Label: LUTATHERA- lutetium lu 177 dotatate injection




- NDC Code(s): 69488-003-01
- Packager: Advanced Accelerator Applications USA, Inc
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated March 7, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LUTATHERA safely and effectively. See full prescribing information for LUTATHERA.
LUTATHERA® (lutetium Lu 177 dotatate) injection, for intravenous use
Initial U.S. Approval: 2018RECENT MAJOR CHANGES
INDICATIONS AND USAGE
LUTATHERA is a radiolabeled somatostatin analog indicated for the treatment of somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs), including foregut, midgut, and hindgut neuroendocrine tumors in adults. (1)
DOSAGE AND ADMINISTRATION
- Verify pregnancy status of females of reproductive potential prior to initiating LUTATHERA. (2.1)
- Administer 7.4 GBq (200 mCi) every 8 weeks (± 1 week) for a total of 4 doses. (2.2)
- Administer long-acting octreotide 30 mg intramuscularly 4 to 24 hours after each LUTATHERA dose and short-acting octreotide for symptomatic management. (2.3)
- Continue long-acting octreotide 30 mg intramuscularly every 4 weeks after completing LUTATHERA until disease progression or for 18 months following treatment initiation. (2.3)
- Administer antiemetics before recommended amino acid solution. (2.3)
- Initiate recommended intravenous amino acid solution 30 minutes before LUTATHERA infusion; continue during and for at least 3 hours after LUTATHERA infusion. Do not decrease dose of amino acid solution if LUTATHERA dose is reduced. (2.3)
DOSAGE FORMS AND STRENGTHS
Injection: 370 MBq/mL (10 mCi/mL) in single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Risk From Radiation Exposure: Minimize radiation exposure during and after treatment with LUTATHERA consistent with institutional good radiation safety practices and patient management procedures. (2.1, 5.1)
- Myelosuppression: Monitor blood cell counts. Withhold dose, reduce dose, or permanently discontinue based on the severity. (2.4, 5.2)
- Secondary Myelodysplastic Syndrome (MDS) and Leukemia: Median time to onset: MDS is 29 months; acute leukemia is 55 months. (5.3)
- Renal Toxicity: Advise patients to hydrate and to urinate frequently before, on the day of and the day after administration of LUTATHERA. Monitor serum creatinine and calculated creatinine clearance. Withhold dose, reduce dose, or permanently discontinue based on the severity. (2.3, 2.4, 5.4)
- Hepatotoxicity: Monitor transaminases, bilirubin, serum albumin and INR. (2.4, 5.5)
- Hypersensitivity Reactions: Monitor patients closely for signs and symptoms of hypersensitivity reactions, including anaphylaxis. Permanently discontinue LUTATHERA based on severity. (2.3, 2.4, 5.6)
- Neuroendocrine Hormonal Crisis: Monitor for flushing, diarrhea, hypotension, bronchoconstriction or other signs and symptoms. (5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females and males of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.8, 8.1, 8.3)
- Risk of Infertility: LUTATHERA may cause infertility. (5.9, 8.3)
ADVERSE REACTIONS
Most common Grade 3-4 adverse reactions (≥ 4% with a higher incidence in LUTATHERA arm) are lymphopenia, increased GGT, vomiting, nausea, increased AST, increased ALT, hyperglycemia and hypokalemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Safety Instructions
2.2 Recommended Dosage
2.3 Premedications and Concomitant Medications
2.4 Dosage Modifications for Adverse Reactions
2.5 Preparation and Administration
2.6 Radiation Dosimetry
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk from Radiation Exposure
5.2 Myelosuppression
5.3 Secondary Myelodysplastic Syndrome and Leukemia
5.4 Renal Toxicity
5.5 Hepatotoxicity
5.6 Hypersensitivity Reactions
5.7 Neuroendocrine Hormonal Crisis
5.8 Embryo-Fetal Toxicity
5.9 Risk of Infertility
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Somatostatin Analogs
7.2 Glucocorticoids
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
11.1 Physical Characteristics
11.2 External Radiation
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Progressive, Well-Differentiated Advanced or Metastatic Somatostatin Receptor-Positive Midgut Carcinoid Tumors
14.2 Somatostatin Receptor-Positive Gastroenteropancreatic Neuroendocrine Tumors
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Safety Instructions
LUTATHERA is a radiopharmaceutical; handle with appropriate safety measures to minimize radiation exposure [see Warnings and Precautions (5.1)]. Use waterproof gloves and effective radiation shielding when handling LUTATHERA. Radiopharmaceuticals, including LUTATHERA, should be used by or under the control of healthcare providers who are qualified by specific training and experience in the safe use and handling of radiopharmaceuticals, and whose experience and training have been approved by the appropriate governmental agency authorized to license the use of radiopharmaceuticals.
Verify pregnancy status of females of reproductive potential prior to initiating LUTATHERA [see Use in Specific Populations (8.1, 8.3)].
Monitor patients closely for signs and symptoms of hypersensitivity reactions during and following the LUTATHERA administration for a minimum of 2 hours in a setting where cardiopulmonary resuscitation medication and equipment are available [see Warnings and Precautions (5.6)].
2.2 Recommended Dosage
The recommended LUTATHERA dosage is 7.4 GBq (200 mCi) every 8 weeks (± 1 week) for a total of 4 doses. Administer premedications and concomitant medications as recommended [see Dosage and Administration (2.3)].
2.3 Premedications and Concomitant Medications
Somatostatin Analogs
- Before initiating LUTATHERA treatment: Discontinue long-acting somatostatin analogs (e.g., long-acting octreotide) at least 4 weeks prior to initiating LUTATHERA. Administer short-acting octreotide as needed; discontinue at least 24 hours prior to initiating LUTATHERA [see Drug Interactions (7.1)].
- During LUTATHERA treatment: Administer long-acting octreotide 30 mg intramuscularly between 4 to 24 hours after each LUTATHERA dose. Do not administer long-acting octreotide within 4 weeks prior to each subsequent LUTATHERA dose. Short-acting octreotide may be given for symptomatic management during LUTATHERA treatment but must be withheld at least 24 hours before each LUTATHERA dose.
- Following LUTATHERA treatment: Continue long-acting octreotide 30 mg intramuscularly every 4 weeks after completing LUTATHERA until disease progression or for 18 months following treatment initiation at the discretion of the physician.
Antiemetics
Administer antiemetics before the recommended amino acid solution.
Amino Acid Solution
Initiate an intravenous infusion of a sterile amino acid solution containing L-lysine and L-arginine (Table 1) 30 minutes before the start of the LUTATHERA infusion. Use a three-way valve to administer the amino acid solution using the same venous access as LUTATHERA or administer the amino acid solution through a separate venous access in the patient’s other arm. Continue the amino acid solution infusion during and for at least 3 hours after completion of the LUTATHERA infusion. Do not decrease the dose of the amino acid solution if a reduced dose of LUTATHERA is administered [see Warnings and Precautions (5.4)].
