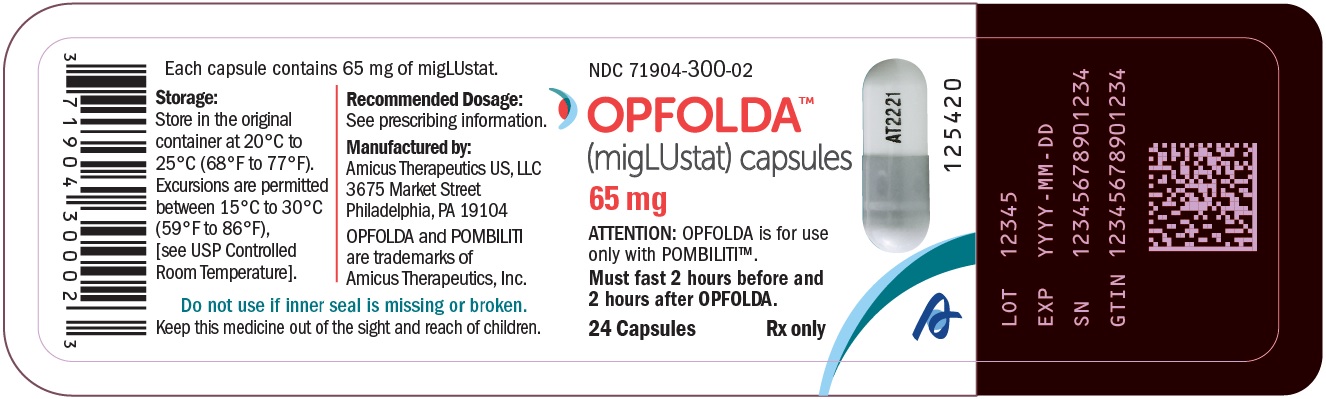
Label: OPFOLDA- miglustat capsule



- NDC Code(s): 71904-300-01, 71904-300-02, 71904-300-03
- Packager: AMICUS THERAPEUTICS US, LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 6, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OPFOLDA™ safely and effectively. See full prescribing information for OPFOLDA.
OPFOLDA (miglustat) capsules, for oral use
Initial U.S. Approval: 2003INDICATIONS AND USAGE
OPFOLDA is an enzyme stabilizer indicated, in combination with Pombiliti, a hydrolytic lysosomal glycogen-specific enzyme, for the treatment of adult patients with late-onset Pompe disease (lysosomal acid alpha-glucosidase [GAA] deficiency) weighing ≥40 kg and who are not improving on their current enzyme replacement therapy (ERT). (1)
DOSAGE AND ADMINISTRATION
- Verify pregnancy status in females of reproductive potential prior to initiating treatment. (2.1)
- Administer OPFOLDA in combination with Pombiliti. (2.2)
- Recommended OPFOLDA dosage (based on actual body weight), administered orally every other week, is: (2.2)
- 260 mg for patients weighing ≥50 kg.
- 195 mg for patients weighing ≥40 kg to <50 kg.
- Start OPFOLDA in combination with Pombiliti 2 weeks after the last ERT dose. (2.2)
- Take OPFOLDA with an unsweetened beverage approximately 1 hour before the start of Pombiliti infusion; do not consume other beverages or food for at least 2 hours prior to and 2 hours after taking OPFOLDA. (2.2)
- Missed dose: If the OPFOLDA dosage is missed, Pombiliti should not be administered and treatment should be rescheduled at least 24 hours after OPFOLDA was last taken. If OPFOLDA in combination with Pombiliti are both missed, re-start treatment as soon as possible. (2.2)
- See full prescribing information for recommended OPFOLDA dosage in patients with renal impairment. (2.3)
DOSAGE FORMS AND STRENGTHS
Capsules: 65 mg (3)
WARNINGS AND PRECAUTIONS
- Embryo-Fetal Toxicity: May cause embryo-fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for at least 60 days after the last dose. (4, 5.1, 8.1, 8.3)
- Risks Associated with Pombiliti: Refer to the Pombiliti Prescribing Information for a description of additional risks for Pombiliti. (5.2)
ADVERSE REACTIONS
Most common adverse reactions ≥5% are headache, diarrhea, fatigue, nausea, abdominal pain, and pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amicus Therapeutics at 1-877-4AMICUS or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- Lactation: Breastfeeding not recommended. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Pregnancy Evaluation Prior to Initiating Treatment
2.2 Recommended Dosage and Administration
2.3 Recommended Dosage in Patients with Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
5.2 Risks Associated with Pombiliti
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Pregnancy Evaluation Prior to Initiating Treatment
Verify the pregnancy status of females of reproductive potential prior to initiating OPFOLDA in combination with Pombiliti [see Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage and Administration
OPFOLDA must be administered in combination with Pombiliti (see Figure 1 for the dosing timeline). Refer to the Pombiliti Prescribing Information for Pombiliti dosage and administration recommendations.
The recommended dosage of OPFOLDA is based on actual body weight. For patients weighing:
- ≥50 kg, the recommended dosage is 260 mg orally every other week.
- ≥40 kg to <50 kg, the recommended dosage is 195 mg orally every other week.
Start OPFOLDA in combination with Pombiliti 2 weeks after the last ERT dose. Take OPFOLDA approximately 1 hour before intravenous administration of Pombiliti. Swallow the OPFOLDA capsules whole only with unsweetened beverages (e.g., water, tea or coffee with no cream, sugar, or sweeteners). Do not consume other beverages or food for at least 2 hours prior to and 2 hours after administration of OPFOLDA.
