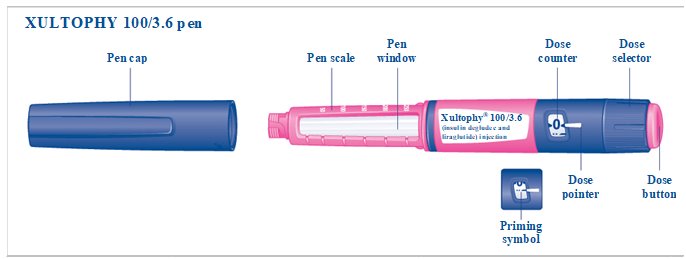
Label: XULTOPHY 100/3.6 (- insulin degludec and liraglutide injection, solution

- NDC Code(s): 0169-2911-15, 0169-2911-90, 0169-2911-97
- Packager: Novo Nordisk
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated July 10, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use XULTOPHY 100/3.6 safely and effectively. See full prescribing information for XULTOPHY 100/3.6.
XULTOPHY® 100/3.6 (insulin degludec and liraglutide) injection, for subcutaneous use
Initial U.S. Approval: 2016WARNING: RISK OF THYROID C-CELL TUMORS
See full prescribing information for complete boxed warning.
- •
- Liraglutide, one of the components of XULTOPHY 100/3.6, causes thyroid C-cell tumors at clinically relevant exposures in both genders of rats and mice. It is unknown whether XULTOPHY 100/3.6 causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined (5.1, 13.1).
- •
- XULTOPHY 100/3.6 is contraindicated in patients with a personal or family history of MTC or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk of MTC and the symptoms of thyroid tumors (4, 5.1).
INDICATIONS AND USAGE
XULTOPHY 100/3.6 is a combination of insulin degludec, a long-acting human insulin analog, and liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist, indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use (1):
- •
- Not recommended as first-line therapy for patients inadequately controlled on diet and exercise.
- •
- Not recommended for use in combination with any other product containing liraglutide or another GLP-1 receptor agonist.
- •
- Not for the treatment of type 1 diabetes mellitus or diabetic ketoacidosis.
- •
- Has not been studied in combination with prandial insulin.
DOSAGE AND ADMINISTRATION
- •
- Administer once daily at same time each day with or without food (2.1).
- •
- XULTOPHY 100/3.6 pen delivers doses from 10 to 50 units with each injection (2.1, 2.2); each XULTOPHY 100/3.6 dosage unit contains 1 unit of insulin degludec and 0.036 mg of liraglutide (2.1).
- •
- Maximum daily dosage is 50 units (50 units of insulin degludec and 1.8 mg of liraglutide) (2.1).
- •
- Recommended starting dosage in patients naïve to basal insulin or GLP-1 receptor agonist is 10 units (10 units of insulin degludec and 0.36 mg of liraglutide) injected subcutaneously once-daily (2.2).
- •
- Discontinue therapy with liraglutide or basal insulin prior to initiation of XULTOPHY 100/3.6 (2.2).
- •
- Recommended starting dosage in patients currently on basal insulin or GLP-1 receptor agonist is 16 units (16 units of insulin degludec and 0.58 mg of liraglutide) injected subcutaneously once-daily (2.2).
- •
- See Full Prescribing Information for titration recommendations (2.3).
- •
- Inject XULTOPHY 100/3.6 subcutaneously into the thigh, upper arm, or abdomen (2.5).
- •
- Rotate injection sites to reduce risk of lipodystrophy and localized cutaneous amyloidosis (2.5).
- •
- Do not administer intravenously or by an infusion pump (2.5).
- •
- Do not dilute or mix with any other insulin products or solutions (2.5).
DOSAGE FORMS AND STRENGTHS
Injection 100 units/mL of insulin degludec and 3.6 mg/mL of liraglutide available in:
- •
- 3 mL single-patient-use pen (3).
CONTRAINDICATIONS
- •
- Patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2 (4).
- •
- During episodes of hypoglycemia (4).
- •
- Patients with a serious hypersensitivity reaction to insulin degludec, liraglutide, or any of the excipients in XULTOPHY 100/3.6 (4).
WARNINGS AND PRECAUTIONS
- •
- Pancreatitis: Postmarketing reports, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis have been reported for liraglutide. Discontinue promptly if pancreatitis is suspected (5.2).
- •
- Never share a XULTOPHY 100/3.6 pen between patients, even if the needle is changed (5.3).
- •
- Hyperglycemia or hypoglycemia with changes in insulin regimen: Make changes to a patient’s insulin regimen (e.g., insulin strength, manufacturer, type, injection site or method of administration) under close medical supervision with increased frequency of blood glucose monitoring (5.4).
- •
- Overdose due to medication errors: XULTOPHY 100/3.6 contains two drugs. Instruct patients to check label before injection since accidental mix-ups with insulin containing products can occur. Do not exceed the maximum dose or administer with other GLP-1 receptor agonists (5.5).
- •
- Hypoglycemia: May be life-threatening. Increase monitoring with changes to: dosage, concomitant drugs, meal pattern, physical activity; and in patients with renal impairment or hepatic impairment or hypoglycemia unawareness (5.6, 6.1).
- •
- Acute Kidney Injury: Has been reported postmarketing for liraglutide, usually in association with nausea, vomiting, diarrhea, or dehydration, which may sometimes require hemodialysis. Advise patients of the potential risk of dehydration due to gastrointestinal adverse reactions and take precautions to avoid fluid depletion (5.7).
- •
- Hypersensitivity Reactions: Severe, life-threatening, generalized allergy, including anaphylaxis, angioedema, bronchospasm, hypotension, and shock can occur. If a hypersensitivity reaction occurs, discontinue and treat per standard of care (5.8).
- •
- Acute Gallbladder Disease: If cholelithiasis or cholecystitis are suspected, gallbladder studies are indicated (5.9).
- •
- Hypokalemia: May be life-threatening. Monitor potassium levels in patients at risk for hypokalemia and treat if indicated (5.10).
- •
- Fluid retention and congestive heart failure with use of thiazolidinediones (TZDs): Observe for signs and symptoms of heart failure; consider dosage reduction or discontinuation if heart failure occurs (5.11).
ADVERSE REACTIONS
- •
- Most common adverse reactions (incidence ≥5%) in clinical trials are nasopharyngitis, headache, nausea, diarrhea, increased lipase and upper respiratory tract infection (6).
- •
- Immunogenicity-related events, including urticaria, were more common among liraglutide-treated patients (0.8%) than among comparator-treated patients (0.4%) in clinical trials (12.6).
To report SUSPECTED ADVERSE REACTIONS, contact Novo Nordisk Inc. at 1-800-727-6500 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- •
- Drugs that affect glucose metabolism: Adjustment of XULTOPHY 100/3.6 dosage may be needed; closely monitor blood glucose (7.1).
- •
- Anti-Adrenergic drugs (e.g., beta-blockers, clonidine, guanethidine, and reserpine): Hypoglycemia signs and symptoms may be reduced or absent (7.1).
- •
- Effects of delayed gastric emptying on oral medications: May impact absorption of concomitantly administered oral medications (7.2).
USE IN SPECIFIC POPULATIONS
Pregnancy: XULTOPHY 100/3.6 should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus (8.1).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK OF THYROID C-CELL TUMORS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage Information
2.2 Recommended Starting Dosage
2.3 Titration of XULTOPHY 100/3.6
2.4 Recommendations Regarding Missed Doses
2.5 Important Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Thyroid C-cell Tumors
5.2 Pancreatitis
5.3 Never Share a XULTOPHY 100/3.6 Pen Between Patients
5.4 Hyperglycemia or Hypoglycemia with Changes in Insulin Regimen
5.5 Overdose due to Medication Errors
5.6 Hypoglycemia
5.7 Acute Kidney Injury
5.8 Hypersensitivity Reactions
5.9 Acute Gallbladder Disease
5.10 Hypokalemia
5.11 Fluid Retention and Congestive Heart Failure with Concomitant Use of a PPAR Gamma Agonist
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Medications that Can Affect Glucose Metabolism
7.2 Effects of Delayed Gastric Emptying on Oral Medications
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Gastroparesis
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Overview of Clinical Studies
14.2 Patients with Type 2 Diabetes Mellitus Uncontrolled on OAD Treatment
14.3 Patients currently on basal insulin or GLP-1 receptor agonist
14.4 Cardiovascular Outcomes Trials in Patients with Type 2 Diabetes Mellitus and Atherosclerotic Cardiovascular Disease Conducted with Liraglutide 1.8 mg and Insulin Degludec
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Recommended Storage
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK OF THYROID C-CELL TUMORS
- •
- Liraglutide, one of the components of XULTOPHY 100/3.6, causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors at clinically relevant exposures in both genders of rats and mice. It is unknown whether XULTOPHY 100/3.6 causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined [see Warnings and Precautions (5.1) and Nonclinical Toxicology (13)].
- •
- XULTOPHY 100/3.6 is contraindicated in patients with a personal or family history of MTC and in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk for MTC with the use of XULTOPHY 100/3.6 and inform them of symptoms of thyroid tumors (e.g. a mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with XULTOPHY 100/3.6 [see Contraindications (4), Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
XULTOPHY 100/3.6 is a combination of insulin degludec and liraglutide and is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Limitations of Use:
- •
- XULTOPHY 100/3.6 is not recommended as first-line therapy for patients who have inadequate glycemic control on diet and exercise because of the uncertain relevance of the rodent C-cell tumor findings to humans [see Warnings and Precautions (5.1)].
- •
- XULTOPHY 100/3.6 is not recommended for use in combination with any other product containing liraglutide or another (glucagon-like peptide-1) GLP-1 receptor agonist [see Warnings and Precautions (5.5)].
- •
- XULTOPHY 100/3.6 is not indicated for use in patients with type 1 diabetes mellitus or for the treatment of diabetic ketoacidosis.
- •
- XULTOPHY 100/3.6 has not been studied in combination with prandial insulin.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage Information
- •
- XULTOPHY 100/3.6 is a combination of insulin degludec and liraglutide.
- •
- Administer XULTOPHY 100/3.6 by subcutaneous injection once-daily at the same time each day with or without food.
- •
- The XULTOPHY 100/3.6 pen delivers doses from 10 to 50 units with each injection. Table 1 presents the units of insulin degludec and the milligrams of liraglutide in each dosage of XULTOPHY 100/3.6 [see Dosage and Administration (2.2)].
- •
- The maximum dosage of XULTOPHY 100/3.6 is 50 units daily (50 units of insulin degludec and 1.8 mg of liraglutide) [see Warnings and Precautions (5.5)].
2.2 Recommended Starting Dosage
In patients naïve to basal insulin or a GLP-1 receptor agonist
- •
- The recommended starting dosage of XULTOPHY 100/3.6 is 10 units (10 units of insulin degludec and 0.36 mg of liraglutide) injected subcutaneously once-daily (see Table 1).
In patients currently on basal insulin or a GLP-1 receptor agonist
- •
- Discontinue therapy with basal insulin or GLP-1 receptor agonist prior to initiation of XULTOPHY 100/3.6.
- •
- The recommended starting dosage of XULTOPHY 100/3.6 is 16 units (16 units of insulin degludec and 0.58 mg of liraglutide) injected subcutaneously once-daily (see Table 1).
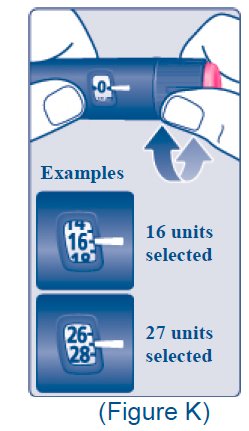
Table 1: Units of Insulin Degludec and Milligrams of Liraglutide in Each Dosage of XULTOPHY 100/3.6
XULTOPHY 100/3.6
(dose counter display)*
insulin degludec
component dose
liraglutide
component dose
Comment
▪▪ ─
---
---
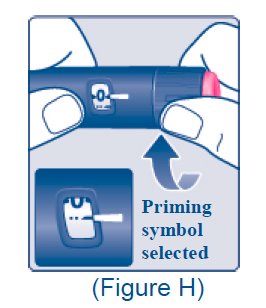
Priming symbol
10
10 units
0.36 mg
Recommended starting dose for patients naïve to basal insulin or GLP-1 receptor agonist
11
11 units
0.4 mg
12
12 units
0.43 mg
13
13 units
0.47 mg
14
14 units
0.5 mg
15
15 units
0.54 mg
16
16 units
0.58 mg
- Recommended starting dose for patients currently on basal insulin or GLP-1 receptor agonist
17
17 units
0.61 mg
18
18 units
0.65 mg
19
19 units
0.68 mg
20
20 units
0.72 mg
21
21 units
0.76 mg
22
22 units
0.79 mg
23
23 units
0.83 mg
24
24 units
0.86 mg
25
25 units
0.9 mg
26
26 units
0.94 mg
27
27 units
0.97 mg
28
28 units
1.01 mg
29
29 units
1.04 mg
30
30 units
1.08 mg
31
31 units
1.12 mg
32
32 units
1.15 mg
33
33 units
1.19 mg
34
34 units
1.22 mg
35
35 units
1.26 mg
36
36 units
1.3 mg
37
37 units
1.33 mg
38
38 units
1.37 mg
39
39 units
1.4 mg
40
40 units
1.44 mg
41
41 units
1.48 mg
42
42 units
1.51 mg
43
43 units
1.55 mg
44
44 units
1.58 mg
45
45 units
1.62 mg
46
46 units
1.66 mg
47
47 units
1.69 mg
48
48 units
1.73 mg
49
49 units
1.76 mg
50
50 units
1.8 mg
Maximum daily dosage [see Warnings and Precautions (5.5)]
*The dose counter on the XULTOPHY 100/3.6 pen displays numbers for the even units and displays lines for the odd units.
2.3 Titration of XULTOPHY 100/3.6
- •
- After starting the recommended starting dosage of XULTOPHY 100/3.6 [see Dosage and Administration (2.2)], titrate the dosage upwards or downwards by two units (see Table 2) once weekly or twice weekly (every three to four days), based on the patient’s metabolic needs, blood glucose monitoring results, and glycemic control goal until the desired fasting plasma glucose is achieved.
- •
- To minimize the risk of hypoglycemia or hyperglycemia, additional titration may be needed with changes in physical activity, meal patterns (i.e., macronutrient content or timing of food intake), or renal or hepatic function; during acute illness; or when used with other medications [see Warnings and Precautions (5.4) and Drug Interactions (7)].
Table 2: Recommended Titration of XULTOPHY 100/3.6 (Once or Twice Weekly)
Self-Monitored Fasting Plasma Glucose
XULTOPHY 100/3.6 Dosage Adjustment
Above target range
+ 2 units (2 units of insulin degludec and 0.072 mg of liraglutide)
Within target range
0 units
Below target range
- 2 units (2 units of insulin degludec and 0.072 mg of liraglutide)
2.4 Recommendations Regarding Missed Doses
- •
- Instruct patients who miss a dose of XULTOPHY 100/3.6 to resume the once-daily dosage regimen as prescribed with the next scheduled dose. Do not administer an extra dose or increase the dose to make up for the missed dose.
- •
- If more than three days have elapsed since the last XULTOPHY 100/3.6 dose, reinitiate XULTOPHY 100/3.6 at the recommended starting dose to mitigate any gastrointestinal symptoms associated with reinitiation of treatment [see Dosage and Administration (2.1, 2.2, 2.3)].
