or CLcr < 40 mL/min; and patients with hepatic transaminases > 2.5 × upper limit of normal unless attributable to lymphoma.
GAZYVA was given by intravenous infusion as a flat dose of 1,000 mg on Days 1, 8 and 15 of Cycle 1 and Day 1 of subsequent treatment cycles.
GAZYVA and bendamustine were given in six 28-day cycles. Bendamustine was administered at 90 mg/m2/day on Days 1 and 2 of each cycle, with prednisone 100 mg orally or equivalent on Day 1 of Cycle 1.
GAZYVA and CHOP were given in six 21-day cycles. Subsequently, two additional cycles of GAZYVA were given for a total of 8 GAZYVA cycles. CHOP consisted of cyclophosphamide 750 mg/m2 intravenously, doxorubicin 50 mg/m2, and vincristine 1.4 mg/m2 (maximum dose, 2 mg) on Day 1 and prednisone 100 mg orally on Days 1-5.
GAZYVA and CVP were given in eight 21-day cycles. CVP consisted of cyclophosphamide 750 mg/m2 intravenously and vincristine 1.4 mg/m2 (maximum dose, 2 mg) on Day 1 and prednisone 100 mg orally on Days 1-5.
Patients had a median age of 59 years, 81% were White and 53% were female; 7% had Stage II, 35% had Stage III, and 56% had Stage IV disease, with 44% having bulky disease (≥ 7 cm) overall; 79% had a FLIPI score of > 2; and 97% had an ECOG performance status of 0–1. The chemotherapy was bendamustine in 57%, CHOP in 33%, and CVP in 10% of patients.
Efficacy was based on PFS per IRC, with a median observation time of 38 months. Upon interim analysis, the risk of progression or death was significantly reduced in the GAZYVA containing arm compared to the rituximab product containing arm (Table 15). Kaplan-Meier curves for PFS are shown in Figure 5. Overall response and complete remission rates were similar.
Table 15 Efficacy in Previously Untreated Follicular Lymphoma (GALLIUM) Endpoint per IRC GAZYVA + chemotherapy followed by GAZYVA monotherapy
n = 601Rituximab product + chemotherapy followed by rituximab product monotherapy
n = 601Progression-Free Survival *
Number of events (%)108 (18%) 141 (23%) HR = 0.72 [95% CI: 0.56, 0.93], p-value = 0.0118 † Overall Response Rate ‡ 91% 88% Complete Remission Rate ‡ 28% 27% Figure 5
Kaplan-Meier Curves of Progression Free Survival in Patients with Previously Untreated FL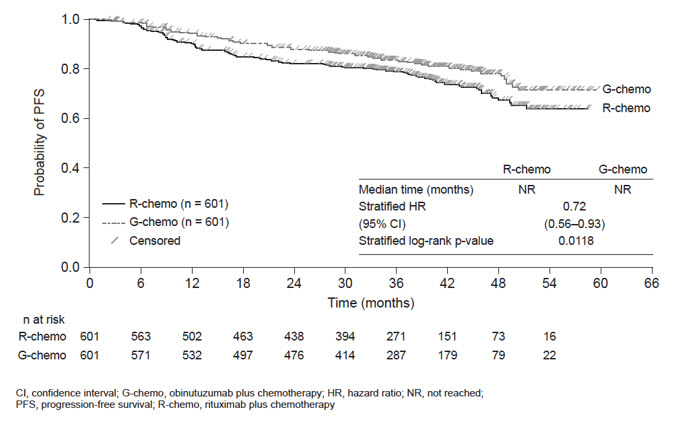
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise patients to seek immediate medical attention for any of the following:
- Signs and symptoms of infusion-related reactions including dizziness, nausea, chills, fever, vomiting, diarrhea, breathing problems, or chest pain [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
- Symptoms of tumor lysis syndrome such as nausea, vomiting, diarrhea, and lethargy [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
- Signs of infections including fever and cough [see Warnings and Precautions (5.6) and Adverse Reactions (6.1)].
- Symptoms of hepatitis including worsening fatigue or yellow discoloration of skin or eyes [see Warnings and Precautions (5.1)].
- New or changes in neurological symptoms such as confusion, dizziness or loss of balance, difficulty talking or walking, or vision problems [see Warnings and Precautions (5.2)].
- Signs and symptoms of bleeding or thrombosis [see Warnings and Precautions (5.8 and 5.9)].
Advise patients of the need for:
- Periodic monitoring of blood counts [see Warnings and Precautions (5.7, 5.8 and 5.9) and Adverse Reactions (6.1)].
- Avoid vaccinations with live viral vaccines [see Warnings and Precautions (5.10)].
- Patients with a history of hepatitis B infection (based on the blood test) should be monitored and sometimes treated for their hepatitis [see Warnings and Precautions (5.1)].
Advise pregnant women of potential fetal B-cell depletion. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.11), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with GAZYVA and for 6 months after the last dose [see Use in Specific Populations (8.3)].
Advise women not to breastfeed during treatment with GAZYVA and for 6 months after the last dose [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
- PRINCIPAL DISPLAY PANEL - 40 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
GAZYVA
obinutuzumab injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-070 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OBINUTUZUMAB (UNII: O43472U9X8) (OBINUTUZUMAB - UNII:O43472U9X8) OBINUTUZUMAB 1000 mg in 40 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) POLOXAMER 188 (UNII: LQA7B6G8JG) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-070-01 1 in 1 CARTON 11/01/2013 1 40 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:50242-070-86 1 in 1 CARTON 11/04/2013 2 40 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125486 11/01/2013 Labeler - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations Roche Diagnostics GmbH 315028860 ANALYSIS(50242-070) , MANUFACTURE(50242-070) , PACK(50242-070) , LABEL(50242-070) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd 485244961 LABEL(50242-070) , PACK(50242-070) Establishment Name Address ID/FEI Business Operations Roche Diagnostics GmbH 323105205 API MANUFACTURE(50242-070) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 080129000 ANALYSIS(50242-070) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 146373191 ANALYSIS(50242-070)