Missed Dose
If the OPFOLDA dosage is missed, Pombiliti should not be administered and treatment should be rescheduled at least 24 hours after OPFOLDA was last taken. If OPFOLDA in combination with Pombiliti are both missed, re-start treatment as soon as possible.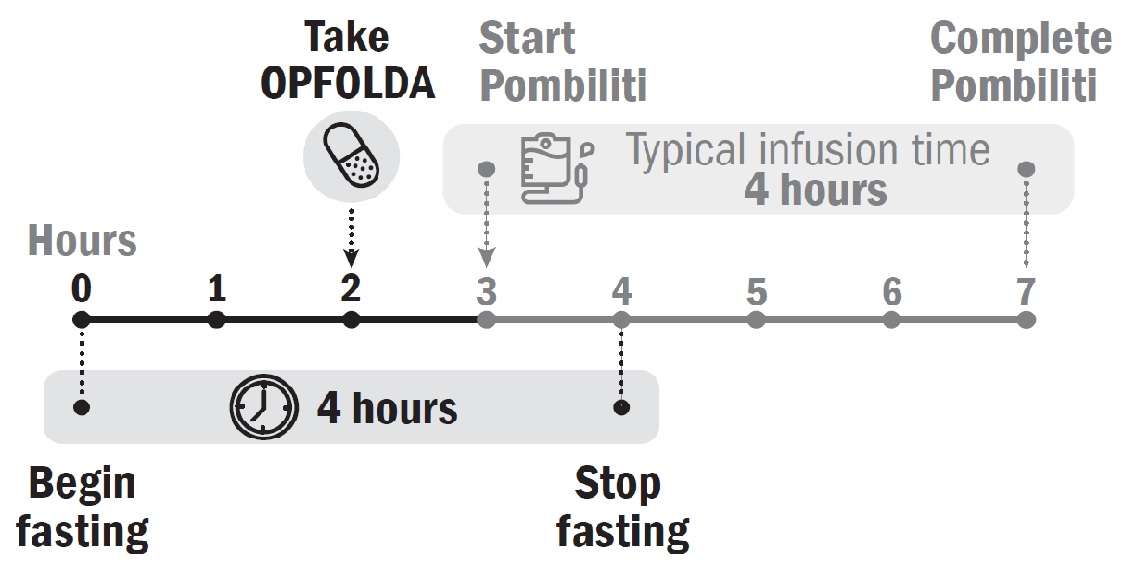
2.3 Recommended Dosage in Patients with Renal Impairment
The recommended dosage of OPFOLDA in patients with moderate or severe renal impairment is shown in Table 1 [see Clinical Pharmacology (12.3)].
Table 1. Recommended OPFOLDA Dosage in Patients with Moderate or Severe Renal Impairment
∗ Renal function classified by CLcr (creatinine clearance) based on the Cockcroft-Gault equation. Patient Weight Moderate Renal Impairment∗(CLcr 30-59 mL/minute) Severe Renal Impairment∗ (CLcr 15-29 mL/minute) ≥50 kg 195 mg 195 mg ≥40 kg to <50 kg 130 mg 130 mg For patients with mild renal impairment (creatinine clearance based on the Cockcroft-Gault equation, CLcr 60-89 mL/minute), the recommended OPFOLDA dosage is the same as for patients with normal renal function.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
OPFOLDA in combination with Pombiliti is contraindicated in Pregnancy [see Use in Specific Populations (8.1)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies, OPFOLDA in combination with Pombiliti may cause embryo-fetal harm when administered to a pregnant female and is contraindicated during pregnancy. In a rabbit embryo-fetal development study, great vessel and cardiac malformations were increased in offspring of pregnant rabbits treated with oral miglustat in combination with cipaglucosidase alfa-atga at 3-fold and 16-fold, respectively, the maximum recommended human dose (MRHD) based on plasma AUC exposure.
Verify the pregnancy status in females of reproductive potential prior to initiating treatment with OPFOLDA in combination with Pombiliti. Advise females of reproductive potential to use effective contraception during treatment with OPFOLDA in combination with Pombiliti and for at least 60 days after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions from the Pooled Clinical Trials Including Trial 1
The pooled safety analysis from 3 clinical trials included 151 adult patients with late-onset Pompe disease (LOPD) treated with OPFOLDA in combination with Pombiliti including:
- 85 patients in the randomized, double-blind, active-controlled trial in adults (Trial 1) [see Clinical Studies (14)],
- 37 patients in the open-label extension trial where patients switched from a non‑U.S.‑approved alglucosidase alfa product [see Clinical Studies (14)] to OPFOLDA in combination with Pombiliti,
- 29 patients in an open-label trial.
The total median duration of exposure in these trials was 21 months, with 120 patients having at least 12 months exposure to OPFOLDA in combination with Pombiliti. In these trials, 78% (n=117) of the patients received previous ERT (ERT‑experienced) with a mean treatment duration of 7.7 years.
The most common adverse reactions (≥5%) reported in the pooled safety population of patients treated with OPFOLDA in combination with Pombiliti in the 3 clinical trials were headache, diarrhea, fatigue, nausea, abdominal pain, and pyrexia.
Adverse Reactions from Trial 1
Trial 1 (a randomized, double-blind, active-controlled trial) included 123 adult patients with LOPD who were randomized in a 2:1 ratio to receive treatment with OPFOLDA in combination with Pombiliti or a non-U.S.-approved alglucosidase alfa product with placebo [see Clinical Studies (14)].
The duration of exposure was similar for both treatment groups (overall mean exposure of 12 months). Most patients (77%) were ERT‑experienced, and a majority of patients in both treatment groups had >5 years of prior treatment with ERT (69% and 63% of patients in the OPFOLDA in combination with Pombiliti group and the non-U.S.-approved alglucosidase alfa product with placebo group, respectively).
The most common adverse reactions (≥5%) reported in the patients who received OPFOLDA in combination with Pombiliti in Trial 1 were headache and diarrhea.
Table 2 summarizes frequent adverse reactions that occurred in patients treated with OPFOLDA in combination with Pombiliti in Trial 1. Trial 1 was not designed to demonstrate a statistically significant difference in the incidence of adverse reactions in the OPFOLDA in combination with Pombiliti and the non-U.S.-approved alglucosidase alfa product with placebo groups.