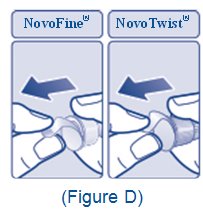
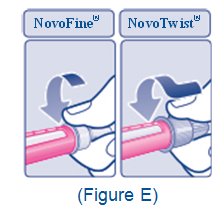
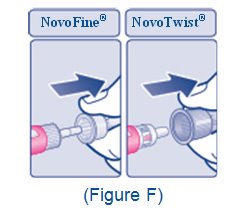
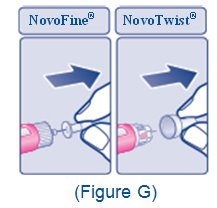
2.5 Important Administration Instructions
- •
- The XULTOPHY 100/3.6 pen is for single-patient-use only [see Warnings and Precautions (5.3)].
- •
- Train patients on proper use and injection technique before initiating XULTOPHY 100/3.6.
- •
- Always check the label on the XULTOPHY 100/3.6 pen before administration [see Warnings and Precautions (5.5)].
- •
- Inspect visually for particulate matter and discoloration prior to administration. Only use XULTOPHY 100/3.6 if the solution appears clear and colorless.
- •
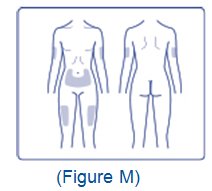
- Inject XULTOPHY 100/3.6 subcutaneously into the thigh, upper arm, or abdomen.
- •
- Rotate injection sites within the same region from one injection to the next to reduce the risk of lipodystrophy and localized cutaneous amyloidosis. Do not inject into areas of lipodystrophy or localized cutaneous amyloidosis [see Warnings and Precautions (5.4), Adverse Reactions (6.1, 6.3)].
- •
- During changes to a patient’s insulin regimen, increase the frequency of blood glucose monitoring [see Warnings and Precautions (5.4)].
- •
- Use XULTOPHY 100/3.6 with caution in patients with visual impairment who may rely on audible clicks to dial their dose.
- •
- The XULTOPHY 100/3.6 pen dials in one-unit increments.
- •
- Do not administer XULTOPHY 100/3.6 intravenously or in an insulin infusion pump.
- •
- Do not dilute or mix XULTOPHY 100/3.6 with any other insulin or solutions.
- •
- Do not split the dose of XULTOPHY 100/3.6.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
XULTOPHY 100/3.6 is contraindicated:
- •
- In patients with a personal or family history of medullary thyroid carcinoma (MTC) or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2) [see Warnings and Precautions (5.1)].
- •
- During episodes of hypoglycemia [see Warnings and Precautions (5.6)].
- •
- In patients with hypersensitivity to insulin degludec, liraglutide, or any of the excipients in XULTOPHY 100/3.6. Serious hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with liraglutide, one of the components of XULTOPHY 100/3.6 [see Warnings and Precautions (5.8)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Thyroid C-cell Tumors
Liraglutide, one of the components of XULTOPHY 100/3.6, causes dose-dependent and treatment-duration-dependent thyroid C-cell tumors (adenomas and/or carcinomas) at clinically relevant exposures in both genders of rats and mice [see Nonclinical Toxicology (13.1)]. Malignant thyroid C-cell carcinomas were detected in rats and mice. It is unknown whether XULTOPHY 100/3.6 will cause thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined.
Cases of MTC in patients treated with liraglutide have been reported in the postmarketing period; the data in these reports are insufficient to establish or exclude a causal relationship between MTC and liraglutide use in humans.
XULTOPHY 100/3.6 is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2. Counsel patients regarding the potential risk for MTC with the use of XULTOPHY 100/3.6 and inform them of symptoms of thyroid tumors (e.g. a mass in the neck, dysphagia, dyspnea, persistent hoarseness).
Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with XULTOPHY 100/3.6. Such monitoring may increase the risk of unnecessary procedures, due to low test specificity for serum calcitonin and a high background incidence of thyroid disease. Significantly elevated serum calcitonin may indicate MTC and patients with MTC usually have calcitonin values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
5.2 Pancreatitis
Based on spontaneous postmarketing reports, acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with liraglutide, one of the components of XULTOPHY 100/3.6. In glycemic control trials of liraglutide, there have been 13 cases of pancreatitis among liraglutide-treated patients and 1 case in a comparator (glimepiride) treated patient (2.7 vs. 0.5 cases per 1000 patient-years). Nine of the 13 cases with liraglutide were reported as acute pancreatitis and four were reported as chronic pancreatitis. In one case in a liraglutide-treated patient, pancreatitis, with necrosis, was observed and led to death; however clinical causality could not be established. Some patients had other risk factors for pancreatitis, such as a history of cholelithiasis or alcohol abuse.
After initiation of XULTOPHY 100/3.6, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back and which may or may not be accompanied by vomiting). If pancreatitis is suspected, XULTOPHY 100/3.6 should promptly be discontinued and appropriate management should be initiated. If pancreatitis is confirmed, restarting XULTOPHY 100/3.6 is not recommended.
Liraglutide, one of the components of XULTOPHY 100/3.6, has been studied in a limited number of patients with a history of pancreatitis. It is unknown if patients with a history of pancreatitis are at higher risk for development of pancreatitis on liraglutide.
5.3 Never Share a XULTOPHY 100/3.6 Pen Between Patients
XULTOPHY 100/3.6 pen must never be shared between patients, even if the needle is changed. Sharing of the pen poses a risk for transmission of blood-borne pathogens.
5.4 Hyperglycemia or Hypoglycemia with Changes in Insulin Regimen
Changes in an insulin regimen (e.g., insulin strength, manufacturer, type, injection site or method of administration) may affect glycemic control and predispose to hypoglycemia [see Warnings and Precautions (5.6)] or hyperglycemia. Repeated insulin injections into areas of lipodystrophy or localized cutaneous amyloidosis have been reported to result in hyperglycemia; and a sudden change in the injection site (to an unaffected area) has been reported to result in hypoglycemia [see Adverse Reactions (6.1, 6.3)]. Make any changes to a patient’s insulin regimen under close medical supervision with increased frequency of blood glucose monitoring. Advise patients who have repeatedly injected into areas of lipodystrophy or localized cutaneous amyloidosis to change the injection site to unaffected areas and closely monitor for hypoglycemia. Adjustments in concomitant oral anti-diabetic treatment may be needed. When initiating XULTOPHY 100/3.6, follow dosing recommendations [see Dosage and Administration (2.1, 2.2, 2.3)].
5.5 Overdose due to Medication Errors
XULTOPHY 100/3.6 contains two drugs: insulin degludec and liraglutide. Administration of more than 50 units of XULTOPHY 100/3.6 daily can result in overdose of the liraglutide component. Do not exceed the 1.8 mg maximum recommended dose of liraglutide or use with other GLP-1 receptor agonists.
Accidental mix-ups between insulin products have been reported. To avoid medication errors between XULTOPHY 100/3.6 (an insulin containing product) and other insulin products, instruct patients to always check the label before each injection.
5.6 Hypoglycemia
Hypoglycemia is the most common adverse reaction of insulin containing products, including XULTOPHY 100/3.6 [see Adverse Reactions (6.1)]. Severe hypoglycemia can cause seizures, may be life-threatening or cause death. Hypoglycemia can impair concentration ability and reaction time; this may place the patient and others at risk in situations where these abilities are important (e.g., driving or operating other machinery). XULTOPHY 100/3.6 (an insulin-containing product) or any insulin, should not be used during episodes of hypoglycemia [see Contraindications (4)].
Hypoglycemia can happen suddenly and symptoms may differ in each patient and change over time in the same patient. Symptomatic awareness of hypoglycemia may be less pronounced in patients with longstanding diabetes, in patients with diabetic neuropathy, in patients using drugs that block the sympathetic nervous system (e.g., beta-blockers) [see Drug Interactions (7.1)], or who experience recurrent hypoglycemia.
The long-acting effect of insulin degludec may delay recovery from hypoglycemia compared to shorter acting insulins.
Risk Factors for Hypoglycemia
The risk of hypoglycemia generally increases with intensity of glycemic control. The risk of hypoglycemia after an injection is related to the duration of action of the insulin [see Clinical Pharmacology (12.2)] and, in general, is highest when the glucose lowering effect of the insulin is maximal. As with all insulin containing products, the glucose lowering effect time course of XULTOPHY 100/3.6 may vary among different patients or at different times in the same patients and depends on many conditions, including the area of injection as well as the injection site blood supply and temperature.
Other factors which may increase the risk of hypoglycemia include changes in meal pattern (e.g., macronutrient content or timing of meals), changes in level of physical activity, or changes to concomitant drugs [see Drug Interactions (7.1)]. Patients with renal or hepatic impairment may be at higher risk of hypoglycemia [see Use in Specific Populations (8.6, 8.7)].
Risk Mitigation Strategies for Hypoglycemia
Patients and caregivers must be educated to recognize and manage hypoglycemia. Self-monitoring of blood glucose plays an essential role in the prevention and management of hypoglycemia. In patients at higher risk for hypoglycemia and patients who have reduced symptomatic awareness of hypoglycemia, increased frequency of blood glucose monitoring is recommended.
5.7 Acute Kidney Injury
There have been postmarketing reports of acute kidney injury and worsening of chronic kidney failure, which may sometimes require hemodialysis in patients treated with liraglutide, one of the components of XULTOPHY 100/3.6 [see Adverse Reactions (6.2)]. Some of these events were reported in patients without known underlying kidney disease. A majority of the reported events occurred in patients who had experienced nausea, vomiting, diarrhea, or dehydration. Some of the reported events occurred in patients receiving one or more medications known to affect renal function or hydration status. Altered kidney function has been reversed in many of the reported cases with supportive treatment and discontinuation of potentially causative agents, including liraglutide. Advise patients of the potential risk of dehydration due to gastrointestinal adverse reactions and take precautions to avoid fluid depletion.
5.8 Hypersensitivity Reactions
Severe, life-threatening, generalized allergy, including anaphylaxis, angioedema, bronchospasm, hypotension, and shock can occur with insulins, including XULTOPHY 100/3.6. Allergic reactions (manifested with signs and symptoms such as urticaria, rash, pruritus) have been reported with XULTOPHY 100/3.6. There have been postmarketing reports of serious hypersensitivity reactions (e.g., anaphylactic reactions and angioedema) in patients treated with liraglutide, one of the components of XULTOPHY 100/3.6 [see Adverse Reactions (6.2)]. If a hypersensitivity reaction occurs, discontinue XULTOPHY 100/3.6; treat promptly per standard of care, and monitor until signs and symptoms resolve.
Anaphylaxis and angioedema have been reported with other GLP-1 receptor agonists. Use caution in a patient with a history of anaphylaxis or angioedema with another GLP-1 receptor agonist because it is unknown whether such patients will be predisposed to these reactions with XULTOPHY 100/3.6. XULTOPHY 100/3.6 is contraindicated in patients who have had hypersensitivity reactions to insulin degludec, liraglutide or one of the excipients of these products [see Contraindications (4)].
5.9 Acute Gallbladder Disease
Acute events of gallbladder disease such as cholelithiasis or cholecystitis have been reported in GLP-1 receptor agonist trials and postmarketing. In a cardiovascular outcomes trial (LEADER trial) [see Clinical Studies (14.4)], 3.1% of patients treated with liraglutide, one of the components of XULTOPHY 100/3.6, versus 1.9% of placebo treated patients reported an acute event of gallbladder disease, such as cholelithiasis or cholecystitis [see Adverse Reactions (6.1)]. If cholelithiasis is suspected, gallbladder studies and appropriate clinical follow-up are indicated.
5.10 Hypokalemia
All insulin-containing products, including XULTOPHY 100/3.6, cause a shift in potassium from the extracellular to intracellular space, possibly leading to hypokalemia. Untreated hypokalemia may cause respiratory paralysis, ventricular arrhythmia, and death. Monitor potassium levels in patients at risk for hypokalemia if indicated (e.g., patients using potassium-lowering medications, patients taking medications sensitive to serum potassium concentrations).
5.11 Fluid Retention and Congestive Heart Failure with Concomitant Use of a PPAR Gamma Agonist
Thiazolidinediones (TZDs), which are peroxisome proliferator-activated receptor (PPAR)-gamma agonists can cause dose related fluid retention, when used in combination with insulin containing products, including XULTOPHY 100/3.6. Fluid retention may lead to or exacerbate congestive heart failure. Patients treated with insulin containing products, including XULTOPHY 100/3.6 and a PPAR-gamma agonist should be observed for signs and symptoms of congestive heart failure. If congestive heart failure develops, it should be managed according to current standards of care and discontinuation or dose reduction of the PPAR-gamma agonist must be considered.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described below or elsewhere in the prescribing information:
- •
- Risk of Thyroid C-cell Tumors [see Warnings and Precautions (5.1)]
- •
- Pancreatitis [see Warnings and Precautions (5.2)]
- •
- Hypoglycemia [see Warnings and Precautions (5.6)]
- •
- Acute Kidney Injury [see Warnings and Precautions (5.7)]
- •
- Hypersensitivity Reactions [see Warnings and Precautions (5.8)]
- •
- Acute Gallbladder Disease [see Warnings and Precautions (5.9)]
- •
- Hypokalemia [see Warnings and Precautions (5.10)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Xultophy 100/3.6
The data in Table 3 reflect the exposure of 1881 patients to XULTOPHY 100/3.6 and a mean duration of exposure of 33 weeks in trials NCT01336023, NCT01618162, NCT02773368, NCT01676116, NCT01392573, NCT01952145 [see Clinical Studies (14.2 and 14.3)]. The mean age was 57 years and 3% were older than 75 years; 53% were male, 75% were White, 6% were Black or African American and 16% were Hispanic or Latino. The mean body mass index (BMI) was 31.8 kg/m2. The mean duration of diabetes was 9 years and the mean HbA1c at baseline was 8.2%. A history of neuropathy, ophthalmopathy, nephropathy and cardiovascular disease at baseline was reported in 25%, 12%, 7% and 6% respectively. The mean estimated glomerular filtration rate (eGFR) at baseline was 88.3 mL/min/1.73 m2 and 6% of the patients had an eGFR less than 60 mL/min/1.73 m2.
Table 3: Adverse Reactions Occurring in ≥5% of XULTOPHY 100/3.6-Treated Patients with Type 2 Diabetes Mellitus XULTOPHY 100/3.6
N = 1881
%
Nasopharyngitis
10
Headache
9
Nausea
8
Diarrhea
8
Increased Lipase
7
Upper respiratory tract infection
6
Hypoglycemia
Hypoglycemia was the most commonly observed adverse reaction in patients treated with insulin and insulin containing products, including XULTOPHY 100/3.6 [see Warnings and Precautions (5.6)]. The number of reported hypoglycemia episodes depended on the definition of hypoglycemia used, insulin dose, intensity of glucose control, background therapies, and other intrinsic and extrinsic patient factors. For these reasons, comparing rates of hypoglycemia in clinical trials for XULTOPHY 100/3.6 with the incidence of hypoglycemia for other products may be misleading and also, may not be representative of hypoglycemia rates that will occur in clinical practice.
In the phase 3 clinical program [see Clinical Studies (14)], events of severe hypoglycemia were defined as an episode requiring assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions (Table 4). Hypoglycemia episodes with a glucose level below 54 mg/dL associated with or without symptoms is shown in Table 4. No clinically important differences in risk of severe hypoglycemia between XULTOPHY 100/3.6 and comparators were observed in clinical trials.
