PRIFTIN- rifapentine tablet, film coated
sanofi-aventis U.S. LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use PRIFTIN® safely and effectively. See full prescribing information for PRIFTIN.
PRIFTIN (rifapentine) tablets, for oral use Initial U.S. Approval: 1998 RECENT MAJOR CHANGES
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONSKnown hypersensitivity to any rifamycin. (4.1) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common adverse reactions with regimen for active pulmonary tuberculosis (3% and greater) are anemia, lymphopenia, hemoptysis, neutropenia, cough, thrombocytosis, increased sweating, increased ALT, increased AST, back pain, rash, anorexia, arthralgia, increased blood urea, and headache. The most common adverse reaction (3% and greater) with the regimen for latent tuberculosis infection is hypersensitivity reaction. (6.1) DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 6/2020 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.1 Active Pulmonary Tuberculosis
PRIFTIN® (rifapentine) is indicated in adults and children 12 years and older for the treatment of active pulmonary tuberculosis (TB) caused by Mycobacterium tuberculosis. PRIFTIN must always be used in combination with one or more antituberculosis (anti-TB) drugs to which the isolate is susceptible [see Dosage and Administration (2.1) and Clinical Studies (14.1)].
Limitations of Use
Do not use PRIFTIN monotherapy in either the initial or the continuation phases of active antituberculous treatment.
PRIFTIN should not be used once weekly in the continuation phase regimen in combination with isoniazid (INH) in HIV-infected patients with active pulmonary tuberculosis because of a higher rate of failure and/or relapse with rifampin (RIF)-resistant organisms [see Warnings and Precautions (5.4) and Clinical Studies (14.1)].
PRIFTIN has not been studied as part of the initial phase treatment regimen in HIV-infected patients with active pulmonary tuberculosis.
1.2 Latent Tuberculosis Infection
PRIFTIN is indicated in adults and children 2 years and older for the treatment of latent tuberculosis infection caused by Mycobacterium tuberculosis in patients at high risk of progression to tuberculosis disease (including those in close contact with active tuberculosis patients, recent conversion to a positive tuberculin skin test, HIV-infected patients, or those with pulmonary fibrosis on radiograph) [see Clinical Studies (14.2)].
Limitations of Use
Active tuberculosis disease should be ruled out before initiating treatment for latent tuberculosis infection.
PRIFTIN must always be used in combination with isoniazid as a 12-week once-weekly regimen for the treatment of latent tuberculosis infection [see Dosage and Administration (2.2) and Clinical Studies (14.2)].
- PRIFTIN in combination with isoniazid is not recommended for individuals presumed to be exposed to rifamycin-resistant or isoniazid-resistant M. tuberculosis.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Active Pulmonary Tuberculosis
PRIFTIN is only recommended for the treatment of active pulmonary tuberculosis caused by drug-susceptible organisms as part of regimens consisting of a 2-month initial phase followed by a 4-month continuation phase.
PRIFTIN should not be used in the treatment of active pulmonary tuberculosis caused by rifampin-resistant strains.
Initial phase (2 Months): PRIFTIN should be administered at a dose of 600 mg twice weekly for two months as directly observed therapy (DOT), with an interval of no less than 3 consecutive days (72 hours) between doses, in combination with other antituberculosis drugs as part of an appropriate regimen which includes daily companion drugs such as isoniazid (INH), ethambutol (EMB) and pyrazinamide (PZA).
Continuation phase (4 Months): Following the initial phase (2 months), continuation phase (4 months) treatment consists of PRIFTIN 600 mg once weekly for 4 months in combination with isoniazid or another appropriate antituberculosis agent for susceptible organisms administered as directly observed therapy.
2.2 Dosage in Latent Tuberculosis Infection
PRIFTIN should be administered once weekly in combination with isoniazid for 12 weeks as directly observed therapy.
Adults and children 12 years and older: The recommended dose of PRIFTIN should be determined based on weight of the patient up to a maximum of 900 mg once weekly (see Table 1). The recommended dose of isoniazid is 15 mg/kg (rounded to the nearest 50 mg or 100 mg) up to a maximum of 900 mg once weekly for 12 weeks.
Children 2 to 11 years: The recommended dose of PRIFTIN should be determined based on weight of the patient up to a maximum of 900 mg once weekly (see Table 1). The recommended dose of isoniazid is 25 mg/kg (rounded to the nearest 50 mg or 100 mg) up to a maximum of 900 mg once weekly for 12 weeks.
| Weight range | PRIFTIN dose | Number of PRIFTIN tablets |
|---|---|---|
| 10–14 kg | 300 mg | 2 |
| 14.1–25 kg | 450 mg | 3 |
| 25.1–32 kg | 600 mg | 4 |
| 32.1–50 kg | 750 mg | 5 |
| >50 kg | 900 mg | 6 |
2.3 Administration
Take PRIFTIN with meals. Administration of PRIFTIN with a meal increases oral bioavailability and may reduce the incidence of gastrointestinal upset, nausea, and/or vomiting [see Clinical Pharmacology (12.3)].
For patients who cannot swallow tablets, the tablets may be crushed and added to a small amount of semi-solid food, all of which should be consumed immediately [see Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
PRIFTIN is supplied as 150 mg round normal convex dark-pink film-coated tablets debossed "Priftin" on top and "150" on the bottom.
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Elevations of liver transaminases may occur in patients receiving PRIFTIN [see Adverse Reactions (6.1)]. Patients on PRIFTIN should be monitored for symptoms of liver injury.
Patients with abnormal liver tests and/or liver disease or patients initiating treatment for active pulmonary tuberculosis should only be given PRIFTIN in cases of necessity and under strict medical supervision. In such patients, obtain serum transaminase levels prior to therapy and every 2 to 4 weeks while on therapy. Discontinue PRIFTIN if evidence of liver injury occurs.
5.2 Hypersensitivity and Related Reactions
Hypersensitivity reactions may occur in patients receiving PRIFTIN. Signs and symptoms of these reactions may include hypotension, urticaria, angioedema, acute bronchospasm, conjunctivitis, thrombocytopenia, neutropenia or flu-like syndrome (weakness, fatigue, muscle pain, nausea, vomiting, headache, fever, chills, aches, rash, itching, sweats, dizziness, shortness of breath, chest pain, cough, syncope, palpitations). There have been reports of anaphylaxis [see Patient Counseling Information (17)].
Monitor patients receiving PRIFTIN therapy for signs and/or symptoms of hypersensitivity reactions. If these symptoms occur, administer supportive measures and discontinue PRIFTIN.
5.3 Severe Cutaneous Adverse Reactions
Severe cutaneous adverse reactions (SCARs) such as Stevens-Johnson syndrome (SJS) and drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome have been reported in association with the use of rifapentine (PRIFTIN) treatment regimens in patients with active and latent tuberculosis. Discontinue PRIFTIN at the first appearance of skin rash, mucosal lesions, or any other sign of hypersensitivity [see Patient Counseling Information (17)].
5.4 Relapse in the Treatment of Active Pulmonary Tuberculosis
PRIFTIN has not been evaluated as part of the initial phase treatment regimen in HIV-infected patients with active pulmonary TB.
Do not use PRIFTIN as a once-weekly continuation phase regimen in HIV-infected patients with active pulmonary tuberculosis because of a higher rate of failure and/or relapse with rifampin-resistant organisms [see Clinical Studies (14.1)].
Higher relapse rates may occur in patients with cavitary pulmonary lesions and/or positive sputum cultures after the initial phase of active tuberculosis treatment and in patients with evidence of bilateral pulmonary disease. Monitor for signs and symptoms of TB relapse in these patients [see Clinical Studies (14.1)].
Poor adherence to therapy is associated with high relapse rate. Emphasize the importance of compliance with therapy [see Patient Counseling Information (17)].
5.5 Drug Interactions
Rifapentine is an inducer of CYP450 enzymes. Concomitant use of rifapentine with other drugs metabolized by these enzymes, such as protease inhibitors, certain reverse transcriptase inhibitors, and hormonal contraception may cause a significant decrease in plasma concentrations and loss of therapeutic effect [see Drug Interactions (7.1, 7.2, 7.3, 7.4) and Clinical Pharmacology (12.3)].
5.6 Discoloration of Body Fluids
PRIFTIN may produce a red-orange discoloration of body tissues and/or fluids (e.g., skin, teeth, tongue, urine, feces, saliva, sputum, tears, sweat, and cerebrospinal fluid). Contact lenses or dentures may become permanently stained.