Table 2. Adverse Reactions that Occurred in Adults with LOPD at an Incidence of ≥2% in Trial 1
LOPD: late-onset Pompe disease ∗ Headache included migraine and migraine with aura. † Rash included erythematous rash and macular rash. ‡ Abdominal pain included upper and lower abdominal pain. § Tachycardia included sinus tachycardia. ¶ Urticaria included mechanical urticaria and urticarial rash. Adverse Reaction OPFOLDA in Combination
with Pombiliti
(n=85)
N (%)A Non-U.S.-Approved
Alglucosidase alfa Product
with Placebo
(n=38)
N (%)Headache∗ 7 (8.2) 3 (7.9) Diarrhea 5 (5.9) 2 (5.3) Dizziness 4 (4.7) 2 (5.3) Dyspnea 3 (3.5) 0 Abdominal distention 3 (3.5) 2 (5.3) Pyrexia 3 (3.5) 1 (2.6) Rash† 3 (3.5) 0 Abdominal pain‡ 2 (2.4) 4 (10.5) Nausea 2 (2.4) 5 (13.2) Chills 2 (2.4) 0 Dysgeusia 2 (2.4) 0 Flushing 2 (2.4) 0 Muscle spasms 2 (2.4) 0 Pruritus 2 (2.4) 2 (5.3) Tachycardia§ 2 (2.4) 0 Urticaria¶ 2 (2.4) 0 Additional adverse reactions reported in at least 2% of patients treated with OPFOLDA in combination with Pombiliti across the 3 clinical trials include: myalgia, arthralgia, increased blood pressure, pain, tremor, dyspepsia, asthenia, constipation, infusion site swelling, flank pain, malaise, paresthesia, and decreased platelet count.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies, OPFOLDA in combination with Pombiliti may cause embryo-fetal harm when administered to a pregnant female and is contraindicated during pregnancy. In a rabbit embryo-fetal development study, great vessel and cardiac malformations were increased in offspring of pregnant rabbits treated with miglustat in combination with cipaglucosidase alfa-atga at 3-fold and 16-fold, respectively, the MRHD of OPFOLDA and Pombiliti based on plasma AUC exposure. A No Observed Adverse Effect Level (NOAEL) was not identified for the combination. In a pre- and post-natal development study in rats, increases in pup mortality were seen following maternal treatment with miglustat in combination with cipaglucosidase alfa-atga (400 mg/kg), or with cipaglucosidase alfa-atga (400 mg/kg) alone. The NOAEL for cipaglucosidase alfa-atga alone is 150 mg/kg (5-fold the Pombiliti MRHD margin). A NOAEL for the combination was not identified. Margins at the lowest observed adverse effect level (LOAEL), relative to exposures at the MRHD of OPFOLDA and Pombiliti were 4-fold and 21-fold, respectively, based on plasma AUC exposure (see Data).
There are no available human data on OPFOLDA in combination with Pombiliti use in pregnant females to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
Animal Data
Reproductive toxicity studies of cipaglucosidase alfa-atga in rats and rabbits included pretreatment with diphenhydramine (DPH) to prevent or minimize hypersensitivity reactions.In a rabbit embryo-fetal development study, 25 mg/kg oral miglustat alone, or in combination with intravenous cipaglucosidase alfa-atga 175 mg/kg, was administered every other day to pregnant females during organogenesis (Gestation Day [GD] 7 through GD 19). Additional experimental groups received cipaglucosidase alfa-atga (30, 70, or 175 mg/kg) with the same dosing frequency during organogenesis. Clusters of great vessel and cardiac malformations were increased in offspring of pregnant rabbits treated with the combination of miglustat and cipaglucosidase alfa-atga at 3-fold and 16-fold the MRHD of OPFOLDA and Pombiliti, respectively, based on plasma AUC exposure. A NOAEL for the combination was not identified. One fetus treated with miglustat alone (25 mg/kg) and one fetus treated with cipaglucosidase alfa-atga alone (175 mg/kg), each showed a similar cluster of these great vessel and cardiac malformations.
In a rat embryo-fetal development study, 60 mg/kg oral miglustat alone, or in combination with intravenous cipaglucosidase alfa-atga 400 mg/kg, was administered every other day to pregnant rats during organogenesis (GD 6 through GD 18). Additional experimental groups received cipaglucosidase alfa-atga (75, 150, or 400 mg/kg) with the same dosing frequency during organogenesis. No evidence of adverse effects was noted in pregnant rats or their offspring in any experimental group. The margin at the NOAEL for miglustat (60 mg/kg) was 4-fold the OPFOLDA MRHD based on plasma AUC exposure. The margin at the NOAEL for cipaglucosidase alfa-atga (400 mg/kg) was 21-fold the Pombiliti MRHD based on plasma AUC exposure.
In a pre-and post-natal development study in rats, 60 mg/kg oral miglustat alone, or in combination with intravenous cipaglucosidase alfa-atga 400 mg/kg, was administered to pregnant females every other day from GD 6 through GD 18, and from Lactation Day (LD) 1 through LD 19. Additional experimental groups received cipaglucosidase alfa-atga (75, 150, or 400 mg/kg) with the same dosing frequency during pregnancy and lactation. Maternal and pup mortality were increased with the combination, and pup mortality was also increased with cipaglucosidase alfa-atga 400 mg/kg alone. The NOAEL for cipaglucosidase alfa-atga alone is 150 mg/kg (5-fold the Pombiliti MRHD margin). A NOAEL was not identified for the combination, for which LOAEL margins at the MRHD of OPFOLDA and Pombiliti were 4-fold and 21-fold, respectively, based on plasma AUC exposure.