Table 1. Amino Acid Solution Item
Specification
aequivalent to 14.4 to 20 g L-lysine.
bequivalent to 14.9 to 20.7 g L-arginine.L-lysine HCl
Between 18 and 25 ga
L-arginine HCl
Between 18 and 25 gb
Volume
1 to 2 L
Osmolality
< 1200 mOsmol/kg
Hypersensitivity Prophylaxis
Premedicate patients who have had prior Grade 1 or 2 hypersensitivity reactions to LUTATHERA. Do not re-challenge patients who experience Grade 3 or 4 hypersensitivity reactions to LUTATHERA [see Warnings and Precautions (5.6)].
2.4 Dosage Modifications for Adverse Reactions
Recommended dose modifications of LUTATHERA for adverse reactions are provided in Table 2.
Table 2. Recommended Dosage Modifications of LUTATHERA for Adverse Reactions aGrading of severity is defined in the most current Common Terminology Criteria for Adverse Events (CTCAE).
bIncluding allergic reaction and anaphylaxis.
cNo dose modification required for hematological toxicities Grade 3 or Grade 4 solely due to lymphopenia.Adverse reaction
Severity of adverse reactiona
Dose modification
Thrombocytopenia
[see Warnings and Precautions (5.2)]First occurrence of Grade 2, 3, or 4
Withhold dose until complete or partial resolution (Grade 0 to 1).
Resume LUTATHERA at 3.7 GBq (100 mCi) in patients with complete or partial resolution. If reduced dose does not result in Grade 2, 3, or 4 thrombocytopenia, administer LUTATHERA at 7.4 GBq (200 mCi) as next dose.
Permanently discontinue LUTATHERA for Grade 2 or higher thrombocytopenia requiring a dosing interval beyond 16 weeks.
Recurrent Grade 2, 3, or 4
Permanently discontinue LUTATHERA.
Anemia and Neutropenia
[see Warnings and Precautions (5.2)]First occurrence of Grade 3 or 4
Withhold dose until complete or partial resolution (Grade 0, 1, or 2).
Resume LUTATHERA at 3.7 GBq (100 mCi) in patients with complete or partial resolution. If reduced dose does not result in Grade 3 or 4 anemia or neutropenia, administer LUTATHERA at 7.4 GBq (200 mCi) as next dose.
Permanently discontinue LUTATHERA for Grade 3 or higher anemia or neutropenia requiring a dosing interval beyond 16 weeks.
Recurrent Grade 3 or 4
Permanently discontinue LUTATHERA.
Renal Toxicity
[see Warnings and Precautions (5.4)]First occurrence of:
- Creatinine clearance less than 40 mL/min; calculated using Cockcroft-Gault formula with actual body weight, or
- 40% increase from baseline serum creatinine, or
- 40% decrease from baseline creatinine clearance; calculated using Cockcroft-Gault formula with actual body weight.
Withhold dose until resolution or return to baseline.
Resume LUTATHERA at 3.7 GBq (100 mCi) in patients with resolution or return to baseline. If reduced dose does not result in renal toxicity, administer LUTATHERA at 7.4 GBq (200 mCi) as next dose.
Permanently discontinue LUTATHERA for renal toxicity requiring a dosing interval beyond 16 weeks.
Recurrent renal toxicity
Permanently discontinue LUTATHERA.
Hepatotoxicity
[see Warnings and Precautions (5.5)]First occurrence of:
- Bilirubinemia greater than 3 times the upper limit of normal (Grade 3 or 4), or
- Serum albumin less than 30 g/L with international normalized ratio (INR) > 1.5.
Withhold dose until resolution or return to baseline.
Resume LUTATHERA at 3.7 GBq (100 mCi) in patients with resolution or return to baseline. If reduced LUTATHERA dose does not result in hepatotoxicity, administer LUTATHERA at 7.4 GBq (200 mCi) as next dose.
Permanently discontinue LUTATHERA for hepatotoxicity requiring a dosing interval beyond 16 weeks.
Recurrent hepatotoxicity
Permanently discontinue LUTATHERA.
Hypersensitivity Reactionsb
[see Warnings and Precautions (5.6)]First occurrence of Grade 3 or 4 Permanently discontinue LUTATHERA. Any Other Adverse Reactionsc
[see Adverse Reactions (6.1)]First occurrence of Grade 3 or 4
Withhold dose until complete or partial resolution (Grade 0 to 2).
Resume LUTATHERA at 3.7 GBq (100 mCi) in patients with complete or partial resolution. If reduced dose does not result in Grade 3 or 4 toxicity, administer LUTATHERA at 7.4 GBq (200 mCi) as next dose.
Permanently discontinue LUTATHERA for Grade 3 or higher adverse reactions requiring a dosing interval beyond 16 weeks.
Recurrent Grade 3 or 4
Permanently discontinue LUTATHERA.
2.5 Preparation and Administration
Preparation Instructions
- Use aseptic technique and radiation shielding when handling or administering the LUTATHERA solution. Use tongs when handling the vial to minimize radiation exposure.
- Inspect the product visually under a shielded screen for particulate matter and discoloration prior to administration. Discard the vial if particulates and/or discoloration are present.
- Do not inject the LUTATHERA solution directly into any other intravenous solution.
- Confirm the amount of radioactivity of LUTATHERA delivered to the patient with an appropriate dose calibrator prior to and after each LUTATHERA administration.
- Dispose of any unused medicinal product or waste material in accordance with local and federal laws.
Administration Instructions
- The gravity method, peristaltic pump method, or the syringe pump method may be used for the administration of the recommended dosage. Do not administer LUTATHERA as an intravenous bolus.
- When using the gravity or peristaltic pump method, LUTATHERA should be infused directly from its original container.
- Use the peristaltic pump or syringe pump method when administering a reduced dose of LUTATHERA following a dosage modification for an adverse reaction. When using the gravity method for a reduced dose, adjust the LUTATHERA dose before the administration to avoid the delivery of an incorrect volume of LUTATHERA.
Intravenous Methods of Administration
Instructions for the Gravity Method
- Insert a 2.5 cm, 20-gauge needle (short needle) into the LUTATHERA vial and connect via a catheter to 500 mL 0.9% Sodium Chloride Injection, USP (used to transport the LUTATHERA solution during the infusion). Ensure that the short needle does not touch the LUTATHERA solution in the vial and do not connect this short needle directly to the patient. Do not allow the 0.9% Sodium Chloride Injection, USP to flow into the LUTATHERA vial prior to the initiation of the LUTATHERA infusion and do not inject the LUTATHERA solution directly into the 0.9% Sodium Chloride Injection, USP.
- Insert a second needle that is 9 cm, 18-gauge (long needle) into the LUTATHERA vial ensuring that this long needle touches and is secured to the bottom of the LUTATHERA vial during the entire infusion. Connect the long needle to the patient by an intravenous catheter that is pre-filled with 0.9% Sodium Chloride Injection, USP and that is used for the LUTATHERA infusion into the patient.