5.7 Clostridioides Difficile–Associated Diarrhea
Clostridioides difficile–associated diarrhea (CDAD) has been reported with the use of nearly all systemic antibacterial agents, including PRIFTIN, with severity ranging from mild diarrhea to fatal colitis. Treatment with antibacterial agents can alter the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary because CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, discontinue antibacterial use not directed against C. difficile if possible. Institute appropriate measures such as fluid and electrolyte management, protein supplementation, antibacterial treatment of C. difficile, and surgical evaluation as clinically indicated.
6 ADVERSE REACTIONS
The following serious and otherwise important adverse drug reactions are discussed in greater detail in other sections of labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Hypersensitivity [see Contraindications (4.1) and Warnings and Precautions (5.2)]
- Severe Cutaneous Adverse Reactions [see Warnings and Precautions (5.3)]
- Discoloration of Body Fluids [see Warnings and Precautions (5.6)]
- Clostridioides Difficile–Associated Diarrhea [see Warnings and Precautions (5.7)]
- Porphyria [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Active Pulmonary Tuberculosis
PRIFTIN was studied in a randomized, open label, active-controlled trial of HIV-negative patients with active pulmonary tuberculosis. The population consisted primarily of male subjects with a mean age of 37 ± 11 years. In the initial 2-month phase of treatment, 361 patients received PRIFTIN 600 mg twice a week in combination with daily isoniazid, pyrazinamide, and ethambutol and 361 subjects received rifampin in combination with isoniazid, pyrazinamide and ethambutol all administered daily. Ethambutol was discontinued when drug susceptibly testing was known. During the 4-month continuation phase, 317 patients in the PRIFTIN group continued to receive PRIFTIN 600 mg dosed once weekly with isoniazid and 304 patients in the rifampin group received twice weekly rifampin and isoniazid. Both treatment groups received pyridoxine (Vitamin B6) over the 6-month treatment period.
Because PRIFTIN was administered as part of a combination regimen, the adverse reaction profile reflects the entire regimen.
Twenty-two deaths occurred in the study, eleven in the rifampin combination therapy group and eleven in the PRIFTIN combination therapy group. 18/361 (5%) rifampin combination therapy patients discontinued the study due to an adverse reaction compared to 11/361 (3%) PRIFTIN combination therapy patients. Three patients (two rifampin combination therapy patients and one PRIFTIN combination therapy patient) were discontinued in the initial phase due to hepatotoxicity. Concomitant medications for all three patients included isoniazid, pyrazinamide, ethambutol, and pyridoxine. All three recovered without sequelae.
Five patients had adverse reactions associated with PRIFTIN overdose. These reactions included hematuria, neutropenia, hyperglycemia, ALT increased, hyperuricemia, pruritus, and arthritis.
Table 2 presents selected treatment-emergent adverse reactions associated with the treatment regimens which occurred in at least 1% of patients during treatment and post treatment through the first three months of follow-up.
| Initial Phase* | Continuation Phase† | |||
|---|---|---|---|---|
| System Organ Class Adverse Reaction | PRIFTIN Combination (N=361) N (%) | Rifampin Combination (N=361) N (%) | PRIFTIN Combination (N=317) N (%) | Rifampin Combination (N=304) N (%) |
|
||||
| Blood and lymphatics | ||||
| Anemia | 41 (11.4) | 41 (11.4) | 5 (1.6) | 10 (3.3) |
| Lymphopenia | 38 (10.5) | 37 (10.2) | 10 (3.2) | 9 (3.0) |
| Neutropenia | 22 (6.1) | 21 (5.8) | 27 (8.5) | 24 (7.9) |
| Leukocytosis | 6 (1.7) | 13 (3.6) | 5 (1.6) | 2 (0.7) |
| Thrombocytosis | 20 (5.5) | 13 (3.6) | 1 (0.3) | 0 (0.0) |
| Thrombocytopenia | 6 (1.7) | 6 (1.7) | 4 (1.3) | 6 (2) |
| Lymphadenopathy | 4 (1.1) | 2 (0.6) | 0 (0.0) | 2 (0.7) |
| Eye | ||||
| Conjunctivitis | 8 (2.2) | 2 (0.6) | 1 (0.3) | 1 (0.3) |
| Gastrointestinal | ||||
| Dyspepsia | 6 (1.7) | 11 (3) | 4 (1.3) | 6 (2) |
| Vomiting | 6 (1.7) | 14 (3.9) | 3 (0.9) | 3 (1) |
| Nausea | 7 (1.9) | 3 (0.8) | 2 (0.6) | 1 (0.3) |
| Diarrhea | 5 (1.4) | 2 (0.6) | 2 (0.6) | 0 (0.0) |
| General | ||||
| Back Pain | 15 (4.2) | 11 (3) | 11 (3.5) | 4 (1.3) |
| Abdominal Pain | 3 (0.8) | 3 (0.8) | 4 (1.3) | 4 (1.3) |
| Fever | 5 (1.4) | 7 (1.9) | 1 (0.3) | 1 (0.3) |
| Anorexia | 14 (3.9) | 18 (5) | 8 (2.5) | 6 (2) |
| Hepatic and biliary | ||||
| ALT Increased | 18 (5) | 23 (6.4) | 7 (2.2) | 10 (3.3) |
| AST Increased | 15 (4.2) | 18 (5) | 7 (2.2) | 8 (2.6) |
| Investigations | ||||
| Blood urea increased | 4 (1.1) | 3 (0.8) | 10 (3.2) | 15 (4.9) |
| Musculoskeletal | ||||
| Arthralgia | 13 (3.6) | 13 (3.6) | 3 (0.9) | 5 (1.6) |
| Neurologic | ||||
| Headache | 11 (3) | 13 (3.6) | 3 (0.9) | 7 (2.3) |
| Dizziness | 5 (1.4) | 5 (1.4) | 1 (0.3) | 1 (0.3) |
| Respiratory | ||||
| Hemoptysis | 27 (7.5) | 20 (5.5) | 6 (1.9) | 6 (2) |
| Coughing | 21 (5.8) | 8 (2.2) | 9 (2.8) | 11 (3.6) |
| Skin | ||||
| Rash | 15 (4.2) | 26 (7.2) | 8 (2.5) | 8 (2.6) |
| Sweating Increased | 19 (5.3) | 18 (5) | 5 (1.6) | 4 (1.3) |
| Pruritus | 10 (2.8) | 16 (4.4) | 3 (0.9) | 0 (0.0) |
| Rash Maculopapular | 6 (1.7) | 3 (0.8) | 0 (0.0) | 1 (0.3) |
The following selected treatment-emergent adverse reactions were reported in less than 1% of the PRIFTIN combination therapy patients during treatment and post treatment through the first three months of follow-up.
Blood and Lymphatics: lymphocytosis, hematoma, purpura, thrombosis.
Cardiovascular: syncope, tachycardia, palpitation, orthostatic hypotension, pericarditis.
Metabolic & Nutritional: alkaline phosphatase increased.
Gastrointestinal: gastritis, esophagitis, pancreatitis, salivary gland enlargement.
General: asthenia, facial edema.
Hepatobiliary: bilirubinemia, hepatomegaly, jaundice.
Infectious Disease: infection fungal.
Musculoskeletal: myalgia, myositis.
Neurologic: somnolence, dysphonia.
Pregnancy, Puerperium and Perinatal Conditions: abortion.
Psychiatric: anxiety, confusion.
Reproductive Disorders: vaginitis, vaginal hemorrhage, leukorrhea.
Respiratory: dyspnea, pneumonitis, pulmonary fibrosis, asthma, bronchospasm, laryngeal edema, laryngitis.
Skin: urticaria, skin discoloration.
In another randomized, open-label trial, 1075 HIV non-infected and infected patients with active pulmonary tuberculosis who had completed an initial 2-month phase of treatment with 4 drugs were randomly assigned to receive either PRIFTIN 600 mg and isoniazid once weekly or rifampin and isoniazid twice weekly for the 4-month continuation phase. Five hundred and two non–HIV-infected and 36 HIV-infected patients were randomized to receive the PRIFTIN regimen and 502 HIV-noninfected and 35 HIV-infected patients were randomized to receive the rifampin regimen.
The death rate was 6.5% for the PRIFTIN combination regimen compared to 6.7% for the rifampin combination regimen.