8.2 Lactation
Risk Summary
There are no data on the presence of miglustat, alone or in combination with cipaglucosidase alfa-atga, in human milk, the effects on the breastfed infant, or the effects on milk production. Miglustat is present in animal milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. Based on findings in animal studies, the use of OPFOLDA in combination with Pombiliti may lead to serious adverse reactions in breastfed infants. Advise females that breastfeeding is not recommended while on treatment with OPFOLDA in combination with Pombiliti.
Evaluation of milk in rats from the pre- and post-natal development study of miglustat in combination with cipaglucosidase alfa-atga (60 mg/kg and 400 mg/kg, respectively) showed excretion of miglustat and cipaglucosidase alfa-atga in rat milk. In this study, the ratio of miglustat exposure in rat milk to the miglustat exposure in rat plasma was 1.7, and the ratio of cipaglucosidase alfa-atga exposure in rat milk to the cipaglucosidase alfa-atga exposure in rat plasma was <4%.
The concentration of drug in animal milk does not necessarily predict the concentration of drug in human milk.
8.3 Females and Males of Reproductive Potential
OPFOLDA in combination with Pombiliti may cause embryo-fetal harm when administered to a pregnant female [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating treatment with OPFOLDA in combination with Pombiliti.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with OPFOLDA in combination with Pombiliti and for at least 60 days after the last dose.Infertility
Females
Based on preimplantation loss observed in female rats treated with oral miglustat (60 mg/kg) in combination with intravenous cipaglucosidase alfa-atga (400 mg/kg) every other day for 14 days prior to mating and continuing through GD 7, OPFOLDA in combination with Pombiliti may impair human female fertility. A NOAEL for the combination was not identified. The LOAEL margins are 4-fold and 21-fold the MRHD for OPFOLDA and Pombiliti, respectively. It is not known whether this preimplantation loss in female rats would be sustained if dosing with the combination were discontinued prior to mating [see Nonclinical Toxicology (13.1)].Males
Based on reversible increases in preimplantation loss in male rats treated with the combination every other day for 28 days prior to mating, OPFOLDA in combination with Pombiliti may impair human male fertility. A NOAEL for the combination was not identified. The LOAEL margins are 4-fold and 21-fold the MRHD for OPFOLDA and Pombiliti, respectively [see Nonclinical Toxicology (13.1)].For additional information about male fertility with the use of Pombiliti, see the Pombiliti Prescribing Information.
8.4 Pediatric Use
Safety and effectiveness of OPFOLDA in combination with Pombiliti have not been established in pediatric patients with late-onset Pompe disease.
8.5 Geriatric Use
Of the total number of patients treated with OPFOLDA in combination with Pombiliti in clinical trials for LOPD, 17 (11%) were 65 to 74 years of age, and none were 75 years of age and older [see Clinical Studies (14)].
Clinical trials of OPFOLDA in combination with Pombiliti did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
8.6 Renal Impairment
Plasma concentrations of miglustat increased in patients with renal impairment [see Clinical Pharmacology (12.3)]. No dosage adjustment of OPFOLDA is recommended in patients with mild (CLcr 60 to 89 mL/minute, estimated by Cockcroft-Gault) renal impairment. Reduce the OPFOLDA dosage in patients with moderate (CLcr 30 to 59 mL/minute) or severe (CLcr 15 to 29 mL/minute) renal impairment [see Dosage and Administration (2.3)]. The pharmacokinetics of miglustat have not been evaluated in patients with end stage renal disease.
-
11 DESCRIPTION
Miglustat is an N-alkylated iminosugar, a synthetic analog of D‑glucose. The pharmacologic class is enzyme stabilizer.
The chemical name is 1,5-(butylimino)-1,5-dideoxy-D-glucitol with the molecular weight of 219.28 g/mol, the molecular formula C10H21NO4, and the following chemical structure:
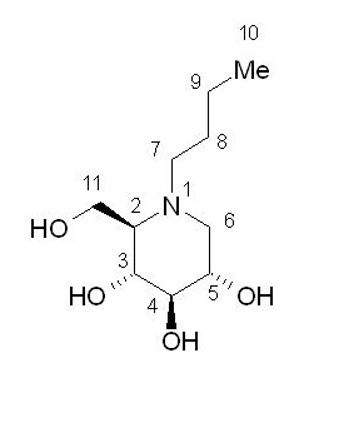
Miglustat is a white to off-white crystalline solid (powder). It is highly soluble in water (>1,000 mg/mL as a free base).
OPFOLDA (miglustat) capsules each contain 65 mg of miglustat for oral administration. The inactive ingredients are: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, pregelatinized maize starch, and sucralose. The capsule shells include black iron oxide, gelatin, and titanium dioxide. Edible printing ink includes black iron oxide, and shellac.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Miglustat binds with, stabilizes, and reduces inactivation of cipaglucosidase alfa-atga in the blood after infusion. The bound miglustat is dissociated from cipaglucosidase alfa-atga after it is internalized and transported into lysosomes. Miglustat alone has no pharmacological activity in cleaving glycogen.
12.2 Pharmacodynamics
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of miglustat have not been fully characterized.
12.3 Pharmacokinetics
Miglustat maximum concentration (Cmax) and area under the plasma concentration-time curve (AUC) increases proportionally over a dosage range from 130 mg to 260 mg (0.5 to 1 times the approved recommended dosage of 260 mg in patients weighing ≥50 kg). At the recommended 260 mg dose, the mean (SD) Cmax was approximately 3 (0.9) mcg/mL and the mean AUC was approximately 25 (6.5) mcg∗hr/mL.