Table 4: Hypoglycemia Episodes Reported in XULTOPHY 100/3.6-Treated Patients with T2DM
†Episode requiring assistance of another person to actively administer carbohydrate, glucagon, or other resuscitative actions. Patients naïve to basal insulin or GLP-1 receptor agonist
Patients currently on GLP-1 receptor agonist
Patients currently on basal insulin
- XULTOPHY 100/3.6
NCT01336023
XULTOPHY 100/3.6
NCT01618162
XULTOPHY 100/3.6
NCT02773368
XULTOPHY 100/3.6
NCT01676116
XULTOPHY 100/3.6
NCT01392573
XULTOPHY 100/3.6
NCT01952145
Total Subjects (N)
825
288
209
291
199
278
Severe Hypoglycemia (%)†
0.2
0.7
0.5
0.3
0.5
0.0
Hypoglycemia with a glucose level <54 mg/dL (%)*
27.6
37.2
14.4
27.1
22.1
24.8
*Episodes of hypoglycemia with a glucose level below 54 mg/dL that are associated with or without symptoms of hypoglycemia.
Gastrointestinal Adverse Reactions
Gastrointestinal adverse reactions including nausea, diarrhea, vomiting, constipation, dyspepsia, gastritis, abdominal pain, flatulence, eructation, gastroesophageal reflux disease, abdominal distension and decreased appetite have been reported in patients treated with XULTOPHY 100/3.6. Gastrointestinal adverse reactions may occur more frequently at the beginning of XULTOPHY 100/3.6 therapy and diminish within a few days or weeks on continued treatment.
Papillary thyroid carcinoma
Liraglutide
In glycemic control trials of liraglutide, there were 7 reported cases of papillary thyroid carcinoma in patients treated with liraglutide and 1 case in a comparator-treated patient (1.5 vs. 0.5 cases per 1000 patient years). Most of these papillary thyroid carcinomas were <1 cm in greatest diameter and were diagnosed in surgical pathology specimens after thyroidectomy prompted by findings on protocol-specified screening with serum calcitonin or thyroid ultrasound.
Cholelithiasis and cholecystitis
Liraglutide
In glycemic control trials of liraglutide, the incidence of cholelithiasis was 0.3% in both liraglutide-treated and placebo-treated patients. The incidence of cholecystitis was 0.2% in both liraglutide treated and placebo-treated patients.
In a cardiovascular outcomes trial (LEADER trial) [see Clinical Studies (14.4)], the incidence of cholelithiasis was 1.5% (3.9 cases per 1000 patient years of observation) in liraglutide-treated and 1.1% (2.8 cases per 1000 patient years of observation) in placebo-treated patients, both on a background of standard of care. The incidence of acute cholecystitis was 1.1% (2.9 cases per 1000 patient years of observation) in liraglutide-treated and 0.7% (1.9 cases per 1000 patient years of observation) in placebo-treated patients. The majority of events required hospitalization or cholecystectomy.
Initiation of insulin containing products and intensification of glucose control
Intensification or rapid improvement in glucose control has been associated with a transitory, reversible ophthalmologic refraction disorder, worsening of diabetic retinopathy, and acute painful peripheral neuropathy. However, long-term glycemic control decreases the risk of diabetic retinopathy and neuropathy.
Lipodystrophy
Long-term use of insulin containing products, including XULTOPHY 100/3.6, can cause lipodystrophy at the site of repeated injections. Lipodystrophy includes lipohypertrophy (thickening of adipose tissue) and lipoatrophy (thinning of adipose tissue), and may affect absorption [see Dosage and Administration (2.5)].
Peripheral Edema
XULTOPHY 100/3.6 may cause sodium retention and edema, particularly if previously poor metabolic control is improved rapidly by intensified therapy.
Weight Gain
Weight gain can occur with insulin containing products, including XULTOPHY 100/3.6, and has been attributed to the anabolic effects of insulin. In study A, after 26 weeks of treatment, patients converting to XULTOPHY 100/3.6 from liraglutide had a mean increase in body weight of 2 kg.
Injection Site reactions
As with any insulin and GLP-1 receptor agonist-containing products, patients taking XULTOPHY 100/3.6 may experience injection site reactions, including injection site hematoma, pain, hemorrhage, erythema, nodules, swelling, discoloration, pruritis, warmth, and injection site mass. In the clinical program, the proportion of injection site reactions occurring in patients treated with XULTOPHY 100/3.6 was 2.6%. These reactions were usually mild and transitory and they normally disappear during continued treatment.
Hypersensitivity Reactions
Severe, life-threatening, generalized allergy, including anaphylaxis, generalized skin reactions, angioedema, bronchospasm, hypotension, and shock have occurred with insulin, including XULTOPHY 100/3.6 and may be life threatening [see Warnings and Precautions (5.8)]. Hypersensitivity (manifested with swelling of tongue and lips, diarrhea, nausea, tiredness, and itching) and urticaria were reported.
Laboratory tests
Bilirubin
Liraglutide
In the five glycemic control trials of at least 26 weeks duration, mildly elevated serum bilirubin concentrations (elevations to no more than twice the upper limit of the reference range) occurred in 4.0% of liraglutide-treated patients, 2.1% of placebo-treated patients and 3.5% of active-comparator-treated patients. This finding was not accompanied by abnormalities in other liver tests. The significance of this isolated finding is unknown.
Calcitonin
XULTOPHY 100/3.6
Calcitonin, a biological marker of MTC, was measured throughout the XULTOPHY 100/3.6 clinical development program. Among patients with pretreatment calcitonin <20 ng/L, calcitonin elevations to >20 ng/L occurred in 0.7% of XULTOPHY 100/3.6-treated patients, 0.7% of placebo-treated patients, and 1.1% and 0.7% of active-comparator-treated patients (basal insulins and GLP-1s respectively). The clinical significance of these findings is unknown.
Liraglutide
Calcitonin, a biological marker of MTC, was measured throughout the liraglutide clinical development program. At the end of the glycemic control trials, adjusted mean serum calcitonin concentrations were higher in liraglutide-treated patients compared to placebo-treated patients but not compared to patients receiving active comparator. Between group differences in adjusted mean serum calcitonin values were approximately 0.1 ng/L or less. Among patients with pretreatment calcitonin <20 ng/L, calcitonin elevations to >20 ng/L occurred in 0.7% of liraglutide-treated patients, 0.3% of placebo-treated patients, and 0.5% of active-comparator-treated patients. The clinical significance of these findings is unknown.
Lipase and Amylase
Liraglutide
In one glycemic control trial in renal impairment patients, a mean increase of 33% for lipase and 15% for amylase from baseline was observed for liraglutide-treated patients while placebo-treated patients had a mean decrease in lipase of 3% and a mean increase in amylase of 1%.
In a cardiovascular outcomes trial (LEADER trial) [see Clinical Studies (14.4)], serum lipase and amylase were routinely measured. Among liraglutide-treated patients, 7.9% had a lipase value at any time during treatment of greater than or equal to 3 times the upper limit of normal compared with 4.5% of placebo-treated patients, and 1% of liraglutide-treated patients had an amylase value at any time during treatment of greater than or equal to 3 times the upper limit of normal versus 0.7% of placebo-treated patients.
The clinical significance of elevations in lipase or amylase with liraglutide is unknown in the absence of other signs and symptoms of pancreatitis [see Warnings and Precautions (5.2)].
Vital signs
Mean increases from baseline in heart rate of 2 to 3 beats per minute have been observed with XULTOPHY 100/3.6 which is attributable to the liraglutide component.
6.2 Postmarketing Experience
The following additional adverse reactions have been reported during post-approval use. Because these events are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Insulin degludec
Localized cutaneous amyloidosis at the injection site has occurred. Hyperglycemia has been reported with repeated
insulin injections into areas of localized cutaneous amyloidosis; hypoglycemia has been reported with a sudden change to an unaffected injection site.
Liraglutide
- •
- Gastrointestinal: Acute pancreatitis, hemorrhagic and necrotizing pancreatitis sometimes resulting in death, ileus
- •
- General Disorders and Administration Site Conditions: Allergic reactions: rash and pruritus
- •
- Hepatobiliary: Elevations of liver enzymes, hyperbilirubinemia, cholestasis, cholecystitis, cholelithiasis requiring cholecystectomy, hepatitis
- •
- Immune system: Angioedema and anaphylactic reactions
- •
- Metabolism and nutrition: Dehydration resulting from nausea, vomiting and diarrhea
- •
- Neoplasms: Medullary thyroid carcinoma
- •
- Nervous system: Dysgeusia, dizziness
- •
- Renal and urinary: Increased serum creatinine, acute renal failure or worsening of chronic renal failure, sometimes requiring hemodialysis.
- •
- Skin and subcutaneous tissue: Cutaneous amyloidosis
-
7 DRUG INTERACTIONS
7.1 Medications that Can Affect Glucose Metabolism
A number of medications affect glucose metabolism and may require dose adjustment of XULTOPHY 100/3.6 and particularly close monitoring [see Dosage and Administration (2.2); Warnings and Precautions (5.6)].
Drugs That May Increase the Risk of Hypoglycemia
Drugs:
Antidiabetic agents, ACE inhibitors, angiotensin II receptor blocking agents, disopyramide, fibrates, fluoxetine, monoamine oxidase inhibitors, pentoxifylline, pramlintide, salicylates, somatostatin analogs (e.g., octreotide), and sulfonamide antibiotics
Intervention:
Dosage reductions and increased frequency of glucose monitoring may be required when XULTOPHY 100/3.6 is co-administered with these drugs.
Drugs That May Decrease the Blood Glucose Lowering Effect of XULTOPHY 100/3.6
Drugs:
Atypical antipsychotics (e.g., olanzapine and clozapine), corticosteroids, danazol, diuretics, estrogens, glucagon, isoniazid, niacin, oral contraceptives, phenothiazines, progestogens (e.g., in oral contraceptives), protease inhibitors, somatropin, sympathomimetic agents (e.g., albuterol, epinephrine, terbutaline), and thyroid hormones.
Intervention:
Dosage increases and increased frequency of glucose monitoring may be required when XULTOPHY 100/3.6 is co-administered with these drugs.
Drugs That May Increase or Decrease the Blood Glucose Lowering Effect of XULTOPHY 100/3.6
Drugs:
Alcohol, beta-blockers, clonidine, and lithium salts. Pentamidine may cause hypoglycemia, which may sometimes be followed by hyperglycemia.
Intervention:
Dosage adjustment and increased frequency of glucose monitoring may be required when XULTOPHY 100/3.6 is co-administered with these drugs.
Drugs That May Blunt Signs and Symptoms of Hypoglycemia
Drugs:
Beta-blockers, clonidine, guanethidine, and reserpine
Intervention:
Increased frequency of glucose monitoring may be required when XULTOPHY 100/3.6 is co-administered with these drugs.
7.2 Effects of Delayed Gastric Emptying on Oral Medications
Liraglutide-containing products, including XULTOPHY 100/3.6, cause a delay of gastric emptying, and thereby have the potential to impact the absorption of concomitantly administered oral medications. In clinical pharmacology trials, liraglutide did not affect the absorption of the tested orally administered medications to any clinically relevant degree [see Clinical Pharmacology (12.3)]. Nonetheless, caution should be exercised when oral medications are concomitantly administered with liraglutide containing products.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal reproduction studies, there may be risks to the fetus from exposure to liraglutide during pregnancy. XULTOPHY 100/3.6 should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
There are no available data with XULTOPHY 100/3.6, insulin degludec or liraglutide in pregnant women to inform a drug associated risk for major birth defects and miscarriage. There are clinical considerations regarding the risks of poorly controlled diabetes in pregnancy [see Clinical Considerations].
For insulin degludec, rats and rabbits were exposed in animal reproduction studies at 5 times (rat) and 10 times (rabbit) the human exposure at a dose of 0.75 U/kg/day. No adverse outcomes were observed for pregnant animals and offspring [see Data].
For liraglutide, animal reproduction studies identified increased adverse developmental outcomes from exposure during pregnancy. Liraglutide exposure was associated with early embryonic deaths and an imbalance in some fetal abnormalities in pregnant rats administered liraglutide during organogenesis at doses that approximate clinical exposures at the maximum recommended human dose (MRHD) of 1.8 mg/day. In pregnant rabbits administered liraglutide during organogenesis, decreased fetal weight and an increased incidence of major fetal abnormalities were seen at exposures below the human exposures at the MRHD [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The estimated background risk of major birth defects is 6 to 10% in women with pre-gestational diabetes with a peri-conceptional HbA1c >7 and has been reported to be as high as 20 to 25% in women with a peri-conceptional HbA1c >10. The estimated background risk of miscarriage for the indicated population is unknown.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/fetal Risk
Hypoglycemia and hyperglycemia occur more frequently during pregnancy in patients with pre-gestational diabetes. Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes mellitus increases the fetal risk for major birth defects, stillbirth, macrosomia related morbidity.
Data
Animal Data
Insulin degludec
Insulin degludec was investigated in studies covering fertility, embryo-fetal development and pre- and post-natal development in rats and during the period of embryo-fetal development in rabbits. Human insulin (NPH insulin) was included as comparator. In these studies insulin degludec was given subcutaneously at up to 21 U/kg/day in rats and 3.3 U/kg/day in rabbits, resulting in 5 times (rat) and 10 times (rabbit) the human exposure (AUC) at a human subcutaneous dose of 0.75 U/kg/day. Overall the effects of insulin degludec were similar to those observed with human insulin.
Liraglutide
Female rats given subcutaneous doses of 0.1, 0.25 and 1.0 mg/kg/day liraglutide beginning 2 weeks before mating through gestation day 17 had estimated systemic exposures 0.8-, 3-, and 11-times the human exposure at the MRHD based on plasma AUC comparison. The number of early embryonic deaths in the 1 mg/kg/day group increased slightly. Fetal abnormalities and variations in kidneys and blood vessels, irregular ossification of the skull, and a more complete state of ossification occurred at all doses. Mottled liver and minimally kinked ribs occurred at the highest dose. The incidence of fetal malformations in liraglutide-treated groups exceeding concurrent and historical controls were misshapen oropharynx and/or narrowed opening into larynx at 0.1 mg/kg/day and umbilical hernia at 0.1 and 0.25 mg/kg/day.
Pregnant rabbits given subcutaneous doses of 0.01, 0.025 and 0.05 mg/kg/day liraglutide from gestation day 6 through day 18 inclusive, had estimated systemic exposures less than the human exposure at the MRHD of 1.8 mg/day at all doses, based on plasma AUC. Liraglutide decreased fetal weight and dose dependently increased the incidence of total major fetal abnormalities at all doses. The incidence of malformations exceeded concurrent and historical controls at 0.01 mg/kg/day (kidneys, scapula), ≥ 0.01 mg/kg/day (eyes, forelimb), 0.025 mg/kg/day (brain, tail and sacral vertebrae, major blood vessels and heart, umbilicus), ≥ 0.025 mg/kg/day (sternum) and at 0.05 mg/kg/day (parietal bones, major blood vessels). Irregular ossification and/or skeletal abnormalities occurred in the skull and jaw, vertebrae and ribs, sternum, pelvis, tail, and scapula; and dose-dependent minor skeletal variations were observed. Visceral abnormalities occurred in blood vessels, lung, liver, and esophagus. Bilobed or bifurcated gallbladder was seen in all treatment groups, but not in the control group.
