Label: CUVITRU (immune globulin subcutaneous- human injection, solution










-
NDC Code(s):
0944-2850-01,
0944-2850-02,
0944-2850-03,
0944-2850-04, view more0944-2850-05, 0944-2850-06, 0944-2850-07, 0944-2850-08, 0944-2850-09, 0944-2850-10
- Packager: Takeda Pharmaceuticals America, Inc.
- Category: PLASMA DERIVATIVE
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated March 23, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CUVITRU safely and effectively. See full prescribing information for CUVITRU.
CUVITRU, Immune Globulin Subcutaneous (Human), 20% Solution
Initial U.S. Approval: 2016WARNING: THROMBOSIS
See full prescribing information for complete boxed warning.
- Thrombosis may occur with immune globulin products, including CUVITRU. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling vascular catheters, hyperviscosity and cardiovascular risk factors.
- For patients at risk of thrombosis, administer CUVITRU at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk of hyperviscosity.
INDICATIONS AND USAGE
CUVITRU is an Immune Globulin Subcutaneous (Human), 20% solution indicated as replacement therapy for primary humoral immunodeficiency (PI) in adult and pediatric patients two years of age and older. (1)
DOSAGE AND ADMINISTRATION
For subcutaneous infusion only.
Administer at regular intervals from daily up to every two weeks (biweekly). (2.2)
Individualize dose based on the patient's pharmacokinetic and clinical response. (2.2)
Monitor serum IgG trough levels regularly to guide subsequent dose adjustments and dosing intervals as needed. (2.2)
Switching from Immune Globulin Intravenous (Human) treatment (IGIV) or adult patients switching from HYQVIA [Immune Globulin Infusion 10% (Human) with Recombinant Human Hyaluronidase]:
- Begin treatment one week after the patient's last IGIV or HYQVIA infusion. (2.2)
- Establish initial weekly dose by converting the monthly IGIV or HYQVIA dose into equivalent weekly dose and increasing it using a dose adjustment factor. (2.2)
Initial Weekly dose = Previous IGIVor HYQVIA dose (in grams) × 1.30 No. of weeks between IGIVor HYQVIA doses. - Frequent dosing (2-7 times per week): Divide the calculated weekly dose by the desired number of times per week. (2.2)
- Biweekly dosing: Multiply the calculated weekly dose by 2. (2.2)
Switching from Immune Globulin Subcutaneous (Human) treatment (IGSC): - Weekly dose (in grams) should be the same as the weekly dose of prior IGSC treatment (in grams). (2.2)
- Frequent dosing (2-7 times per week): Divide the calculated weekly dose by the desired number of times per week. (2.2)
- Biweekly dosing: Multiply the calculated weekly dose by 2. (2.2)
Infusion sites: up to 4 infusion sites simultaneously, with at least 4 inches between sites avoiding bony prominences. Rotate sites with each administration. (2.3)
DOSAGE FORMS AND STRENGTHS
200 mg/mL (20%) protein solution for subcutaneous infusion (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- IgA deficient patients with antibodies to IgA are at greater risk of developing severe hypersensitivity and anaphylactic reaction. (5.1)
- Monitor renal function, including blood urea nitrogen, serum creatinine, and urine output in patients at risk of acute renal failure. (5.2)
- Thrombosis may occur. Monitor for signs and symptoms of thrombosis and assess blood viscosity for those at risk for hyperviscosity. (5.3)
- Aseptic Meningitis Syndrome (AMS) can occur. (5.4)
- Monitor for clinical signs and symptoms of hemolysis. (5.5)
- Monitor patients for pulmonary adverse reactions (transfusion-related acute lung injury, TRALI). (5.6)
- Product is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and theoretically, the Creutzfeldt-Jakob disease agent. (5.7)
ADVERSE REACTIONS
The most common adverse reactions observed in ≥5% of patients were: local adverse reactions, systemic adverse reactions including headache, nausea, fatigue, diarrhea, and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals U.S.A., Inc. at 1-877-TAKEDA-7 (1-877-825-3327) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Passive transfer of antibodies may transiently interfere with the immune responses to live virus vaccines, such as measles, mumps, rubella, and varicella. (7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: THROMBOSIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Preparation and Handling
2.2 Dose
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
5.2 Renal Dysfunction/Failure
5.3 Thrombosis
5.4 Aseptic Meningitis Syndrome (AMS)
5.5 Hemolysis
5.6 Transfusion-Related Acute Lung Injury (TRALI)
5.7 Transmissible Infectious Agents
5.8 Monitoring: Laboratory Tests
5.9 Interference with Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: THROMBOSIS
- Thrombosis may occur with immune globulin products, including CUVITRU. Risk factors may include advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling vascular catheters, hyperviscosity and cardiovascular risk factors.
- For patients at risk of thrombosis, administer CUVITRU at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk of hyperviscosity.
-
1 INDICATIONS AND USAGE
CUVITRU is an Immune Globulin Subcutaneous (Human) (IGSC), 20% Solution indicated as replacement therapy for primary humoral immunodeficiency (PI) in adult and pediatric patients two years of age and older. This includes, but is not limited to, common variable immunodeficiency (CVID), X-linked agammaglobulinemia, congenital agammaglobulinemia, Wiskott-Aldrich syndrome, and severe combined immunodeficiencies1,2.
-
2 DOSAGE AND ADMINISTRATION
For subcutaneous administration only. Do not administer intravenously or intramuscularly.
2.1 Preparation and Handling
- Inspect the drug product visually for particulate matter and discoloration prior to administration. CUVITRU is a clear and colorless or pale yellow or light brown solution and clear of particulate matter. Do not use if the solution is cloudy, turbid, or if it contains particulates.
- Do not mix CUVITRU with other products.
- Do not shake.
- Do not dilute.
- CUVITRU comes in a single-dose vial. Any vial that has been entered should be used promptly. Partially used vials should be discarded. CUVITRU contains no preservative.
- Record the name and lot number of the product in the recipient's records.
2.2 Dose
- CUVITRU can be administered at regular intervals from daily up to every two weeks (biweekly).
- Individualize the dose based on the patient's pharmacokinetic and clinical response.
- Monitor serum IgG trough levels regularly to guide subsequent dose adjustments and dosing intervals as needed (see Dose Adjustment).
For patients switching from Immune Globulin Intravenous (Human) treatment (IGIV) or adult patients switching from HYQVIA [Immune Globulin Infusion 10% (Human) with Recombinant Human Hyaluronidase]:
- Begin treatment with CUVITRU one week after the patient's last IGIV or HYQVIA infusion.
- Establish the initial weekly dose by converting the monthly IGIV or HYQVIA dose into an equivalent weekly dose and increasing it using a dose adjustment factor.
- To calculate the initial weekly dose, divide the previous IGIV or HYQVIA dose in grams by the number of weeks between intravenous doses; then multiply this dose by the dose adjustment factor of 1.30.
Initial Weekly dose = Previous IGIV or HYQVIA dose (in grams) × 1.30 Number of weeks between IGIV or HYQVIA doses - To convert the dose (in grams) to milliliters (mL), multiply the calculated dose (in grams) by 5.
- Doses divided over the course of a week, or once weekly, or biweekly, achieve similar exposure when administered regularly at steady-state.
- To determine the dose for alternative regular dosing intervals:
- ‒
- Frequent dosing (2-7 times per week): Divide the calculated weekly dose by the desired number of times per week.
- ‒
- Biweekly dosing: Multiply the calculated weekly dose by 2.
To guide dose adjustments, see section Dose Adjustments (Table 1).
For patients switching from Immune Globulin Subcutaneous (Human) treatment (IGSC):
- The weekly dose of CUVITRU (in grams) is recommended to be the same as the weekly dose of prior IGSC treatment (in grams).
- Doses divided over the course of a week, once weekly, or biweekly, achieve similar exposure when administered regularly at steady-state.
- To determine the dose for alternative regular dosing intervals:
- ‒
- Frequent dosing (2-7 times per week): Divide the calculated weekly dose by the desired number of times per week.
- ‒
- Biweekly dosing: Multiply the calculated weekly dose by 2.
- To convert the dose (in grams) to milliliters (mL), multiply the calculated dose (in grams) by 5.
To guide dose adjustments, see section Dose Adjustments (Table 1).
For patients at risk for measles exposure:
- If a patient has been exposed to measles, it may be prudent to administer a dose of Immune Globulin Intravenous as soon as possible and within 6 days of exposure. A dose of 400 mg/kg should provide a serum level > 240 mIU/mL of measles antibodies for at least two weeks.