Latent Tuberculosis Infection
Main study
PRIFTIN in combination with isoniazid given once weekly for 3 months (3RPT/INH) was compared to isoniazid given once daily for 9 months (9INH) in an open-label, randomized trial in patients with a positive tuberculin skin test, and at high risk for progression from latent tuberculosis infection to active tuberculosis disease. PRIFTIN was dosed by weight, and isoniazid mg/kg dose was determined according to age [see Dosage and Administration (2.2)] to a maximum of 900 mg each.
A total of 4040 patients received at least one dose of the 3RPT/INH regimen, including 348 children 2 to 17 years of age and 105 HIV-infected individuals. A total of 3759 received at least one dose of the 9INH regimen, including 342 children 2 to 17 years of age and 95 HIV-infected individuals.
Patients were followed for 33 months from the time of enrollment. Treatment-emergent adverse reactions were defined as those occurring during treatment and 60 days after the last dose of treatment. One hundred and sixty- one (4%) 3RPT/INH subjects had a rifamycin hypersensitivity reaction, defined as either: a) one of the following: hypotension, urticaria, angioedema, acute bronchospasm, or conjunctivitis occurring in relation to study drug or b) at least four of the following symptoms occurring in relation to the study drug, with at least one symptom being CTCAE Grade 2 or higher: weakness, fatigue, nausea, vomiting, headache, fever, aches, sweats, dizziness, shortness of breath, flushing or chills. No specific definition was used for isoniazid hypersensitivity; 18 (0.5%) 9INH subjects were classified as having a hypersensitivity reaction. Hepatotoxicity was defined as AST ≥3 × upper limit of normal in the presence of specific signs and symptoms of hepatitis, or AST >5 × upper limit of normal regardless of signs or symptoms. One hundred and thirteen (3%) 9INH subjects and 24 (0.6%) 3RPT/INH subjects developed hepatotoxicity.
One hundred and ninety-six subjects (4.9%) in the 3RPT/INH arm discontinued treatment due to a treatment related adverse reaction patients and 142 (3.8%) in the 9INH arm discontinued treatment due to a treatment related adverse reaction. In the 3RPT/INH group, the most frequent treatment related adverse reaction resulting in treatment discontinuation was hypersensitivity reaction, occurring in 120 (3%) patients. In the 9INH group, the most frequent treatment related adverse reaction resulting in treatment discontinuation was hepatotoxicity, occurring in 76 (2%) patients.
Seventy-one deaths occurred, 31/4040, 0.77% in the 3RPT/INH group and 40/3759 (1.06%) in the 9INH group) during the 33-month study period. During the treatment emergent period, 11 deaths occurred, 4 in the 3RPT/INH group and 7 in the 9INH group. None of the reported deaths were considered related to treatment with study drugs or were attributed to tuberculosis disease.
Table 3 presents select adverse reactions that occurred during the treatment emergent period in the main study in LTBI patients treated with 3RPT/INH or 9INH at a frequency greater than 0.5%.
| System Organ Class Adverse Reaction | 3RPT/INH (N=4040) N (%) | 9INH (N=3759) N (%) |
|---|---|---|
|
||
| Immune system disorders | ||
| Hypersensitivity | 161 (4) | 18 (0.5) |
| Hepatobiliary disorders | ||
| Hepatitis | 24 (0.6) | 113 (3) |
| Nervous system disorders | ||
| Headache | 26 (0.6) | 17 (0.5) |
| Skin and subcutaneous tissue disorders | ||
| Skin reaction | 31 (0.8) | 21 (0.6) |
Pediatric substudy
Six hundred and ninety children 2 to 17 years of age received at least one dose of study drugs in the main study. An additional 342 children 2 to 17 years of age received at least one dose in the pediatric extension study (total 1032 children; 539 received 3RPT/INH and 493 received 9INH).
No children in either treatment arm developed hepatotoxicity. Using the same definition for rifamycin hypersensitivity reaction as in the main study, 7 (1.3%) of children in the 3RPT/INH group experienced a rifamycin hypersensitivity reaction. Adverse reactions in children 2 to 11 years of age and 12 to 17 years of age were similar.
HIV substudy
Two hundred HIV-infected patients with latent tuberculosis infection received at least one dose of study drugs in the main study and an additional 193 patients received at least one dose in the extension study (total of 393; 207 received 3RPT/INH and 186 received 9INH). Compared to the HIV-negative patients enrolled in the main study, a higher proportion of HIV-infected patients in each treatment arm experienced a treatment emergent adverse reaction, including a higher incidence of hepatotoxicity. Hepatotoxicity occurred in 3/207 (1.5%) patients in the 3RPT/INH arm and in 14/186 (7.5%) in the 9INH arm. Rifamycin hypersensitivity occurred in only one HIV-infected patient.
Eleven deaths occurred during the 33-month follow up period (6/207 in the 3RPT/INH group and 5/186 in the 9INH group) including one death in the 9INH arm during the treatment emergent period. None of the reported deaths were considered related to treatment with study drugs or tuberculosis disease.
Selected treatment-emergent adverse reactions reported during treatment and 60 days post treatment in less than 0.5% of the 3RPT/INH combination-therapy group in the main study are presented below by body system.
Eye Disorders: conjunctivitis.
Blood and Lymphatic System Disorders: leukopenia, anemia, lymphadenopathy, neutropenia.
Gastrointestinal Disorders: nausea, diarrhea, vomiting, abdominal pain, constipation, dry mouth, dyspepsia, esophageal irritation, gastritis, pancreatitis.
General Disorders and Administration Site Conditions: fatigue, pyrexia, asthenia, chest pain, chills, feeling jittery.
Infections and Infestations: pharyngitis, viral infection, vulvovaginal candidiasis.
Metabolism and Nutrition Disorders: hyperglycemia, gout, hyperkalemia, decreased appetite, hyperlipidemia.
Musculoskeletal and Connective Tissue Disorders: arthralgia, myalgia, back pain, rhabdomyolysis.
Nervous System Disorders: dizziness, convulsion, paresthesia, headache, neuropathy peripheral, syncope.
Psychiatric Disorders: depression, anxiety, disorientation, suicidal ideation.
Renal and Urinary Disorders: azotemia.
Reproductive System and Breast Disorders: vulvovaginal pruritus.
Respiratory, Thoracic and Mediastinal Disorders: cough, dyspnea, oropharyngeal pain, asthma, bronchial hyperactivity, epistaxis.
Skin and Subcutaneous Tissue Disorders: rash, hyperhidrosis, pruritus, urticaria.
6.2 Postmarketing Experience
The following adverse reactions have been identified from postmarketing surveillance of rifapentine. Because these reactions are reported from a population of unknown size, it is not always possible to estimate their frequency or establish a causal relationship to drug exposure.
Skin and subcutaneous tissue disorders: Severe cutaneous adverse reactions (SCARs) such as Stevens-Johnson syndrome (SJS) and drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome [see Warnings and Precautions (5.3)].
7 DRUG INTERACTIONS
7.1 Protease Inhibitors and Reverse Transcriptase Inhibitors
Rifapentine is an inducer of CYP450 enzymes. Concomitant use of PRIFTIN with other drugs metabolized by these enzymes, such as protease inhibitors and certain reverse transcriptase inhibitors, may cause a significant decrease in plasma concentrations and loss of therapeutic effect of the protease inhibitor or reverse transcriptase inhibitor [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
7.2 Fixed-Dose Combination of Efavirenz, Emtricitabine, and Tenofovir
Once-weekly coadministration of 900 mg PRIFTIN with the antiretroviral fixed-dose combination of efavirenz 600 mg, emtricitabine 200 mg and tenofovir disoproxil fumarate 300 mg in HIV-infected patients did not result in any substantial change in steady state exposures of efavirenz, emtricitabine, and tenofovir. No clinically significant change in CD4 cell counts or viral loads were noted [see Clinical Pharmacology (12.3)].
7.3 Hormonal Contraceptives
PRIFTIN may reduce the effectiveness of hormonal contraceptives. Patients using hormonal contraception should be advised to use an alternative non-hormonal contraceptive method or add a barrier method of contraception during treatment with PRIFTIN [see Use in Specific Populations (8.3) and Clinical Pharmacology (12.3)].
7.4 Cytochrome P450 3A4 and 2C8/9
Rifapentine is an inducer of cytochromes P450 3A4 and P450 2C8/9. Therefore, PRIFTIN may increase the metabolism of other coadministered drugs that are metabolized by these enzymes. Induction of enzyme activities by PRIFTIN occurred within 4 days after the first dose. Enzyme activities returned to baseline levels 14 days after discontinuing PRIFTIN.