- Use a clamp or an infusion pump to regulate the flow of the 0.9% Sodium Chloride Injection, USP via the short needle into the LUTATHERA vial at a rate of 50 mL/hour to 100 mL/hour for 5 to 10 minutes and then 200 mL/hour to 300 mL/hour for an additional 25 to 30 minutes (the 0.9% Sodium Chloride Injection, USP entering the vial through the short needle will carry the LUTATHERA solution from the vial to the patient via the intravenous catheter connected to the long needle over a total duration of 30 to 40 minutes).
- During the infusion, ensure that the level of solution in the LUTATHERA vial remains constant.
- Disconnect the vial from the long needle line and clamp the 0.9% Sodium Chloride Injection, USP line once the level of radioactivity is stable for at least five minutes.
- Follow the infusion with an intravenous flush of 25 mL of 0.9% Sodium Chloride Injection, USP through the intravenous catheter to the patient.
Instructions for the Peristaltic Pump Method
- Insert a filtered 2.5 cm, 20-gauge needle (short venting needle) into the LUTATHERA vial. Ensure that the short needle does not touch the LUTATHERA solution in the vial and do not connect this short needle directly to the patient or to the peristaltic pump.
- Insert a second needle that is 9 cm, 18 gauge (long needle) into the LUTATHERA vial ensuring that the long needle touches and is secured to the bottom of the LUTATHERA vial during the entire infusion. Connect the long needle and a 0.9% Sodium Chloride Injection, USP to a 3-way stopcock valve via appropriate tubing.
- Connect the output of the 3-way stopcock valve to tubing installed on the input side of the peristaltic pump according to manufacturer’s instructions.
- Prime the line by opening the 3-way stopcock valve and pumping the LUTATHERA solution through the tubing until it reaches the exit of the valve.
- Prime the intravenous catheter that will be connected to the patient by opening the 3-way stopcock valve to the 0.9% Sodium Chloride Injection, USP and pumping the 0.9% Sodium Chloride Injection, USP until it exits the end of the catheter tubing.
- Connect the primed intravenous catheter to the patient and set the 3-way stopcock valve such that the LUTATHERA solution is in line with the peristaltic pump.
- Infuse an appropriate volume of LUTATHERA solution over a 30-40 min period to deliver the desired radioactivity.
- When the desired LUTATHERA radioactivity has been delivered, stop the peristaltic pump and then change the position of the 3-way stopcock valve so that the peristaltic pump is in line with the 0.9% Sodium Chloride Injection, USP. Restart the peristaltic pump and infuse an intravenous flush of 25 mL of 0.9% Sodium Chloride Injection, USP through the intravenous catheter to the patient.
Instructions for the Syringe Pump Method
- Withdraw an appropriate volume of LUTATHERA solution to deliver the desired radioactivity by using a disposable syringe fitted with a syringe shield and a disposable sterile needle that is 9 cm, 18 gauge (long needle). To aid the withdrawal of the solution, a filtered 2.5 cm, 20-gauge needle (short venting needle) can be used to reduce the resistance from the pressurized vial. Ensure that the short needle does not touch the LUTATHERA solution in the vial.
- Fit the syringe into the shielded pump and include a 3-way stopcock valve between the syringe and an intravenous catheter pre-filled with 0.9% Sodium Chloride Injection, USP and used for LUTATHERA administration to the patient.
- Infuse an appropriate volume of LUTATHERA solution over a 30-40 min period to deliver the desired radioactivity.
- When the desired LUTATHERA radioactivity has been delivered, stop the syringe pump and then change the position of the 3-way stopcock valve to flush the syringe with 25 mL of 0.9% Sodium Chloride Injection, USP. Restart the syringe pump.
- After the flush of the syringe has been completed, perform an intravenous flush with 25 mL of 0.9% Sodium Chloride Injection, USP through the intravenous catheter to the patient.
2.6 Radiation Dosimetry
The mean and standard deviation (SD) of the estimated radiation absorbed doses for adults receiving LUTATHERA are shown in Table 3. The maximum penetration of lutetium-177 in tissue is 2.2 mm and the mean penetration is 0.67 mm.
Table 3. Estimated Radiation Absorbed Dose for LUTATHERA in NETTER-1 aN = 18 (two patients excluded because the liver absorbed dose was biased by the uptake of the liver metastases).
bN = 9 (female patients only).
cN = 11 (male patients only).Absorbed dose per unit activity
(Gy/GBq)
(N = 20)
Calculated absorbed dose for 4 x 7.4 GBq
(29.6 GBq cumulative activity)
(Gy)
Organ
Mean
SD
Mean
SD
Adrenals
0.037
0.016
1.1
0.5
Brain
0.027
0.016
0.8
0.5
Breasts
0.027
0.015
0.8
0.4
Gallbladder wall
0.042
0.019
1.2
0.6
Heart wall
0.032
0.015
0.9
0.4
Kidneys
0.654
0.295
19.4
8.7
Livera
0.299
0.226
8.9
6.7
Lower large intestine wall
0.029
0.016
0.9
0.5
Lungs
0.031
0.015
0.9
0.4
Muscle
0.029
0.015
0.8
0.4
Osteogenic cells
0.151
0.268
4.5
7.9
Ovariesb
0.031
0.013
0.9
0.4
Pancreas
0.038
0.016
1.1
0.5
Red marrow
0.035
0.029
1.0
0.8
Skin
0.027
0.015
0.8
0.4
Small intestine
0.031
0.015
0.9
0.5
Spleen
0.846
0.804
25.1
23.8
Stomach wall
0.032
0.015
0.9
0.5
Testesc
0.026
0.018
0.8
0.5
Thymus
0.028
0.015
0.8
0.5
Thyroid
0.027
0.016
0.8
0.5
Total body
0.052
0.027
1.6
0.8
Upper large intestine wall
0.032
0.015
0.9
0.4
Urinary bladder wall
0.437
0.176
12.8
5.3
Uterusb
0.032
0.013
1.0
0.4
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk from Radiation Exposure
LUTATHERA contributes to a patient’s overall long-term cumulative radiation exposure. Long-term cumulative radiation exposure is associated with an increased risk for cancer.
Radiation can be detected in the urine for up to 30 days following LUTATHERA administration. Minimize radiation exposure to patients, medical personnel, and household contacts during and after treatment with LUTATHERA consistent with institutional good radiation safety practices, patient management procedures, Nuclear Regulatory Commission patient-release guidance, and instructions to the patient for follow-up radiation protection at home [see Dosage and Administration (2.1), Clinical Pharmacology (12.3)].