- If a patient is at risk of future measles exposure and receives a dose of CUVITRU of less than 230 mg/kg subcutaneously per week, the dose should be increased to at least 230 mg/kg per week, or the weekly equivalent of 230 mg/kg for dosing intervals other than weekly.
Dose Adjustment:
To guide dose adjustment, calculate the difference between the patient's target serum IgG trough level and the IgG trough level during subcutaneous treatment. Find this difference in Table 1 and the corresponding amount (in mL) by which to increase (or decrease) the weekly/biweekly dose based on the patient's body weight. If the difference between measured and target trough levels is less than 100 milligram/dL, then no adjustment is necessary. However, the patient's clinical response should be the primary consideration in dose adjustment.
Table 1 Change in Volume to Be Administered Weekly/Biweekly for Intended IgG Trough Level Change* Body Weight Difference from Target Serum IgG Trough Levels Dosing Frequency 30 kg 50 kg 70 kg 90 kg 110 kg - *
- Derived using a linear approximation of trough levels and weekly dose per kg body mass with a slope of 52.1 kg/dL.
100 mg/dL Weekly 3 mL 5 mL 7 mL 9 mL 11 mL Biweekly 6 mL 10 mL 13 mL 17 mL 21 mL 200 mg/dL Weekly 6 mL 10 mL 13 mL 17 mL 21 mL Biweekly 12 mL 19 mL 27 mL 35 mL 42 mL 300 mg/dL Weekly 9 mL 14 mL 20 mL 26 mL 32 mL Biweekly 17 mL 29 mL 40 mL 52 mL 63 mL Example 1: A patient with a body weight of 70 kg who is on a weekly treatment has a measured IgG trough level of 600 milligrams/dL, and the target trough level is 800 milligrams/dL. The desired target trough level difference is 200 milligrams/dL (800 milligrams/dL minus 600 milligrams/dL). The weekly dose of CUVITRU should be increased by 13 mL.
Example 2: A patient with a body weight of 50 kg who is on a biweekly treatment has a measured IgG trough of 900 milligrams/dL, and the target trough level is 700 milligrams/dL. The desired target trough level difference is 200 milligrams/dL (900 milligrams/dL minus 700 milligrams/dL). The biweekly dose of CUVITRU should be decreased by 19 mL.
2.3 Administration
Table 2 Infusion Volume and Rate* Infusion Parameters First 2 Infusions Subsequent Infusions Patients
<40 kgPatients
≥40 kgPatients
<40 kgPatients
≥40 kg- *
- If the initial infusions are well tolerated then subsequent infusions can begin at the maximum tolerated rate
Volume
(mL/site)≤20 ≤60 ≤60 Rate (mL/hr/site) 10 - 20 ≤60 Selection of Infusion Site: Suggested areas for subcutaneous infusion of CUVITRU are abdomen, thighs, upper arms, or lateral hip. CUVITRU may be infused into multiple infusion sites. Use up to 4 sites simultaneously. Infusion sites should be at least four inches apart, avoiding bony prominences. Rotate sites with each administration.
Volume per Site: To calculate the number of sites to be used, divide the total volume to be infused by the maximum volume/site (up to 60 mL/site) to be infused. Simultaneous subcutaneous infusion at multiple sites can be facilitated by use of a multi-needle administration set.
Infusion rate: For the first two infusions of CUVITRU, the recommended infusion rate is 10-20 mL/hr/site. For subsequent infusions, the infusion rate may be increased to 60 mL/hr/site as tolerated (e.g., 60 mL/hr/site × 2 sites = 120 mL/hr). For patients utilizing 4 infusion sites, the maximum infusion rate for all sites combined is 240 mL/hr.
Instructions for Administration:
Use aseptic technique when preparing and administering CUVITRU for infusion.
- 1.
- Inspect the vials: Inspect for clarity, color, and expiration date(s) [see Dosage and Administration (2.1)].
- 2.
-
Prepare for infusion:
- Gather supplies: CUVITRU vial(s), ancillary supplies, sharps container and infusion pump.
- Prepare a clean work area.
- Wash hands.
- 3.
-
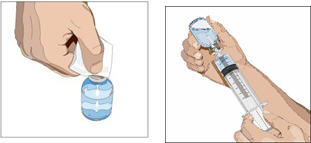
Prepare the CUVITRU:
- Wipe each stopper with a sterile alcohol wipe and allow to dry.
- Transfer into syringe(s), preferably using a vented spike.
- Start the infusion promptly after drawing CUVITRU into the syringe. It is suggested to complete the administration within 2 hours due to the potential formation of particles caused by siliconized syringes.
- 4.
-
Prepare the infusion pump and tubing:
- Follow manufacturer directions for priming the tubing and pump usage.
- Attach the syringe filled with CUVITRU to the needle set.
- Prime the needle set up to the needle hub.
- 5.
-
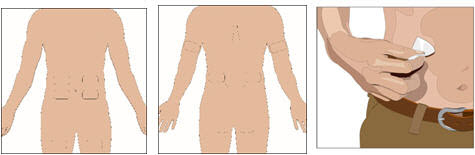
Prepare the infusion site(s):
- Potential sites for infusion include the abdomen, thighs, upper arms, or lateral hip.
- Avoid: bony areas, visible blood vessels, scars and any areas of inflammation (irritation) or infection.
- The number and location of infusion sites depends on the volume of the total dose.
- Infusion sites should be at least 4 inches apart.
- Rotate sites of the body between successive infusions.
- Cleanse the infusion site(s) with a sterile alcohol wipe beginning at the center of each infusion site and moving outward in a circular motion. Allow the infusion site(s) to dry.
- 6.
-
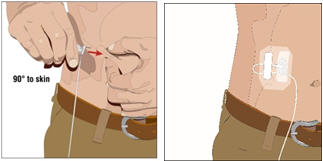
Insert and secure the subcutaneous needle set:
- Pinch at least one inch of skin between two fingers. Insert the needle at a 90-degree angle into the subcutaneous tissue and secure the needle with sterile tape.
- If more than one site is used, repeat steps.
- Secure the needle in place with a sterile protective dressing.
- 7.
-
Start the infusion of CUVITRU based on prescriber's order:
Follow the manufacturer's instructions to turn on the infusion pump. - 8.
-
Remove subcutaneous needle(s) from the infusion site(s):
After the infusion is complete, remove the needle set and cover with a protective dressing. Discard any partially used vial(s) and disposable supplies in accordance with local requirements. - 9.
-
Document the infusion:
Remove the peel-off label from each vial of CUVITRU used and affix to the patient's treatment record or infusion log. In addition, record the time, date, dose, infusion site location and any reactions after each infusion.
For self-administration, provide the patient with instructions and training for infusion in the home or other appropriate setting.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- CUVITRU is contraindicated in patients who have had an anaphylactic or severe systemic hypersensitivity reaction to the subcutaneous administration of human immune globulin.
- CUVITRU is contraindicated in IgA-deficient patients with antibodies against IgA and a history of hypersensitivity to human immune globulin treatment.
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Severe hypersensitivity reactions may occur, even in patients who had tolerated previous treatment with human immune globulin. If a hypersensitivity reaction occurs, discontinue CUVITRU infusion immediately and institute appropriate treatment.
CUVITRU contains trace amounts of IgA (average concentration of 80 mcg/mL). Patients with known antibodies to IgA have a greater risk of developing potentially severe hypersensitivity reactions, including anaphylaxis, with administration of CUVITRU.
5.2 Renal Dysfunction/Failure
Acute renal dysfunction/failure, acute tubular necrosis, proximal tubular nephropathy, osmotic nephrosis, and death may occur upon use of immune globulin treatment, especially those containing sucrose3,4. CUVITRU does not contain sucrose. Ensure that patients are not volume depleted prior to the initiation of infusion of CUVITRU. In patients who are at risk of developing renal dysfunction because of pre-existing renal insufficiency or predisposition to acute renal failure (such as diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs), monitor renal function and consider lower, more frequent dosing[see Dosage and Administration (2.1)].
Periodic monitoring of renal function and urine output is particularly important in patients predisposed to be at increased risk for developing acute renal failure. Assess renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, before the initial infusion of CUVITRU and again at appropriate intervals thereafter. If renal function deteriorates, consider discontinuation of CUVITRU [see Dosage and Administration (2.3)].
5.3 Thrombosis
Thrombosis may occur following treatment with immune globulin products.5,6 Risk factors may include advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients at risk of thrombosis, administer CUVITRU at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity [see Boxed Warning, Dosage and Administration (2.1), Patient Counseling Information (17)].
