Label: ELMIRON- pentosan polysulfate sodium capsule, gelatin coated

- NDC Code(s): 50458-098-01
- Packager: Janssen Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated November 10, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
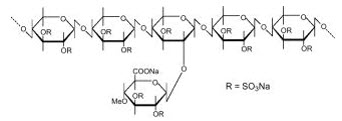
Pentosan polysulfate sodium is a semi-synthetically produced heparin-like macromolecular carbohydrate derivative, which chemically and structurally resembles glycosaminoglycans. It is a white odorless powder, slightly hygroscopic and soluble in water to 50% at pH 6. It has a molecular weight of 4000 to 6000 Dalton with the following structural formula:
ELMIRON ® is supplied in white opaque hard gelatin capsules containing 100 mg pentosan polysulfate sodium, microcrystalline cellulose, and magnesium stearate. It also contains pharmaceutical glaze (modified) in SD-45, synthetic black iron oxide, FD&C Blue No. 2 aluminum lake, FD&C Red No. 40 aluminum lake, FD&C Blue No. 1 aluminum lake, D&C Yellow No. 10 aluminum lake, n-butyl alcohol, propylene glycol, SDA-3A alcohol, and titanium dioxide. It is formulated for oral use.
-
CLINICAL PHARMACOLOGY
General
Pentosan polysulfate sodium is a low molecular weight heparin-like compound. It has anticoagulant and fibrinolytic effects. The mechanism of action of pentosan polysulfate sodium in interstitial cystitis is not known.
Pharmacokinetics
Absorption
In a clinical pharmacology study in which healthy female volunteers received a single oral 300 or 450 mg dose of pentosan polysulfate sodium containing radiolabeled drug as a solution under fasted conditions, maximal levels of plasma radioactivity were seen approximately at a median of 2 hours (range 0.6-120 hours) after dosing. Based on urinary excretion of radioactivity, a mean of approximately 6% of a radiolabeled oral dose of pentosan polysulfate sodium is absorbed and reaches the systemic circulation.
Distribution
Preclinical studies with parenterally administered radiolabeled pentosan polysulfate sodium showed distribution to the uroepithelium of the genitourinary tract with lesser amounts found in the liver, spleen, lung, skin, periosteum, and bone marrow. Erythrocyte penetration is low in animals.
Metabolism
The fraction of pentosan polysulfate sodium that is absorbed is metabolized by partial desulfation in the liver and spleen, and by partial depolymerization in the kidney to a large number of metabolites. Both the desulfation and depolymerization can be saturated with continued dosing.
Excretion
Following administration of an oral solution of a 300 or 450 mg dose of pentosan polysulfate sodium containing radiolabeled drug to groups of healthy subjects, plasma radioactivity declined with mean half-lives of 27 and 20 hours, respectively. A large proportion of the orally administered dose of pentosan polysulfate sodium (mean 84% in the 300 mg group and 58% in the 450 mg group) is excreted in feces as unchanged drug. A mean of 6% of an oral dose is excreted in the urine, mostly as desulfated and depolymerized metabolites. Only a small fraction of the administered dose (mean 0.14%) is recovered as intact drug in urine.
Special Populations
The pharmacokinetics of pentosan polysulfate sodium has not been studied in geriatric patients or in patients with hepatic or renal impairment. See also PRECAUTIONS-Hepatic Insufficiency.
Drug-Drug Interactions
In a study in which healthy subjects received pentosan polysulfate sodium 100 mg capsule or placebo every 8 hours for 7 days, and were titrated with warfarin to an INR of 1.4 to 1.8, the pharmacokinetic parameters of R-warfarin and S-warfarin were similar in the absence and presence of pentosan polysulfate sodium. INR for warfarin + placebo and warfarin + pentosan polysulfate sodium were comparable. See also PRECAUTIONS on the use of ELMIRON ® in patients receiving other therapies with anticoagulant effects.
Pharmacodynamics
The mechanism by which pentosan polysulfate sodium achieves its effects in patients is unknown. In preliminary clinical models, pentosan polysulfate sodium adhered to the bladder wall mucosal membrane. The drug may act as a buffer to control cell permeability preventing irritating solutes in the urine from reaching the cells.