5.2 Myelosuppression
In NETTER-1, myelosuppression occurred more frequently in patients receiving LUTATHERA with long-acting octreotide compared to patients receiving high-dose long-acting octreotide (all Grades/Grade 3 or 4): anemia (81%/0) versus (54%/1%); thrombocytopenia (53%/1%) versus (17%/0); and neutropenia (26%/3%) versus (11%/0). In NETTER-1, platelet nadir occurred at a median of 5.1 months following the first dose. Of the 59 patients who developed thrombocytopenia, 68% had platelet recovery to baseline or normal levels. The median time to platelet recovery was 2 months. Fifteen of the nineteen patients in whom platelet recovery was not documented had post-nadir platelet counts. Among these 15 patients, 5 improved to Grade 1, 9 to Grade 2, and 1 to Grade 3.
Monitor blood cell counts. Withhold dose, reduce dose, or permanently discontinue LUTATHERA based on the severity of myelosuppression [see Dosage and Administration (2.4)].
5.3 Secondary Myelodysplastic Syndrome and Leukemia
In NETTER-1, with a median follow-up time of 76 months in the main study, myelodysplastic syndrome (MDS) was reported in 2.3% of patients receiving LUTATHERA with long-acting octreotide compared to no patients receiving high-dose long-acting octreotide.
In ERASMUS, 16 patients (2.0%) developed MDS and 4 (0.5%) developed acute leukemia. The median time to onset was 29 months (9 to 45 months) for MDS and 55 months (32 to 125 months) for acute leukemia.
5.4 Renal Toxicity
In ERASMUS, 8 patients (< 1%) developed renal failure 3 to 36 months following LUTATHERA. Two of these patients had underlying renal impairment or risk factors for renal failure (e.g., diabetes or hypertension) and required dialysis.
Administer the recommended amino acid solution before, during and after LUTATHERA [see Dosage and Administration (2.3)] to decrease the reabsorption of lutetium Lu 177 dotatate through the proximal tubules and decrease the radiation dose to the kidneys. Advise patients to hydrate and to urinate frequently before, on the day of, and the day after administration of LUTATHERA.
Monitor serum creatinine and calculated creatinine clearance. Withhold dose, reduce dose, or permanently discontinue LUTATHERA based on the severity of renal toxicity [see Dosage and Administration (2.4)].
Patients with baseline renal impairment may be at increased risk of toxicity due to increased radiation exposure [see Use in Specific Populations (8.6)].
5.5 Hepatotoxicity
In ERASMUS, 2 patients (< 1%) were reported to have hepatic tumor hemorrhage, edema, or necrosis, with one patient experiencing intrahepatic congestion and cholestasis. Patients with hepatic metastasis may be at increased risk of hepatotoxicity due to radiation exposure.
Monitor transaminases, bilirubin, serum albumin and international normalized ratio (INR) during treatment. Withhold dose, reduce dose, or permanently discontinue LUTATHERA based on the severity of hepatotoxicity [see Dosage and Administration (2.4)].
5.6 Hypersensitivity Reactions
Hypersensitivity reactions, including angioedema, occurred in patients treated with LUTATHERA [see Adverse Reactions (6.2)]. Monitor patients closely for signs and symptoms of hypersensitivity reactions, including anaphylaxis, during and following LUTATHERA administration for a minimum of 2 hours in a setting where cardiopulmonary resuscitation medication and equipment are available. Discontinue the infusion upon the first observation of any signs or symptoms consistent with a severe hypersensitivity reaction and initiate appropriate therapy.
Premedicate patients with a history of Grade 1 or 2 hypersensitivity reactions to LUTATHERA before subsequent doses [see Dosage and Administration (2.3)]. Permanently discontinue LUTATHERA in patients who experience Grade 3 or 4 hypersensitivity reactions [see Dosage and Administration (2.4)].
5.7 Neuroendocrine Hormonal Crisis
Neuroendocrine hormonal crises, manifesting with flushing, diarrhea, bronchospasm and hypotension, occurred in < 1% of patients in ERASMUS and typically occurred during or within 24 hours following the initial LUTATHERA dose. Two (< 1%) patients were reported to have hypercalcemia.
Monitor patients for flushing, diarrhea, hypotension, bronchoconstriction or other signs and symptoms of tumor-related hormonal release. Administer intravenous somatostatin analogs, fluids, corticosteroids, and electrolytes as indicated.
5.8 Embryo-Fetal Toxicity
Based on its mechanism of action, LUTATHERA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on LUTATHERA use in pregnant women. No animal studies using lutetium Lu 177 dotatate have been conducted to evaluate its effect on female reproduction and embryo-fetal development; however, radioactive emissions, including those from LUTATHERA, can cause fetal harm.
Verify pregnancy status of females of reproductive potential prior to initiating LUTATHERA [see Dosage and Administration (2.1)].
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LUTATHERA and for 7 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with LUTATHERA and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
5.9 Risk of Infertility
LUTATHERA may cause infertility in males and females. The recommended cumulative dose of 29.6 GBq of LUTATHERA results in a radiation absorbed dose to the testes and ovaries within the range where temporary or permanent infertility can be expected following external beam radiotherapy [see Dosage and Administration (2.6), Use in Specific Populations (8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Myelosuppression [see Warnings and Precautions (5.2)]
- Secondary Myelodysplastic Syndrome and Leukemia [see Warnings and Precautions (5.3)]
- Renal Toxicity [see Warnings and Precautions (5.4)]
- Hepatotoxicity [see Warnings and Precautions (5.5)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.6)]
- Neuroendocrine Hormonal Crisis [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in Warnings and Precautions reflect exposure to LUTATHERA in 111 patients with advanced, progressive midgut neuroendocrine tumors (NETTER-1). Safety data in Warnings and Precautions were also obtained in an additional 22 patients in a non-randomized pharmacokinetic sub-study of NETTER-1 and in a subset of patients (811 of 1214) with advanced somatostatin receptor-positive tumors enrolled in ERASMUS [see Warnings and Precautions (5)].
NETTER-1
The safety data of LUTATHERA with octreotide was evaluated in NETTER-1 [see Clinical Studies (14.1)]. Patients with progressive, somatostatin receptor-positive midgut carcinoid tumors received LUTATHERA 7.4 GBq (200 mCi) administered every 8 to 16 weeks concurrently with the recommended amino acid solution and with long-acting octreotide (30 mg administered by intramuscular injection within 24 hours of each LUTATHERA dose) (N = 111), or high-dose octreotide (defined as long-acting octreotide 60 mg by intramuscular injection every 4 weeks) (N = 112) [see Clinical Studies (14.1)]. Among patients receiving LUTATHERA with octreotide, 79% received a cumulative dose > 22.2 GBq (> 600 mCi) and 76% of patients received all four planned doses. Six percent (6%) of patients required a dose reduction and 13% of patients discontinued LUTATHERA. Five patients discontinued LUTATHERA for renal-related events and 4 discontinued for hematological toxicities.
Table 4 and Table 5 summarize the incidence of adverse reactions and laboratory abnormalities, respectively. The most common Grade 3-4 adverse reactions occurring with a greater frequency among patients receiving LUTATHERA with octreotide compared to patients receiving high-dose octreotide include: lymphopenia (44%), increased gamma-glutamyltransferase (GGT) (20%), vomiting (7%), nausea and increased aspartate aminotransferase (AST) (5% each), and increased alanine aminotransferase (ALT), hyperglycemia and hypokalemia (4% each).