5.4 Aseptic Meningitis Syndrome (AMS)
AMS has been reported with the use of immune globulin, including CUVITRU [See Postmarketing Experience (6.2)]. The syndrome usually begins within several hours to two days following immune globulin treatment. AMS may occur more frequently in female patients.
AMS is characterized by the following signs and symptoms: severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, nausea and vomiting [see Patient Counseling Information (17)]. Cerebrospinal fluid (CSF) studies frequently reveal pleocytosis up to several thousand cells per mm3, predominantly from the granulocytic series, and elevated protein levels up to several hundred milligram/dL, but negative culture results. Conduct a thorough neurological examination, including CSF studies, on patients exhibiting such signs and symptoms, to rule out other causes of meningitis. Discontinuation of treatment has resulted in remission of AMS within several days without sequelae.
5.5 Hemolysis
CUVITRU can contain blood group antibodies that may act as hemolysins and induce in vivo coating of red blood cells (RBC) with immune globulin. This may cause a positive direct antiglobulin test [DAT (Coomb's test)]7,8. Delayed hemolytic anemia can develop subsequent to CUVITRU therapy due to enhanced RBC sequestration; acute hemolysis, consistent with intravascular hemolysis, can occur7-11.
The following risk factors may be related to the development of hemolysis: high doses (e.g., ≥2 grams/kg, single administration or divided over several days) and non-O blood group7, 11. Underlying inflammatory state in an individual patient may increase the risk of hemolysis7 but its role is uncertain10,12.
Monitor patients for clinical signs and symptoms of hemolysis, particularly patients with risk factors noted above. Consider appropriate laboratory testing in higher risk patients, including measurement of hemoglobin or hematocrit prior to infusion and within approximately 36 to 96 hours post infusion.
5.6 Transfusion-Related Acute Lung Injury (TRALI)
Non-cardiogenic pulmonary edema (TRALI) has been reported in patients following treatment with immune globulin products. TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever. Symptoms typically occur within 1 to 6 hours after treatment.
Monitor patients for pulmonary adverse reactions. If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil and anti-HLA antibodies in both the product and patient serum. TRALI may be managed using oxygen therapy with adequate ventilatory support.
5.7 Transmissible Infectious Agents
Because CUVITRU is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and theoretically, the Creutzfeldt-Jakob disease agent. This also applies to unknown or emerging viruses and other pathogens. No confirmed cases of viral transmission or vCJD have been associated with CUVITRU.
All infections thought by a physician to possibly have been transmitted by this product should be reported by the physician or other healthcare provider to Takeda Pharmaceuticals U.S.A., Inc., at 1-877-TAKEDA-7 (1-877-825-3327).
5.8 Monitoring: Laboratory Tests
- Periodic monitoring of renal function and urine output is particularly important in patients predisposed to be at increased risk of developing acute renal failure. Assess renal function, including measurement of BUN and serum creatinine, before the initial infusion of CUVITRU and at appropriate intervals thereafter.
- Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies, because of the potentially increased risk of thrombosis3,5.
- If signs and/or symptoms of hemolysis are present after an infusion of CUVITRU, perform appropriate laboratory testing for confirmation.
- If TRALI is suspected, perform appropriate tests for the presence of anti-neutrophil antibodies and anti-HLA antibodies in both the product and patient's serum.
5.9 Interference with Laboratory Tests
- After infusion of IgG, the transitory rise of the various passively transferred antibodies in the patient's blood may yield false positive serological test results, with the potential for misleading interpretation. Passive transmission of antibodies to erythrocyte antigens (e.g., A, B, and D) may cause a positive direct or indirect antiglobulin (Coombs') test.
- Administration of CUVITRU can lead to false positive readings in assays that depend on detection of beta-D-glucans for diagnosis of fungal infections; this may persist during the weeks following infusion of the product.
-
6 ADVERSE REACTIONS
The most common adverse reactions observed in ≥5% of subjects were: local adverse reactions, systemic adverse reactions including headache, nausea, fatigue, diarrhea, and vomiting.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
CUVITRU was administered subcutaneously in two prospective, open-label, non-controlled, multi-center studies to evaluate efficacy, safety, tolerability, and pharmacokinetics in subjects with primary immunodeficiency (PI). One study was performed in North America and the other was performed in Europe.
North American Study
In a clinical study conducted in North America, 67 out of 74 subjects treated with CUVITRU completed the study including 20/21 subjects aged 2 to <16 years old. Of the 7 subjects that discontinued treatment with CUVITRU, one subject withdrew due to fatigue (assessed as not related), 1 subject withdrew because of non-compliance, and 5 subjects withdrew for personal reasons.
A total of 4327 CUVITRU infusions were administered during the clinical study. No serious adverse reactions occurred during treatment with CUVITRU; 278 non-serious adverse reactions occurred at a rate per infusion of 0.06. Among the 4327 CUVITRU infusions, 99.3% (276/278) of adverse reactions were mild or moderate and transient in nature. Of the 278 non-serious adverse reactions (excluding infections), 83% (231/278) were rated as mild, 16% (45/278) were rated as moderate, and 1% (2/278, hemoptysis and abdominal pain) were severe.
Adverse reactions occurring in ≥5% of subjects (defined as adverse events occurring during or within 72 hours of infusion or any causally related event occurring within the study period) are shown in Table 3. No subject discontinued treatment due to local adverse reactions.
Table 3 Adverse Reactions* in ≥5% of Subjects Associated with Infusions of CUVITRU Adverse Reactions By Subject
N (%)†
(N=74 Subjects)By Infusion
N (Rate)‡
(N=4327 infusions)Local Adverse Reactions 23 (31.1%) 96 (0.022) Systemic Adverse Reactions 41 (55.4%) 182 (0.042) Headache 10 (13.5%) 50 (0.012) Nausea 9 (12.2%) 16 (0.004) Fatigue 6 (8.1%) 9 (0.002) Diarrhea 5 (6.8%) 5 (0.001) Vomiting 4 (5.4%) 5 (0.001) Systemic adverse reactions to immune globulin intravenous in part 1 of the study occurred at a rate of 0.302 relative to a rate of 0.042 during treatment with CUVITRU. The CUVITRU systemic adverse reaction rate was approximately 7-fold lower than the immune globulin intravenous rate.
Local Adverse Reactions: The most frequent local adverse reactions are listed by frequency in Table 4. Of the total 96 local adverse reactions, 100% were either mild (92.5%) or moderate (7.5%) in severity. No severe local adverse reactions were reported. During the clinical study, no local adverse reactions were observed in 68.9% (51/74) of subjects and in 98.2% (4247/4327) of infusions. The overall rate of local adverse reactions (excluding infections) during the clinical study was 0.022 (0.021 mild and 0.002 moderate). The rate of local adverse reactions was not associated with increased rate of infusion or volume per site.
European Study
Efficacy and safety during treatment with CUVITRU were evaluated in 48 subjects in a clinical trial in Europe. CUVITRU was administered for a median treatment duration of 358 days (range: 127.0-399 days) and a mean (± SD) of 347.4 ± 47.9 days. CUVITRU treatment: 45/48 subjects treated with CUVITRU completed the study, including 23/25 subjects aged 2 to <18 years old.
A total of 2349 CUVITRU infusions were administered during this clinical study. No serious adverse reactions occurred during treatment with CUVITRU. In total, 176 local adverse reactions and 205 systemic adverse reactions were reported (adverse reaction defined as adverse events occurring during or within 72 hours of infusion or any causally related event occurring within the study period), excluding infections. Of the 205 systemic reactions, the majority (134) were mild, 70 were moderate and one event was severe (headache, assessed as temporally associated, not causally related). The rate of systemic adverse reactions (excluding infections) during treatment with CUVITRU was 0.087, and the rate of related systemic AEs (excluding infections) per infusion was 0.032. During treatment with IGIV 10% in study part 1, the rate of related systemic AEs (excluding infections) was 0.173 events per infusion. The CUVITRU related systemic adverse event rate was approximately 5-fold lower than the immune globulin intravenous rate.
Table 5 EU Study: Adverse Reactions* in ≥5% of Subjects Associated with Infusions of CUVITRU Adverse Reactions By Subject
N (%)†
(N=48 Subjects)By Infusion
N (Rate)‡
(N=2349 infusions)Local Adverse Reactions 18 (37.5%) 176 (0.075) Systemic Adverse Reactions 33 (68.8%) 205 (0.087) Headache 14 (29.2%) 59 (0.025) Diarrhea 9 (18.8%) 58 (0.025) Cough 5 (10.4%) 7 (0.003) Fatigue 6 (12.5) 8 (0.003) Arthralgia 3 (6.3%) 5 (0.002) Oropharyngeal pain 3 (6.3%) 3 (0.001) Local Adverse Reactions: Among the 176 local adverse reactions in total (adverse reaction defined as adverse events occurring during or within 72 hours of infusion or any causally related event occurring within the study period), none was severe. In total, 175 adverse reactions (99.4%) were mild, and 1 adverse reaction (0.6%) was moderate. The overall rate of local adverse reactions was 0.075 per infusion. The most common reactions (by subject) were infusion site pain, infusion site erythema and infusion site pruritus.