Absorption
The mean time to reach the maximum concentration (Tmax) of miglustat ranged from 2 hours to 3 hours.Effect of Food
Co-administration of miglustat with food is predicted to result in delayed absorption and decreased Cmax in healthy subjects [see Dosage and Administration (2.2)].Distribution
The apparent volume of distribution of miglustat was approximately 94 L in adult patients with LOPD.Elimination
The apparent clearance of miglustat was approximately 10 L/hr. The terminal elimination half‑life was approximately 6 hours.Specific Populations
No clinically significant differences in the pharmacokinetics of miglustat were observed based on age (18 to 74 years) and sex. The effect of hepatic impairment on the pharmacokinetics of miglustat is unknown.Patients with Renal Impairment
The AUC0-24hr of miglustat increased by 21%, 32%, and 41% in patients with mild (CLcr 60 to 89 mL/minute, estimated by Cockcroft-Gault), moderate (CLcr 30 to 59 mL/minute), and severe (CLcr 15 to 29 mL/minute) renal impairment, respectively, compared to patients with normal renal function. The effect of end stage renal disease on the pharmacokinetics of miglustat is unknown.Drug Interaction Studies
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Miglustat is not a known substrate or inhibitor of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, or CYP4A11.Transporter Systems: Miglustat is not an inhibitor or a substrate of P-gP.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenicity of OPFOLDA is based on studies from another miglustat product. Below are findings from the carcinogenicity studies with the other miglustat product.Two-year carcinogenicity studies have been conducted with miglustat in CD-1 mice at oral doses up to 500 mg/kg/day and in Sprague Dawley rats at oral doses up to 180 mg/kg/day. Oral administration of miglustat for 104 weeks produced mucinous adenocarcinomas of the large intestine at 210, 420, and 500 mg/kg/day (about 3, 5, and 6-fold the MRHD of OPFOLDA 260 mg, respectively, based on body surface area [BSA]) in male mice and at 420 and 500 mg/kg/day (about 5 and 6-fold the MRHD of OPFOLDA 260 mg, respectively, based on BSA) in female mice. The adenocarcinomas were considered rare in CD-1 mice and occurred in the presence of inflammatory and hyperplastic lesions in the large intestine of both males and females. In rats, oral administration of miglustat for 100 weeks produced increased incidences of interstitial cell adenomas of the testis at 30, 60, and 180 mg/kg/day (about 1, 2, and 4-fold the MRHD of OPFOLDA 260 mg, respectively, based on BSA).
Mutagenesis
The mutagenicity of OPFOLDA is based on studies from another miglustat product. Below are findings from the carcinogenicity studies with the other miglustat product.Miglustat was not mutagenic or clastogenic in a battery of in vitro and in vivo assays including the bacterial reverse mutation (Ames), chromosomal aberration (in human lymphocytes), gene mutation in mammalian cells (Chinese hamster ovary), and mouse micronucleus assays, in studies with another miglustat product.
Impairment of Fertility
In a fertility study in rats, oral miglustat (60 mg/kg) was administered alone, or in combination with intravenous cipaglucosidase alfa-atga (400 mg/kg), to male and female rats every other day. Dosing in males was initiated 28 days prior to cohabitation with untreated females. Dosing in females was initiated 14 days prior to cohabitation with untreated males and continued through GD 7. Additional experimental groups received intravenous cipaglucosidase alfa-atga alone (75, 150, or 400 mg/kg/day) with the same frequency over the same pre-mating interval (males) or pre-mating and pregnancy interval (females).There was no effect on male or female rat fertility in any experimental group. Treatment of male rats with the combination was associated with increased preimplantation loss that was reversible. Treatment of female rats with the combination, or with miglustat alone, resulted in preimplantation loss; whether this would be reversible if treatment were discontinued prior to cohabitation is unknown. NOAELs were not identified for the combination in either male or female rats. The LOAEL margins for these doses represent 4-fold and 21-fold the MRHD of OPFOLDA and Pombiliti, respectively, based on plasma AUC exposures.
The impact of OPFOLDA on fertility is based, in part, on studies from another miglustat product. Below are findings from the fertility studies with the other miglustat product.
Fertility studies conducted with another miglustat product showed that male rats, given 20 mg/kg/day miglustat (exposure less than that expected for the MRHD of OPFOLDA 260 mg, based on BSA comparisons, mg/m2) by oral gavage, 14 days prior to mating, had decreased spermatogenesis with altered sperm morphology and motility and decreased fertility. Decreased spermatogenesis was reversible following 6 weeks of miglustat withdrawal in this study. A higher dose of 60 mg/kg/day (2 times the MRHD of OPFOLDA, based on BSA comparisons, mg/m2) resulted in seminiferous tubule and testicular atrophy/degeneration. Female rats were given oral miglustat gavage doses of 20, 60, and 180 mg/kg/day beginning 14 days before mating and continuing through gestation in this study. Effects observed at 20 mg/kg/day (systemic exposure less than the human therapeutic systemic exposure, based on BSA comparisons) included decreased corpora lutea, increased post-implantation loss, and decreased live births.
13.2 Animal Toxicology and/or Pharmacology
Animal toxicology findings of OPFOLDA are based, in part, from studies from another miglustat product. In studies conducted for this other miglustat product, the following effects were noted following daily administration.
In a 52-week monkey toxicity study of oral miglustat (750 and 2,000 mg/kg/day), histopathology findings in the absence of clinical signs in the central nervous system (brain, spine) including vascular mineralization and mineralization and necrosis of white matter were observed at both ≥750 and 2000 mg/kg/day. In a 4-week rat study of miglustat by oral gavage (180, 840, and 4200 mg/kg/day), vacuolization of white matter was observed at ≥180 mg/kg/day (5 times the MRHD of OPFOLDA 260 mg, based on BSA comparisons, mg/m2). Vacuolization can sometimes occur as an artifact of tissue processing. In a 4-week dog toxicity study of miglustat by oral gavage (35, 70, 105, and 140 mg/kg/day), findings included tremor and absent corneal reflexes at 105 mg/kg/day (9 times the MRHD of OPFOLDA 260 mg, based on BSA comparisons, mg/m2). In a 2-week rat toxicity study of miglustat by oral gavage (85, 165, 495, and 825 mg/kg/day), ataxia, diminished/absent pupillary, palpebral, or patellar reflexes were observed at ≥495 mg/kg/day (43 times the MRHD of OPFOLDA 260 mg, based on BSA comparisons, mg/m2). In a 52-week rat toxicity study of miglustat by oral gavage (180, 420, 840, and 1,680 mg/kg/day), cataracts were observed at ≥180 mg/kg/day.