-
CLINICAL TRIALS
ELMIRON ® was evaluated in two clinical trials for the relief of pain in patients with chronic interstitial cystitis (IC). All patients met the NIH definition of IC based upon the results of cystoscopy, cytology, and biopsy. One blinded, randomized, placebo-controlled study evaluated 151 patients (145 women, 5 men, 1 unknown) with a mean age of 44 years (range 18 to 81). Approximately equal numbers of patients received either placebo or ELMIRON ® 100 mg three times a day for 3 months. Clinical improvement in bladder pain was based upon the patient's own assessment. In this study, 28/74 (38%) of patients who received ELMIRON ® and 13/74 (18%) of patients who received placebo showed greater than 50% improvement in bladder pain (p = 0.005).
A second clinical trial, the physician's usage study, was a prospectively designed retrospective analysis of 2499 patients who received ELMIRON ® 300 mg a day without blinding. Of the 2499 patients, 2220 were women, 254 were men, and 25 were of unknown sex. The patients had a mean age of 47 years and 23% were over 60 years of age. By 3 months, 1307 (52%) of the patients had dropped out or were ineligible for analysis, overall, 1192 (48%) received ELMIRON ® for 3 months; 892 (36%) received ELMIRON ® for 6 months; and 598 (24%) received ELMIRON ® for one year.
Patients had unblinded evaluations every 3 months for the patient's rating of overall change in pain in comparison to baseline and for the difference calculated in "pain/discomfort" scores. At baseline, pain/discomfort scores for the original 2499 patients were severe or unbearable in 60%, moderate in 33% and mild or none in 7% of patients. The extent of the patients' pain improvement is shown in Table 1.
At 3 months, 722/2499 (29%) of the patients originally in the study had pain scores that improved by one or two categories. By 6 months, in the 892 patients who continued taking ELMIRON ®, an additional 116/2499 (5%) of patients had improved pain scores. After 6 months, the percent of patients who reported the first onset of pain relief was less than 1.5% of patients who originally entered in the study (see Table 2).
Table 1: Pain Scores in Reference to Baseline in Open Label Physician's Usage Study (N=2499) * Efficacy Parameter 3 months † 6 months † Patient Rating of Overall Change in Pain (Recollection of difference between current pain and baseline pain) ‡ N=1161
Median = 3
Mean = 3.44
CI: (3.37, 3.51)N=724
Median = 4
Mean = 3.91
CI: (3.83, 3.99)Change in Pain/Discomfort Score (Calculated difference in scores at the time point and baseline) § N=1440
Median = 1
Mean = 0.51
CI: (0.45, 0.57)N=904
Median = 1
Mean = 0.66
CI: (0.61, 0.71)Table 2: Number (%) of Patients with New Relief of Pain/Discomfort * in the Open-Label Physician's Usage Study (N=2499) at 3 months †
(n=1192)at 6 months ‡
(n=892)Considering only the patients who continued treatment 722/1192 (61%) 116/892 (13%) Considering all the patients originally enrolled in the study 722/2499 (29%) 116/2499 (5%) - INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Retinal Pigmentary Changes
Pigmentary changes in the retina, reported in the literature as pigmentary maculopathy, have been identified with long-term use of ELMIRON ® (see ADVERSE REACTIONS). Although most of these cases occurred after 3 years of use or longer, cases have been seen with a shorter duration of use. While the etiology is unclear, cumulative dose appears to be a risk factor. Visual symptoms in the reported cases included difficulty reading, slow adjustment to low or reduced light environments, and blurred vision. The visual consequences of these pigmentary changes are not fully characterized. Caution should be used in patients with retinal pigment changes from other causes in which examination findings may confound the appropriate diagnosis, follow-up, and treatment. Detailed ophthalmologic history should be obtained in all patients prior to starting treatment with ELMIRON ®. If there is a family history of hereditary pattern dystrophy, genetic testing should be considered. For patients with pre-existing ophthalmologic conditions, a comprehensive baseline retinal examination (including color fundoscopic photography, ocular coherence tomography (OCT), and auto-fluorescence imaging) is recommended prior to starting therapy. A baseline retinal examination (including OCT and auto-fluorescence imaging) is suggested for all patients within six months of initiating treatment and periodically while continuing treatment. If pigmentary changes in the retina develop, then risks and benefits of continuing treatment should be re-evaluated, since these changes may be irreversible. Follow-up retinal examinations should be continued given that retinal and vision changes may progress even after cessation of treatment.