Table 6 EU Study: Most Frequent Local Adverse Reactions Reported in ≥5% of Subjects Events Total Number of Adverse Reactions By Subject
N (%)*
(N =48 Subjects)By Infusion
N (Rate)†
(N=2349 infusions)Mild Moderate Pain 34 0 10 (20.8%) 34 (0.014) Erythema 54 0 10 (20.8%) 54 (0.023) Pruritus 30 0 7 (14.6%) 30 (0.013) Swelling 46 0 4 (8.3%) 46 (0.020) 6.2 Postmarketing Experience
Because postmarketing reporting of adverse reactions is voluntary and from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
In addition to the adverse reactions listed above in clinical trials, the following adverse reactions have been reported in the post-marketing experience:
Infections and Infestations: Aseptic meningitis
Postmarketing Experience of Immune Globulin Products
The following adverse reactions have been identified and reported during the postmarketing use of immune globulin products administered subcutaneously:
Immune system disorders Anaphylactic reaction Cardiac disorders Tachycardia Nervous system disorders Tremor and paresthesia Respiratory, Thoracic and Mediastinal disorders Dyspnea and laryngospasm General disorders and administration site conditions Injection site reaction (such as induration and warmth) and chest discomfort -
7 DRUG INTERACTIONS
Passive transfer of antibodies may transiently impair the immune responses to live attenuated virus vaccines such as mumps, rubella and varicella for up to 6 months and for a year or more to measles (rubeola). Inform the immunizing physician of recent therapy with CUVITRU so that appropriate precautions can be taken [see Patient Counseling Information (17)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
No human data are available to indicate the presence or absence of drug-associated risk. Animal reproduction studies have not been conducted with CUVITRU. It is also not known whether CUVITRU can cause fetal harm when administered to a pregnant woman or can affect reproductive capacity. Immune globulins cross the placenta from maternal circulation increasingly after 30 weeks of gestation. CUVITRU should be given to a pregnant woman only if clearly indicated. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
No human data are available to indicate the presence or absence of drug-associated risk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CUVITRU and any potential adverse effects on the breastfed infant from CUVITRU or from the underlying maternal condition.
8.4 Pediatric Use
CUVITRU was evaluated in 21 pediatric subjects with PI (2 to 16 years of age) in a multicenter clinical study. The safety and efficacy profiles were similar to adult subjects. No pediatric-specific dose requirements were necessary to achieve the desired serum IgG levels.
Safety and effectiveness of CUVITRU has not been evaluated in neonates or infants <2 years old.
8.5 Geriatric Use
CUVITRU was evaluated in 9 subjects over the age of 65 years in a multicenter clinical study. No differences in safety or efficacy were observed for this group when compared to the rest of the study population.
Monitor patients who are at an increased risk for developing renal failure or thrombotic events. Do not exceed the recommended dose, and infuse at the minimum infusion rate practicable [see Boxed Warning, Warnings and Precautions (5.2, 5.4), Dosage and Administration (2.3)].
-
11 DESCRIPTION
CUVITRU is a ready-for-use, sterile, liquid preparation of highly purified and concentrated immunoglobulin G (IgG) antibodies. The distribution of the IgG subclasses is similar to that of normal plasma. The Fc and Fab functions are maintained in CUVITRU.
CUVITRU has a purity ≥98% IgG and a pH of 4.6 to 5.1. The osmolality is 280-292 milli-osmoles per kilogram. CUVITRU contains 200 milligram/mL protein. The average immunoglobulin A (IgA) concentration is 80 mcg/mL. CUVITRU contains a broad spectrum of IgG antibodies against bacterial and viral agents. Glycine (0.25M) serves as a stabilizing and buffering agent, and there are no added sugars, sodium or preservatives.
CUVITRU is manufactured from large pools of human plasma. IgG preparations are purified from plasma pools using a modified Cohn-Oncley cold ethanol fractionation process, as well as cation and anion exchange chromatography.
Screening against potentially infectious agents begins with the donor selection process and continues throughout plasma collection and plasma preparation. Each individual plasma donation used in the manufacture of CUVITRU is collected only at FDA approved blood establishments and is tested by FDA licensed serological tests for Hepatitis B Surface Antigen (HBsAg), and for antibodies to Human Immunodeficiency Virus (HIV-1/HIV-2) and Hepatitis C Virus (HCV) in accordance with U.S. regulatory requirements. As an additional safety measure, mini-pools of the plasma are tested for the presence of HIV-1 and HCV by FDA licensed Nucleic Acid Testing (NAT) and found to be negative.
To further improve the margin of safety, validated virus inactivation/removal steps have been integrated into the manufacturing and formulation processes, namely solvent/detergent (S/D) treatment13, 35 nm nanofiltration, and a low pH incubation at elevated temperature (30°C to 32°C). The S/D process includes treatment with an organic mixture of tri-n-butyl phosphate, octoxynol 9 and polysorbate 80 at 18°C to 25°C for a minimum of 60 minutes. S/D treatment inactivates the lipid-enveloped viruses investigated to below detection limits within minutes13. The ethanol fractionation process provides an additional virus clearance capacity.
In vitro virus spiking studies have been used to validate the capability of the manufacturing process to inactivate and remove viruses. To establish the minimum applicable virus clearance capacity of the manufacturing process, these virus clearance studies were performed under extreme conditions (e.g., at minimum S/D concentrations, incubation time and temperature for the S/D treatment).
Virus clearance studies for CUVITRU performed in accordance with good laboratory practices are summarized in Table 7.
Table 7 Three Dedicated Independent Virus Inactivation/Removal Steps Mean Log10 Reduction Factors* (RFs) For Each Virus and Manufacturing Step Virus type Enveloped RNA Enveloped DNA Non-enveloped RNA Non-enveloped DNA Family Retroviridae Flaviviridae Herpesviridae Picornaviridae Parvoviridae Virus HIV-1 BVDV WNV PRV HAV EMCV MMV Abbreviations: HIV-1, Human Immunodeficiency Virus Type 1; BVDV, Bovine Viral Diarrhea Virus (model for Hepatitis C Virus and other lipid enveloped RNA viruses); WNV, West Nile Virus; PRV, Pseudorabies Virus (model for lipid enveloped DNA viruses, including Hepatitis B Virus); EMCV, Encephalomyocarditis Virus (model for non-lipid enveloped RNA viruses, including Hepatitis A virus [HAV]); MMV, Mice Minute Virus (model for non-lipid enveloped DNA viruses, including B19 virus [B19V]); n.d. (not done), n.a. (not applicable). - *
- For the calculation of these RF data from virus clearance study reports, applicable manufacturing conditions were used. Log10 RFs on the order of 4 or more are considered effective for virus clearance in accordance with the Committee for Medicinal Products for Human Use (CHMP, formerly CPMP) guidelines.
- †
- No RF obtained due to immediate neutralization of HAV by the anti-HAV antibodies present in the product.
Fractionation >5.1 1.3 >6.1 >4.9 3.9 4.2 4.9 SD treatment >4.5 >6.2 n.a. >4.8 n.d. n.d. n.d 35 nm nanofiltration >4.5 >5.1 >6.2 >5.6 5.7 1.4 2.0 Low pH treatment >5.8 >5.5 >6.0 >6.5 n.d.† >6.3 3.1 Overall log reduction factor (ORF) >19.9 >18.1 >18.3 >21.8 9.6† >11.9 10.1 -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CUVITRU supplies a broad spectrum of opsonizing and neutralizing IgG antibodies against a wide variety of bacterial and viral agents. CUVITRU also contains a spectrum of antibodies capable of interacting with and altering the activity of cells of the immune system as well as antibodies capable of reacting with cells such as erythrocytes. The role of these antibodies and the mechanisms of action of IgG in CUVITRU have not been fully elucidated.
12.2 Pharmacodynamics
Human normal immunoglobulin contains mainly immunoglobulin G (IgG) with a broad spectrum of antibodies against infectious agents. Human normal immunoglobulin contains the IgG antibodies present in the normal population. It has a distribution of immunoglobulin G subclasses closely proportional to that in native human plasma.