In a 2-week dog toxicity study of oral miglustat (85, 165, 495, and 825 mg/kg/day), gastrointestinal necrosis, inflammation, and hemorrhage were observed at ≥85 mg/kg/day (8 times the MRHD of OPFOLDA 260 mg based on BSA comparisons, mg/m2). In a 26-week rat toxicity study of miglustat by oral gavage (300, 600, and 1,200 mg/kg/day), similar GI toxicity occurred at 1200 mg/kg/day. In a 52-week monkey toxicity study of miglustat by oral gavage (750 and 2,000 mg/kg/day), similar GI toxicity occurred at ≥750 mg/kg/day.
-
14 CLINICAL STUDIES
Trial 1 was a randomized, double‑blind, active‑controlled, international, multi-center clinical trial (NCT#03729362) in patients ≥18 years old diagnosed with LOPD. Patients were randomized 2:1 to receive OPFOLDA (260 mg orally for those ≥50 kg or 195 mg orally for those ≥40 kg to <50 kg) in combination with Pombiliti (20 mg/kg by intravenous infusion) or a non-U.S.-approved alglucosidase alfa product with placebo every other week for 52 weeks. The efficacy population included a total of 123 patients of whom 95 (77%) had received prior treatment with U.S.-approved alglucosidase alfa or a non-U.S.-approved alglucosidase alfa product (ERT‑experienced) and 28 (23%) were ERT‑naïve. More than two thirds (n=64, 67%) of ERT‑experienced patients had been on ERT treatment for more than 5 years prior to entering Trial 1 (mean of 7.4 years).
Demographics, baseline sitting forced vital capacity (FVC) (% predicted), and 6-minute walk distance (6MWD) were generally similar between the 2 treatment groups (see Table 5 for baseline sitting FVC [percent predicted] values). Of the 123 randomized patients, 56 were males, baseline mean age was 47 years old (range from 19 to 74 years old), and mean age at diagnosis was 39 years old (range from 1 to 66). The racial groups for the patients consisted of 104 White (85%), 6 Japanese (5%), 6 Other racial group (5%), 4 Asian (3%), 1 Native Hawaiian or other Pacific Islander (1%), 1 American Indian or Alaska Native (1%), and 1 Black or African American (1%).
Key efficacy endpoints included assessment of sitting FVC (% predicted) and 6MWD (Table 3 and Table 4).
Sitting FVC (Percent‑predicted) at 52 Weeks
Patients treated with OPFOLDA in combination with Pombiliti showed a mean change in sitting FVC from baseline at Week 52 of ‑1.1% as compared with patients treated with a non-U.S.-approved alglucosidase alfa product with placebo of ‑3.3%; the estimated treatment difference was 2.3% (95% CI: 0.02, 4.62).The ERT‑experienced patients treated with OPFOLDA in combination with Pombiliti showed a numerically favorable change in sitting FVC from baseline at Week 52 (Table 3 and Figure 2).
Table 3. Summary of Sitting FVC in Adults with LOPD by ERT Status at 52 Weeks in Trial 1
FVC: forced vital capacity; LOPD: late-onset Pompe disease; ERT: enzyme replacement therapy; SD: standard deviation; Diff.: difference; SE: standard error; CI: confidence interval ∗ OPFOLDA in combination with Pombiliti is not approved for use in ERT-naïve patients with LOPD [see Indications and Usage (1)]. The ERT naïve patient subgroup enrolled too few patients to conclusively interpret the data. For the ERT-naïve group, the treatment difference was estimated using a 2-sample t-test. † A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and OPFOLDA in combination with Pombiliti for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT . ‡ For the ERT-experienced group, the treatment difference of the mean was estimated by analysis of covariance which included treatment, gender, baseline FVC, age, weight, and height in the model. Nominal p=0.006. Missing data at Week 52 was imputed using last observed values. Efficacy Endpoint ERT-experienced ERT-naïve* Sitting FVC
(% predicted)OPFOLDA in Combination with Pombiliti A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo OPFOLDA in Combination with Pombiliti A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo Baseline
n
Mean (SD)
Median
n=65
67.9 (19.1)
68.0
n=30
67.5 (21.0)
69.0
n=20
80.2 (18.7)
82.3
n=8
79.6 (21.0)
88.5Change from baseline at Week 52
n
Mean (SD)
Median
n=55
0.1 (5.9)
0.5
n=26
-3.5 (4.7)
-2.5
n=19
-4.7 (6.2)
-4.5
n=7
-2.4 (6.3)
-3.0Change to Week 52
Diff. of means (SE)
(95% CI)
3.5 (1.3)
(1.0, 6.0)‡
-1.9 (2.7)
(-7.3, 3.6)Figure 2. Mean Change (± SE) in Sitting FVC (% predicted) from Baseline to Week 52 in ERT-experienced Adults with LOPD in Trial 1*
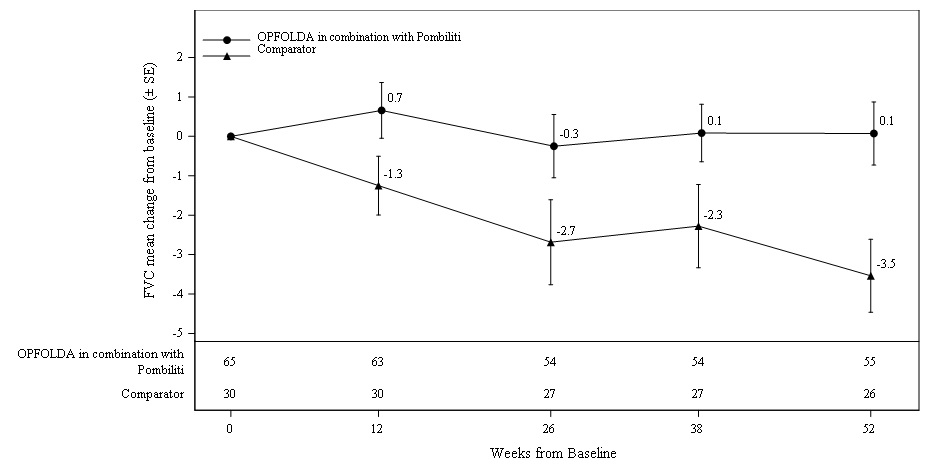
SE: standard error; FVC: forced vital capacity; ERT: enzyme replacement therapy; LOPD: late-onset Pompe disease
* A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and OPFOLDA in combination with Pombiliti for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT.6-Minute Walk Distance (6MWD) at 52 Weeks
Patients treated with OPFOLDA in combination with Pombiliti walked on average 21 meters farther from baseline as compared to those treated with a non-U.S.-approved alglucosidase alfa product with placebo who walked 8 meters farther from baseline; the estimated treatment difference was 14 meters (95% CI: -1, 28).The ERT-experienced patients treated with OPFOLDA in combination with Pombiliti showed a numerically favorable change in 6MWD from baseline at Week 52 (Table 4 and Figure 3).