Rifampin has been reported to accelerate the metabolism and may reduce the activity of the following drugs; hence, PRIFTIN may also increase the metabolism and decrease the activity of these drugs. Dosage adjustments of the drugs in Table 4 or of other drugs metabolized by cytochrome P450 3A4 or P450 2C8/9 may be necessary if they are given concurrently with PRIFTIN.
| Drug Class | Examples of Drugs Within Class |
|---|---|
| Antiarrhythmics | Disopyramide, mexiletine, quinidine, tocainide |
| Antibiotics | Chloramphenicol, clarithromycin, dapsone, doxycycline; Fluoroquinolones (such as ciprofloxacin) |
| Oral Anticoagulants | Warfarin |
| Anticonvulsants | Phenytoin |
| Antimalarials | Quinine |
| Azole Antifungals | Fluconazole, itraconazole, ketoconazole |
| Antipsychotics | Haloperidol |
| Barbiturates | Phenobarbital |
| Benzodiazepines | Diazepam |
| Beta-Blockers | Propranolol |
| Calcium Channel Blockers | Diltiazem, nifedipine, verapamil |
| Cardiac Glycoside Preparations | Digoxin |
| Corticosteroids | Prednisone |
| Fibrates | Clofibrate |
| Oral Hypoglycemics | Sulfonylureas (e.g., glyburide, glipizide) |
| Hormonal Contraceptives/Progestins | Ethinyl estradiol, levonorgestrel |
| Immunosuppressants | Cyclosporine, tacrolimus |
| Methylxanthines | Theophylline |
| Narcotic analgesics | Methadone |
| Phosphodiesterase-5 (PDE-5) Inhibitors | Sildenafil |
| Thyroid preparations | Levothyroxine |
| Tricyclic antidepressants | Amitriptyline, nortriptyline |
7.5 Other Interactions
The conversion of PRIFTIN to 25-desacetyl rifapentine is mediated by an esterase enzyme. There is minimal potential for PRIFTIN metabolism to be inhibited or induced by another drug, based upon the characteristics of the esterase enzymes.
Since PRIFTIN is highly bound to albumin, drug displacement interactions may also occur [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal data, PRIFTIN may cause fetal harm when administered to a pregnant woman. Available data from clinical trials, case reports, epidemiology studies and postmarketing experience with PRIFTIN use in pregnant women are insufficient to establish a drug-associated risk of major birth defects, adverse maternal or fetal outcomes. In two clinical trials, a total of 59 patients who were treated with rifapentine in combination with other anti-tuberculosis drugs became pregnant. Overall, the reported rate of miscarriage following rifapentine exposure in these two clinical trials did not represent an increase over the background rate of miscarriage reported in the general population (see Data). There are risks associated with active tuberculosis during pregnancy. When administered during the last few weeks of pregnancy, PRIFTIN may be associated with maternal postpartum hemorrhage and bleeding in the exposed neonates (see Clinical Considerations). In animal reproduction and developmental toxicity studies, adverse developmental outcomes (including cleft palate or mal-positioned aortic arches) were observed following administration of rifapentine to pregnant rats and rabbits at doses approximately 0.6 and 0.3 to 1.3 times, respectively, of the recommended human dose based on body surface area comparisons (see Data). Based on animal data, advise pregnant women of the risk for fetal harm. As rifapentine is always used in combination with other antituberculosis drugs such as isoniazid, ethambutol, and pyrazinamide, refer to the prescribing information of the other drug(s) for more information on their associated risks of use during pregnancy.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15%–20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo-fetal risk
Active tuberculosis in pregnancy is associated with adverse maternal and neonatal outcomes including maternal anemia, cesarean delivery, preterm birth, low birth weight, birth asphyxia, and perinatal infant death.
Labor or delivery
When administered during the last few weeks of pregnancy, PRIFTIN may increase the risk for maternal postpartum hemorrhage and bleeding in the exposed neonate. Monitor prothrombin time of pregnant women and neonates who are exposed to PRIFTIN during the last few weeks of pregnancy. Treatment with Vitamin K may be indicated.
Data
Human data
Fourteen patients with active tuberculosis treated with multiple antituberculosis drugs including PRIFTIN became pregnant during clinical studies. Six delivered normal infants, four had first trimester spontaneous abortions (of these, one patient abused ethanol and another patient was HIV-infected), one had an elective abortion, and outcome was unknown in three patients. These data are, however, limited by the quality of reporting and confounded by comorbid medical conditions and multiple antituberculosis drug exposures.
In the trial that compared the safety and effectiveness of PRIFTIN in combination with isoniazid to isoniazid alone for the treatment of latent tuberculosis infection, a total of 45 (2.5%) women in the PRIFTIN/isoniazid arm and 71 (4.1%) women in the isoniazid arm became pregnant. Among the 46 total pregnancies in the PRIFTIN/isoniazid arm, there were 31 live births, 6 elective abortions, 7 spontaneous abortions, and 2 unknown outcomes. Of the 31 live infants, 21 were reported healthy while in the other ten cases no further details were available. The rate of spontaneous abortion in the PRIFTIN/isoniazid arm (15%) and the rate of spontaneous abortion in the isoniazid arm (19%) did not represent an increase over the background rate of 15 to 20 percent reported in the general population. Further interpretation of these results is limited by the quality of adverse event reporting.
Animal data
Animal studies in rats and rabbits revealed malformations and other adverse developmental outcomes in both species. Pregnant rats given oral rifapentine during organogenesis (gestational days 5 through 15) at 40 mg/kg/day (0.6 times the human dose of 600 mg based on body surface area comparisons) produced pups with cleft palates and mal-positioned aortic arches, delayed ossification, increased number of ribs, a decrease in litter size and mean litter weight, an increase in number of stillbirths, and an increase in mortality during lactation.
When rifapentine was administered orally to mated female rats late in gestation, at 20 mg/kg/day (0.3 times the human dose based on body surface area), pup weights and gestational survival (live pups born/pups born) were reduced compared to controls. Increased resorptions and postimplantation loss, decreased mean fetal weights, increased numbers of stillborn pups, and slightly increased pup mortality during lactation were also noted. When pregnant rabbits received oral rifapentine at 10 mg/kg to 40 mg/kg (0.3 times to 1.3 times the human dose based on body surface area) during organogenesis (GD6 to GD18), major fetal malformations occurred including: ovarian agenesis, pes varus, arhinia, microphthalmia, and irregularities of the ossified facial tissues. At 40 mg/kg/day, there were increases in postimplantation loss and the incidence of stillborn pups.
8.2 Lactation
Risk Summary
There are no data on the presence of rifapentine or its metabolite in human or animal milk, the effects on the breastfed infant, or the effects on milk production.
Since PRIFTIN may produce a red-orange discoloration of body fluids, there is a potential for discoloration of breast milk. Monitor infants exposed to rifapentine through breast milk for signs of hepatotoxicity (see Clinical Considerations). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PRIFTIN and any potential adverse effects on the breastfed infant from PRIFTIN or from the underlying maternal condition.
Clinical Considerations
Monitor infants exposed to rifapentine through breast milk for signs of hepatotoxicity to include irritability, prolonged unexplained crying, yellowing of the eyes, loss of appetite, vomiting, and changes in color of the urine (darkening) or stool (lightening, pale or light brown).
8.3 Females and Males of Reproductive Potential
Contraception
Use of PRIFTIN may reduce the efficacy of hormonal contraceptives. Advise patients using hormonal contraceptives to use an alternative non-hormonal contraceptive method or add a barrier method of contraception during treatment with PRIFTIN [see Warnings and Precautions (5.5) and Drug Interactions (7.3)].
8.4 Pediatric Use
The safety and effectiveness of PRIFTIN in the treatment of active pulmonary tuberculosis have not been established in pediatric patients under the age of 12.
The safety and effectiveness of PRIFTIN in combination with isoniazid once-weekly regimen has been evaluated in pediatric patients (2 to17 years of age) for the treatment of latent tuberculosis infection. In clinical studies, the safety profile in children was similar to that observed in adult patients [see Adverse Reactions (6.1) and Clinical Studies (14.2)].