Adequate doses of CUVITRU may restore abnormally low immunoglobulin G levels to the normal range.
12.3 Pharmacokinetics
Pharmacokinetic (PK) parameters of subcutaneously administered CUVITRU were evaluated in 60 subjects with primary immunodeficiency (PI) during a clinical study in North America [see Clinical Studies (14)]. Subjects were treated intravenously for 13 weeks with a comparator product [GAMMAGARD LIQUID, Immune Globulin (Human), 10%] and then switched to weekly subcutaneous CUVITRU infusions. Initially, subjects were treated for up to 12 to 16 weeks at a subcutaneous dose that was 145% of the intravenous dose. A comparison of the area under the curve (AUC) for subcutaneous versus intravenous infusions was performed on 15 subjects aged 12 years and older. Subsequently, all subjects were treated with this dose for 12 weeks after which the dose was individualized for all subjects using the trough IgG levels, as described below. After approximately 4 months treatment at this subcutaneous dose, a PK evaluation was conducted on all subjects.
At this dose adjustment, the geometric mean ratio of the AUC for subcutaneous CUVITRU versus intravenous administration immune globulin 10% was 109%. The peak IgG level occurred at a geometric mean of 79 hours after subcutaneous CUVITRU administration.
In part 4 of the study, pharmacokinetic parameters for CUVITRU were assessed for 60 subjects aged 2 years and older. The pharmacokinetic parameters of CUVITRU administered subcutaneously are shown in Table 8. The median peak IgG levels were lower (1809 mg/dL) during subcutaneous treatment with CUVITRU compared to IGIV 10% administration (2602 mg/dL for 3 week intervals and 2521 mg/dL for 4 week intervals), consistent with the lower weekly dose compared with the dose administered every 3 or 4 weeks intravenously. In contrast, the geometric mean trough levels were higher with CUVITRU (1474 mg/dL), compared with those when given intravenously (1158 mg/dL for 3 week intervals and 1019 mg/dL for 4 week intervals), a result of both higher monthly dose and more frequent dosing. Weekly subcutaneous administration resulted in relatively stable steady-state serum IgG levels compared with IGIV administered at 3 to 4 week intervals. Pharmacokinetic parameters for CUVITRU did not significantly differ between age groups. The pharmacokinetic parameters of CUVITRU for the different age groups are shown in Table 9.
Table 8 Pharmacokinetic Parameters Parameter Median (95% Cl)
N=60AUC [g*days/L] 115 (110 to 121) Apparent clearance [mL/kg/day] 1.86 (1.80 to 2.17) Cmax [mg/dL] 1809 (1745 to 2068) Cmin [mg/dL] 1477 (1323 to 1535) Tmax [hours] 105 (71 to 119) Table 9 Pharmacokinetic Parameters by Age Group Parameter Age Groups 2 to <5
(n=1)
Median
(95% CI)5 to <12
(n=10)
Median
(95% CI)12 to <16
(n=5)
Median
(95% CI)16 to <65
(n=37)
Median
(95% CI)≥65
(n=7)
Median
(95% CI)AUC [g*days/L] 106
(N/A)110
(87 to 121)116
(N/A)114
(103 to 127)139
(90 to 158)Apparent clearance [mL/kg/day] 1.86
(N/A)1.85
(1.31 to 2.37)1.80
(N/A)1.98
(1.81 to 2.23)1.72
(0.90 to 2.36)Cmax [mg/dL] 1619
(N/A)1725
(1518 to 1892)1732
(N/A)1940
(1778 to 2220)2419
(1480 to 3201)Cmin [mg/dL] 1527
(N/A)1351
(1130 to 1635)1554
(N/A)1423
(1231 to 1524)1585
(976 to 2000)Tmax [hours] 70
(N/A)164
(70 to 168)68
(N/A)74
(68 to 119)119
(24 to 164) -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies were conducted to evaluate the carcinogenic or mutagenic effects of CUVITRU or its effects on fertility.
13.2 Animal Toxicology and/or Pharmacology
Animal studies were conducted to evaluate possible toxicity of CUVITRU.
In a local tolerance study in mini-pigs, 50 mL of CUVITRU were administered subcutaneously to anesthetized mini-pigs. Subcutaneous administration of CUVITRU was well tolerated in this study.
In local tolerance studies in rabbits, mild to moderate inflammatory reactions were observed locally after subcutaneous administration of 2.5 mL/kg of CUVITRU. These reactions are considered to be a consequence of the animals' immune response to the human IgG preparation and thus model specific and of minor relevance for the assessment of clinical local tolerability.
-
14 CLINICAL STUDIES
North America Study
A prospective, open-label, non-controlled, multi-center clinical study was conducted in North America to determine the efficacy, tolerability and PK of CUVITRU in 77 adult and pediatric subjects with PI. Efficacy was determined in 53 adults aged 16 years or older, 6 adolescents aged 12 to <16 years, and 15 children aged 2 to <12 years. CUVITRU was administered to 74 subjects with a mean dose of 222 mg/kg/week ± 71 mg/kg/week for a median treatment duration of 380.5 days (range: 30 - 629 days) and a mean (± SD) of 413.1 ± 116.5 days. The median duration of treatment did not vary significantly between age groups. The total exposure to CUVITRU was 83.70 subject-years and 4327 infusions.
Initially subjects received immune globulin 10% intravenously (IGIV) every 3 or 4 weeks at a monthly dose equivalent to that received prior to the study for 13 weeks. The objective of part 1 of the study was to determine AUCIV of total IgG following IGIV administration. In part 2 of the study, subjects received CUVITRU subcutaneously at an adjusted dose of 145% of the IGIV dose. The objective of part 2 was to determine AUCSC of total IgG following weekly CUVITRU administration and to calculate an adjusted dose to be used in part 3. The dose adjustment factor was assesed to be 145% of the IGIV 10% dose by comparing the AUCSC with the AUCIV, 0-τ (standardized to 1 week) of part 1 for the first 15 subjects that completed part 2. Subjects who completed part 1 after this assessment was available, went directly into part 3. In part 3 of the study, subjects were treated weekly for 12 weeks at the adjusted dose. The ratio of serum IgG trough levels for part 1 and 3 were compared to the expected trough level determined in part 2 to establish the individually adapted dose for part 4 for each subject. In part 4 of the study, subjects were infused weekly with CUVITRU at the individually adapted dose for 40 weeks. During part 4, an additional pharmacokinetic assessment was performed. Follow-up with the subject either by diary system or by investigator occurred 3-5 days after every infusion in each study part to document adverse events. Adverse events were assessed using the subject's eDiary – all subjects received eDiary tablet to continuously record home treatments, adverse events, and additional information as they occurred.
One acute serious bacterial infection (ASBI) of pneumonia was reported in a 78-year old subject who had specific antibody deficiency and allergic bronchopulmonary aspergillosis while receiving CUVITRU. The point estimate of the annualized rate of ASBIs was 0.012 (upper limit of 99% CI: 0.024) during CUVITRU treatment. This annual rate of ASBIs was lower than 1.0 ASBIs /year (p<0.0001), the threshold specified as providing substantial evidence of efficacy.
The summary of infections and associated events for subjects during subcutaneous treatment with CUVITRU is summarized in Table 10.
Table 10 Summary of Infections and Associated Events Parameters Results Number of subjects
Total number of subject-years on treatment
Annual rate of any infections (per subject-year)74
83.70
2.41 (95% CI: 1.89 to 3.03)Days on antibiotics (rate per subject-year) 57.59 (95% CI: 40.71 to 78.59) Days off work/school/unable to perform normal daily activities due to illness or infection (rate per subject-year) 1.16 (95% CI: 0.70 to 1.79) Number of hospitalizations due to infections (rate per subject-year) 0.012 (95% CI: 0.006 to 0.022) Number of days in hospital due to infections (rate per subject-year) 0.06 (95% CI: 0.03 to 0.11) In the clinical study, across all age groups, the median maximum infusion rate was 60 mL/hr/site. This infusion rate was achieved in 57.3% (2480/4327) of completed CUVITRU infusions. CUVITRU infusion rate of 60 mL/h/site was achieved in 28.6% (6/21) of pediatric subjects (2 years to <16 years of age), in 88.7% (47/53) of adults (16 years of age and older) and in 71.6% (53/74) of all subject. For more than half of CUVITRU infusions (2393/4327), a volume of 30 to 39 mL (1096/4327 infusions) or 40 to 49 mL (1297/4327 infusions) was infused per site. For 320/4327of CUVITRU infusions, a volume of 60 mL/site or more was infused. Infusion paramenters resulted in a median of 2 infusion sites (range: 1 to 4) per CUVITRU administration. During CUVITRU treatment, 84.9% (3662/4314) of infusions were administered using 1 infusion site (18.5%; 798/4314) or 2 infusion sites (66.4%; 2864/4314) across all ages. The median duration of infusions was less than 1 hour (0.95 h; range: 0.2-6.4 hours). During all treatment periods, 99.8% of infusions were completed without a reduction, interruption, or discontinuation for tolerability reasons. Infusion characteristics did not significantly differ between adult and pediatric subjects.