In pregnant female rats given subcutaneous doses of 0.1, 0.25 and 1.0 mg/kg/day liraglutide from gestation day 6 through weaning or termination of nursing on lactation day 24, estimated systemic exposures were 0.8-, 3-, and 11-times human exposure at the MRHD of 1.8 mg/day, based on plasma AUC. A slight delay in parturition was observed in the majority of treated rats. Group mean body weight of neonatal rats from liraglutide-treated dams was lower than neonatal rats from control group dams. Bloody scabs and agitated behavior occurred in male rats descended from dams treated with 1 mg/kg/day liraglutide. Group mean body weight from birth to postpartum day 14 trended lower in F2 generation rats descended from liraglutide-treated rats compared to F2 generation rats descended from controls, but differences did not reach statistical significance for any group.
8.2 Lactation
Risk Summary
There are no data on the presence of liraglutide or insulin degludec in human milk, the effects on the breastfed infant, or the effects on milk production. In lactating rats, insulin degludec and liraglutide, the two components of XULTOPHY 100/3.6, were present in milk.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for XULTOPHY 100/3.6 and any potential adverse effects on the breastfed infant from XULTOPHY 100/3.6 or from the underlying maternal condition.
Data
Insulin degludec
In lactating rats, insulin degludec was present in milk at a concentration lower than that in plasma.
Liraglutide
In lactating rats, liraglutide was present unchanged in milk at concentrations approximately 50% of maternal plasma concentrations.
8.4 Pediatric Use
Safety and effectiveness of XULTOPHY 100/3.6 have not been established in pediatric patients.
8.5 Geriatric Use
Of the total number of 1881 subjects in clinical studies of XULTOPHY 100/3.6, 375 (19.9%) were 65 years and over, while 52 (2.8%) were 75 years and over [see Clinical Studies (14)]. No overall differences in safety or effectiveness of XULTOPHY 100/3.6 were observed between patients 65 years of age and older and younger patients.
Age had no clinically relevant effect on the pharmacokinetics of XULTOPHY 100/3.6 [see Clinical Pharmacology (12.3)].
In geriatric patients with diabetes, the initial dosing, dose increments, and maintenance dosage should be conservative to avoid hypoglycemic reactions. Hypoglycemia may be more difficult to recognize in geriatric patients.
8.6 Renal Impairment
XULTOPHY 100/3.6
There is limited experience with XULTOPHY 100/3.6 in patients with mild and moderate kidney impairment and when used in these patients, additional glucose monitoring and XULTOPHY 100/3.6 dose adjustments may be required on an individual basis. XULTOPHY 100/3.6 has not been studied in patients with severe kidney impairment [see Warnings and Precautions (5.7) and Clinical Pharmacology (12.3)].
Insulin degludec
No clinically relevant difference in the pharmacokinetics of insulin degludec was identified in a study comparing healthy subjects and subjects with kidney impairment including subjects with end stage kidney disease.
Liraglutide
The safety and efficacy of liraglutide was evaluated in a 26 week clinical study that included patients with moderate kidney impairment (eGFR 30 to 60 mL/min/1.73 m2). In the liraglutide treatment arm of a cardiovascular outcomes trial (LEADER trial) [see Clinical Studies (14.4)], 1932 (41.4%) patients had mild kidney impairment, 999 (21.4%) patients had moderate renal impairment and 117 (2.5%) patients had severe kidney impairment at baseline. No overall differences in safety or efficacy were seen in these patients compared to patients with normal kidney function.
There is limited experience with liraglutide in patients with end stage kidney disease. There have been postmarketing reports of acute kidney failure and worsening of chronic kidney failure, which may sometimes require hemodialysis [see Warnings and Precautions (5.7) and Adverse Reactions (6.3)].
8.7 Hepatic Impairment
XULTOPHY 100/3.6
XULTOPHY 100/3.6 has not been studied in patients with hepatic impairment.
Insulin degludec
No clinically relevant difference in the pharmacokinetics of insulin degludec, one of the components of XULTOPHY 100/3.6, was identified in a study comparing healthy subjects and subjects with hepatic impairment (mild, moderate, and severe hepatic impairment) [see Clinical Pharmacology (12.3)].
Liraglutide
There is limited experience in patients with mild, moderate or severe hepatic impairment with liraglutide, one of the components of XULTOPHY 100/3.6 [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Hypoglycemia (from insulin and liraglutide) and gastrointestinal adverse reactions (from liraglutide) may develop if a patient is dosed with more XULTOPHY 100/3.6 than required.
An excess of insulin-containing products like XULTOPHY 100/3.6 relative to food intake, energy expenditure, or both may lead to severe and sometimes prolonged and life-threatening hypoglycemia and hypokalemia [see Warnings and Precautions (5.6, 5.10)]. Mild episodes of hypoglycemia usually can be treated with oral glucose. Lowering the drug dosage, and adjustments in meal patterns, or exercise may be needed. More severe episodes of hypoglycemia with coma, seizure, or neurologic impairment may be treated with intramuscular/subcutaneous glucagon for emergency use or concentrated intravenous glucose. After apparent clinical recovery from hypoglycemia, continued observation and additional carbohydrate intake may be necessary to avoid reoccurrence of hypoglycemia. Hypokalemia must be corrected appropriately.
Overdoses have been reported in clinical trials and postmarketing use of liraglutide, one of the components of XULTOPHY 100/3.6. Effects have included severe nausea and severe vomiting. In the event of overdosage, appropriate supportive treatment should be initiated according to the patient’s clinical signs and symptoms.
-
11 DESCRIPTION
Insulin degludec
Insulin degludec is a long-acting basal human insulin analog. Insulin degludec is produced by a process that includes expression of recombinant DNA in Saccharomyces cerevisiae followed by chemical modification.
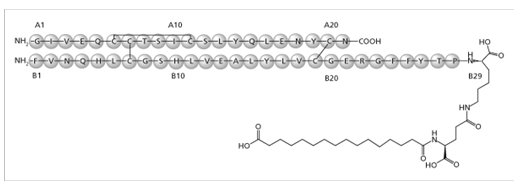
Insulin degludec differs from human insulin in that the amino acid threonine in position B30 has been omitted and a side-chain consisting of glutamic acid and a C16 fatty acid has been attached (chemical name: LysB29(Nε-hexadecandioyl-γ-Glu) des(B30) human insulin). Insulin degludec has a molecular formula of C274H411N65O81S6 and a molecular weight of 6.104 kDa. It has the following structure:
Figure 1: Structural Formula of Insulin degludec
Liraglutide
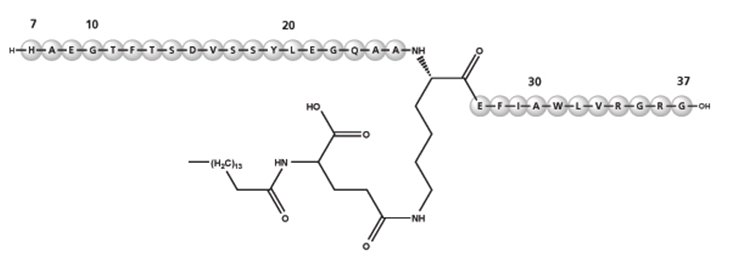
Liraglutide is an analog of human GLP-1 and acts as a GLP-1 receptor agonist. The peptide precursor of liraglutide, produced by a process that includes expression of recombinant DNA in Saccharomyces cerevisiae, has been engineered to be 97% homologous to native human GLP-1 by substituting arginine for lysine at position 34. Liraglutide is made by attaching a C16 fatty acid (palmitic acid) with a glutamic acid spacer on the remaining lysine residue at position 26 of the peptide precursor. The molecular formula of liraglutide is C172H265N43O51 and the molecular weight is 3.751 kDa. It has the following structure:
Figure 2: Structural Formula of Liraglutide
XULTOPHY 100/3.6 (insulin degludec and liraglutide) injection, for subcutaneous use, is a combination of a long-acting basal human insulin analog, insulin degludec, and a GLP-1 receptor agonist, liraglutide.
XULTOPHY 100/3.6 is a sterile, aqueous, clear, and colorless solution. Each pre-filled pen contains 3 mL equivalent to 300 units insulin degludec and 10.8 mg liraglutide. Each mL contains 100 units insulin degludec and 3.6 mg liraglutide.
XULTOPHY 100/3.6 contains the following inactive ingredients per mL: glycerol (19.7 mg), phenol (5.7 mg), zinc (55 mcg), and Water for Injection, USP. XULTOPHY 100/3.6 has a pH of approximately 8.15. Hydrochloric acid or sodium hydroxide may be added to adjust pH.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
XULTOPHY 100/3.6
XULTOPHY 100/3.6 is a combination product consisting of insulin degludec and liraglutide.
Insulin degludec
The primary activity of insulin degludec is the regulation of glucose metabolism. Insulin and its analogs lower blood glucose by stimulating peripheral glucose uptake, especially by skeletal muscle and fat, and by inhibiting hepatic glucose production. Insulin also inhibits lipolysis and proteolysis, and enhances protein synthesis.
Liraglutide
Liraglutide is a GLP-1 receptor agonist that increases glucose-dependent insulin release, decreases glucagon secretion, and slows gastric emptying.
12.2 Pharmacodynamics
Following a single dose administration, XULTOPHY 100/3.6 has a duration of action reflecting the combination of the individual glucodynamic action profiles of insulin degludec and liraglutide.
Following once-daily administration, XULTOPHY 100/3.6 lowers fasting plasma glucose levels and postprandial glucose levels.
Cardiac Electrophysiology (QTc):
XULTOPHY 100/3.6
The effect of XULTOPHY 100/3.6 on QTc has not been studied.
Liraglutide
The effect of liraglutide, one of the components of XULTOPHY 100/3.6, on cardiac repolarization was tested in a QTc study. Liraglutide, at steady state concentrations with daily doses up to 1.8 mg, did not produce QTc prolongation.
12.3 Pharmacokinetics
Overall, the pharmacokinetics of insulin degludec and liraglutide were not affected in a clinically relevant manner when administered as XULTOPHY 100/3.6.
Absorption
In patients with type 2 diabetes mellitus (mean body weight 87.5 kg) reaching the maximum daily dose (50 units/1.8 mg) of XULTOPHY 100/3.6, the estimated mean steady-state exposure (AUC 0-24 h) of insulin degludec was 113 h*nmol/L and of liraglutide 1227 h*ng/mL based on population pharmacokinetic analysis. The corresponding maximum concentrations were 5196 pmol/L for insulin degludec and 55 ng/mL for liraglutide. Steady state concentrations of insulin degludec and liraglutide are reached after 2-3 days of daily administration.
Distribution
Insulin degludec and liraglutide are extensively bound to plasma proteins >99% and >98%, respectively.
Elimination
The half-life of insulin degludec is approximately 25 hours and the half-life of liraglutide is approximately 13 hours.
Metabolism
Insulin degludec
Degradation of insulin degludec is similar to that of human insulin; all metabolites formed are inactive.
Liraglutide
During the initial 24 hours following administration of a single [3H]-liraglutide dose to healthy subjects, the major component in plasma was intact liraglutide. Liraglutide is endogenously metabolized in a similar manner to large proteins without a specific organ as a major route of elimination.
Specific Populations
Geriatric Patients
Age had no clinically relevant effect on the pharmacokinetics of XULTOPHY 100/3.6 based on results from a population pharmacokinetic analysis including adult patients up to 83 years treated with XULTOPHY 100/3.6 [see Use in Specific Populations (8.5)].
Male and Female Patients
There were no clinically relevant differences in the pharmacokinetics of XULTOPHY 100/3.6 in male and female patients, based on results from a population pharmacokinetic analysis.
Racial or Ethnic Groups
There were no clinically relevant differences in the pharmacokinetics of XULTOPHY 100/3.6 between the racial and ethnic groups investigated, based on results from a population pharmacokinetic analysis.
Body weight
The effect of body weight on the exposure level of the components of XULTOPHY 100/3.6 was investigated in the population pharmacokinetic analysis. Exposure levels decreased with increase in baseline body weight for both insulin degludec and liraglutide.
Patients with Renal Impairment
XULTOPHY 100/3.6
There is limited experience with XULTOPHY 100/3.6 in patients with mild and moderate renal impairment. XULTOPHY 100/3.6 has not been studied in patients with severe renal impairment [see Warnings and Precautions (5.7)].
Insulin degludec
Insulin degludec has been studied in a pharmacokinetic study in 32 subjects (n=6/group) with normal or impaired renal function/end-stage renal disease following administration of a single dose (0.4 U/kg) of insulin degludec. Renal function was defined using creatinine clearance (Clcr) as follows: >80 mL/min (normal), 50-80 mL/min (mild), 30-50 mL/min (moderate) and <30 mL/min (severe). Subjects requiring dialysis were classified as having end-stage renal disease (ESRD). Total exposure (AUCIDeg,0-120h,SD) of insulin degludec was similar in subjects with normal and impaired renal function. No clinically relevant difference in the pharmacokinetics of insulin degludec was identified between healthy subjects and subjects with renal impairment. Hemodialysis did not affect clearance of insulin degludec (CL/FIDeg,SD) in subjects with ESRD [see Warnings and Precautions (5.7)].
Liraglutide
The single-dose pharmacokinetics of liraglutide were evaluated in subjects with varying degrees of renal impairment. Subjects with mild (estimated creatinine clearance 50-80 mL/min) to severe (estimated creatinine clearance <30 mL/min) renal impairment and subjects with end-stage renal disease requiring dialysis were included in the trial. Compared to healthy subjects, liraglutide AUC in mild, moderate, and severe renal impairment and in end-stage renal disease was on average 35%, 19%, 29% and 30% lower, respectively [see Warnings and Precautions (5.7)].
Patients with Hepatic Impairment
XULTOPHY 100/3.6
XULTOPHY 100/3.6 has not been studied in patients with hepatic impairment.
Insulin degludec
Insulin degludec has been studied in a pharmacokinetic study in 24 subjects (n=6/group) with normal or impaired hepatic function (mild, moderate, and severe hepatic impairment) following administration of a single dose (0.4 U/kg) of insulin degludec. Hepatic function was defined using Child-Pugh Scores ranging from 5 (mild hepatic impairment) to 15 (severe hepatic impairment). No clinically relevant differences in the pharmacokinetics of insulin degludec were identified between healthy subjects and subjects with hepatic impairment [see Hepatic Impairment (8.7)].
Liraglutide
The single-dose pharmacokinetics of liraglutide were evaluated in subjects with varying degrees of hepatic impairment. Subjects with mild (Child Pugh score 5-6) to severe (Child Pugh score > 9) hepatic impairment were included in the trial. Compared to healthy subjects, liraglutide AUC in subjects with mild, moderate and severe hepatic impairment was on average 11%, 14% and 42% lower, respectively.
Drug Interaction Studies
In vitro assessment of drug-drug interactions
In vitro data suggest that the potential for pharmacokinetic drug interactions related to CYP interaction and protein binding is low for both the liraglutide and insulin degludec components of XULTOPHY 100/3.6.
The delay of gastric emptying with liraglutide one of the components of XULTOPHY 100/3.6 may influence absorption of concomitantly administered oral medicinal products. Interaction studies did not show any clinically relevant delay of absorption.
In vivo assessment of drug-drug interactions
Liraglutide
The drug-drug interaction studies were performed at steady state with liraglutide 1.8 mg/day. Before administration of concomitant treatment, subjects underwent a 0.6 mg weekly dose increase to reach the maximum dose of 1.8 mg/day. Administration of the interacting drugs was timed so that Cmax of liraglutide (8-12 h) would coincide with the absorption peak of the co-administered drugs.
Digoxin
A single dose of digoxin 1 mg was administered 7 hours after the dose of liraglutide at steady state. The concomitant administration with liraglutide resulted in a reduction of digoxin AUC by 16%; Cmax decreased by 31%. Digoxin median time to maximal concentration (Tmax) was delayed from 1 h to 1.5 h.