Table 4. Adverse Reactions Occurring at Higher Incidence in Patients Receiving LUTATHERA with Long-Acting Octreotide Compared to High-Dose Long-Acting Octreotide (Between Arm Difference of ≥ 5% All Grades or ≥ 2% Grades 3-4)a aNational Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. Only displays adverse reactions occurring at a higher incidence in LUTATHERA-treated patients [between arm difference of ≥ 5% (all Grades) or ≥ 2% (Grades 3-4)].
bIncludes the terms: Glomerular filtration rate decreased, acute kidney injury, acute prerenal failure, azotemia, renal disorder, renal failure, renal impairment.
cIncludes the terms: Dysuria, micturition urgency, nocturia, pollakiuria, renal colic, renal pain, urinary tract pain and urinary incontinence.Adverse reactiona
LUTATHERA with Long-acting Octreotide
(30 mg)
(N = 111)Long-acting Octreotide
(60 mg)
(N = 112)All Grades
%Grades 3-4
%All Grades
%Grades 3-4
%Gastrointestinal disorders
Nausea
65
5
12
2
Vomiting
53
7
10
0
Abdominal pain
26
3
19
3
Diarrhea
26
3
18
1
Constipation
10
0
5
0
General disorders
Fatigue
38
1
26
2
Peripheral edema
16
0
9
1
Pyrexia
8
0
3
0
Metabolism and nutrition disorders
Decreased appetite
21
0
11
3
Nervous system disorders
Headache 17 0 5 0 Dizziness 17 0 8 0 Dysgeusia 8 0 2 0 Vascular disorders Flushing 14 1 9 0 Hypertension 12 2 7 2 Musculoskeletal and connective tissue disorders
Back pain
13
2
10
0
Pain in extremity
11
0
5
0
Myalgia
5
0
0
0
Neck pain
5
0
0
0
Renal and urinary disorders
Renal failureb
13
3
4
1
Radiation-related urinary tract adverse reactionsc
8
0
3
0
Psychiatric disorders
Anxiety
12
1
5
0
Skin and subcutaneous tissue disorders
Alopecia
12
0
2
0
Respiratory, thoracic and mediastinal disorders
Cough
11
1
6
0
Cardiac disorders
Atrial fibrillation
5
1
0
0
Table 5. Laboratory Abnormalities Occurring at Higher Incidence in Patients Receiving LUTATHERA with Long-Acting Octreotide Compared to High-Dose Long-Acting Octreotide (Between Arm Difference of ≥ 5% All Grades or ≥ 2% Grades 3-4)a,b aValues are worst grade observed after randomization.
bNational Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. Only displays laboratory abnormalities occurring at a higher incidence in LUTATHERA-treated patients [between arm difference of ≥ 5% (all Grades) or ≥ 2% (Grades 3-4)].Laboratory abnormalityb LUTATHERA with Long-acting Octreotide
(30 mg)
(N = 111)Long-acting Octreotide
(60 mg)
(N = 112)All Grades
%Grades 3-4
%All Grades
%Grades 3-4
%Hematology
Lymphopenia
90
44
39
5
Anemia
81
0
55
1
Leukopenia
55
2
20
0
Thrombocytopenia
53
1
17
0
Neutropenia
26
3
11
0
Renal/Metabolic
Creatinine increased
85
1
73
0
Hyperglycemia
82
4
67
2
Hyperuricemia
34
6
30
6
Hypocalcemia
32
0
14
0
Hypokalemia
26
4
21
2
Hyperkalemia
19
0
11
0
Hypernatremia
17
0
7
0
Hypoglycemia
15
0
8
0
Hepatic
GGT increased
66
20
67
16
Alkaline phosphatase increased
65
5
55
9
AST increased
50
5
35
0
ALT increased
43
4
34
0
Blood bilirubin increased
30
2
28
0
ERASMUS
Safety data are available from 1214 patients in ERASMUS, an international, single-institution, single-arm, open-label trial of patients with somatostatin receptor-positive tumors (neuroendocrine and other primaries). Patients received LUTATHERA 7.4 GBq (200 mCi) administered every 6 to 13 weeks with or without octreotide. Retrospective medical record review was conducted on a subset of 811 patients to document serious adverse reactions. Eighty-one (81%) percent of patients in the subset received a cumulative dose ≥ 22.2 GBq (≥ 600 mCi). With a median follow-up time of more than 4 years, the following rates of serious adverse reactions were reported: myelodysplastic syndrome (2%), acute leukemia (1%), renal failure (2%), hypotension (1%), cardiac failure (2%), myocardial infarction (1%), and neuroendocrine hormonal crisis (1%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of LUTATHERA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Immune System Disorders: hypersensitivity reactions, including angioedema
-
7 DRUG INTERACTIONS
7.1 Somatostatin Analogs
Somatostatin and its analogs competitively bind to somatostatin receptors and may interfere with the efficacy of LUTATHERA. Discontinue long-acting somatostatin analogs at least 4 weeks and short-acting octreotide at least 24 hours prior to each LUTATHERA dose. Administer short- and long-acting octreotide during LUTATHERA treatment as recommended [see Dosage and Administration (2.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, LUTATHERA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on LUTATHERA use in pregnant women. No animal studies using lutetium Lu 177 dotatate have been conducted to evaluate its effect on female reproduction and embryo-fetal development; however, radioactive emissions, including those from LUTATHERA, can cause fetal harm. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of lutetium Lu 177 dotatate in human milk, or its effects on the breastfed child or milk production. No lactation studies in animals were conducted. Because of the potential risk for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with LUTATHERA and for 2.5 months after the last dose.
8.3 Females and Males of Reproductive Potential
Based on mechanism of action, LUTATHERA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiating LUTATHERA [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with LUTATHERA and for 7 months after the last dose.
Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with LUTATHERA and for 4 months after the last dose [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)].
Infertility
The recommended cumulative dose of 29.6 GBq of LUTATHERA results in a radiation absorbed dose to the testes and ovaries within the range where temporary or permanent infertility can be expected following external beam radiotherapy [see Dosage and Administration (2.6)].
8.4 Pediatric Use
The safety and effectiveness of LUTATHERA have not been established in pediatric patients.
8.5 Geriatric Use
Of the 1325 patients treated with LUTATHERA in clinical trials, 438 patients (33%) were 65 years and older. The response rate and the number of patients with a serious adverse event were similar to those of younger subjects.
8.6 Renal Impairment
No dose adjustment is recommended for patients with baseline mild to moderate (creatinine clearance 30 to 89 mL/min by Cockcroft-Gault formula) renal impairment. However, patients with baseline mild or moderate renal impairment may be at greater risk of toxicity, including renal toxicity, due to increased radiation exposure. Perform more frequent assessments of renal function in patients with baseline mild to moderate impairment. The pharmacokinetic profile and safety of LUTATHERA in patients with baseline severe renal impairment (creatinine clearance < 30 mL/min by Cockcroft-Gault formula) or end-stage renal disease have not been studied [see Warnings and Precautions (5.4)].