-
PRECAUTIONS
General
ELMIRON ® is a weak anticoagulant (1/15 the activity of heparin). At a daily dose of 300 mg (n=128), rectal hemorrhage was reported as an adverse event in 6.3% of patients. Bleeding complications of ecchymosis, epistaxis, and gum hemorrhage have been reported (see ADVERSE REACTIONS). Patients undergoing invasive procedures or having signs/symptoms of underlying coagulopathy or other increased risk of bleeding (due to other therapies such as coumarin anticoagulants, heparin, t-PA, streptokinase, high dose aspirin, or nonsteroidal anti-inflammatory drugs) should be evaluated for hemorrhage. Patients with diseases such as aneurysms, thrombocytopenia, hemophilia, gastrointestinal ulcerations, polyps, or diverticula should be carefully evaluated before starting ELMIRON ®.
A similar product that was given subcutaneously, sublingually, or intramuscularly (and not initially metabolized by the liver) is associated with delayed immunoallergic thrombocytopenia with symptoms of thrombosis and hemorrhage. Caution should be exercised when using ELMIRON ® in patients who have a history of heparin induced thrombocytopenia.
Alopecia is associated with pentosan polysulfate and with heparin products. In clinical trials of ELMIRON ®, alopecia began within the first 4 weeks of treatment. Ninety-seven percent (97%) of the cases of alopecia reported were alopecia areata, limited to a single area on the scalp.
Hepatic Insufficiency
ELMIRON ® has not been studied in patients with hepatic insufficiency. Because there is evidence of hepatic contribution to the elimination of ELMIRON ®, hepatic impairment may have an impact on the pharmacokinetics of ELMIRON ®. Caution should be exercised when using ELMIRON ® in this patient population.
Mildly (< 2.5 × normal) elevated transaminase, alkaline phosphatase, γ-glutamyl transpeptidase, and lactic dehydrogenase occurred in 1.2% of patients. The increases usually appeared 3 to 12 months after the start of ELMIRON ® therapy, and were not associated with jaundice or other clinical signs or symptoms. These abnormalities are usually transient, may remain essentially unchanged, or may rarely progress with continued use. Increases in PTT and PT (< 1% for both) or thrombocytopenia (0.2%) were noted.
Information for Patients
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Patients should take the drug as prescribed, in the dosage prescribed, and no more frequently than prescribed.
Patients should be informed that changes in vision should be reported and evaluated. Retinal examinations including optical coherence tomography (OCT) and auto-fluorescence imaging are suggested for all patients within six months of starting ELMIRON ® and periodically during long-term treatment (see WARNINGS).
Patients should be reminded that ELMIRON ® has a weak anticoagulant effect. This effect may increase bleeding times.
Laboratory Test Findings
Pentosan polysulfate sodium did not affect prothrombin time (PT) or partial thromboplastin time (PTT) up to 1200 mg per day in 24 healthy male subjects treated for 8 days. Pentosan polysulfate sodium also inhibits the generation of factor Xa in plasma and inhibits thrombin-induced platelet aggregation in human platelet rich plasma ex vivo. (See PRECAUTIONS-Hepatic Insufficiency Section for additional information.)
Carcinogenicity, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies of ELMIRON ® in F344/N rats and B6C3F1 mice have been conducted. In these studies, ELMIRON ® was orally administered once daily via gavage, 5 days per week, for up to 2 years. The dosages administered to mice were 56, 168 or 504 mg/kg. The dosages administered to rats were 14, 42, or 126 mg/kg for males, and 28, 84, or 252 mg/kg for females. The dosages tested were up to 60 times the maximum recommended human dose (MRHD) in rats, and up to 117 times the MRHD in mice, on a mg/kg basis. The results of these studies in rodents showed no clear evidence of drug-related tumorigenesis or carcinogenic risk.