Lisinopril
A single dose of lisinopril 20 mg was administered 5 minutes after the dose of liraglutide at steady state. The co-administration with liraglutide resulted in a reduction of lisinopril AUC by 15%; Cmax decreased by 27%. Lisinopril median Tmax was delayed from 6 h to 8 h with liraglutide.
Atorvastatin
Liraglutide did not change the overall exposure (AUC) of atorvastatin following a single dose of atorvastatin 40 mg, administered 5 hours after the dose of liraglutide at steady state. Atorvastatin Cmax was decreased by 38% and median Tmax was delayed from 1 to 3 hours with liraglutide.
Acetaminophen
Liraglutide did not change the overall exposure (AUC) of acetaminophen following a single dose of acetaminophen 1000 mg, administered 8 hours after the dose of liraglutide at steady state. Acetaminophen Cmax was decreased by 31% and median Tmax was delayed up to 15 minutes.
Griseofulvin
Liraglutide did not change the overall exposure (AUC) of griseofulvin following co-administration of a single dose of griseofulvin 500 mg with liraglutide at steady state. Griseofulvin Cmax increased by 37% while median Tmax did not change.
Oral Contraceptives
A single dose of an oral contraceptive combination product containing 0.03 mg ethinylestradiol and 0.15 mg levonorgestrel was administered under fed conditions and 7 hours after the dose of liraglutide at steady state. Liraglutide lowered ethinylestradiol and levonorgestrel Cmax by 12% and 13%, respectively. There was no effect of liraglutide on the overall exposure (AUC) of ethinylestradiol. Liraglutide increased the levonorgestrel AUC0-∞ by 18%. Liraglutide delayed Tmax for both ethinylestradiol and levonorgestrel by 1.5 hours.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of XULTOPHY 100/3.6, insulin degludec, or liraglutide.
XULTOPHY 100/3.6
Administration of XULTOPHY 100/3.6 may cause formation of antibodies against insulin degludec and/or liraglutide. In rare cases, the presence of such antibodies may necessitate adjustment of the XULTOPHY 100/3.6 dose in order to correct a tendency to hyper- or hypoglycemia. During the 26-52 week clinical trials where antibodies were measured in patients receiving XULTOPHY 100/3.6, 90/812 (11%) patients were positive for insulin degludec specific antibodies at end of treatment vs. 22/936 (2%) at baseline, 250/811 (31%) patients were positive for antibodies cross-reacting with human insulin at end of treatment vs. 136/930 (15%) at baseline. 17/815 (2%) of patients developed treatment induced anti-liraglutide antibodies. No patients had pre-existing anti-liraglutide antibodies. Antibody formation has not been associated with reduced efficacy of XULTOPHY 100/3.6.
Liraglutide
A subset of liraglutide-treated patients (1104 of 2501, 44%) in five adult double-blind clinical trials of 26 weeks duration or longer were tested for the presence of anti-liraglutide antibodies at the end of treatment [see Clinical Studies (14.1)] and 102/1104 (9%) of liraglutide-treated patients developed anti-liraglutide antibodies. Of these 102 liraglutide-treated patients, 56 (5%) patients developed antibodies that cross-reacted with native GLP-1. These cross-reacting antibodies were not tested for neutralizing effect against native GLP-1, and thus the potential for clinically significant neutralization of native GLP-1 was not assessed. Antibodies that had a neutralizing effect on liraglutide in an in vitro assay occurred in 12 (1% ) of the liraglutide-treated patients. There was no identified clinically significant effect of anti-liraglutide antibodies on effectiveness of liraglutide.
In five double-blind glycemic control trials of liraglutide, events from a composite of adverse events potentially related to immunogenicity (e.g. urticaria, angioedema) occurred among 0.8% of liraglutide-treated patients and among 0.4% of comparator-treated patients. Urticaria accounted for approximately one-half of the events in this composite for liraglutide-treated patients. Patients who developed anti-liraglutide antibodies were not more likely to develop events from the immunogenicity events composite than were patients who did not develop anti-liraglutide antibodies.
In a cardiovascular outcomes trial (LEADER trial) [see Clinical Studies (14.4)], anti-liraglutide antibodies were detected in 11 out of the 1247 (0.9%) liraglutide-treated patients with antibody measurements. Of the 11 liraglutide-treated patients who developed anti-liraglutide antibodies, none were observed to develop neutralizing antibodies to liraglutide, and 5 patients (0.4%) developed cross-reacting antibodies against native GLP-1.
TRESIBA (insulin degludec)
In a 52-week study of adult insulin-naïve type 2 diabetes mellitus patients, of the patients who received insulin degludec, 13/760 (2%) patients were positive at baseline for anti-insulin degludec antibodies and 46/744 (6%) patients developed anti-insulin degludec antibodies at least once during the study. The 58/60 (97%) patients who were positive for anti-insulin degludec antibodies were also positive for anti-human insulin antibodies.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
XULTOPHY 100/3.6
No studies have been conducted with the XULTOPHY 100/3.6 combination to evaluate carcinogenesis, mutagenesis or impairment of fertility. The following data are based upon studies with insulin degludec and liraglutide individually.
Insulin degludec
Standard 2-year carcinogenicity studies in animals have not been performed to evaluate the carcinogenic potential of insulin degludec.
In a 52-week study including human insulin (NPH insulin) as comparator, Sprague-Dawley rats were dosed subcutaneously with insulin degludec at 3.3, 6.7, and 10 U/kg/day, resulting in 5 times the human exposure (AUC) when compared to a human subcutaneous dose of 0.75 U/kg/day. Human insulin was dosed at 6.7 U/kg/day. No treatment-related increases in incidences of hyperplasia, benign or malignant tumors were recorded in female mammary glands from rats dosed with insulin degludec and no treatment related changes in the female mammary gland cell proliferation were found using BrdU incorporation. Further, no treatment related changes in the occurrence of hyperplastic or neoplastic lesions were seen in any animals dosed with insulin degludec when compared to vehicle or human insulin.
Genotoxicity testing of insulin degludec was not performed.
In a combined fertility and embryo-fetal study in male and female rats, treatment with insulin degludec up to 21 U/kg/day (approximately 5 times the human subcutaneous dose of 0.75 U/kg/day, based on U/body surface area) prior to mating and in female rats during gestation had no effect on mating performance and fertility.
Liraglutide
A 104-week carcinogenicity study was conducted in male and female CD-1 mice at doses of 0.03, 0.2, 1.0, and 3.0 mg/kg/day liraglutide administered by bolus subcutaneous injection yielding systemic exposures 0.2-, 2-, 10- and 45-times the human exposure, respectively, at the MRHD of 1.8 mg/day based on plasma AUC comparison. A dose-related increase in benign thyroid C-cell adenomas was seen in the 1.0 and the 3.0 mg/kg/day groups with incidences of 13% and 19% in males and 6% and 20% in females, respectively. C-cell adenomas did not occur in control groups or 0.03 and 0.2 mg/kg/day groups. Treatment-related malignant C-cell carcinomas occurred in 3% of females in the 3.0 mg/kg/day group. Thyroid C-cell tumors are rare findings during carcinogenicity testing in mice. A treatment-related increase in fibrosarcomas was seen on the dorsal skin and subcutis, the body surface used for drug injection, in males in the 3 mg/kg/day group. These fibrosarcomas were attributed to the high local concentration of drug near the injection site. The liraglutide concentration in the clinical formulation (6 mg/mL) is 10-times higher than the concentration in the formulation used to administer 3 mg/kg/day liraglutide to mice in the carcinogenicity study (0.6 mg/mL).
A 104-week carcinogenicity study was conducted in male and female Sprague Dawley rats at doses of 0.075, 0.25 and 0.75 mg/kg/day liraglutide administered by bolus subcutaneous injection with exposures 0.5-, 2- and 8-times the human exposure, respectively, resulting from the MRHD based on plasma AUC comparison. A treatment-related increase in benign thyroid C-cell adenomas was seen in males in 0.25 and 0.75 mg/kg/day liraglutide groups with incidences of 12%, 16%, 42%, and 46% and in all female liraglutide-treated groups with incidences of 10%, 27%, 33%, and 56% in 0 (control), 0.075, 0.25, and 0.75 mg/kg/day groups, respectively. A treatment-related increase in malignant thyroid C-cell carcinomas was observed in all male liraglutide-treated groups with incidences of 2%, 8%, 6%, and 14% and in females at 0.25 and 0.75 mg/kg/day with incidences of 0%, 0%, 4%, and 6% in 0 (control), 0.075, 0.25, and 0.75 mg/kg/day groups, respectively. Thyroid C-cell carcinomas are rare findings during carcinogenicity testing in rats.
Studies in mice demonstrated that liraglutide-induced C-cell proliferation was dependent on the GLP-1 receptor and that liraglutide did not cause activation of the REarranged during Transfection (RET) proto-oncogene in thyroid C-cells.
Human relevance of thyroid C-cell tumors in mice and rats is unknown and has not been determined by clinical studies or nonclinical studies [see Boxed Warning and Warnings and Precautions (5.1)].
Liraglutide was negative with and without metabolic activation in the Ames test for mutagenicity and in a human peripheral blood lymphocyte chromosome aberration test for clastogenicity. Liraglutide was negative in repeat-dose in vivo micronucleus tests in rats.
In rat fertility studies using subcutaneous doses of 0.1, 0.25 and 1.0 mg/kg/day liraglutide, males were treated for 4 weeks prior to and throughout mating and females were treated 2 weeks prior to and throughout mating until gestation day 17. No direct adverse effects on male fertility was observed at doses up to 1.0 mg/kg/day, a high dose yielding an estimated systemic exposure 11- times the human exposure at the MRHD, based on plasma AUC. In female rats, an increase in early embryonic deaths occurred at 1.0 mg/kg/day. Reduced body weight gain and food consumption were observed in females at the 1.0 mg/kg/day dose.
-
14 CLINICAL STUDIES
14.1 Overview of Clinical Studies
A total of 3908 patients with type 2 diabetes mellitus participated in 6 randomized, parallel and active or placebo-controlled phase 3 trials of 26 weeks duration.
Three studies were conducted in patients inadequately controlled on one or more OADs (e.g. metformin, pioglitazone, sulfonylurea, or sodium-glucose cotransporter-2 inhibitors (SGLT2i) (Tables 5-7). Three studies were conducted in patients converting from liraglutide or basal insulin: one study was conducted in patients converting from liraglutide (with doses up to 1.8 mg), (Table 8), one study was conducted in patients converting from any basal insulin (Table 9), and one study was conducted in patients converting from insulin glargine U-100 (Table 10).
In all trials, XULTOPHY 100/3.6 was titrated twice weekly by increments or decrements of 2 units (2 units insulin degludec/0.072 mg liraglutide), towards a pre-specified fasting blood glucose target. The same titration algorithm was applied for basal insulin comparators.
14.2 Patients with Type 2 Diabetes Mellitus Uncontrolled on OAD Treatment
NCT01336023: The efficacy and safety of XULTOPHY 100/3.6 compared to insulin degludec and liraglutide, all administered once-daily, was studied in a 26-week randomized, open-label, three-arm parallel trial in 1660 patients with type 2 diabetes mellitus inadequately controlled on 1-2 OADs (metformin or metformin with or without pioglitazone).
The mean age of the trial population was 55 years and mean duration of diabetes was 7 years. 51% were male. 62% were White, 7% were Black or African American and 15% were Hispanic or Latino ethnicity, 5% of patients had eGFR < 60mL/min/1.73m2; no patients had eGFR < 30mL/min/1.73m2. The mean BMI was 31.2 kg/m2.
The starting dose of XULTOPHY 100/3.6 was 10 units (10 units insulin degludec/0.36 mg liraglutide). The starting dose of insulin degludec was 10 units. XULTOPHY 100/3.6 and insulin degludec were titrated twice weekly towards a target fasting blood glucose goal of 72-90 mg/dL. Patients in the liraglutide arm followed a fixed dose escalation scheme with a starting dose of 0.6 mg and a dose increase of 0.6 mg weekly until a daily dose of 1.8 mg was reached. Patients continued on pre-trial treatment with metformin or metformin and pioglitazone throughout the trial.
At the end of 26 weeks, treatment with XULTOPHY 100/3.6, insulin degludec, and liraglutide resulted in a reduction in HbA1c from baseline of 1.81%, 1.35% and 1.21%, respectively, (see Table 5). The end of trial dose of XULTOPHY 100/3.6 was 38 units (38 units insulin degludec/1.37 mg liraglutide).
Table 5: Results of a 26-Week Trial with XULTOPHY 100/3.6 in Patients with Type 2 Diabetes Mellitus Inadequately Controlled on Metformin Alone or in Combination with Pioglitazone
- XULTOPHY 100/3.6
- + metformin ± pioglitazone
- Insulin degludec
- + metformin
- ± pioglitazone
- Liraglutide
- + metformin
- ± pioglitazone
Total (N)
- 833
- 413
- 414
HbA1c (%)
Baseline
- 8.3
- 8.3
- 8.3
End of Trial (LS Mean)#
- 6.5
- 6.9
- 7.1
Change from baseline (LS Mean)#
- -1.81
- -1.35
- -1.21
Estimated treatment difference [95% CI]#
- --
- -0.46%
- [-0.59; -0.34]A
- -0.60%
- [-0.72; -0.47]A
Percentage of patients achieving HbA1c <7%##
- 74.1%
- 60.5%
- 56.0%
Fasting Plasma Glucose (FPG) (mg/dL)
Baseline
- 166
- 169
- 163
End of Trial (LS Mean)#
- 104
- 107
- 133
Change from baseline (LS Mean)#
- -61.8
- -59.2
- -32.4
Ap<0.01. Primary endpoint was tested for non-inferiority of XULTOPHY 100/3.6 to insulin degludec based on pre-specified non-inferiority margin of 0.3% and for superiority of XULTOPHY 100/3.6 to liraglutide.
#Estimated using an ANCOVA with treatment, baseline HbA1c stratum, sub-study, concomitant diabetes treatment and country as factors and baseline response as covariate. Multiple imputation modelled “return to baseline” of the treatment effect for subjects having missing week 26 data.
##Patients with missing HbA1c value at week 26 data were considered non-responders.
There were 11.8% of subjects in the XULTOPHY 100/3.6 arm, 12.3% in the insulin degludec arm and 15.5% in the liraglutide arm for whom HbA1c data was missing at week 26.
NCT01618162: The efficacy and safety of XULTOPHY 100/3.6 compared to placebo were studied in a 26-week randomized, double-blind, treat-to-target trial in 435 patients with type 2 diabetes mellitus inadequately controlled on sulfonylurea alone or in combination with metformin.
The mean age of the trial population was 60 years, and mean duration of diabetes was 9 years. 52% were male. 75% were White, 7% were Black or African American and 9% were Hispanic or Latino ethnicity, 11% of patients had eGFR < 60mL/min/1.73m2; no patients had eGFR < 30mL/min/1.73m2. The mean BMI was 31.5 kg/m2.
XULTOPHY 100/3.6 was started at 10 units (10 units insulin degludec/0.36 mg liraglutide) and titrated twice weekly towards a target fasting blood glucose goal of 72-108 mg/dL. Patients continued on pre-trial treatment with sulfonylurea, with or without metformin throughout the trial.
Treatment with XULTOPHY 100/3.6 for 26 weeks resulted in a statistically significant reduction in mean HbA1c compared to placebo (see Table 6). The end of trial dose of XULTOPHY 100/3.6 was 28 units (28 units insulin degludec/1.01 mg liraglutide).