In a pharmacokinetic study conducted in 2 to 11-year-old pediatric patients with latent tuberculosis infection, PRIFTIN was administered once weekly based on weight (15 mg/kg to 30 mg/kg, up to a maximum of 900 mg). Exposures (AUC) in children 2 to 11 years old with latent tuberculosis infection were higher (average 31%) than those observed in adults receiving PRIFTIN 900 mg once weekly [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical studies with PRIFTIN did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from younger subjects. In a pharmacokinetic study with PRIFTIN, no substantial differences in the pharmacokinetics of rifapentine and 25-desacetyl metabolite were observed in the elderly compared to younger adults [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
While there is no experience with the treatment of acute overdose with PRIFTIN, clinical experience with rifamycins suggests that gastric lavage to evacuate gastric contents (within a few hours of overdose), followed by instillation of an activated charcoal slurry into the stomach, may help adsorb any remaining drug from the gastrointestinal tract.
Rifapentine and 25-desacetyl rifapentine are 97.7% and 93.2% plasma protein bound, respectively. Rifapentine and related compounds excreted in urine account for only 17% of the administered dose, therefore, neither hemodialysis nor forced diuresis is expected to enhance the systemic elimination of unchanged rifapentine from the body of a patient with PRIFTIN overdose.
11 DESCRIPTION
PRIFTIN (rifapentine) for oral administration contains 150 mg of the active ingredient rifapentine per tablet.
The 150 mg tablets also contain, as inactive ingredients: calcium stearate, disodium EDTA, FD&C Blue No. 2 aluminum lake, hydroxypropyl cellulose, hypromellose USP, microcrystalline cellulose, polyethylene glycol, pregelatinized starch, propylene glycol, sodium ascorbate, sodium lauryl sulfate, sodium starch glycolate, synthetic red iron oxide, and titanium dioxide.
Rifapentine is a rifamycin derivative antimicrobial and has a similar profile of microbiological activity to rifampicin. The molecular weight is 877.04.
The molecular formula is C47H64N4O12.
The chemical name for rifapentine is rifamycin, 3-[[(4-cyclopentyl-1-piperazinyl)imino]methyl]-or 3-[N-(4-Cyclopentyl-1-piperazinyl)formimidoyl]rifamycin or 5,6,9,17,19,21-hexahydroxy-23-methoxy-2,4,12,16,18,20,22-heptamethyl-8-[N-(4-cyclopentyl-l-piperazinyl)-formimidoyl]-2,7-(epoxypentadeca[1,11,13]trienimino)naphtho[2,1-b]furan-1,11(2H)-dione 21-acetate. It has the following structure:
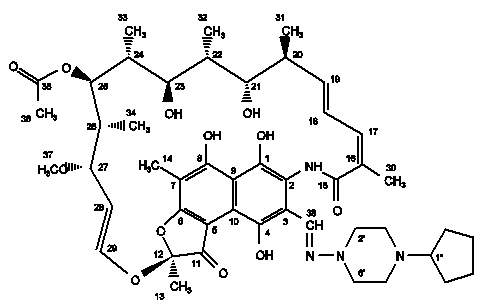
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Rifapentine, a cyclopentyl rifamycin, is an antimycobacterial agent [see Clinical Pharmacology, Microbiology (12.4)].
12.3 Pharmacokinetics
When oral doses of PRIFTIN were administered once daily or once every 72 hours to healthy volunteers for 10 days, single dose AUC (0–∞) of rifapentine was similar to its steady-state AUCss (0–24h) or AUCss (0–72h) values, suggesting no significant auto-induction effect on steady-state pharmacokinetics of rifapentine. Steady-state conditions were achieved by day 10 following daily administration of PRIFTIN 600 mg. No plasma accumulation of rifapentine and 25-desacetyl rifapentine (active metabolite) is expected after once weekly administration of PRIFTIN.
The pharmacokinetic parameters of rifapentine and 25-desacetyl rifapentine on day 10 following oral administration of 600 mg PRIFTIN every 72 hours to healthy volunteers are described in Table 5.
| Parameter | Rifapentine | 25-desacetyl Rifapentine |
|---|---|---|
| Mean ± SD (n=12) | ||
| Cmax (µg/mL) | 15.05 ± 4.62 | 6.26 ± 2.06 |
| AUC (0–72h) (µg∙h/mL) | 319.54 ± 91.52 | 215.88 ± 85.96 |
| T1/2 (h) | 13.19 ± 1.38 | 13.35 ± 2.67 |
| Tmax (h) | 4.83 ± 1.80 | 11.25 ± 2.73 |
| Cl/F (L/h) | 2.03 ± 0.60 | -- |
The pharmacokinetic parameters of rifapentine and 25-desacetyl rifapentine following single-dose oral administration of 900 mg PRIFTIN in combination with 900 mg isoniazid in fed conditions are described in Table 6.
| Parameter | Rifapentine | 25-desacetyl Rifapentine |
|---|---|---|
| Cmax (µg/mL) | 25.8 ± 5.83 | 13.3 ± 4.83 |
| AUC (µg∙h/mL) | 817 ± 128 | 601 ± 187 |
| T1/2(h) | 16.6 ± 5.02 | 17.5 ± 7.42 |
| Tmax (h)* | 8 (3–10) | 24 (10–36) |
| Cl/F (L/h) | 1.13 ± 0.174 | NA† |
Absorption
The absolute bioavailability of PRIFTIN has not been determined. The relative bioavailability (with an oral solution as a reference) of PRIFTIN after a single 600 mg dose to healthy adult volunteers was 70%. The maximum concentrations were achieved from 5 hours to 6 hours after administration of the 600 mg PRIFTIN dose.
The administration of PRIFTIN with a high fat meal increased rifapentine Cmax and AUC by 40% to 50% over that observed when PRIFTIN was administered under fasting conditions.
The administration of PRIFTIN (900 mg single dose) and isoniazid (900 mg single dose) with a low fat, high carbohydrate breakfast, led to a 47% and 51% increase in rifapentine Cmax and AUC, respectively. In contrast, the ingestion of the same meal decreased isoniazid Cmax and AUC by 46% and of 23%, respectively.
Distribution
In a population pharmacokinetic analysis in 351 tuberculosis patients who received 600 mg PRIFTIN in combination with isoniazid, pyrazinamide and ethambutol, the estimated apparent volume of distribution was 70.2 ± 9.1 L. In healthy volunteers, rifapentine and 25-desacetyl rifapentine were 97.7% and 93.2% bound to plasma proteins, respectively. Rifapentine was mainly bound to albumin. Similar extent of protein binding was observed in healthy volunteers, asymptomatic HIV-infected subjects and hepatically impaired subjects.
Metabolism/Excretion
Following a single 600 mg oral dose of radiolabeled rifapentine to healthy volunteers (n=4), 87% of the total 14C-rifapentine was recovered in the urine (17%) and feces (70%). Greater than 80% of the total 14C-rifapentine dose was excreted from the body within 7 days. Rifapentine was hydrolyzed by an esterase enzyme to form a microbiologically active 25-desacetyl rifapentine. Rifapentine and 25-desacetyl rifapentine accounted for 99% of the total radioactivity in plasma. Plasma AUC(0–∞) and Cmax values of the 25-desacetyl rifapentine metabolite were one-half and one-third those of the rifapentine, respectively. Based upon relative in vitro activities and AUC(0–∞) values, rifapentine and 25-desacetyl rifapentine potentially contributes 62% and 38% to the clinical activities against M. tuberculosis, respectively.
Specific Populations
Gender: In a population pharmacokinetics analysis of sparse blood samples obtained from 351 tuberculosis patients who received 600 mg PRIFTIN in combination with isoniazid, pyrazinamide and ethambutol, the estimated apparent oral clearance of PRIFTIN for males and females was 2.51 ± 0.14 L/h and 1.69 ± 0.41 L/h, respectively. The clinical significance of the difference in the estimated apparent oral clearance is not known.
Elderly: Following oral administration of a single 600 mg dose of PRIFTIN to elderly (65 years and older) male healthy volunteers (n=14), the pharmacokinetics of rifapentine and 25-desacetyl metabolite were similar to that observed for young (18 to 45 years) healthy male volunteers (n=20).
Pediatric: In a pharmacokinetic study in pediatric patients (age 2 to 12 years), a single oral dose of 150 mg PRIFTIN was administered to those weighing less than 30 kg (n=11) and a single oral dose of 300 mg was administered to those weighing greater than 30 kg (n=12). The mean estimates of AUC and Cmax were approximately 30% to 50% lower in these pediatric patients than those observed in healthy adults administered single oral doses of 600 mg and 900 mg.