Pentosan polysulfate sodium was not clastogenic or mutagenic when tested in the mouse micronucleus test or the Ames test ( S. typhimurium). The effect of pentosan polysulfate sodium on spermatogenesis has not been investigated.
Pregnancy
Reproduction studies have been performed in mice and rats with intravenous daily doses of 15 mg/kg, and in rabbits with 7.5 mg/kg. These doses are 0.42 and 0.14 times the daily oral human doses of ELMIRON ® when normalized to body surface area. These studies did not reveal evidence of impaired fertility or harm to the fetus from ELMIRON ®. Direct in vitro bathing of cultured mouse embryos with pentosan polysulfate sodium (PPS) at a concentration of 1 mg/mL may cause reversible limb bud abnormalities. Adequate and well-controlled studies have not been performed in pregnant women. Because animal studies are not always predictive of human response, this drug should be used in pregnancy only if clearly needed.
-
ADVERSE REACTIONS
ELMIRON ® was evaluated in clinical trials in a total of 2627 patients (2343 women, 262 men, 22 unknown) with a mean age of 47 [range 18 to 88 with 581 (22%) over 60 years of age]. Of the 2627 patients, 128 patients were in a 3-month trial and the remaining 2499 patients were in a long-term, unblinded trial.
Deaths occurred in 6/2627 (0.2%) patients who received the drug over a period of 3 to 75 months. The deaths appear to be related to other concurrent illnesses or procedures, except in one patient for whom the cause was not known.
Serious adverse events occurred in 33/2627 (1.3%) patients. Two patients had severe abdominal pain or diarrhea and dehydration that required hospitalization. Because there was not a control group of patients with interstitial cystitis who were concurrently evaluated, it is difficult to determine which events are associated with ELMIRON ® and which events are associated with concurrent illness, medicine, or other factors.
Adverse Experience in Placebo-Controlled Clinical Trials of ELMIRON ® 100 mg Three Times a Day for 3 Months Body System/Adverse Experience ELMIRON ®
n=128Placebo
n=130- *
- Within a body system, the individual events do not sum to equal overall number of patients because a patient may have more than one event.
CNS Overall Number of Patients * 3 5 Insomnia 1 0 Headache 1 3 Severe Emotional Lability/Depression 2 1 Nystagmus/Dizziness 1 1 Hyperkinesia 1 1 GI Overall Number of Patients * 7 7 Nausea 3 3 Diarrhea 3 6 Dyspepsia 1 0 Jaundice 0 1 Vomiting 0 2 Skin/Allergic Overall Number of Patients * 2 4 Rash 0 2 Pruritus 0 2 Lacrimation 1 1 Rhinitis 1 1 Increased Sweating 1 0 Other Overall Number of Patients * 1 3 Amenorrhea 0 1 Arthralgia 0 1 Vaginitis 1 1 Total Events 17 27 Total Number of Patients Reporting Adverse Events 13 19 The adverse events described below were reported in an unblinded clinical trial of 2499 interstitial cystitis patients treated with ELMIRON ®. Of the original 2499 patients, 1192 (48%) received ELMIRON ® for 3 months; 892 (36%) received ELMIRON ® for 6 months; and 598 (24%) received ELMIRON ® for one year, 355 (14%) received ELMIRON ® for 2 years, and 145 (6%) for 4 years.
Frequency (1 to 4%): Alopecia (4%), diarrhea (4%), nausea (4%), headache (3%), rash (3%), dyspepsia (2%), abdominal pain (2%), liver function abnormalities (1%), dizziness (1%).
Frequency (≤ 1%):
Digestive: Vomiting, mouth ulcer, colitis, esophagitis, gastritis, flatulence, constipation, anorexia, gum hemorrhage.
Hematologic: Anemia, ecchymosis, increased prothrombin time, increased partial thromboplastin time, leukopenia, thrombocytopenia.
Hypersensitive Reactions: Allergic reaction, photosensitivity.
Respiratory System: Pharyngitis, rhinitis, epistaxis, dyspnea.
Skin and Appendages: Pruritus, urticaria.