8.7 Hepatic Impairment
No dose adjustment is recommended for patients with baseline mild or moderate hepatic impairment. The pharmacokinetic profile and safety of LUTATHERA in patients with baseline severe hepatic impairment (total bilirubin > 3 times upper limit of normal, regardless of AST level) have not been studied.
-
11 DESCRIPTION
Lutetium Lu 177 dotatate is a radiolabeled somatostatin analog. The drug substance lutetium Lu 177 dotatate is a cyclic peptide linked with the covalently bound chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid to a radionuclide.
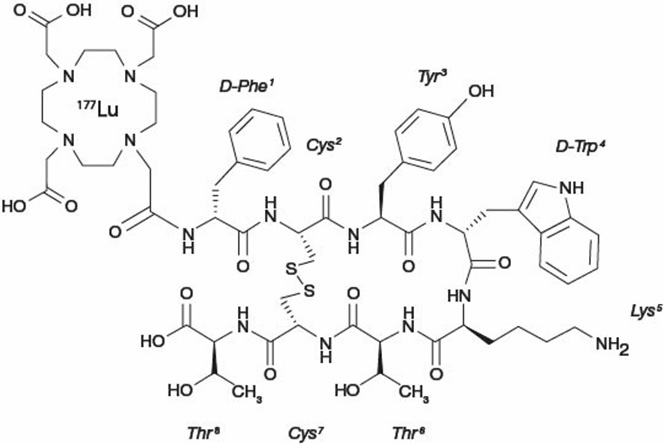
Lutetium Lu 177 dotatate is described as lutetium (Lu 177)-N-[(4,7,10-Tricarboxymethyl-1,4,7,10-tetraazacyclododec-1-yl) acetyl]-D-phenylalanyl-L-cysteinyl-L-tyrosyl-D-tryptophanyl-L-lysyl-L-threoninyl-L-cysteinyl-L-threonine-cyclic (2-7) disulfide. The molecular weight is 1609.6 Daltons and the structural formula is as follows:
LUTATHERA (lutetium Lu 177 dotatate) 370 MBq/mL (10 mCi/mL) Injection is a sterile, clear, colorless to slightly yellow solution for intravenous use. Each single-dose vial contains acetic acid (0.48 mg/mL), sodium acetate (0.66 mg/mL), gentisic acid (0.63 mg/mL), sodium hydroxide (0.64 mg/mL), ascorbic acid (2.8 mg/mL), diethylene triamine pentaacetic acid (0.05 mg/mL), sodium chloride (6.85 mg/mL), and Water for Injection (ad 1 mL). The pH range of the solution is 4.5 to 6.
11.1 Physical Characteristics
Lutetium-177 decays to stable hafnium-177 with a half-life of 6.647 days, by emitting beta-minus radiation with a maximum energy of 0.498 MeV (79%) and photonic radiation (γ) of 0.208 MeV (11%) and 0.113 MeV (6.2%). The main radiations of lutetium-177 are detailed in Table 6.
Table 6. Lutetium-177 Main Radiations Radiation
Energy (keV)
Iβ-%
Iγ%
β-
176.5
12.2
β-
248.1
0.05
β-
384.9
9.1
β-
497.8
78.6
γ
71.6
0.15
γ
112.9
6.20
γ
136.7
0.05
γ
208.4
11.0
γ
249.7
0.21
γ
321.3
0.22
11.2 External Radiation
Table 7 summarizes the radioactive decay properties of lutetium-177.
Table 7. Physical Decay Chart: Lutetium-177 Physical Half-Life = 6.647 Days Hours
Fraction remaining
Hours
Fraction remaining
0
1.000
48 (2 days)
0.812
1
0.996
72 (3 days)
0.731
2
0.991
168 (7 days)
0.482
5
0.979
336 (14 days)
0.232
10
0.958
720 (30 days)
0.044
24 (1 day)
0.901
1080 (45 days)
0.009
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lutetium Lu 177 dotatate binds to somatostatin receptors with highest affinity for subtype 2 somatostatin receptors (SSTR2). Upon binding to somatostatin receptor expressing cells, including malignant somatostatin receptor-positive tumors, the compound is internalized. The beta-minus emission from lutetium-177 induces cellular damage by formation of free radicals in somatostatin receptor-positive cells and in neighboring cells.
12.2 Pharmacodynamics
Lutetium Lu 177 dotatate exposure-response relationships and the time course of pharmacodynamics response are unknown.
Cardiac Electrophysiology
The ability of LUTATHERA to prolong the QTc interval at the recommended dose was assessed in an open-label study in 20 patients with somatostatin receptor-positive midgut carcinoid tumors. No large changes in the mean QTc interval (i.e., > 20 ms) were detected.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of lutetium Lu 177 dotatate have been characterized in patients with progressive, somatostatin receptor-positive neuroendocrine tumors. The mean blood exposure (area under the curve) of lutetium Lu 177 dotatate at the recommended dose is 41 ng.h/mL [coefficient of variation (CV) 36%]. The mean maximum blood concentration (Cmax) for lutetium Lu 177 dotatate is 10 ng/mL (CV 50%), which generally occurred at the end of the LUTATHERA infusion.
Distribution
The mean volume of distribution (Vz) for lutetium Lu 177 dotatate is 460 L (CV 54%).
The non-radioactive lutetium Lu 175 dotatate is 43% bound to human plasma proteins.
Within 4 hours after administration, lutetium Lu 177 dotatate distributes in kidneys, tumor lesions, liver, spleen, and, in some patients, pituitary gland and thyroid. The co-administration of amino acids reduced the median radiation dose to the kidneys by 47% (34% to 59%) and increased the mean beta-phase blood clearance of lutetium Lu 177 dotatate by 36%.
Elimination
The mean clearance (CL) is 4.5 L/h (CV 31%) and the mean terminal half-life is 71 (±28) hours for lutetium 177 dotatate.
Metabolism
Lutetium Lu 177 dotatate does not undergo hepatic metabolism.
Excretion
Lutetium Lu 177 dotatate is primarily eliminated renally with cumulative excretion of 44% within 5 hours, 58% within 24 hours, and 65% within 48 hours following LUTATHERA administration. Prolonged elimination of lutetium Lu 177 dotatate in the urine is expected; however, based on the half-life of lutetium-177 and terminal half-life of lutetium Lu 177 dotatate, greater than 99% of the administered radioactivity will be eliminated within 14 days after administration of LUTATHERA [see Warnings and Precautions (5.1)].
Drug Interaction Studies
In Vitro Studies
CYP450 enzymes: The non-radioactive lutetium Lu 175 dotatate is not an inhibitor or inducer of cytochrome P450 (CYP) 1A2, 2B6, 2C9, 2C19 or 2D6 in vitro.