A study compared the pharmacokinetics of rifapentine in pediatric patients (age 2 years to 11 years) with latent tuberculosis infection (n=80) receiving PRIFTIN once weekly based on weight (15 mg/kg to 30 mg/kg, up to a maximum of 900 mg, see Table 1) to that of adults (n=77) receiving PRIFTIN 900 mg once weekly. Children who could not swallow whole tablets were administered crushed tablets mixed in soft food. Overall, the geometric mean AUC of rifapentine in this age group was 31% higher compared to adult patients receiving 900 mg PRIFTIN once weekly (720 versus 551 mcg∙h/mL). The geometric mean AUC of rifapentine was 60% higher in children administered whole tablets (884 versus 551 mcg∙h/mL) and 19% higher in children administered crushed tablets (656 versus 551 mcg∙h/mL), as compared to exposures in adults. Pediatric patients administered crushed PRIFTIN tablets had 26% lower rifapentine exposures compared to those pediatric patients who were given whole tablets.
Population pharmacokinetic analysis showed that rifapentine clearance adjusted to body weight decreased with increasing age of pediatric patients (2 to 18 years).
In another pharmacokinetics study of PRIFTIN in healthy adolescents (age 12 to 15 years), 600 mg PRIFTIN was administered to those weighing ≥45 kg (n=10) and 450 mg was administered to those weighing less than 45 kg (n=2). The pharmacokinetics of rifapentine was similar to those observed in healthy adults.
Renal Impaired Patients: The pharmacokinetics of rifapentine has not been evaluated in renal impaired patients. Although only about 17% of an administered dose is excreted via the kidneys, the clinical significance of impaired renal function on the disposition of rifapentine and its 25-desacetyl metabolite is not known.
Hepatic Impaired Patients: Following oral administration of a single 600 mg dose of PRIFTIN to mild to severe hepatic impaired patients (n=15), the pharmacokinetics of rifapentine and 25-desacetyl metabolite were similar in patients with various degrees of hepatic impairment and to that observed in another study for healthy volunteers (n=12).
Asymptomatic HIV-Infected Volunteers: Following oral administration of a single 600 mg dose of PRIFTIN to asymptomatic HIV-infected volunteers (n=15) under fasting conditions, mean Cmax and AUC(0–∞) of rifapentine were lower (20%–32%) than that observed in other studies in healthy volunteers (n=55). In a cross-study comparison, mean Cmax and AUC values of the 25-desacetyl rifapentine, when compared to healthy volunteers were higher (6%–21%) in one study (n=20), but lower (15%–16%) in a different study (n=40). The clinical significance of this observation is not known. Food (850 total calories: 33 g protein, 55 g fat, and 58 g carbohydrate) increases the mean AUC and Cmax of rifapentine observed under fasting conditions in asymptomatic HIV-infected volunteers by about 51% and 53%, respectively.
Drug-Drug Interactions
Isoniazid: Coadministration of PRIFTIN (900 mg single dose) and isoniazid (900 mg single dose), in fasted condition, did not result in any significant change in the exposure of rifapentine and isoniazid compared to when administered alone in fasted condition.
Rifapentine is an inducer of cytochrome P450 3A4 and 2C8/9. Therefore, it may increase the metabolism and decrease the activity of other coadministered drugs that are metabolized by these enzymes. Dosage adjustments of the coadministered drugs may be necessary if they are given concurrently with PRIFTIN [see Drug Interactions (7.4)].
Indinavir: In a study in which 600 mg PRIFTIN was administered twice weekly for 14 days followed by PRIFTIN twice weekly plus 800 mg indinavir 3 times a day for an additional 14 days, indinavir Cmax decreased by 55% while AUC reduced by 70%. Clearance of indinavir increased by 3-fold in the presence of PRIFTIN while half-life did not change. But when indinavir was administered for 14 days followed by coadministration with PRIFTIN for an additional 14 days, indinavir did not affect the pharmacokinetics of rifapentine [see Warnings and Precautions (5.5) and Drug Interactions (7.1)].
Fixed-dose combination of efavirenz, emtricitabine and tenofovir: Once-weekly coadministration of 900 mg PRIFTIN with the antiretroviral fixed-dose combination of efavirenz 600 mg, emtricitabine 200 mg and tenofovir disoproxil fumarate 300 mg in HIV-infected patients did not result in any substantial change in steady state exposures of efavirenz, emtricitabine, and tenofovir (Table 7). A 15% decrease in efavirenz Cmin and AUC and a 13% decrease in tenofovir Cmin were observed with repeated weekly doses of PRIFTIN (Table 7). No clinically significant change in CD4 cell counts or viral loads were noted.
| Efavirenz Point Estimates (90% CI) | Emtricitabine Point Estimates (90% CI) | Tenofovir Point Estimates (90% CI) |
|
|---|---|---|---|
| Cmax | 0.92 (0.82–1.03) | 0.95 (0.81–1.10) | 1.00 (0.82–1.22) |
| Cmin | 0.85 (0.79–0.93) | 0.97 (0.90–1.05) | 0.87(0.73–1.05) |
| AUC0–24 | 0.86 (0.79–0.93) | 0.93 (0.89–0.98) | 0.91(0.85–0.98) |
12.4 Microbiology
Mechanism of Action
Rifapentine, a cyclopentyl rifamycin, inhibits DNA-dependent RNA polymerase in susceptible strains of Mycobacterium tuberculosis but does not affect mammalian cells at concentrations that are active against these bacteria. At therapeutic levels, rifapentine inhibits RNA transcription by preventing the initiation of RNA chain formation. It forms a stable complex with bacterial DNA-dependent RNA polymerase, leading to repression of RNA synthesis and cell death. Rifapentine and its 25-desacetyl metabolite accumulate in human monocyte-derived macrophages and are bactericidal to both intracellular and extracellular M. tuberculosis bacilli.
Mechanism of Resistance
The mechanism of resistance to rifapentine appears to be similar to that of rifampin. Bacterial resistance to rifapentine is caused by an alteration in the target site, the beta subunit of the DNA-dependent RNA polymerase, caused by a one-step mutation in the rpoß gene. The incidence of rifapentine resistant mutants in an otherwise susceptible population of M. tuberculosis strains is approximately one in 107 to 108 bacilli. Rifapentine resistance appears to be associated with monotherapy. Therefore, rifapentine should always be used in combination with other antituberculosis drugs.
Cross Resistance
M. tuberculosis organisms resistant to other rifamycins are likely to be resistant to rifapentine. A high level of cross-resistance between rifamycin and rifapentine has been demonstrated with M. tuberculosis strains. Cross-resistance between rifapentine and non-rifamycin antimycobacterial agents has not been identified in clinical isolates.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Hepatocellular carcinomas were increased in male NMRI mice (Harlan Winklemann) which were treated orally with rifapentine for two years at or above doses of 5 mg/kg/day (0.04 times the recommended human dose based on body surface area conversions). In a two year rat study, there was an increase in nasal cavity adenomas in Wistar rats treated orally with rifapentine at 40 mg/kg/day (0.6 times human dose based on body surface area conversions).
Rifapentine was negative in the following genotoxicity tests: in vitro gene mutation assay in bacteria (Ames test); in vitro point mutation test in Aspergillus nidulans; in vitro gene conversion assay in Saccharomyces cerevisiae; host-mediated (mouse) gene conversion assay with Saccharomyces cerevisiae; in vitro Chinese hamster ovary cell/hypoxanthine-guanine phosphoribosyltransferase (CHO/HGPRT) forward mutation assay; in vitro chromosomal aberration assay utilizing rat lymphocytes; and in vivo mouse bone marrow micronucleus assay.
The 25-desacetyl metabolite of rifapentine was positive in the in vitro mammalian chromosome aberration test in V79 Chinese hamster cells, but was negative in the in vitro gene mutation assay in bacteria (Ames test), the in vitro Chinese hamster ovary cell/hypoxanthine-guanine phosphoribosyltransferase (CHO/HGPRT) forward mutation assay, and the in vivo mouse bone marrow micronucleus assay. Fertility and reproductive performance were not affected by oral administration of rifapentine to male and female rats at doses of up to 20 mg/kg/day (one-third of the human dose based on body surface area conversions).
14 CLINICAL STUDIES
14.1 Active Pulmonary Tuberculosis
PRIFTIN was studied in two randomized, open-label controlled clinical trials in the treatment of active pulmonary tuberculosis.