Table 6: Results of a 26-Week Trial with XULTOPHY 100/3.6 in Patients with Type 2 Diabetes Mellitus Inadequately Controlled on Sulfonylurea Alone or in Combination with Metformin
XULTOPHY 100/3.6
- + sulfonylurea###
± metformin
Placebo
- + sulfonylurea###
± metformin
Total (N)
289
146
HbA1c (%)
Baseline
7.9
7.9
End of Trial (LS Mean)#
6.5
7.3
Change from baseline (LS Mean)#
-1.42
-0.62
Estimated treatment difference [95% CI]#
-0.81% [-0.98; -0.63]A
Percentage of patients achieving HbA1c <7%##
70.9%
26.7%
FPG (mg/dL)
Baseline
164
165
End of Trial (LS Mean)#
118
152
Change from baseline (LS Mean)#
-46.2
-12.1
Ap<0.01. Primary endpoint was tested for superiority of XULTOPHY 100/3.6 to placebo.
#Estimated using an ANCOVA with treatment, region and pre-trial medication as fixed factors and baseline response as covariate. Multiple imputation modelled “jump-to-control” of the treatment effect for subjects having missing week 26 data.
##Patients with missing HbA1c value at week 26 data were considered non-responders.
There were 12.8% of subjects in the XULTOPHY 100/3.6 arm and 24.7% in the placebo arm for whom HbA1c data was missing at week 26.
###Dose of sulfonylurea was ≥ half of the maximum approved dose.
NCT02773368: The efficacy and safety of XULTOPHY 100/3.6 compared to insulin glargine U-100, both administered once-daily, were studied in a 26-week randomized, open-label, two-arm parallel trial in 420 patients with type 2 diabetes mellitus inadequately controlled on a SGLT2i alone or in combination with other OADs (with or without metformin, pioglitazone, and/or dipeptidyl peptidase-4 [DPP4] inhibitor). At randomization, the DPP4 inhibitor was discontinued.
The mean age of the trial population was 57 years and mean duration of diabetes was 10 years. 59% were male. 82% were White, 1% were Black or African American and 16% were Hispanic or Latino ethnicity, 3% of patients had eGFR < 60mL/min/1.73m2; none of the patients had eGFR < 30mL/min/1.73m2. The mean BMI was 31.2 kg/m2.
The starting dose of XULTOPHY 100/3.6 was 10 units (10 units insulin degludec/0.36 mg liraglutide). The starting dose of insulin glargine U-100 was 10 units. XULTOPHY 100/3.6 and insulin glargine U-100 were titrated twice weekly to target a fasting blood glucose goal of 72-90 mg/dL. Patients could not increase the dose of XULTOPHY 100/3.6 and insulin glargine U-100 by more than 4 units per week, and there was no maximum allowed dose of insulin glargine. The patients continued on pre-trial treatment with SGLT2i, with or without other OADs throughout the entire trial. The targeted fasting blood glucose goal was achieved by 49.0% of patients randomized to XULTOPHY 100/3.6 and 41.9% of patients randomized to insulin glargine at 26 weeks.
At the end of 26 weeks, XULTOPHY 100/3.6 resulted in a reduction in HbA1c from baseline of 1.97% and insulin glargine U-100 resulted in a reduction of 1.59% (see Table 7). At the end of trial, the average dose of XULTOPHY 100/3.6 was 36 units (36 units insulin degludec/1.3 mg liraglutide) and the dose of insulin glargine was 54 units; it is unclear that these observed differences in insulin doses are clinically important. The difference in HbA1c effect observed at 26 weeks may not necessarily reflect the effect in the care setting where insulin glargine may be more rapidly titrated.
Table 7: Results of a 26-Week Trial in Patients with Type 2 Diabetes Mellitus Inadequately
Controlled on SGLT2i Alone or in Combination with Metformin, Pioglitazone and/or DPP4 Inhibitor
XULTOPHY 100/3.6
+ SGLT2i
± metformin± pioglitazone
Insulin Glargine U-100
+ SGLT2i
± metformin± pioglitazone
Total (N)
210
210
HbA1c (%)
Baseline
8.2
8.4
End of Trial (LS Mean)#
6.3
6.7
Change from baseline (LS Mean)#
-1.97
-1.59
Estimated treatment difference [95% CI]#
-0.38% [-0.54; -0.23]A
Percentage of patients achieving HbA1c <7%##
79.5%
68.6%
FPG (mg/dL)
Baseline
171
172
End of Trial (LS Mean)###
108
112
Change from baseline (LS Mean)###
-63.8
-59.9
APrimary endpoint was tested for non-inferiority of XULTOPHY 100/3.6 to insulin glargine U-100 on a non-inferiority margin of 0.3%.
#Estimated using an ANCOVA with treatment, pre-trial OAD group and region as factors and corresponding baseline value as covariate. For HbA1c (%), missing week 26 measurements from subjects who discontinued early were multiply-imputed using information from subjects who also discontinued early but still had a week 26 measurement (retrieved dropouts).
##Patients with missing HbA1c value at week 26 data were considered non-responders.
There were 6.2% of subjects in the XULTOPHY 100/3.6 arm and 2.4% in the insulin glargine U-100 arm for whom HbA1c data was missing at week 26.
###Missing week 26 FPG measurements from subjects who discontinued early were imputed using multiple imputation with a mean for each subject equal to their respective baseline value.
14.3 Patients currently on basal insulin or GLP-1 receptor agonist
Converting to XULTOPHY 100/3.6 from GLP-1 receptor agonist
NCT01676116: The efficacy and safety of XULTOPHY 100/3.6 (once-daily) compared to unchanged pre-trial liraglutide up to a dose of 1.8 mg daily, were studied in a 26-week randomized, open-label, treat-to-target (fasting blood glucose goal of 72-90 mg/dL) trial. The trial included 348 patients with type 2 diabetes mellitus inadequately controlled on liraglutide and metformin alone or in combination with pioglitazone, sulfonylurea or both. Oral anti-diabetic drugs (OADs) were continued at pre-trial doses throughout the trial, and 22% of subjects were treated with sulfonylureas (SU) in combination with metformin with or without pioglitazone. The mean age of the population was 58 years, and mean duration of diabetes was 10 years. 49% were male. 91% were White, 8% Black or African American. 11% were Hispanic or Latino ethnicity, 6% of patients had eGFR < 60mL/min/1.73m2; no patient had eGFR < 30mL/min/1.73m2. The mean BMI was 33.1 kg/m2.
The starting dose of XULTOPHY 100/3.6 was 16 units (16 units insulin degludec/0.58 mg liraglutide), and the average starting dose of liraglutide was 1.7 mg. XULTOPHY 100/3.6 was titrated twice weekly to target a fasting blood glucose goal of 72-90 mg/dL. The end of trial dose of XULTOPHY 100/3.6 was 44 units (44 units insulin degludec/1.58 mg liraglutide). The primary endpoint, change in HbA1c, was tested for superiority of XULTOPHY 100/3.6 to unchanged liraglutide therapy.
At the end of 26 weeks, there was a reduction in HbA1c from baseline of 1.31% for XULTOPHY 100/3.6 and 0.36% for liraglutide (see Table 8).
Table 8: Results of a 26-Week Trial with XULTOPHY 100/3.6 in Patients with Type 2 Diabetes Mellitus Inadequately Controlled on Liraglutide up to 1.8 mg Daily
XULTOPHY 100/3.6 + metformin±pioglitazone±SU
liraglutide 1.8 mg+ metformin±pioglitazone±SU
Total (N)
232
116
HbA1c (%)
Baseline
7.8
7.8
End of Trial (LS Mean)#
6.4
7.4
Change from baseline (LS Mean)#
-1.31
-0.36
Estimated treatment difference [95% CI]
-0.95 [-1.15; -0.75]A
Percentage of patients achieving HbA1c <7%##
74.6%
30.2%
FPG (mg/dL)
Baseline
161
169
End of Trial (LS Mean)#
112
153
Change from baseline (LS Mean)#
-51.1
-10.9
- ATest for superiority evaluated at 5.0% level for significance, (p<0.0001)
- #Estimated using an ANCOVA with treatment, pre-trial liraglutide and region as fixed factors and baseline response as covariate. Multiple imputation modelled “return to baseline” of the treatment effect for subjects having missing week 26 data.
- ##Patients with missing HbA1c data at week 26 were considered as non-responders.
- There were 5.2% of subjects in the XULTOPHY 100/3.6 arm and 19.0% in the liraglutide arm for whom HbA1c data was missing at week 26.
Figure 3: Mean HbA1c (%) By Treatment Week in Patients with Type 2 Diabetes Mellitus Inadequately Controlled on Liraglutide
IDegLira=XULTOPHY 100/3.6
Converting from basal insulin
NCT01392573: The efficacy and safety of XULTOPHY 100/3.6 compared to insulin degludec, both once daily and added on to metformin, were studied in a 26-week randomized, double-blind, trial in 398 patients with type 2 diabetes mellitus inadequately controlled on basal insulin and metformin alone or in combination with sulfonylurea/glinides. Basal insulin and sulfonylurea/glinides were discontinued at randomization.
The mean age of the trial population was 57 years, and mean duration of diabetes was 11 years. 55% were male. 77% were White, 5% Black or African American. 10% were Hispanic or Latino ethnicity, 7% of patients had eGFR < 60mL/min/1.73m2; no patient had eGFR < 30mL/min/1.73m2. The mean BMI was 33.7 kg/m2. The mean dose of metformin and basal insulin in patients entering the trial was approximately 2 g and 29 units, respectively.
The starting dose of XULTOPHY 100/3.6 and insulin degludec was 16 units (16 units insulin degludec/0.58 mg liraglutide) and 16 units, respectively. XULTOPHY 100/3.6 and degludec were to be titrated twice weekly to target a fasting blood glucose goal of 72-90 mg/dL. Patients could not increase their dose by more than 4 units per week, and the maximum dose of insulin degludec was limited to 50 units. The targeted fasting blood glucose goal was achieved in 24.0% of patients randomized to insulin degludec and in 31.6% of the patients randomized to XULTOPHY 100/3.6 at 26 weeks.
At the end of 26 weeks, reductions in HbA1c from baseline of 1.94% for XULTOPHY 100/3.6 and 1.05% for insulin degludec limited to 50 units daily were observed (see Table 9). The mean difference (95% CI) in HbA1c reduction between XULTOPHY 100/3.6 and insulin degludec was -0.89 [-1.10; -0.68] and statistically significant. The trial was designed to show the contribution of the liraglutide component to glycemic lowering and the insulin degludec dosing algorithm was selected to isolate the effect of the GLP-1 component. At the end of the trial, the doses of insulin degludec were equivalent between treatment groups. The mean final dose of XULTOPHY 100/3.6 and insulin degludec was 46 units (for XULTOPHY 100/3.6: 46 units insulin degludec/1.66 mg liraglutide). The difference in glucose lowering effect observed in the trial may not necessarily reflect the effect that will be observed in the care setting where insulin degludec dosage can be different than that used in the trial.
Table 9: Results of a 26-Week Trial in Patients with Type 2 Diabetes Mellitus Inadequately Controlled on Basal Insulin
XULTOPHY 100/3.6 + metformin
Insulin degludec* + metformin
Total (N)
199
199
HbA1c (%)
Baseline
8.7
8.8
End of Trial (LS Mean)#
6.9
7.7
Change from baseline (LS Mean)#
-1.94
-1.05
Estimated treatment difference [95% CI]#
-0.89 [-1.10; -0.68]A
Percentage of patients achieving HbA1c <7%##
57.3%
22.6%
FPG (mg/dL)
Baseline
175
172
End of Trial (LS Mean)#
110
118
Change from baseline (LS Mean)#
-63.5
-55.5
Ap<0.01. The trial was designed to show the contribution of the liraglutide component to glycemic lowering and the insulin degludec dosing algorithm was selected to isolate the effect of the GLP-1 component. At the end of the trial, the doses of insulin degludec were equivalent between treatment groups. The mean final dose of XULTOPHY 100/3.6 and insulin degludec was 46 units (for XULTOPHY 100/3.6: 46 units insulin degludec/1.66 mg liraglutide). The difference in glucose lowering effect observed in the trial may not necessarily reflect the effect that will be observed in the care setting where alternative insulin degludec dosage can be used.
*Maximum dose 50 units
- #Estimated using an ANCOVA with treatment, country, and previous antidiabetic treatment as fixed factors and baseline response as covariate. Multiple imputation modelled “jump to control” of the treatment effect for subjects having missing week 26 data.
- ##Patients with missing HbA1c data at week 26 were considered non-responders.
- There were 11.1% of subjects in the XULTOPHY 100/3.6 arm and 13.1% in the insulin degludec arm for whom HbA1c data was
- missing at week 26.
NCT01952145: The efficacy and safety of XULTOPHY 100/3.6 compared to insulin glargine U-100, both once daily and added on to metformin, were studied in a 26-week randomized, open-label, two-arm parallel trial in 557 patients with type 2 diabetes mellitus inadequately controlled on insulin glargine U-100 and metformin.
The mean age of the trial population was 59 years and mean duration of diabetes was 12 years. 50% were male. 95% were White, 2% Black or African American. 43% were Hispanic or Latino ethnicity, 6% of patients had eGFR < 60mL/min/1.73m2; one patient had eGFR < 30mL/min/1.73m2. The mean BMI was 32 kg/m2. The mean dose of insulin glargine U-100 in patients entering the trial was 32 units.
XULTOPHY 100/3.6 and insulin glargine were to be titrated twice weekly to target a fasting blood glucose goal of 72-90 mg/dL. The starting dose of XULTOPHY 100/3.6 was 16 units (16 units insulin degludec/0.58 mg liraglutide). The average starting dose of insulin glargine U-100 was 32 units. Patients could not increase the dose of the two products by more than 4 units per week and there was no maximum allowed dose of insulin glargine. The targeted fasting plasma blood glucose goal was achieved in 39.6% of patients randomized to insulin glargine and 32.9% of the patients randomized to XULTOPHY 100/3.6 at 26 weeks.
At the end of 26 weeks, treatment with XULTOPHY 100/3.6 resulted in a reduction in HbA1c from baseline of 1.67% and was 1.16% for insulin glargine U-100 (see Table 10) and excluded the pre-specified non-inferiority margin of 0.3%. At the end of the trial, the average dose of XULTOPHY 100/3.6 was 41 units (41 units insulin degludec/1.48 mg liraglutide) and the dose of glargine was 66 units, it is unclear that these observed differences in insulin doses are clinically important. The difference in HbA1c effect observed at 26 weeks may not necessarily reflect the effect in the care setting where insulin glargine may be more rapidly titrated.
Table 10: Results of a 26-Week Trial in Patients with Type 2 Diabetes Mellitus Inadequately Controlled on Insulin Glargine U-100
XULTOPHY 100/3.6
+ metformin
Insulin glargine U-100
+ metformin
Total (N)
278
279
HbA1c (%)
Baseline
8.4
8.2
End of Trial (LS Mean)#
6.6
7.1
Change from baseline (LS Mean)#
-1.67
-1.16
Estimated treatment difference [95% CI]
-0.51 [-0.67; -0.34] A
Percentage of patients achieving HbA1c <7%##
68.3%
46.2%
FPG (mg/dL)
Baseline
161
160
End of Trial (LS Mean)#
110
110
Change from baseline (LS Mean)#
-49.9
-49.6
- Ap<0.01. Primary endpoint was tested for noninferiority of XULTOPHY 100/3.6 to insulin glargine U-100. The difference in glucose lowering effect observed in the trial may not necessarily reflect the effect that will be observed in the care setting where alternative insulin glargine dosage can be used.