Throughout the study, health-related quality of life was assessed using the Pediatric Quality of Life Inventory™ (PEDS-QL) questionnaire14 (pediatric subjects) or the self-administered SF-36 survey15(adult subjects). Quality of life was analyzed separately for the age groups 2 to 4 and 5 to 7 years (PEDS-QL, observer: parent), 8 to 12 and 13 years (PEDS-QL, observer: subject) and 14 years and older (SF-36, observer: subject). Treatment satisfaction was measured using the Life Quality Index questionnaire (LQI)16,17 and the Treatment Satisfaction Questionnaire for Medication (TSQM-9)18. The LQI was assessed for the age group 2 years to 12 years (observer: parent) and the age group 13 years and older (observer: subject) in three domains: Treatment Interference, Therapy-related Problems and Therapy Settings. The TSQM-9 was assessed in subjects aged 2 to 12 years (observer: parent) and 13 years and older (observer: subject) in 3 domains: Effectiveness, Convenience and Global Satisfaction. Differences between scores during the intravenous study part and subcutaneous 20% study part were calculated for selected domains of the instruments, see Table 11.
Table 11 Selected Patient Reported Outcomes: Differences Between the Intravenous and Subcutaneous Treatment Scale Difference p-value SF-36 Physical Component Score 0.89 0.067 SF-36 Mental Component Score 1.31 0.976 Total Score (PedsQL) 1.09 0.449 Treatment Interference (LQI) 1.50 0.008 Convenience (TSQM-9) 11.11 <0.001 European Study
A prospective, open-label, non-controlled, multi-center study conducted in 16 sites in Europe to evaluate the efficacy, safety, tolerability, and PK parameters of CUVITRU in subjects with PI aged 2 years and older at time of screening. The study consisted of 2 parts. In study part 1, subjects were treated with IGSC 16% for 12 weeks or with IGIV 10% for 13 weeks. Administration, dosage frequency, and dose were dependent on the pre-study treatment. However, the dose range had to be within 0.3-1.0 g/kg BW/4 weeks.
During study part 2, subjects received weekly CUVITRU infusions for 51 weeks at the dose used during study part 1, adjusted to a weekly equivalent dose if necessary. PK assessments were performed before the end of study part 1 and after approximately 5 months in study part 2 in subjects aged ≥12 years. For younger subjects (aged 2 to <12 years) only IgG trough levels were assessed to avoid multiple blood draws. The geometric mean of CUVITRU trough levels was 827 mg/dL [95% CI: 748-913]. Human and population PK parameters for CUVITRU were calculated from levels of Immunoglobulin G (IgG) measured during each part of the study.
CUVITRU was administered at the same weekly-equivalent dose as with the previously used IG product (mean (± SD) dose: 0.125 ± 0.042 g/kg/week). CUVITRU administered at this dose was shown to be effective in PI subjects aged ≥2 years.
One acute serious bacterial infection (ASBI) of pneumonia was reported in a 12-year old subject with a more severe form of hypogammaglobulinemia (XLA) while receiving CUVITRU. The point estimate of the annualized rate of ASBIs was 0.022 (upper limit of 99% CI: 0.049) during CUVITRU treatment. This annual rate of ASBIs was lower than 1.0 ASBIs /year, (p<0.0001), the threshold specified as providing substantial evidence of efficacy.
The summary of infections and associated events for subjects in the EU study during subcutaneous treatment with CUVITRU are summarized in Table 12.
Table 12 Summary of Infections and Associated Events Parameters Results - *
- Rate = number of infections divided by the total number of subject-years under treatment
Number of subjects
Annual rate* of any infections (rate per subject-year)48
4.38 (95% CI: 3.38 to 5.56)Days on antibiotics (rate per subject-year) 18.11 (95% CI: 13.01 to 24.41) Days off work/school/unable to perform normal daily activities due to illness or infection (rate per subject-year) 15.55 (95% CI: 10.06 to 22.75) Number of hospitalizations due to infections (rate per subject-year) 0.04 (95% CI: 0.02 to 0.08) Number of days in hospital due to infections (rate per subject-year) 0.11 (95% CI: 0.05 to 0.21) -
15 REFERENCES
- Orange JS, Hossny EM, Weiler CR, Ballow M, Berger M, Bonilla FA, Buckley R, Chinen J, El-Gamal Y, Mazer BD, Nelson Jr. RP, Patel DD, Secord E, Sorenson RU, Wasserman RL, Cunningham-Rundles C, Use of Intravenous Immunoglobulin in Human Disease: A Review of Evidence by Members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma, and Immunology. J Allergy Clin Immunol 2006; 117:S525-53.
- Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005; 94(suppl 1):S1-63.
- Pierce LR, Jain N. Risks associated with the use of intravenous immunoglobulin. Transfusion Med Rev. 2003;17:241-251.
- Dantal J. Intravenous immunoglobulins: in-depth review of excipients and acute kidney injury risk. Am J Nephrol. 2013;38(4):275-84.
- Katz U, Sheonfeld Y. Review: intravenous immunoglobulin therapy and thromboembolic complications. Lupus 2005;14:802-8.
- Ramírez E, Romero-Garrido JA, López-Granados E, Borobia AM, Pérez T, Medrano N, Rueda C, Tong HY, Herrero A, Frías J. Symptomatic thromboembolic events in patients treated with intravenous-immunoglobulins: results from a retrospective cohort study. Thromb Res. 2014 Jun;133(6):1045-51.
- Wilson JR, Bhoopalam N, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle Nerve 1997;20:1142-1145.
- Kessary-Shoham H, Levy Y, Shoenfeld Y, Lorber M, Gershon H. In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J Autoimmun 1999;13:129-135.
- Daw Z, Padmore R, Neurath D, Cober N, Tokessy M, Desjardins D, et al. Hemolytic transfusion reactions after administration of intravenous immune (gamma) globulin: a case series analysis. Transfusion. 2008; 48:1598-601.
- Copelan EA, Strohm PL, Kennedy MS, Tuschka PJ, Hemolysis following intravenous immune globulin therapy. Transfusion 1986;26:410-412.
- Berg R, Shebl A, Kimber MC, Abraham M, Schreiber GB. Hemolytic events associated with intravenous immune globulin therapy: a qualitative analysis of 263 cases reported to four manufacturers between 2003 and 2012. Transfusion. 2015 Jul;55 Suppl 2:S36-46.
- Kahwaji J, et al.; Acute hemolysis after High-Dose Intravenous Immunoglobulin Therapy in Highly HLA Sensitized Patients. Clin J Am Soc Nephrol; 2009 (4):1993-97.
- Kreil TR, Berting A, Kistner O, Kindermann J. West Nile virus and the safety of plasma derivatives: verification of high safety margins, and the validity of predictions based on model virus data. Transfusion 2003;43:1023-1028.
- Varni JW, Seid M, Rode CA. The PedsQL: measurement model for the pediatric quality of life inventory. Med.Care 1999;37:126-139.
- Ware JE, Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med.Care 1992;30:473-483.
- Daly PB, Evans JH, Kobayashi RH et al. Home-based immunoglobulin infusion therapy: quality of life and patient health perceptions. Ann.Allergy 1991;67:504-510.
- Nicolay U, Haag S, Eichmann F et al. Measuring treatment satisfaction in patients with primary immunodeficiency diseases receiving lifelong immunoglobulin replacement therapy. Qual.Life Res. 2005;14:1683-1691.
- Bharmal M, Payne K, Atkinson MJ et al. Validation of an abbreviated treatment satisfaction questionnaire for medication (TSQM-9) among patients on antihypertensive medications. Health Qual.Life Outcomes 2009;7:36.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
CUVITRU is supplied in single-dose vials containing the labeled amount of functionally active IgG. The components used in the packaging for CUVITRU are not made with natural rubber latex.