The first trial was an open-label, prospective, parallel group, active-controlled trial in HIV-negative patients with active pulmonary tuberculosis. The population mostly comprised Black (approximately 60%) or multiracial (approximately 31%) patients. Treatment groups were comparable for age and sex and consisted primarily of male subjects with a mean age of 37 ± 11 years. In the initial 2-month phase of treatment, 361 patients received PRIFTIN 600 mg twice a week in combination with daily isoniazid, pyrazinamide, and ethambutol and 361 subjects received rifampin 600 mg in combination with isoniazid, pyrazinamide and ethambutol all administered daily. The doses of the companion drugs were the same in both treatment groups during the initial phase: isoniazid 300 mg, pyrazinamide 2000 mg, and ethambutol 1200 mg. For patients weighing less than 50 kg, the doses of rifampin (450 mg), pyrazinamide (1500 mg) and ethambutol (800 mg) were reduced. Ethambutol was discontinued when isoniazid and rifampin susceptibility testing results were confirmed. During the 4-month continuation phase, 317 patients in the PRIFTIN group continued to receive PRIFTIN 600 mg dosed once weekly with isoniazid 300 mg and 304 patients in the rifampin group received twice weekly rifampin and isoniazid 900 mg. For patients weighing less than 50 kg, the doses of rifampin (450 mg) and isoniazid (600 mg) were reduced. Both treatment groups received pyridoxine (Vitamin B6) over the 6-month treatment period. Treatment was directly observed. 65/361 (18%) of patients in the PRIFTIN group and 34/361 (9%) in the rifampin group received overdoses of one or more of the administered study medications during the initial or continuation phase of treatment. Seven of these patients had adverse reactions reported with the overdose (5 in the PRIFTIN group and 2 in the rifampin group).
Table 8 below contains assessments of sputum conversion at end of treatment (6 months) and relapse rates at the end of follow-up (24 months).
| PRIFTIN Combination Treatment % and (n/N*) | Rifampin Combination Treatment % and (n/N*) | |
|---|---|---|
| Status at End of 6 months of Treatment | ||
| Converted | 87% (248/286) | 80% (226/283) |
| Not Converted | 1% (4/286) | 3% (8/283) |
| Lost to Follow-up | 12% (34/286) | 17% (49/283) |
| Status Through 24 Month Follow-up† | ||
| Relapsed | 12% (29/248) | 7% (15/226) |
| Sputum Negative | 57% (142/248) | 64% (145/226) |
| Lost to Follow-up | 31% (77/248) | 29% (66/226) |
Risk of relapse was greater in the group treated with the PRIFTIN combination. Higher relapse rates were associated with a lower rate of compliance as well as a failure to convert sputum cultures at the end of the initial 2-month treatment phase. Relapse rates were also higher for males in both regimens. Relapse in the PRIFTIN group was not associated with development of monoresistance to rifampin.
The second trial was randomized, open-label performed in 1075 HIV-negative and HIV-positive patients with active pulmonary tuberculosis. Patients with culture-positive, drug-susceptible pulmonary tuberculosis who had completed the initial 2-month phase of treatment with 4 drugs (rifampin, isoniazid, pyrazinamide, and either ethambutol or streptomycin) under direct observation were randomly assigned to receive either PRIFTIN 600 mg and isoniazid 15 mg/kg (max 900 mg) once weekly or rifampin 10 mg/kg (max 600 mg) and isoniazid 15 mg/kg (max 900 mg) twice weekly for the 4 month continuation phase. Study drugs were given under direct observation therapy in both groups.
In the PRIFTIN group, 502 HIV-negative and 36 HIV-positive patients were randomized and in the rifampin group 502 HIV-negative and 35 HIV-positive patients were randomized to treatment. Enrollment of HIV-infected patients was stopped when 4 of 36 patients in the PRIFTIN combination group relapsed with isolates that were rifampin resistant.
Table 9 below contains assessments of sputum conversion at the end of treatment (6 months total: 2 months of initial and 4 months of randomized continuation treatment) and relapse rates at the end of follow-up (24 months) in all HIV-negative patients randomized to treatment. Positive culture was based on either one sputum sample with >10 colonies on solid media OR at least 2 positive sputum samples on liquid or solid media. However, only one sputum sample was collected at each visit in a majority of patients.
| PRIFTIN Combination Treatment % (n/N) | Rifampin Combination Treatment % (n/N) | |
|---|---|---|
|
||
| Status at End of 4 Months Continuation Phase | ||
| Treatment Response* | 93.8% (471/502) | 91% (457/502) |
| Not Converted | 1% (5/502) | 1.2% (6/502) |
| Did Not Complete Treatment† | 4.2% (21/502) | 7% (35/502) |
| Deaths | 1% (5/502) | 0.8% (4/502) |
| Status Through 24 Month Follow-up: | ||
| Relapsed | 8.7% (41/471) | 4.8% (22/457) |
| Sputum Negative | 79.4% (374/471) | 80.1% (366/457) |
| Lost to Follow-up | 7.9% (37/471) | 9.8% (45/457) |
| Deaths | 4% (19/471) | 5.3% (24/457) |
In HIV-negative patients, higher relapse rates were seen in patients with a positive sputum culture at 2 months (i.e., at the time of study randomization), cavitation on chest x-ray, and bilateral pulmonary involvement.
Sixty-one HIV-positive patients were assessed for relapse. The rates of relapse were 16.7% (5/30) in the PRIFTIN group and 9.7% (3/31) in the rifampin group. In HIV-positive patients, 4 of the 5 relapses in the PRIFTIN combination group involved M. tuberculosis strains with rifampin monoresistance. No relapse strain in the twice weekly rifampin/isoniazid group acquired drug resistance.
The death rate among all study participants did not differ between the two treatment groups.
14.2 Latent Tuberculosis Infection
A multicenter, prospective, open-label, randomized, active-controlled trial compared the effectiveness of 12 weekly doses of PRIFTIN in combination with isoniazid (3RPT/INH arm) administered by directly observed therapy to 9 months of self-administered daily isoniazid (9INH arm). The trial enrolled patients two years of age or older with positive tuberculin skin test and at high risk for progression to tuberculosis disease. Enrolled patients included those having close contact with a patient with active tuberculosis disease, recent (within two years) conversion to a positive tuberculin skin test, HIV-infection, or fibrosis on chest radiograph. PRIFTIN was dosed by weight, for a maximum of 900 mg weekly. Isoniazid mg/kg dose was determined by age, for a maximum of 900 mg weekly in the 3RPT/INH arm and 300 mg daily in the 9INH arm [see Dosage and Administration (2.2)].
The outcome measure was the development of active tuberculosis disease, defined as culture confirmed tuberculosis in adults and culture-confirmed or clinical tuberculosis in children less than 18 years of age, at 33 months after trial enrollment. Patients who were found after enrollment to be ineligible because they had active tuberculosis disease, were contacts of a source case with culture-negative or drug-resistant tuberculosis disease cases or no information regarding susceptibility of M. tuberculosis, and young children lacking a positive TST on initial and repeat testing were excluded from the analysis.
Active tuberculosis disease developed in 5 of 3074 randomized patients in the 3RPT/INH group (0.16%) versus 10 of 3074 patients in 9INH group (0.32%), for a difference in cumulative rates of 0.17%, 95% CI (-0.43, 0.09) (Table 10).
| Outcome | 3RPT/INH (n=3074) | 9INH (n=3074) | Difference†, 95% CI |
|---|---|---|---|
| Tuberculosis n (%) | 5 (0.16) | 10 (0.32) | -0.16 (-0.42, 0.01) |
| Cumulative TB Rate (%) | 0.17 | 0.35 | -0.17 (-0.43, 0.09) |
| Deaths | 22 (0.72) | 35 (1.14) | -0.42 (-0.91, 0.06) |
| Lost to Follow-Up | 320 (10.41) | 357 (11.61) | -1.20 (-2.77, -0.36) |
The proportion of patients completing treatment was 81.2% in the 3RPT/INH group and 68.3% in the 9INH group for a difference (3RPT/INH-9INH) of 12.8% 95% CI (10.7, 15.0).
In the 9INH treatment group, two of the thirteen culture-confirmed cases were found to be isoniazid-monoresistant. In the 3RPT/INH treatment group, one of the seven cases was rifampin-resistant, isoniazid-susceptible M. bovis infection.
Pediatric substudy
Enrollment of children was extended after the overall target number of patients was attained in the main study. Data from both the main study and the extension were pooled resulting in an eligible population for analysis of 375 children in the 3RPT/INH arm and 367 in the 9INH arm.