- #Estimated using an ANCOVA with treatment and region as fixed factors and baseline response as covariate. Multiple imputation modelled “return to baseline” of the treatment effect for subjects having missing week 26 data.
- ##Patients with missing HbA1c value at week 26 data were considered non-responder.
- There were 10.1% of subjects in the XULTOPHY 100/3.6 arm and 4.7% in the insulin glargine U-100 arm for whom HbA1c data was missing at week 26.
14.4 Cardiovascular Outcomes Trials in Patients with Type 2 Diabetes Mellitus and Atherosclerotic Cardiovascular Disease Conducted with Liraglutide 1.8 mg and Insulin Degludec
The effect of XULTOPHY 100/3.6 on the risk of cardiovascular outcomes in patients with type 2 diabetes mellitus and atherosclerotic cardiovascular disease has not been established. The studies below were conducted with liraglutide 1.8 mg and insulin degludec, individually.
Liraglutide 1.8 mg
The LEADER trial (NCT01179048) randomized 9340 patients with inadequately controlled type 2 diabetes mellitus and cardiovascular disease to liraglutide 1.8 mg or placebo in addition to standard of care treatments for type 2 diabetes mellitus for a median follow up of 3.5 years.
Patients either were 50 years of age or older with established, stable cardiovascular, cerebrovascular, peripheral artery disease, chronic kidney disease or chronic heart failure (80% of patients) or were 60 years of age or older and had other specified risk factors for cardiovascular disease (20% of patients). The population was 64% male, 78% White, 10% Asian, and 8% Black or African American; 12% of the population was Hispanic or Latino. The mean duration of type 2 diabetes mellitus was 13 years, the mean HbA1c was 8.7% and the mean BMI was 33 kg/m2; the mean eGFR at baseline was 79 mL/min/1.73 m2.
In total, 96.8% of the patients completed the trial; vital status was available for 99.7%. The primary endpoint was the time from randomization to first occurrence of a major adverse cardiovascular event (MACE) defined as: cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke. No increased risk for MACE was observed with liraglutide 1.8 mg. The total number of primary component MACE endpoints was 1302 (608[13.0%] with liraglutide 1.8 mg and 694 [14.9%] with placebo).
Insulin degludec
The DEVOTE trial (NCT01959529) randomized 7,637 patients with inadequately controlled type 2 diabetes mellitus and cardiovascular disease to either insulin degludec or insulin glargine U-100. Each was administered once-daily in addition to standard of care treatments for diabetes for a median duration of follow up of 2 years.
Patients either were 50 years of age or older and had established, stable cardiovascular, cerebrovascular, peripheral artery disease, chronic kidney disease or chronic heart failure (85% of patients) or were 60 years of age or older and had other specified risk factors for cardiovascular disease (15% of patients). The population was 63% male, 76% White 11% Black or African American, and 10% Asian; 15% of the population was Hispanic or Latino. The mean HbA1c was 8.4% and the mean BMI was 33.6 kg/m2. The baseline mean eGFR was 68 mL/min/1.73m2.
In total, 98% of the patients completed the trial; vital status was known at the end of the trial for 99%. The primary endpoint was the time from randomization to the first occurrence of MACE, defined as: cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke. No increased risk for MACE was observed with insulin degludec when compared to insulin glargine U-100. The total number of primary MACE endpoints was 681 (325 [8.5%] with insulin degludec and 356 [9.3%] with insulin glargine).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
XULTOPHY 100/3.6 (insulin degludec and liraglutide) injection is an injection supplied as a clear, colorless solution in a 3 mL pre-filled, disposable, single-patient use pen injector. The XULTOPHY 100/3.6 pen dials in one unit increments.
Dosage Unit/Strength
Package size
NDC #
3 mL single-patient-use XULTOPHY 100/3.6 pen
(100 units/mL insulin degludec and 3.6 mg/mL liraglutide)
Package of 5
0169-2911-15
16.2 Recommended Storage
Dispense in the original sealed carton with the enclosed Instructions for Use.
Prior to first use, XULTOPHY 100/3.6 should be stored between 2°C and 8°C (36°F to 46°F) until the expiration date printed on the label. Store prefilled pens in the carton so they will stay clean and protected from light. Do not store in the freezer or directly adjacent to the refrigerator cooling element. Do not freeze. Do not use XULTOPHY 100/3.6 if it has been frozen.
After first use, the XULTOPHY 100/3.6 pen can be stored for 21 days at controlled room temperature (59°F to 86°F; 15°C to 30°C) or in a refrigerator (36°F to 46°F; 2°C to 8°C). Keep all XULTOPHY 100/3.6 pens away from direct heat and light.
Always remove the needle after each injection and store the XULTOPHY 100/3.6 pen without a needle attached. This prevents contamination and/or infection, or leakage of the XULTOPHY 100/3.6 pen, and will ensure accurate dosing. Always use a new needle for each injection to prevent contamination.
The storage conditions are summarized in Table 11:
Table 11: Storage Conditions for XULTOPHY 100/3.6 Pen
Prior to first use
After first use
Refrigerated
36°F to 46°F
(2°C to 8°C)Room Temperature
59°F to 86°F
(15°C to 30°C)Refrigerated
36°F to 46°F
(2°C to 8°C)Until expiration date
21 Days
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use)
Risk of Thyroid C-cell Tumors
Inform patients that liraglutide, one of the components of XULTOPHY 100/3.6, causes benign and malignant thyroid C-cell tumors in mice and rats and that the human relevance of this finding is unknown. Counsel patients to report symptoms of thyroid tumors (e.g., a lump in the neck, hoarseness, dysphagia or dyspnea) to their physician [see Boxed Warning and Warnings and Precautions (5.1)].
Pancreatitis
Inform patients of the potential risk for pancreatitis. Explain that persistent severe abdominal pain that may radiate to the back and which may or may not be accompanied by vomiting, is the hallmark symptom of acute pancreatitis. Instruct patients to discontinue XULTOPHY 100/3.6 promptly and contact their physician if persistent severe abdominal pain occurs [see Warnings and Precautions (5.2)].
Never Share a XULTOPHY 100/3.6 Pen Between Patients
Advise patients that they must never share a XULTOPHY 100/3.6 pen with another person, even if the needle is changed, because doing so carries a risk for transmission of blood-borne pathogens [see Warnings and Precautions (5.3].
Hyperglycemia or Hypoglycemia with Changes in Insulin Regimen
Inform patients that hypoglycemia is the most common adverse reaction with insulin products. Inform patients of the symptoms of hypoglycemia (e.g. impaired ability to concentrate and react). This may present a risk in situations where these abilities are especially important, such as driving or operating other machinery. Advise patients who have frequent hypoglycemia or reduced or absent warning signs of hypoglycemia to use caution when driving or operating machinery.
Advise patients that changes in insulin regimen can predispose to hyperglycemia or hypoglycemia and that changes in insulin regimen should be made under close medical supervision [see Warnings and Precautions (5.4)].
Overdose due to Medication Errors
Inform patients that XULTOPHY 100/3.6 contains two drugs: insulin degludec and liraglutide. Accidental mix-ups between insulin products have been reported. To avoid medication errors between XULTOPHY 100/3.6 (an insulin containing product) and other insulin products, instruct patients to always check the label before each injection.
Advise patients that the administration of more than 50 units of XULTOPHY 100/3.6 daily can result in overdose of the liraglutide component. Instruct patients not to administer concurrently with other glucagon-like peptide-1 receptor agonists [see Warnings and Precautions (5.5].
Acute Kidney Injury
Advise patients of the potential risk of dehydration and acute kidney injury due to gastrointestinal adverse reactions and take precautions to avoid fluid depletion. Inform patients of the potential risk for worsening renal function, which in some cases may require dialysis [see Warnings and Precautions (5.7)].
Hypersensitivity Reactions
Inform patients that serious hypersensitivity reactions have been reported during postmarketing use of liraglutide, one of the components of XULTOPHY 100/3.6.
Advise patients on the symptoms of hypersensitivity reactions and instruct them must stop taking XULTOPHY 100/3.6 and seek medical advice promptly [see Warnings and Precautions (5.8)].
Acute Gallbladder Disease
Inform patients of the potential risk for cholelithiasis or cholecystitis. Instruct patients to contact their physician if cholelithiasis or cholecystitis is suspected for appropriate clinical follow-up [see Warnings and Precautions (5.9)].
Pregnancy
Advise a pregnant woman of the potential risk to a fetus. Advise women to inform their healthcare provider if they are pregnant or intend to become pregnant [see Use in Specific Populations (8.1)].
Missed Dose
Inform patients not to take an extra dose of XULTOPHY 100/3.6 or increase the dose to make up for the missed dose. Instruct patients who miss a dose of XULTOPHY 100/3.6 to resume the once-daily dosage regimen as prescribed with the next scheduled dose. If more than three days have elapsed since the last XULTOPHY 100/3.6 dose, reinitiate XULTOPHY 100/3.6 at the recommended starting dose to mitigate any gastrointestinal symptoms associated with reinitiation of treatment. XULTOPHY 100/3.6 should be titrated at the direction of a healthcare provider [see Dosage and Administration (2.1, 2.2, 2.3)].
© 2023 Novo Nordisk
Novo Nordisk®, TRESIBA® and XULTOPHY® 100/3.6 are registered trademarks of Novo Nordisk A/S.
PATENT Information: http://novonordisk-us.com/products/product-patents.html
Manufactured by:
Novo Nordisk Inc.
800 Scudders Mill Road
Plainsboro, NJ 08536
U.S. License Number 1261
For information about XULTOPHY 100/3.6 contact:
Novo Nordisk Inc.
800 Scudders Mill Road
Plainsboro, NJ 08536
1-800-727-6500 (Se habla español)
www.novonordisk-us.com
-
Medication Guide
XULTOPHY® 100/3.6 (ZUL-to-fye)
(insulin degludec and liraglutide) injection for subcutaneous injectionRead this Medication Guide before you start using XULTOPHY 100/3.6 and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about XULTOPHY 100/3.6?
XULTOPHY 100/3.6 may cause serious side effects, including:
- •
- Possible thyroid tumors, including cancer. Tell your healthcare provider if you get a lump or swelling in your neck, hoarseness, trouble swallowing, or shortness of breath. These may be symptoms of thyroid cancer. In studies with rats and mice, liraglutide, one of the components of XULTOPHY 100/3.6, and medicines that work like liraglutide caused thyroid tumors, including thyroid cancer. It is not known if XULTOPHY 100/3.6 will cause thyroid tumors or a type of thyroid cancer called medullary thyroid carcinoma (MTC) in people.
- •
- Do not use XULTOPHY 100/3.6 if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC), or if you have an endocrine system condition called Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
What is XULTOPHY 100/3.6?
- •
- XULTOPHY 100/3.6 is an injectable prescription medicine that contains 2 diabetes medicines, insulin degludec, 100 units/mL, and liraglutide, 3.6 mg/mL. XULTOPHY 100/3.6 should be used along with diet and exercise to lower blood sugar (glucose) in adults with type 2 diabetes mellitus.
- •
- XULTOPHY 100/3.6 is not recommended as the first choice of medicine for treating diabetes.
- •
- XULTOPHY 100/3.6 is not recommended for use in combination with any other product containing liraglutide or another glucagon-like peptide 1 receptor agonist (GLP-1 receptor agonist).
- •
- XULTOPHY 100/3.6 is not for use in people with type 1 diabetes or people with diabetic ketoacidosis (increased ketones in the blood or urine).
- •
- It is not known if XULTOPHY 100/3.6 can be used with mealtime insulin.
- •
- It is not known if XULTOPHY 100/3.6 is safe and effective for use in children.
Who should not use XULTOPHY 100/3.6?
Do not use XULTOPHY 100/3.6 if:
- •
- you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC) or if you have an endocrine system condition called Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- •
- you are allergic to insulin degludec, liraglutide or any of the ingredients in XULTOPHY 100/3.6. See the end of this Medication Guide for a complete list of ingredients in XULTOPHY 100/3.6.
- •
- you are having an episode of low blood sugar (hypoglycemia).
What should I tell my healthcare provider before using XULTOPHY 100/3.6?
Before using XULTOPHY 100/3.6, tell your healthcare provider about all your medical conditions, including if you:
- •
- have or have had problems with your pancreas, kidneys, or liver.
- •
- have heart failure or other heart problems. If you have heart failure, it may get worse while you take TZDs with XULTOPHY 100/3.6.
- •
- have severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems with digesting food.
- •
- are taking certain medicines called GLP-1 receptor agonists.
- •
- have had an allergic reaction to a GLP-1 receptor agonist medicine.
- •
- are pregnant or plan to become pregnant. It is not known if XULTOPHY 100/3.6 will harm your unborn baby. Tell your healthcare provider if you become pregnant or think you may be pregnant while using XULTOPHY 100/3.6.
- •
- are breastfeeding or plan to breastfeed. It is not known if XULTOPHY 100/3.6 passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby while using XULTOPHY 100/3.6.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. XULTOPHY 100/3.6 may affect the way some medicines work, and some medicines may affect the way XULTOPHY 100/3.6 works. Before using XULTOPHY 100/3.6, talk to your healthcare provider about low blood sugar and how to manage it. Tell your healthcare provider if you are taking other medicines to treat diabetes.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I use XULTOPHY 100/3.6?
- •
- Read the Instructions for Use that comes with XULTOPHY 100/3.6.
- •
- Use XULTOPHY 100/3.6 exactly as your healthcare provider tells you to.
- •
- Do not change your dosing schedule without first talking to your healthcare provider. The dose counter on your XULTOPHY 100/3.6 pen shows the number of units of XULTOPHY 100/3.6 to be injected.
- •
- Your healthcare provider should show you how to use XULTOPHY 100/3.6 before you use it for the first time.
- •
- XULTOPHY 100/3.6 is injected under the skin (subcutaneously) of your thigh, upper arm or stomach (abdomen).
- •
- Do not inject XULTOPHY 100/3.6 into a vein (intravenously) or use in an insulin infusion pump.
- •
- Use XULTOPHY 100/3.6 at the same time each day with or without food.
- •
- If you miss a dose of XULTOPHY 100/3.6, resume your 1 time daily dosing schedule at the next scheduled dose. Do not take 2 doses at the same time or increase your dose to make up for the missed dose. If you miss more than 3 days of XULTOPHY 100/3.6, call your healthcare provider for further instructions about taking XULTOPHY 100/3.6 at the right dose and to help lower your chance of having an upset stomach.
- •
- Do not dilute XULTOPHY 100/3.6 with any other liquids.
- •
- Do not mix XULTOPHY 100/3.6 with any other insulin products or GLP-1 receptor agonists in the same injection.
- •
- Do not split your dose of XULTOPHY 100/3.6. Give your full dose of XULTOPHY 100/3.6 in 1 injection.
- •
- Check the Pen label each time you give your injection to make sure you are using the correct medicine.
- •
- Do not take more than 50 units of XULTOPHY 100/3.6 each day. XULTOPHY 100/3.6 contains two medicines: insulin degludec and liraglutide. If you take too much XULTOPHY 100/3.6, it can cause severe nausea and vomiting. Do not take XULTOPHY 100/3.6 with other GLP-1 receptor agonists. If you take too much XULTOPHY 100/3.6, call your healthcare provider or go to the nearest hospital emergency room right away.