The following presentations of CUVITRU are available:
NDC Number Volume Grams Protein 0944-2850-01 5 mL 1.0 0944-2850-03 10 mL 2.0 0944-2850-05 20 mL 4.0 0944-2850-07 40 mL 8.0 0944-2850-09 50 mL 10.0 Storage and Handling
- Store at refrigeration temperature: 2°C to 8°C [36°F to 46°F] for up to 36 months or
- Room temperature: not to exceed 25°C [77°F]) for up to 24 months.
- Do not return CUVITRU to the refrigerator if you take it out to room temperature.
- Do not freeze.
- Do not shake.
- Keep the vials in the carton in order to protect from light.
- Discard any unused product.
- Do not use past the expiration date.
-
17 PATIENT COUNSELING INFORMATION
- Advise patients to read the FDA-approved patient labeling (Information for Patients and Instructions for Use).
- Prior to starting CUVITRU ask about a history of IgA deficiency, allergic reactions to immune globulin or other blood products. Patients with a history of allergic reactions should not be treated subcutaneously at home until several treatments have been administered and tolerated under medical supervision [see Warnings and Precautions (5.1)].
- Inform patients to immediately report the following signs and symptoms to their healthcare provider:
- Decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath [see Warnings and Precautions (5.2)].
- Instruct patients to immediately report symptoms of thrombosis. These symptoms may include pain and/or swelling of an arm or leg with warmth over the affected area, discoloration of an arm or leg, unexplained shortness of breath, chest pain or discomfort that worsens on deep breathing, unexplained rapid pulse, numbness or weakness on one side of the body [see Warnings and Precautions (5.3)].
- Severe headache, neck stiffness, drowsiness, fever, sensitivity to light, painful eye movements, nausea, and vomiting [see Warnings and Precautions (5.4)].
- Increased heart rate, fatigue, yellowing of the skin or eyes, and dark-colored urine [see Warnings and Precautions (5.5)].
- Trouble breathing, chest pain, blue lips or extremities, or fever that can occur 1 to 6 hours after an infusion of CUVITRU [see Warnings and Precautions (5.6)].
- Inform patients that CUVITRU is made from human plasma and may contain infectious agents that can cause disease (e.g., viruses and, theoretically, the vCJD agent). The risk of CUVITRU transmitting an infectious agent has been reduced by screening plasma donors for prior exposure, testing donated plasma, and inactivating or removing certain viruses during manufacturing. Patients should report any symptoms that concern them which might be caused by virus infections [see Warnings and Precautions (5.7)]
- Inform patients that CUVITRU can interfere with their immune response to live viral vaccines such as measles, mumps, rubella and varicella, and instruct patients to notify their healthcare professional of this potential interaction when they are receiving vaccinations [see Drug Interactions (7)].
Self-administration – If self-administration is deemed to be appropriate by the physician, clear instructions and training on subcutaneous infusion should be given to the patient/caregiver, and the demonstration of their ability to independently administer subcutaneous infusions should be documented.
- Ensure the patient understands the importance of consistent subcutaneous infusions to maintain appropriate steady IgG levels.
- Inform the patient to start the infusion promptly after drawing CUVITRU into the syringe. Ensure the patient understands that it is suggested to complete the administration within 2 hours due to the potential formation of particles caused by siliconized syringes.
- Instruct the patient to keep a treatment diary/log book. This diary/log book should include information about each infusion such as, the time, date, dose, lot number(s), infusion sites, and any reactions.
- Inform the patient that mild to moderate local infusion-site reactions (e.g., pain, redness and itching) are a common side effect of subcutaneous treatment, but to contact their healthcare professional if a local reaction increases in severity or persists for more than a few days.
- SPL UNCLASSIFIED SECTION
-
Information For Patients
CUVITRU ( CUE-vih-troo)
Immune Globulin Subcutaneous (Human), 20% SolutionSubcutaneous Administration
Information for Patients
The following summarizes important information about CUVITRU. Please read it carefully before using this medicine. This information does not take the place of talking with your healthcare provider, and it does not include all of the important information about CUVITRU. If you have any questions after reading this, ask your healthcare provider.
What is the most important information I need to know about CUVITRU?
CUVITRU can cause the following serious reactions:
- Severe allergic reactions causing difficulty in breathing or skin rashes
- Decreased kidney function or kidney failure
- Blood clots in the heart, brain, lungs, or elsewhere in the body
- Severe headache, drowsiness, fever, painful eye movements, or nausea and vomiting
- Dark colored urine, swelling, fatigue, or difficulty breathing
What is CUVITRU?
CUVITRU is a ready-to-use, liquid medicine that contains immunoglobulin G (IgG) antibodies, which protect the body against infection. CUVITRU is used to treat patients with primary immunodeficiency diseases (PI).
There are many forms of PI. The most common types of PI result in an inability to make a very important type of protein called antibodies, which help the body fight off infections from bacteria or viruses. CUVITRU is made from human plasma that is donated by healthy people. CUVITRU contains antibodies collected from these healthy people that replace the missing antibodies in PI patients.
Who should not use CUVITRU?
Do not use CUVITRU if you have a known history of a severe allergic reaction to immune globulin or other blood products. If you have such a history, discuss this with your healthcare provider to determine if CUVITRU can be given to you. Tell your healthcare provider if you have a condition called selective (or severe) immunoglobulin A (IgA) deficiency.
How should I use CUVITRU?
CUVITRU is given under the skin (subcutaneously). Most of the time infusions under the skin are given at home by self infusion or by caregivers. Instructions for giving CUVITRU under the skin (subcutaneously) are provided in the Instructions for Use brochure. Only use CUVITRU by yourself after you have been instructed by your healthcare provider.
What should I avoid while taking CUVITRU?
CUVITRU can make vaccines (like measles/mumps/rubella or chickenpox vaccines) not work as well for you. Before you get any vaccines, tell your healthcare provider that you take CUVITRU.
Tell your healthcare provider if you are pregnant, or plan to become pregnant, or if you are nursing.
What are the possible or reasonably likely side effects of CUVITRU?
The following one or more possible reactions may occur at the site of infusion. These generally go away within a few hours, and are less likely after the first few infusions.
- Mild or moderate pain
- Redness
- Itching
The most common side effects with CUVITRU are:
- Headache
- Nausea
- Fatigue
- Diarrhea
- Vomiting
If any of the following problems occur after starting treatment with CUVITRU, stop the infusion immediately and contact your healthcare provider or call emergency services. These could be signs of a serious problem.
- Hives, swelling in the mouth or throat, itching, trouble breathing, wheezing, fainting or dizziness. These could be signs of a serious allergic reaction.
- Bad headache with nausea, vomiting, stiff neck, fever, and sensitivity to light. These could be signs of irritation of the lining around your brain.
- Reduced urination, sudden weight gain, or swelling in your legs. These could be signs of a kidney problem.
- Pain, swelling, warmth, redness, or a lump in your legs or arms. These could be signs of a blood clot.
- Brown or red urine, fast heart rate, yellow skin or eyes. These could be signs of a liver problem or a blood problem.
- Chest pain or trouble breathing, or blue lips or extremities. These could be signs of a serious heart or lung problem.
- Fever over 100°F. This could be a sign of an infection.
These are not all of the possible side effects with CUVITRU. You can ask your healthcare provider for physician's information leaflet. Tell your healthcare provider about any side effect that bothers you or that does not go away.
Whenever giving yourself treatments at home, you should have another responsible person present to help treat side effects or get help if you have a serious adverse reaction occur. Ask your healthcare provider whether you should have rescue medications, such as antihistamines or epinephrine.
How do I store CUVITRU?
Store CUVITRU refrigerated or at room temperature.
- You can store CUVITRU in the refrigerator (36°F to 46°F [2°C to 8°C]) for up to 36 months or
- You can store CUVITRU at room temperature (up to 77°F [25°C]) for up to 24 months.
- Do not return CUVITRU to the refrigerator if you take it out to room temperature.
- Do not freeze.
- Do not shake.
- Check the expiration date on the carton and vial label. Do not use CUVITRU after the expiration date.
- Protect from light. You can use the original CUVITRU containers to protect it from light.
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898CUVITRU® is a registered trademark of Baxalta Incorporated, a Takeda company.
TAKEDA® and the TAKEDA Logo® are registered trademarks of Takeda Pharmaceutical Company Limited.Issue Date: 3/2023
CUV350
-
Detailed Instructions for Administration for Patients
Do not use CUVITRU at home until you get instructions and training from your healthcare professional.
Prepare CUVITRU vial(s):
- Remove CUVITRU from the box. Allow vials to reach room temperature. This may take up to 90 minutes.
- Do not apply heat or place in microwave.
- Do not shake the vial(s).
- 1.
-
Check the vial(s):
- Do not use beyond expiration date.
- Do not use if the protective cap is missing or broken.