Table 4. Summary of 6MWD in Adults with LOPD by ERT Status at 52 Weeks in Trial 1
6MWD: 6-minute walk distance; LOPD: late-onset Pompe disease; ERT: enzyme replacement therapy; SD: standard deviation; Diff.: difference; SE: standard error; CI: confidence interval ∗ OPFOLDA in combination with Pombiliti is not approved for use in ERT-naïve patients with LOPD [see Indications and Usage (1)]. The ERT-naïve patient subgroup enrolled too few patients to conclusively interpret the data. For the ERT-naïve group, the treatment difference was estimated using a 2-sample t-test. One ERT-naïve subject in the control arm was excluded from this table because their change of 355 meters in 6MWD from baseline at Week 52 was a statistical outlier and not considered clinically plausible. † A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and OPFOLDA in combination with Pombiliti for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT. ‡ For the ERT-experienced group, the treatment difference of the mean was estimated by nonparametric analysis of covariance which included treatment, gender, baseline 6MWD, age, weight, and height in the model. Nominal p=0.047. Missing data at Week 52 was imputed using last observed values. Efficacy Endpoint ERT-experienced ERT-naïve* 6MWD OPFOLDA in Combination with Pombiliti A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo OPFOLDA in Combination with Pombiliti A Non-U.S.-Approved Alglucosidase alfa Product† with Placebo Baseline
n
Mean (SD)
Median
n=65
347 (110)
353
n=30
335 (114)
344
n=20
394 (112)
375
n=7
421 (136)
386Change from baseline at Week 52
n
Mean (SD)
Median
n=61
16 (39)
10
n=29
1 (40)
-9
n=20
33 (49)
24
n=7
38 (29))
34Change to Week 52
Diff. of means (SE)
(95% CI)
17 (8)
(0.2, 33)‡
-5 (20)
(-45, 36)Figure 3. Mean Change (± SE) of 6MWD from Baseline to Week 52 in ERT-experienced Adults with LOPD in Trial 1*
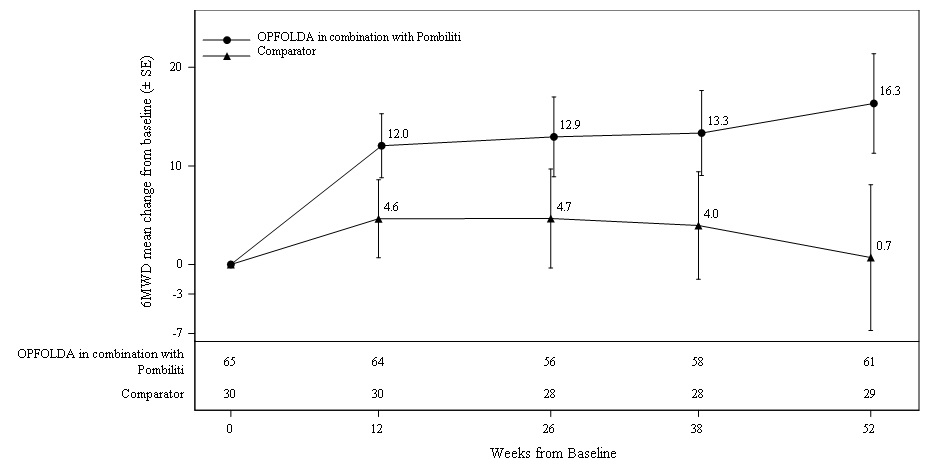
SE: standard error; 6MWD: 6-minute walk distance; ERT: enzyme replacement therapy; LOPD: late-onset Pompe disease
* A U.S.-approved alglucosidase alfa product was not used in this clinical trial. Conclusions cannot be drawn from this clinical trial regarding comparative effectiveness between a U.S.-approved alglucosidase alfa product and OPFOLDA in combination with Pombiliti for the treatment of adult patients with LOPD weighing ≥40 kg and who are not improving on their current ERT. -
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
OPFOLDA (miglustat) capsules are supplied as 65 mg, white opaque hard gelatin capsules with a grey opaque cap with “AT2221” printed in black on the body, and are available in bottles with child resistant caps. See Table 5 for the available OPFOLDA packages.Package Size NDC 4 count bottle 71904-300-01 24 count bottle 71904-300-02 100 count bottle 71904-300-03 Storage and Handling
Store at 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C to 30°C (59°F to 86°F), [see USP Controlled Room Temperature].Store in the original container to protect from light.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA‑approved patient labeling (Patient Information).
OPFOLDA must be administered in combination with Pombiliti. Refer to the Pombiliti Prescribing Information for Pombiliti patient counseling information.