- •
-
Change (rotate) your injection sites within the area you choose with each injection to reduce your risk of getting lipodystrophy (pits in skin or thickened skin) and localized cutaneous amyloidosis (skin with lumps) at the injection sites.
- o
- Do not use the exact same spot for each injection.
- o
- Do not inject where the skin has pits, is thickened, or has lumps.
- o
- Do not inject where the skin is tender, bruised, scaly or hard, or into scars or damaged skin.
- •
- Do not share your XULTOPHY 100/3.6 pen with other people, even if the needle has been changed. You may give other people a serious infection or get a serious infection from them.
Check your blood sugar levels. Ask your healthcare provider what your blood sugars should be and when you should check your blood sugar levels.
Your dose of XULTOPHY 100/3.6 and other diabetes medicines may need to change because of:
- •
- change in level of physical activity or exercise, weight gain or loss, increased stress, illness, change in diet, or because of other medicines you take.
What should I avoid while taking XULTOPHY 100/3.6?
While taking XULTOPHY 100/3.6 do not:
- •
- drive or operate heavy machinery, until you know how XULTOPHY 100/3.6 affects you.
- •
- drink alcohol or use prescription or over-the-counter medicines that contain alcohol.
What are the possible side effects of XULTOPHY 100/3.6?
XULTOPHY 100/3.6 may cause serious side effects that can lead to death, including:
- •
- See “What is the most important information I should know about XULTOPHY 100/3.6?”
- •
- inflammation of your pancreas (pancreatitis). Stop using XULTOPHY 100/3.6 and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
- •
- low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use XULTOPHY 100/3.6 with another medicine that can cause low blood sugar.
Signs and symptoms of low blood sugar may include:
- o
- dizziness or light-headedness
- o
- sweating
- o
- confusion or drowsiness
- o
- headache
- o
- blurred vision
- o
- slurred speech
- o
- shakiness
- o
- fast heartbeat
- o
- anxiety, irritability, or mood changes
- o
- hunger
- o
- weakness
- o
- feeling jittery
- •
- kidney problems (kidney failure). Worsening of kidney failure and sudden kidney failure have happened in people with kidney problems and in people without kidney problems, who have taken liraglutide, one of the ingredients in XULTOPHY 100/3.6. Diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration) which may cause kidney problems to get worse. Tell your healthcare provider if you have diarrhea, nausea, or vomiting. Drink plenty of fluids to help reduce your risk of dehydration during treatment with XULTOPHY 100/3.6.
- •
- serious allergic reactions. Stop using XULTOPHY 100/3.6 and get medical help right away, if you have any symptoms of a serious allergic reaction including:
- o
- hives
- o
- rash
- o
- itching
- o
- fast heartbeat
- o
- fainting or feeling dizzy
- o
- swelling of your face, lips, tongue, or throat
- o
- problems breathing or swallowing
- o
- sudden coughing
- o
- chest pain or tightness
- •
- gallbladder problems. Gallbladder problems have happened in some people who take XULTOPHY 100/3.6. Tell your healthcare provider right away if you get symptoms of gallbladder problems which may include:
- o
- pain in your upper stomach (abdomen)
- o
- fever
- o
- yellowing of skin or eyes (jaundice)
- o
- clay-colored stools
- •
- low potassium in your blood (hypokalemia).
- •
- heart failure. Taking certain diabetes pills called thiazolidinediones or TZDs with XULTOPHY 100/3.6 may cause heart failure in some people. This can happen even if you have never had heart failure or heart problems before. If you already have heart failure, it may get worse while you take TZDs with XULTOPHY 100/3.6. Your healthcare provider should monitor you closely while you are taking TZDs with XULTOPHY 100/3.6. Tell your healthcare provider if you have any new or worse symptoms of heart failure including shortness of breath, tiredness, swelling of your ankles or feet and sudden weight gain. Treatment with TZDs and XULTOPHY 100/3.6 may need to be adjusted or stopped by your healthcare provider if you have new or worse heart failure.
The most common side effects of XULTOPHY 100/3.6 include stuffy or runny nose, sore throat, headache, nausea, diarrhea, increased blood levels of lipase, and upper respiratory tract infection. Talk to your healthcare provider about any side effect that bothers you or does not go away.
These are not all the possible side effects of XULTOPHY 100/3.6.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
Keep XULTOPHY 100/3.6 and all medicines out of the reach of children.
General information about the safe and effective use of XULTOPHY 100/3.6.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use XULTOPHY 100/3.6 for a condition for which it was not prescribed. Do not give XULTOPHY 100/3.6 to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about XULTOPHY 100/3.6 that is written for health professionals.
What are the ingredients in XULTOPHY 100/3.6?
Active Ingredients: insulin degludec and liraglutide
Inactive Ingredients: glycerol, phenol, zinc, and Water for Injection. Hydrochloric acid or sodium hydroxide may be added to adjust pH.
Manufactured by:
Novo Nordisk Inc.
Plainsboro, NJ 08536U.S. License Number 1261
For more information, go to www.novonordisk-us.com or call 1-800-727-6500 (Se habla español).
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: 06/2022
Instructions for Use
XULTOPHY® 100/3.6 (ZUL-to-fye)
(insulin degludec and liraglutide) injection
- •
- Do not share your XULTOPHY 100/3.6 pen with another person. You may give an infection to them or get an infection from them.
- •
- XULTOPHY 100/3.6 pen (“pen”) is a prefilled disposable, single-patient-use pen containing 300 units of insulin degludec and 10.8 mg of liraglutide (insulin degludec and liraglutide) injection. You can inject doses from 10 to 50 units in a single injection (with each unit of insulin degludec, the pen also delivers 0.036 mg of liraglutide). The dose can be increased by 1 unit at a time. The dose equals the number of units shown in the dose counter.
- •
- People who are blind or have vision problems should not use the pen without help from a person trained to use the pen.
Supplies you will need to give your XULTOPHY 100/3.6 injection:
• XULTOPHY 100/3.6 pen
• a new NovoFine or NovoTwist needle
• alcohol swab
• a sharps container for throwing away used pens and needles. See “After your injection” at the end of these instructions.
Preparing your XULTOPHY 100/3.6 pen:
• Wash your hands with soap and water.
• Before you start to prepare your injection, check the XULTOPHY 100/3.6 pen label before each use to make sure it is your XULTOPHY 100/3.6 pen.
• XULTOPHY 100/3.6 should look clear and colorless. Do not use XULTOPHY 100/3.6 if it is cloudy or colored.
• Do not use XULTOPHY 100/3.6 past the expiration date printed on the label or 21 days after you start using the pen.
• Always use a new needle for each injection to help ensure sterility and prevent blocked needles. Do not reuse or share needles with another person. You may give other people a serious infection or get a serious infection from them.
Step 1:
• Pull pen cap straight off (See Figure B).
Step 2:
• Check the liquid in the pen (See Figure C). XULTOPHY 100/3.6 should look clear and colorless. Do not use it if it looks cloudy or colored.
Step 3:
• Select a new needle.
• Pull off the paper tab from the outer needle cap (See Figure D).
Step 4:
• Push the capped needle straight onto the pen and twist the needle on until it is tight (See Figure E).
Step 5:
• Pull off the outer needle cap. Do not throw it away (See Figure F).
Step 6:
• Pull off the inner needle cap and throw it away (See Figure G).
Priming your XULTOPHY 100/3.6 pen:
Step 7:
• Turn the dose selector to select the priming symbol (
 ). (See Figure H).
). (See Figure H).Step 8:
• Hold the pen with the needle pointing up. Tap the top of the pen gently a few times to let any air bubbles rise to the top (See Figure I).
Step 9:
• Hold the pen with the needle pointing up. Press and hold in the dose button until the dose counter shows “0”. The “0” must line up with the dose pointer.
• A drop of XULTOPHY 100/3.6 should be seen at the needle tip (See Figure J).
- o
- If you do not see a drop of XULTOPHY 100/3.6, repeat steps 7 to 9, no more than 6 times, until a drop of XULTOPHY 100/3.6 appears at the needle tip.
- o
- If you still do not see a drop of XULTOPHY 100/3.6, change the needle and repeat steps 7 to 9.
Selecting your dose: Make sure you prime your pen before setting your dose.
Step 10:
XULTOPHY 100/3.6 pen is made to deliver the number of units that your healthcare provider prescribed. Take your dose exactly as your healthcare provider tells you to. Do not change your dosing schedule without first talking to your healthcare provider.
• Turn the dose selector to select the dose you need to inject. The dose pointer should line up with your dose (See Figure K).
- o
- If you select the wrong dose, you can turn the dose selector forwards or backwards to the correct dose.
- o
- The even numbers are printed on the dial.
- o
- The odd numbers are shown as lines.
• The XULTOPHY 100/3.6 pen scale will show you how much XULTOPHY 100/3.6 is left in your pen (See Figure L).
• To see how much XULTOPHY 100/3.6 is left in your pen:
- o
- Turn the dose selector until it stops. The dose counter will line up with the dose that is left in your pen. If the dose counter shows 50, there is a dose of at least 50 units left in your pen.
- o
- If the dose counter shows between 10 and 50, the number shown in the dose counter is the total units left in your pen.
- •
- If there is not enough XULTOPHY 100/3.6 left in your pen for a full dose, do not use it. Use a new XULTOPHY 100/3.6 pen.
Giving your injection:
• Inject your XULTOPHY 100/3.6 exactly as your healthcare provider has shown you. Your healthcare provider should tell you if you need to pinch the skin before injecting.
• XULTOPHY 100/3.6 can be injected under the skin (subcutaneously) of your stomach area (abdomen), upper legs (thighs) or upper arms.
• Change (rotate) your injection sites within the area you choose for each dose to reduce your risk of getting lipodystrophy (pits in skin or thickened skin) and localized cutaneous amyloidosis (skin with lumps) at the injection sites. Do not use the same injection site for each injection. Do not inject where the skin has pits, is thickened, or has lumps. Do not inject where the skin is tender, bruised, scaly or hard, or into scars or damaged skin.
Step 11:
• Choose your injection site (stomach, upper legs or upper arms) and wipe the skin with an alcohol swab (See Figure M). Let the injection site dry before you inject your dose.
Step 12:
• Insert the needle into your skin (See Figure N).
- o
- Make sure you can see the dose counter. Do not cover it with your fingers, this can stop your injection.
Step 13:
• Press and hold down the dose button until the dose counter shows “0” (See Figure O).
- o
- The “0” must line up with the dose pointer. You may hear or feel a click.
Step 14:
• Keep the needle in your skin after the dose counter has returned to “0” and slowly count to 6 (See Figure P).
- o
- When the dose counter returns to “0”, you will not get your full dose until 6 seconds later.
- o
- If the needle is removed before you count to 6, you may see a stream of XULTOPHY 100/3.6 coming from the needle tip.
- o
- If you see a stream of XULTOPHY 100/3.6 coming from the needle tip you will not get your full dose. If this happens you should check your blood sugar levels more often because you may need more XULTOPHY 100/3.6.
Step 15:
• Pull the needle out of your skin (See Figure Q).
- o
- If you see blood after you take the needle out of your skin, press the injection site lightly with a piece of gauze or an alcohol swab. Do not rub the area.
Step 16:
• Carefully remove the needle from the pen after each use and throw it away (See Figure R).
- o
- Do not recap the needle. Recapping the needle can lead to needle stick injury.
Note: If you do not have a sharps container, follow the steps below:
- o
- Carefully slip the needle into the outer needle cap (See Figure S). Safely remove the needle and throw it away as soon as you can.
- o
- Do not store the pen with the needle attached. Storing without the needle attached helps prevent leaking, blocking of the needle, and air from entering the pen.
Step 17:
• Replace the pen cap by pushing it straight on (See Figure T).
After your injection:
• The used XULTOPHY pen may be thrown away in your household trash after you have removed the needle.
• Put your used needles in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles in your household trash.
• If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- o
- made of a heavy-duty plastic
- o
- can be closed with a tight-fitting, puncture resistant lid, without sharps being able to come out
- o
- upright and stable during use
- o
- leak-resistant
- o
- properly labeled to warn of hazardous waste inside the container
• When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about the safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
• Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
How should I store my XULTOPHY 100/3.6 pen?
Before use:
• Store unused XULTOPHY 100/3.6 pens in the refrigerator at 36°F to 46°F (2°C to 8°C).
• Do not freeze XULTOPHY 100/3.6. Do not use XULTOPHY 100/3.6 if it has been frozen.
• Unused pens may be used until the expiration date printed on the label, if kept in the refrigerator.
• If XULTOPHY 100/3.6 is stored outside of refrigeration prior to first use, it should be used or thrown away within 21 days.
• Store the pens in the carton they come in to keep them clean and protected from light.
Pen in use:
• Store the pen you are currently using at room temperature at 59°F to 86°F (15°C to 30°C) or in a refrigerator at 36°F to 46°F (2°C to 8°C).
• Do not freeze XULTOPHY 100/3.6. Do not use XULTOPHY 100/3.6 if it has been frozen.
• Keep XULTOPHY 100/3.6 away from heat and light.
• The XULTOPHY 100/3.6 pen you are using should be thrown away after 21 days, even if it still has XULTOPHY 100/3.6 left in it and the expiration date has not passed.
General Information about the safe and effective use of XULTOPHY 100/3.6.
• Keep XULTOPHY 100/3.6 pens and needles out of the reach of children.
• Always use a new needle for each injection.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Novo Nordisk Inc.
800 Scudders Mill Road
Plainsboro, New Jersey 08536
U.S. License Number 1261
For more information call Novo Nordisk at 1-800-727-6500
Revised: 06/2022
© 2016 – 2022 Novo Nordisk
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
Xultophy®100/3.6
NDC 0169-2911-15
List: 291115
(insulin degludec and liraglutide) injection
For Single Patient Use Only
100 units/mL and 3.6 mg/mL
With each unit of insulin degludec, the pen
also delivers 0.036 mg of liraglutide
5x3 mL Prefilled Pens
For subcutaneous use only
Recommended for use with NovoFine®,
Novofine® Plus or NovoTwist® disposable needles.
Must be refrigerated until first use.
Store at 36°F – 46°F (2°C – 8°C).
Do not freeze.
Protect from light.
Rx Only
Dispense in this sealed carton.
ATTENTION: Dispense the enclosed Medication Guide to each patient.
-
INGREDIENTS AND APPEARANCE
XULTOPHY 100/3.6
(insulin degludec and liraglutide) injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0169-2911 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength INSULIN DEGLUDEC (UNII: 54Q18076QB) (INSULIN DEGLUDEC - UNII:54Q18076QB) INSULIN DEGLUDEC 100 [iU] in 1 mL LIRAGLUTIDE (UNII: 839I73S42A) (LIRAGLUTIDE - UNII:839I73S42A) LIRAGLUTIDE 3.6 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCERIN (UNII: PDC6A3C0OX) 19.7 mg in 1 mL PHENOL (UNII: 339NCG44TV) 5.70 mg in 1 mL ZINC (UNII: J41CSQ7QDS) 55 ug in 1 mL WATER (UNII: 059QF0KO0R) Other Ingredients Ingredient Kind Ingredient Name Quantity May contain HYDROCHLORIC ACID (UNII: QTT17582CB) May contain SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0169-2911-15 5 in 1 CARTON 11/21/2016 1 3 mL in 1 SYRINGE, PLASTIC; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC:0169-2911-97 1 in 1 CARTON 11/21/2016 05/31/2021 2 NDC:0169-2911-90 3 mL in 1 SYRINGE, PLASTIC; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA208583 11/21/2016 Labeler - Novo Nordisk (622920320)