Transporters: The non-radioactive lutetium Lu 175 dotatate is not an inhibitor of P-glycoprotein, BCRP, OAT1, OAT3, OCT1, OCT2, OATP1B1, or OATP1B3 in vitro.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity and mutagenicity studies have not been conducted with lutetium Lu 177 dotatate; however, radiation is a carcinogen and mutagen.
No animal studies were conducted to determine the effects of lutetium Lu 177 dotatate on fertility.
13.2 Animal Toxicology and/or Pharmacology
The primary target organ in animal studies using the non-radioactive lutetium Lu 175 dotatate was the pancreas, a high SSTR2 expressing organ. Pancreatic acinar apoptosis occurred at lutetium Lu 175 dotatate doses ≥ 5 mg/kg in repeat dose toxicology studies in rats. Pancreatic acinar cell atrophy also occurred in repeat dose toxicology studies in dogs at doses ≥ 500 mcg/kg. These findings were consistent with high uptake of the radiolabeled peptide in the pancreas in animal biodistribution studies.
-
14 CLINICAL STUDIES
14.1 Progressive, Well-Differentiated Advanced or Metastatic Somatostatin Receptor-Positive Midgut Carcinoid Tumors
The efficacy of LUTATHERA in patients with progressive, well-differentiated, locally advanced/inoperable or metastatic somatostatin receptor-positive midgut carcinoid tumors was established in NETTER-1 (NCT01578239), a randomized, multicenter, open-label, active-controlled trial. Key eligibility criteria included Ki67 index ≤ 20%, Karnofsky performance status ≥ 60, confirmed presence of somatostatin receptors on all lesions (OctreoScan uptake ≥ normal liver), creatinine clearance ≥ 50 mL/min, no prior treatment with peptide receptor radionuclide therapy (PRRT), and no prior external beam radiation therapy to more than 25% of the bone marrow.
At the time of the primary analysis, 229 patients were randomized (1:1) to receive either LUTATHERA 7.4 GBq (200 mCi) every 8 weeks (±1 week) for up to 4 administrations (maximum cumulative dose of 29.6 GBq) or high-dose long-acting octreotide (defined as 60 mg by intramuscular injection every 4 weeks). Patients in the LUTATHERA arm also received long-acting octreotide 30 mg as an intramuscular injection 4 to 24 hours after each LUTATHERA dose and every 4 weeks after completion of LUTATHERA treatment until disease progression or until week 76 of the study. Patients in both arms could receive short-acting octreotide for symptom management; however, short-acting octreotide was withheld at least 24 hours before each LUTATHERA dose. Randomization was stratified by OctreoScan tumor uptake score (Grade 2, 3 or 4) and the length of time that patients had been on the most recent constant dose of octreotide prior to randomization (≤ 6 or > 6 months). The major efficacy outcome measure was progression-free survival (PFS) as determined by a blinded independent review committee (IRC) per RECIST v1.1. Additional efficacy outcome measures were overall response rate (ORR) by IRC, duration of response (DoR) by IRC, and overall survival (OS).
Demographic and baseline disease characteristics were balanced between the treatment arms. Of the 229 patients, 82% were White, 4% were Black, 3% were Hispanic or Latino, 0.4% were Asian, 0.4% were Other, and 9% were not reported. The median age was 64 years (28 to 87 years); 51% were male, 74% had an ileal primary, and 96% had metastatic disease in the liver. The median Karnofsky performance score was 90 (60 to 100), 74% received a constant dose of octreotide for > 6 months and 12% received prior treatment with everolimus. Sixty-nine percent of patients had Ki67 expression in ≤ 2% of tumor cells, 77% had CgA > 2 times the upper limit of normal (ULN), 65% had 5-HIAA > 2 times ULN, and 65% had alkaline phosphatase ≤ ULN.
At the time of the final OS analysis, which occurred 66 months after the primary PFS analysis, 117 patients were randomized to the LUTATHERA arm and 114 patients were randomized to the octreotide arm. In the final OS analysis, there was no statistically significant difference in OS between the two treatment arms.
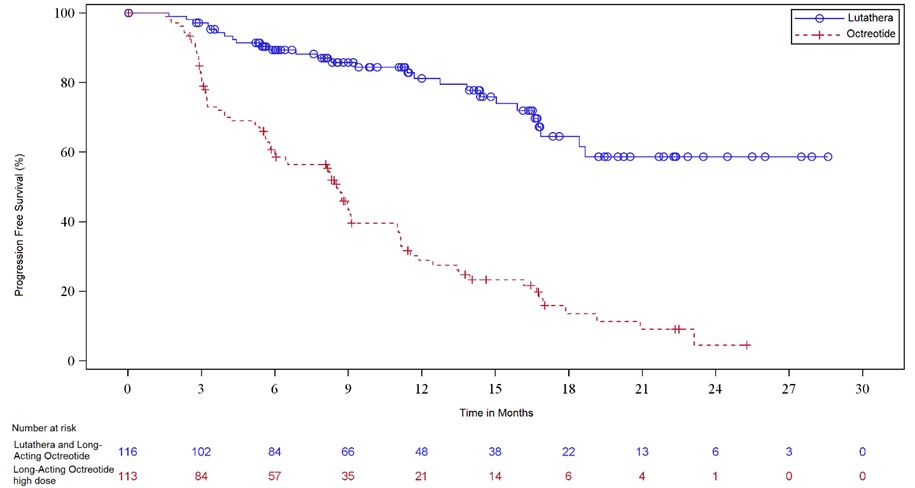
Efficacy results for NETTER-1 are presented in Table 8 and Figure 1.
Table 8. Efficacy Results in NETTER-1 Abbreviations: CI, confidence interval; IRC, independent radiology committee; NE, not evaluable; NR, not reached; ORR, overall response rate; PFS, progression-free survival.
aHazard ratio based on the unstratified Cox model.
bUnstratified log rank test.