Special Senses: Conjunctivitis, tinnitus, optic neuritis, amblyopia, retinal hemorrhage.
Post-Marketing Experience
The following adverse reactions have been identified during post approval use of pentosan polysulfate sodium; because these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- pigmentary changes in the retina (see WARNINGS).
Rectal Hemorrhage
ELMIRON ® was evaluated in a randomized, double-blind, parallel group, Phase 4 study conducted in 380 patients with interstitial cystitis dosed for 32 weeks. At a daily dose of 300 mg (n=128), rectal hemorrhage was reported as an adverse event in 6.3% of patients. The severity of the events was described as "mild" in most patients. Patients in that study who were administered ELMIRON ® 900 mg daily, a dose higher than the approved dose, experienced a higher incidence of rectal hemorrhage, 15%.
Liver Function Abnormality
A randomized, double-blind, parallel group, Phase 2 study was conducted in 100 men (51 ELMIRON ® and 49 placebo) dosed for 16 weeks. At a daily dose of 900 mg, a dose higher than the approved dose, elevated liver function tests were reported as an adverse event in 11.8% (n=6) of ELMIRON ®-treated patients and 2% (n=1) of placebo-treated patients.
-
OVERDOSAGE
Overdose has not been reported. Based upon the pharmacodynamics of the drug, toxicity is likely to be reflected as anticoagulation, bleeding, thrombocytopenia, liver function abnormalities, and gastric distress. (See CLINICAL PHARMACOLOGY and PRECAUTIONS sections.) At a daily dose of 900 mg for 32 weeks (n=127) in a clinical trial, rectal hemorrhage was reported as an adverse event in 15% of patients. At a daily dose of ELMIRON ® 900 mg for 16 weeks in a clinical trial that enrolled 51 patients in the ELMIRON ® group and 49 in the placebo group, elevated liver function tests were reported as an adverse event in 11.8% of patients in the ELMIRON ® group and 2% of patients in the placebo group. In the event of acute overdosage, the patient should be given gastric lavage if possible, carefully observed and given symptomatic and supportive treatment.
-
DOSAGE AND ADMINISTRATION
The recommended dose of ELMIRON ® is 300 mg/day taken as one 100 mg capsule orally three times daily. The capsules should be taken with water at least 1 hour before meals or 2 hours after meals.
Patients receiving ELMIRON ® should be reassessed after 3 months. If improvement has not occurred and if limiting adverse events are not present, ELMIRON ® may be continued for another 3 months.
The clinical value and risks of continued treatment in patients whose pain has not improved by 6 months is not known.
- HOW SUPPLIED
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: 03/2021 MEDICATION GUIDE
ELMIRON ® (EL ma ron)
(pentosan polysulfate sodium)
capsules, for oral useWhat is the most important information I should know about ELMIRON?
Serious side effects have been reported with the use of ELMIRON, including:- Changes in the retina of the eye (pigmentary maculopathy). Taking ELMIRON may be associated with pigment changes in the retina of the eye that may continue even after stopping treatment with ELMIRON. Tell your healthcare provider including your eye doctor right away if you have any vision changes including any of these symptoms:
- difficulty reading
- your vision takes longer to adjust to low or reduced light
- blurred vision
Throughout your treatment, regular eye examinations that include retinal examinations are suggested for early detection of retinal/macular changes. Your doctor will discuss with you when to get your first eye examination and follow up exams, and whether the treatment should be continued. - Increased bleeding. ELMIRON may increase bleeding. Tell your healthcare provider right away if you have any of these symptoms:
- bruising easily
- nosebleeds
- bleeding gums
- blood in your stool
Your risk of bleeding may be increased if you take ELMIRON along with other medicines such as: - warfarin sodium
- heparin
- high doses of aspirin
- anti-inflammatory medicines such as ibuprofen
Tell your healthcare provider if you are taking any of these medicines.
Before you start taking ELMIRON, tell your healthcare provider if you are going to have surgery. Your healthcare provider may stop ELMIRON before you have surgery. Talk to your healthcare provider about when to stop taking ELMIRON and when to start taking it again.What is ELMIRON? - ELMIRON is a prescription medicine used to treat bladder pain or discomfort associated with interstitial cystitis.