- Look at the color: it should be clear and colorless to pale yellow or light brown.
- Do not use if the solution is cloudy or has particles.
- 2.
-
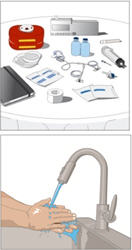
Gather all supplies
- Gather all supplies:
Items include: vial(s) of CUVITRU, infusion supplies: subcutaneous needle set, transfer device(s), syringe(s), sterile tip caps, sterile clear bandage, tape, gauze, sharps container, infusion pump, infusion log. - Clean work area.
- Program the infusion pump according to prescribed infusion rates and manufacturer's instructions.
- Wash hands thoroughly and allow to dry.
- Open supplies as shown by your healthcare professional.
- Gather all supplies:
- 3.
-
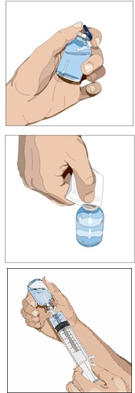
Prepare the syringe(s):
- Remove the cap from the vial.
- Wipe each stopper with a sterile alcohol wipe and allow to dry.
- Attach a sterile syringe to a vented spike.
- Insert the vented spike into the center of the IG vial.
- Turn the vial upside down and pull back on the plunger to pull the IG into the syringe(s).
- Repeat these steps, if using multiple vials to achieve the desired dose.
- Start the infusion promptly after drawing CUVITRU into the syringe(s). It is suggested to complete the administration within 2 hours.
- 4.
-
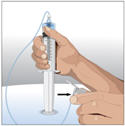
Prepare the infusion pump and tubing:
- Use manufacturer directions for filling the tubing and using the pump.
- Attach the syringe filled with CUVITRU to the needle set.
- Point the syringe tip up and gently push the plunger of the syringe to remove the air and fill the needle set up to the needle hub.
- 5.
-
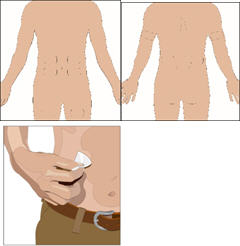
Prepare the infusion site(s):
- Select the number of infusion sites based on the volume of the total dose.
- Choose infusion site(s): upper arms, abdomen, thighs, or lower back.
- Avoid: bony areas, visible blood vessels, scars and any areas of inflammation (irritation) or infection.
- Infuse CUVITRU from 1 to 4 infusion sites at the same time.
- Select sites at least 4 inches apart.
- Rotate sites between future infusions.
- Wipe the infusion site(s) with a sterile alcohol wipe beginning at the center of each infusion site and moving outward in circular motion. Allow the infusion site(s) to dry (at least 30 seconds).
- 6.
-
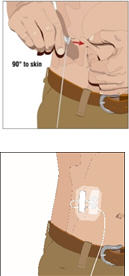
Insert and secure the subcutaneous needle set:
- Remove the needle cover. Firmly grasp and pinch at least 1 inch of skin between two fingers.
- Insert needle with a rapid motion straight into the skin at a 90 degree angle. Tape needle in place with sterile tape (included on transparent dressing).
- If more than one site is used, repeat the steps.
- Secure the needle set in place by applying a sterile protective dressing over the site(s).
- 7.
-
Start the infusion:
- Follow the manufacturer's instructions to turn pump on and start the infusion.
- Check infusion site(s) occasionally throughout the infusion.
- 8.
-
Remove subcutaneous needle(s) from the infusion site(s):
- Remove the needle set by loosening the tape on all edges.
- Pull the needle wings straight up and out.
- Gently press a small piece of gauze over the needle site and cover with a dressing.
- Throw away the needle(s) into the sharps container.
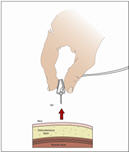
- 9.
-
Record the infusion:
- Remove the peel-off label from the vial(s), which has the product lot number and expiration date, and place the label in your treatment record/infusion log.
- Write down the date, time, dose, site(s) of infusion (to assist in rotating sites) and any reactions after each infusion.
- Throw away the disposable supplies, vials, and unused product as recommended by your healthcare professional.
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898CUVITRU® is a registered trademark of Baxalta Incorporated, a Takeda company.
TAKEDA® and the TAKEDA Logo® are registered trademarks of Takeda Pharmaceutical Company Limited.Issue Date: 3/2023
CUV350
-
PRINCIPAL DISPLAY PANEL - 5 mL Vial Label
NDC: 0944-2850-02
Immune Globulin Subcutaneous (Human) 20%
CUVITRU
Rx Only1g
5 mLSingle-Dose Vial
Solution for Subcutaneous Administration
The vial stopper is not made with natural rubber latex.
Refrigeration: 2°C to 8°C (36°F to 46°F) for up to 36 months.
Do not freeze. Room temperature: not to
exceed 25°C (77°F) for up to 24 months.
Discard unused portion.LE-07-53927
Takeda

- PRINCIPAL DISPLAY PANEL - 5 mL Vial Carton
-
PRINCIPAL DISPLAY PANEL - 10 mL Vial Label
NDC: 0944-2850-04
Immune Globulin Subcutaneous (Human) 20%
CUVITRU
Rx Only2g
10 mLSingle-Dose Vial
Solution for Subcutaneous Administration
The vial stopper is not made with natural rubber
latex. Refrigeration: 2°C to 8°C (36°F to 46°F) for up
to 36 months. Do not freeze. Room temperature:
not to exceed 25°C (77°F) for up to 24 months.
Discard unused portion.LE-07-53931
Takeda

- PRINCIPAL DISPLAY PANEL - 10 mL Vial Carton
-
PRINCIPAL DISPLAY PANEL - 20 mL Vial Label
NDC: 0944-2850-06
Immune Globulin Subcutaneous (Human) 20%
CUVITRU
Single-Dose Vial
4g
20 mLSolution for Subcutaneous Administration
The vial stopper is not made with natural rubber latex.
Refrigeration: 2°C to 8°C (36°F to 46°F) for up to 36 months. Do not freeze.
Room temperature: not to exceed 25°C (77°F) for up to 24 months.
Discard unused portion.
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898
CUVITRU® is a registered trademark of Baxalta Incorporated, a Takeda company.Rx Only
LE-07-53933
Takeda
- PRINCIPAL DISPLAY PANEL - 20 mL Vial Carton
-
PRINCIPAL DISPLAY PANEL - 40 mL Vial Label
NDC: 0944-2850-08
Immune Globulin Subcutaneous (Human) 20%
CUVITRU
Rx OnlySingle-Dose Vial
Solution for Subcutaneous Administration
The vial stopper is not made with natural rubber latex.8g
40 mLRefrigeration: 2°C to 8°C (36°F to 46°F) for up to 36 months.
Do not freeze.Room temperature: not to exceed 25°C (77°F) for up to 24 months.
Discard unused portion.CUVITRU® is a registered trademark of Baxalta Incorporated,
a Takeda company.Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898LE-07-53936
Takeda
- PRINCIPAL DISPLAY PANEL - 40 mL Vial Carton
-
PRINCIPAL DISPLAY PANEL - 50 mL Vial Label
NDC: 0944-2850-10
Immune Globulin Subcutaneous (Human) 20%
CUVITRU
Rx Only10g
50 mLSingle-Dose Vial
Solution for Subcutaneous Administration
The vial stopper is not made with natural rubber latex.Refrigeration: 2°C to 8°C (36°F to 46°F) for up to 36 months.
Do not freeze.Room temperature: not to exceed 25°C (77°F) for up to 24 months.
Discard unused portion.CUVITRU® is a registered trademark of Baxalta Incorporated,
a Takeda company.Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898LE-07-53938
Takeda
- PRINCIPAL DISPLAY PANEL - 50 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
CUVITRU
immune globulin subcutaneous (human) injection, solutionProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC:0944-2850 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 200 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0944-2850-01 1 in 1 CARTON 1 NDC:0944-2850-02 5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:0944-2850-03 1 in 1 CARTON 2 NDC:0944-2850-04 10 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 3 NDC:0944-2850-05 1 in 1 CARTON 3 NDC:0944-2850-06 20 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 4 NDC:0944-2850-07 1 in 1 CARTON 4 NDC:0944-2850-08 40 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 5 NDC:0944-2850-09 1 in 1 CARTON 5 NDC:0944-2850-10 50 mL in 1 BOTTLE, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125596 10/18/2016 Labeler - Takeda Pharmaceuticals America, Inc. (039997266) Establishment Name Address ID/FEI Business Operations Baxalta Belgium Manufacturing SA 370634700 MANUFACTURE(0944-2850)