cFisher’s exact test.LUTATHERA with Long-acting Octreotide
(30 mg)
N = 116Long-acting Octreotide
(60 mg)
N = 113PFS by IRC Events (%) 27 (23%) 78 (69%) Progressive disease, n (%) 15 (13%) 61 (54%) Death, n (%) 12 (10%) 17 (15%) Median in months (95% CI) NR (18.4, NE) 8.5 (6.0, 9.1) Hazard ratioa (95% CI) 0.21 (0.13, 0.32) p-valueb < 0.0001 ORR by IRC ORR, % (95% CI) 13% (7%,19%) 4% (0.1%, 7%) Complete response rate, n (%) 1 (1%) 0 Partial response rate, n (%) 14 (12%) 4 (4%) p-valuec 0.0148 Duration of response, median in months (95% CI) NR (2.8, NE) 1.9 (1.9, NE) Figure 1. Kaplan-Meier Curves for Progression-Free Survival in NETTER-1
14.2 Somatostatin Receptor-Positive Gastroenteropancreatic Neuroendocrine Tumors
The efficacy of LUTATHERA in patients with foregut, midgut, and hindgut gastroenteropancreatic neuroendocrine tumors (GEP-NETs) was assessed in 360 patients in the ERASMUS study. In ERASMUS, LUTATHERA was initially provided as expanded access under a general peptide receptor radionuclide therapy protocol at a single site in the Netherlands. A subsequent LUTATHERA-specific protocol written eight years after study initiation did not describe a specific sample size or hypothesis testing plan but allowed for retrospective data collection. A total of 1214 patients received LUTATHERA in ERASMUS, of whom 578 patients had baseline tumor assessments. Of the 578 patients, 360 (62%) had GEP-NETs and long-term follow-up. Of these 360 patients, 145 (40%) had their tumors prospectively evaluated according to RECIST criteria. LUTATHERA 7.4 GBq (200 mCi) was administered every 6 to 13 weeks for up to 4 doses concurrently with the recommended amino acid solution. The major efficacy outcome was investigator-assessed ORR. The median age in the efficacy subset was 60 years (30 to 85 years), 51% were male, 71% had a baseline Karnofsky performance status ≥ 90, 51% had progressed within 12 months of treatment, and 7% had received prior chemotherapy. Fifty-two percent (52%) of patients received a concomitant somatostatin analog. The median dose of LUTATHERA was 29.6 GBq (800 mCi). The investigator-assessed ORR was 17% (95% CI: 13, 21) based on an analysis that required responders to have had prospective response assessments according to RECIST criteria. Three complete responses were observed (< 1%). Median DoR in the 60 responding patients was 35 months (95% CI: 17, 38).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
LUTATHERA Injection containing 370 MBq/mL (10 mCi/mL) of lutetium Lu 177 dotatate is a sterile, preservative-free and clear, colorless to slightly yellow solution for intravenous use supplied in a clear, colorless Type I glass 30 mL single-dose vial containing 7.4 GBq (200 mCi) ± 10% of lutetium Lu 177 dotatate at the time of injection (NDC# 69488-003-01). The solution volume in the vial ranges between 20.5 to 25 mL to provide a total of 7.4 GBq (200 mCi) of radioactivity.
The product vial is enclosed within a lead shielded container (NDC# 69488-003-01) placed in a plastic sealed container. The product is shipped in a Type A package (NDC# 69488-003-70).
Store below 25°C (77°F). Do not freeze LUTATHERA. Store in the original package to protect from ionizing radiation (lead shielding).
The shelf life is 72 hours from the date and time of calibration. Discard appropriately at 72 hours.
-
17 PATIENT COUNSELING INFORMATION
Risk From Radiation Exposure
Advise patients to minimize radiation exposure to household contacts during and after treatment with LUTATHERA consistent with institutional good radiation safety practices and patient management procedures [see Dosage and Administration (2.1), Warnings and Precautions (5.1)].
Myelosuppression
Advise patients to contact their healthcare provider for any signs or symptoms of myelosuppression or infection, such as fever, chills, dizziness, shortness of breath, or increased bleeding or bruising [see Warnings and Precautions (5.2)].
Secondary Myelodysplastic Syndrome and Leukemia
Advise patients of the potential for secondary cancers, including myelodysplastic syndrome and acute leukemia [see Warnings and Precautions (5.3)].
Renal Toxicity
Advise patients to hydrate and to urinate frequently before, on the day of, and the day after administration of LUTATHERA [see Warnings and Precautions (5.4)]. Advise patients to contact their healthcare provider for any signs or symptoms of renal toxicity [see Warnings and Precautions (5.4)].
Hepatotoxicity
Advise patients of the need for periodic laboratory tests to monitor for hepatotoxicity [see Warnings and Precautions (5.5)]. Advise patients to contact their healthcare provider for any signs or symptoms of hepatotoxicity [see Warnings and Precautions (5.5)].
Hypersensitivity
Advise patients that LUTATHERA may cause hypersensitivity reactions, including angioedema, and to seek immediate medical attention for signs or symptoms of hypersensitivity [see Warnings and Precautions (5.6)].
Neuroendocrine Hormonal Crises
Advise patients to contact their healthcare provider for signs or symptoms that may occur following tumor-related hormonal release, including severe flushing, diarrhea, bronchospasm, and hypotension [see Warnings and Precautions (5.7)].
Embryo-Fetal Toxicity
Advise pregnant women and males and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.8), Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to use effective contraception during treatment with LUTATHERA and for 7 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with LUTATHERA and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise females not to breastfeed during treatment with LUTATHERA and for 2.5 months after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise female and male patients that LUTATHERA may impair fertility [see Warnings and Precautions (5.9), Use in Specific Populations (8.3)].
Distributed by:
Advanced Accelerator Applications USA, Inc., Millburn NJ 07041©2023 Advanced Accelerator Applications USA, Inc.
LUTATHERA® is a registered trademark of Novartis AG and/or its affiliates.T2023-05
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LUTATHERA
lutetium lu 177 dotatate injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69488-003 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LUTETIUM OXODOTREOTIDE LU-177 (UNII: AE221IM3BB) (LUTETIUM OXODOTREOTIDE LU-177 - UNII:AE221IM3BB) LUTETIUM OXODOTREOTIDE LU-177 10 mCi in 1 mL Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) SODIUM ACETATE (UNII: 4550K0SC9B) GENTISIC ACID (UNII: VP36V95O3T) SODIUM HYDROXIDE (UNII: 55X04QC32I) PENTETIC ACID (UNII: 7A314HQM0I) SODIUM CHLORIDE (UNII: 451W47IQ8X) ASCORBIC ACID (UNII: PQ6CK8PD0R) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69488-003-01 1 in 1 PACKAGE 01/26/2018 1 20.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208700 01/26/2018 Labeler - Advanced Accelerator Applications USA, Inc (051714355) Establishment Name Address ID/FEI Business Operations Advanced Accelerator Applications USA, Inc. 080178357 API MANUFACTURE(69488-003) , MANUFACTURE(69488-003) , ANALYSIS(69488-003) , LABEL(69488-003) , PACK(69488-003) Establishment Name Address ID/FEI Business Operations Advanced Accelerator Applications (Italy) srl 516796146 API MANUFACTURE(69488-003) , MANUFACTURE(69488-003) , ANALYSIS(69488-003) , LABEL(69488-003) , PACK(69488-003) Establishment Name Address ID/FEI Business Operations Advanced Accelerator Applications Ibérica SL 464939761 MANUFACTURE(69488-003) Establishment Name Address ID/FEI Business Operations Advanced Accelerator Applications (Italy) S.r.l. 441182729 ANALYSIS(69488-003) Establishment Name Address ID/FEI Business Operations IDB Radiopharmacy B.V. 489201088 API MANUFACTURE(69488-003) Establishment Name Address ID/FEI Business Operations University of Missouri System 030000603 API MANUFACTURE(69488-003) , ANALYSIS(69488-003)