Administration
Advise the patient and caregiver to follow the timeline recommendations for taking OPFOLDA prior to the intravenous infusion with Pombiliti and to follow the fasting recommendation. Advise the patient and caregiver that OPFOLDA should be swallowed only with unsweetened beverages [see Dosage and Administration (2.2)].Embryo-Fetal Toxicity
OPFOLDA in combination with Pombiliti may cause embryo-fetal harm. Advise a female patient and caregiver to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].Advise a female of reproductive potential to use effective contraception during treatment with OPFOLDA in combination with Pombiliti and for at least 60 days after the last dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise a lactating female not to breastfeed during treatment with OPFOLDA in combination with Pombiliti [see Use in Specific Populations (8.2)].Infertility
Advise the male or female of reproductive potential that OPFOLDA in combination with Pombiliti may impair fertility [see Use in Specific Populations (8.3)].
Manufactured for:
Amicus Therapeutics US, LLC
3675 Market Street
Philadelphia, PA 19104OPFOLDA and Pombiliti are trademarks of Amicus Therapeutics, Inc.
For more information, go to pombilitiopfoldahcp.com.
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 9/2023 PATIENT INFORMATION
OPFOLDA™ (op fol dah)
(miglustat)
capsules, for oral useWhat is OPFOLDA?
OPFOLDA is a prescription medicine used in combination with Pombiliti for the treatment of adults with late-onset Pompe disease weighing 88 pounds (40 kg) or more and who are not improving on their current enzyme replacement therapy (ERT).It is not known if OPFOLDA in combination with Pombiliti is safe and effective in children with late-onset Pompe disease.
Do not take OPFOLDA in combination with Pombiliti if you are pregnant. See “Before taking OPFOLDA, tell your healthcare provider about all of your medical conditions, including if you.”
Before taking OPFOLDA, tell your healthcare provider about all of your medical conditions, including if you:
have kidney problems
are pregnant or plan to become pregnant. OPFOLDA in combination with Pombiliti may cause harm to your unborn baby.
Females who are able to become pregnant:- Your healthcare provider will check if you are pregnant before you start treatment with OPFOLDA in combination with Pombiliti.
- You should use effective birth control (contraception) during treatment with OPFOLDA in combination with Pombiliti and for at least 60 days after the last dose.
- Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with OPFOLDA in combination with Pombiliti.
are breastfeeding or plan to breastfeed. It is not known if OPFOLDA alone or in combination with Pombiliti passes into your breast milk. Do not breastfeed during treatment with OPFOLDA in combination with Pombiliti. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.How should I take OPFOLDA?
OPFOLDA must be taken in combination with Pombiliti. OPFOLDA in combination with Pombiliti will be given to you 1 time every other week.Take OPFOLDA exactly as your healthcare provider tells you to.
Take your prescribed dose of OPFOLDA every other week about 1 hour before the start of your Pombiliti infusion.
Swallow OPFOLDA capsules whole only with unsweetened beverages, including water, tea or coffee with no cream, sugar, or sweeteners. Do not drink other beverages or eat food for at least 2 hours before and 2 hours after taking OPFOLDA.
Your healthcare provider will stop your current ERT before starting your treatment with OPFOLDA in combination with Pombiliti.
If you miss a dose of OPFOLDA, you should not receive your Pombiliti infusion. Call your healthcare provider right away to reschedule your Pombiliti infusion.What are the possible side effects of OPFOLDA in combination with Pombiliti?
The most common side effects of OPFOLDA in combination with Pombiliti include:
headache
diarrhea
tiredness
nausea
stomach area pain
fever
OPFOLDA in combination with Pombiliti may cause fertility problems in females and males, which may affect the ability to have children. Talk to your healthcare provider if you have concerns about fertility.
These are not all of the possible side effects of OPFOLDA in combination with Pombiliti.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store OPFOLDA?
Store OPFOLDA at room temperature between 68°F to 77°F (20°C to 25°C).
Keep OPFOLDA capsules in the original container to protect from light.Keep OPFOLDA and all medicines out of the reach of children.
General information about the safe and effective use of OPFOLDA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use OPFOLDA for a condition for which it was not prescribed. Do not give OPFOLDA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about OPFOLDA that is written for health professionals.
What are the ingredients in OPFOLDA?
Active ingredient: miglustat
Inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, pregelatinized maize starch, and sucralose. The capsule shell contains: black iron oxide, gelatin, and titanium dioxide. The edible printing ink contains: black iron oxide and shellac.
Manufactured for:
Amicus Therapeutics US, LLC
3675 Market Street
Philadelphia, PA 19104 USA
OPFOLDA and POMBILITI are trademarks of Amicus Therapeutics, Inc.
For more information, call AMICUS at 1-877-426-4287 or go to pombilitiopfolda.com.
- PRINCIPAL DISPLAY PANEL - NDC: 71904-300-01 - 4 Count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 71904-300-02 - 24 Count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 71904-300-03 - 100 Count Bottle Label
-
INGREDIENTS AND APPEARANCE
OPFOLDA
miglustat capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71904-300 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MIGLUSTAT (UNII: ADN3S497AZ) (MIGLUSTAT - UNII:ADN3S497AZ) MIGLUSTAT 65 mg Product Characteristics Color GRAY (GRAY OPAQUE CAP) , WHITE (WHITE OPAQUE BODY) Score no score Shape CAPSULE Size 18mm Flavor Imprint Code AT2221 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71904-300-01 4 in 1 BOTTLE, PLASTIC 10/11/2023 1 1 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 2 NDC:71904-300-02 24 in 1 BOTTLE, PLASTIC 10/11/2023 2 1 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 3 NDC:71904-300-03 100 in 1 BOTTLE, PLASTIC 10/11/2023 3 1 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215211 10/11/2023 Labeler - AMICUS THERAPEUTICS US, LLC (080932337)