One child in the 9INH group developed tuberculosis (1/367, cumulative rate 0.32%) versus zero tuberculosis cases in the 3RPT/INH group (0/375) at 33 months post enrollment. The proportion of patients completing treatment in the 3RPT/INH and the 9INH groups was 87.5% and 79.6% respectively for a difference of 7.9%, 95% CI (2.5, 13.2).
HIV substudy
Enrollment of HIV-positive patients was extended after the overall target number of patients was attained in the main study. Data from both the main study and the extension were pooled resulting in an eligible population for analysis of 206 patients in the 3RPT/INH group and 193 in the 9INH group. Tuberculosis disease developed in 2/206 patients in the 3RPT/INH group (cumulative rate, 1.01%) and in 6/193 patients in the 9INH group (cumulative rate, 3.45%). The proportion of patients completing treatment in the 3RPT/INH and 9INH groups was 88.8% and 63.7%, respectively for a difference of 25.1%, 95% CI (16.8, 32.9).
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
PRIFTIN is supplied as 150 mg round normal convex dark-pink film-coated tablets debossed "Priftin" on top and "150" on the bottom, packaged in aluminum formable foil blister strips inserted into an aluminum foil laminated pouch.
Carton of 32 tablets (4 strips of 8 tablets) NDC 0088-2100-32
Carton of 24 tablets (3 strips of 8 tablets) NDC 0088-2100-24
17 PATIENT COUNSELING INFORMATION
Advise patient to read FDA-approved patient labeling (Medication Guide).
Treatment Adherence
Emphasize the importance of compliance with the full course of therapy, and the importance of not missing any doses of PRIFTIN or companion medications in the treatment of active pulmonary tuberculosis or the treatment of latent tuberculosis infection.
Hypersensitivity Reactions
Inform patients that PRIFTIN may cause hypersensitivity reactions. Signs and symptoms of this reaction may include a flu-like illness, hypotension, urticaria, angioedema, bronchospasm, conjunctivitis, thrombocytopenia or neutropenia. Anaphylaxis may also occur [see Warnings and Precautions (5.2)].
Inform patients of signs and symptoms of hypersensitivity reactions and advise them to stop the medication and contact their healthcare provider if they experience any of these symptoms.
Severe Cutaneous Adverse Reactions
Advise patients about the signs and symptoms of serious skin manifestations. Instruct patients to stop taking PRIFTIN immediately and promptly report the first signs or symptoms of skin rash, mucosal lesions, or any other sign of hypersensitivity [see Warnings and Precautions (5.3)].
Hepatitis
Instruct patients to stop the medication and notify their physician promptly if they experience any of the following: fever, loss of appetite, malaise, nausea and vomiting, darkened urine, yellowish discoloration of the skin and eyes, and pain or swelling of the joints [see Warnings and Precautions (5.1)].
Drug Interactions
Rifapentine may increase the metabolism and decrease the activity of other drugs that are metabolized by the P450 3A4 and 2C8/9 pathways. Dosage adjustments of the coadministered drugs may be necessary. Advise patients to discuss with their physician any other medications they are taking before starting treatment with PRIFTIN [see Warnings and Precautions (5.5), Drug Interactions (7.1, 7.4)].
Concomitant use of PRIFTIN with protease inhibitors or reverse transcriptase inhibitors may cause a significant decrease in plasma concentrations and loss of therapeutic effect of the protease inhibitor or reverse transcriptase inhibitor [see Warnings and Precautions (5.5) and Drug Interactions (7.4)].
Discoloration of Body Fluids
Inform the patient that PRIFTIN produces a red-orange discoloration of the urine, sweat, sputum, tears, and breast milk. Contact lenses or dentures may be permanently stained [see Warnings and Precautions (5.6)].
Lactation
Monitor infants exposed to rifapentine through breast milk for signs of hepatotoxicity to include irritability, prolonged unexplained crying, yellowing of the eyes, loss of appetite, vomiting, and changes in color of the urine (darkening) or stool (lightening, pale or light brown) [see Use in Specific Populations (8.2)].
Contraception
Advise patients that use of PRIFTIN may reduce the efficacy of hormonal contraceptives. Advise patients using hormonal contraceptives to use an alternative non-hormonal contraceptive method or add a barrier method of contraception during treatment [see Warning and Precautions (5.5), Drug Interactions (7.3), and Use in Specific Populations (8.3)].
| Medication Guide PRIFTIN (prif - tin) (rifapentine) Tablets |
||||||
|---|---|---|---|---|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | ||||||
| Revised: June 2020 | ||||||
| Read this Medication Guide before you start taking PRIFTIN and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. | ||||||
| What is the most important information I should know about PRIFTIN? | ||||||
| PRIFTIN may cause serious side effects, including: | ||||||
|
||||||
|
|
|||||
|
||||||
|
|
|||||
| Signs and symptoms of a flu-like reaction may include: | ||||||
|
|
|
||||
|
||||||
|
|
|
||||
| What is PRIFTIN? | ||||||
| PRIFTIN is a prescription medicine used with other anti-tuberculosis (TB) medicines to: | ||||||
|
||||||
| PRIFTIN should not be used: | ||||||
|
||||||
| PRIFTIN is safe and effective in children older than 2 years of age who have inactive (latent TB), but it is not known if PRIFTIN is safe and effective for use in the treatment of active TB in children under 12 years of age. | ||||||
| Who should not take PRIFTIN? | ||||||
|
||||||
| What should I tell my doctor before taking PRIFTIN? | ||||||
| Before taking PRIFTIN, tell your doctor about all of your medical conditions, including if you: | ||||||
|
||||||
| Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | ||||||
| Using PRIFTIN with other medicines may affect each other causing serious side effects. PRIFTIN may affect the way other medicines work, and other medicines may affect how PRIFTIN works. Especially tell your doctor if you take medicines to treat HIV infection or oral contraceptives. | ||||||
| Ask your doctor or pharmacist for a list of these medicines if you are not sure. | ||||||
| Know the medicines you take. Keep a list of them to show your doctor or pharmacist when you get a new medicine. | ||||||
| How should I take PRIFTIN? | ||||||
|
||||||
| What are possible side effects of PRIFTIN? | ||||||
| PRIFTIN may cause serious side effects, including: | ||||||
|
||||||
| The most common side effects of PRIFTIN include: allergic reactions and flu-like symptoms; abnormalities such as low red blood cells, low white blood cells, coughing up blood, cough, excessive number of platelets in the blood, increased sweating, high liver function tests, back pain, rash, decreased appetite, joint pain, increased blood urea, and headache. | ||||||
| Tell your doctor if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of PRIFTIN. For more information, ask your doctor or pharmacist. | ||||||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||||||
| How should I store PRIFTIN? | ||||||
|
||||||
| General information about the safe and effective use of PRIFTIN. | ||||||
| Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use PRIFTIN for a condition for which it was not prescribed. Do not give PRIFTIN to other people, even if they have the same symptoms you have. It may harm them. | ||||||
| This Medication Guide summarizes the most important information about PRIFTIN. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about PRIFTIN that is written for healthcare professionals. | ||||||
| What are the ingredients in PRIFTIN? | ||||||
| Active ingredient: rifapentine | ||||||
| Inactive ingredients: calcium stearate, disodium EDTA, FD&C Blue No. 2 aluminum lake, hydroxypropyl cellulose, hypromellose USP, microcrystalline cellulose, polyethylene glycol, pregelatinized starch, propylene glycol, sodium ascorbate, sodium lauryl sulfate, sodium starch glycolate, synthetic red iron oxide, and titanium dioxide | ||||||
| Manufactured by: sanofi-aventis U.S. LLC, Bridgewater, NJ 08807 | ||||||
| For more information, go to www.sanofi.us or call 1-800-633-1610, and select option 1. | ||||||
PRINCIPAL DISPLAY PANEL - 150 mg blister
NDC 0088-2100-03
Priftin® 150mg
rifapentine
sanofi-aventis U.S. LLC
Peel at unsealed corners
DO NOT CUT BLISTER


| PRIFTIN
rifapentine tablet, film coated |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (783243835) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi S.p.A | 338454274 | ANALYSIS(0088-2100) , LABEL(0088-2100) , MANUFACTURE(0088-2100) , PACK(0088-2100) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi S.p.A (also known as Sanofi-Aventis S.p.A) | 378937440 | ANALYSIS(0088-2100) , API MANUFACTURE(0088-2100) | |