- It is not known if ELMIRON is safe and effective in children under 16 years of age.
Do not take ELMIRON if you: - are allergic to pentosan polysulfate sodium or any of the ingredients in ELMIRON. See the end of this Medication Guide for a complete list of ingredients in ELMIRON.
Before your take ELMIRON, tell your healthcare provider about all of your medical conditions, including if you: - have a personal or family history of eye problems of the retina.
- have a history of aneurysms.
- have problems with easy bleeding (thrombocytopenia).
- have hemophilia.
- have gastrointestinal problems such as ulcerations, polyps, or diverticula.
- have liver problems
- are pregnant or plan to become pregnant. ELMIRON should be used during pregnancy only if clearly needed. Tell your healthcare provider if you become pregnant while taking ELMIRON. You and your healthcare provider should decide if you should continue to take ELMIRON.
- are breastfeeding or plan to breastfeed. It is not known if ELMIRON passes into your breastmilk. You and your healthcare provider should decide if you will take ELMIRON or breastfeed.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take ELMIRON? - Take ELMIRON exactly as your healthcare provider tells you to take it.
- Take 1 capsule of ELMIRON by mouth 3 times a day with water at least 1 hour before meals or 2 hours after meals. Each capsule contains 100 mg of ELMIRON.
- If you take too much ELMIRON, call your healthcare provider right away or go to the nearest emergency room.
What are the possible side effects of ELMIRON?
Serious side effects have been reported with the use of ELMIRON, including:
Changes in the retina of the eye
Increased bleeding
(See " What is the most important information I should know about ELMIRON?")
The most common side effects of ELMIRON are:- hair loss
- diarrhea
- nausea
- stomach pain
- upset stomach
- headache
- rash
- abnormal liver function tests
- dizziness
These are not all of the possible side effects of ELMIRON.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store ELMIRON? - Store ELMIRON at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep ELMIRON and all medicines out of the reach of children.
General information about the safe and effective use of ELMIRON.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ELMIRON for a condition for which it was not prescribed. Do not give ELMIRON to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ELMIRON that is written for health professionals.What are the ingredients in ELMIRON?
Active ingredient: pentosan polysulfate sodium
Inactive ingredients: microcrystalline cellulose, magnesium stearate, gelatin, pharmaceutical glaze (modified) in SD-45, synthetic black iron oxide, FD&C Blue No. 2 aluminum lake, FD&C Red No. 40 aluminum lake, FD&C Blue No. 1 aluminum lake, D&C Yellow No. 10 aluminum lake, n-butyl alcohol, propylene glycol, SDA-3A alcohol, and titanium dioxide.ELMIRON is a registered trademark of Teva Branded Pharmaceutical Products R&D Inc., used under license.
© 2002, 2021 Janssen Pharmaceutical Companies
Product of Germany
Manufactured for:
Janssen Pharmaceuticals, Inc.
Titusville, New Jersey 08560
For more information, go to www.ORTHOELMIRON.com or call 1-800-526-7736. - PRINCIPAL DISPLAY PANEL - 100 mg Capsule Bottle Label
-
INGREDIENTS AND APPEARANCE
ELMIRON
pentosan polysulfate sodium capsule, gelatin coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50458-098 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PENTOSAN POLYSULFATE SODIUM (UNII: 914032762Y) (PENTOSAN POLYSULFATE - UNII:F59P8B75R4) PENTOSAN POLYSULFATE SODIUM 100 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) ALUMINUM OXIDE (UNII: LMI26O6933) FD&C RED NO. 40 (UNII: WZB9127XOA) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white (WHITE OPAQUE) Score no score Shape CAPSULE Size 18mm Flavor Imprint Code BNP7600 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50458-098-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/26/1996 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020193 09/26/1996 Labeler - Janssen Pharmaceuticals, Inc. (063137772) Establishment Name Address ID/FEI Business Operations Janssen Ortho, LLC 805887986 analysis(50458-098) , manufacture(50458-098) Establishment Name Address ID/FEI Business Operations bene pharmaChem GmbH & Co. KG 332479380 api manufacture(50458-098)
