Label: DECITABINE injection, powder, lyophilized, for solution

- NDC Code(s): 25021-231-20
- Packager: Sagent Pharmaceuticals
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated June 14, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DECITABINE FOR INJECTION safely and effectively. See full prescribing information for DECITABINE FOR INJECTION.
DECITABINE for injection, for intravenous use
Initial U.S. Approval: 2006INDICATIONS AND USAGE
Decitabine for Injection is a nucleoside metabolic inhibitor indicated for treatment of adult patients with myelodysplastic syndromes (MDS) including previously treated and untreated, de novo and secondary MDS of all French-American-British subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia) and intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups. (1)
DOSAGE AND ADMINISTRATION
-
Three Day Regimen
Administer decitabine for injection at a dose of 15 mg/m2 by continuous intravenous infusion over 3 hours repeated every 8 hours for 3 days. Repeat cycle every 6 weeks. (2.1) -
Five Day Regimen
Administer decitabine for injection at a dose of 20 mg/m2 by continuous intravenous infusion over 1 hour repeated daily for 5 days. Repeat cycle every 4 weeks. (2.1)
DOSAGE FORMS AND STRENGTHS
For Injection: 50 mg of decitabine as a lyophilized powder in a single-dose vial for reconstitution. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Most common adverse reactions (> 50%) are neutropenia, thrombocytopenia, anemia, and pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Sagent Pharmaceuticals at 1-866-625-1618 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2023
-
Three Day Regimen
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modifications for Adverse Reactions
2.3 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
5.2 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis and Impairment of Fertility
14 CLINICAL STUDIES
14.1 Controlled Trial in Myelodysplastic Syndrome
14.2 Single-arm Studies in Myelodysplastic Syndrome
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Decitabine for Injection is indicated for treatment of adult patients with myelodysplastic syndromes (MDS) including previously treated and untreated, de novo and secondary MDS of all French-American-British subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia) and intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Pre-Medications and Baseline Testing
- Consider pre-medicating for nausea with antiemetics.
- Conduct baseline laboratory testing: complete blood count (CBC) with platelets, serum hepatic panel, and serum creatinine.
Decitabine for Injection Regimen Options
Three Day Regimen
Administer decitabine for Injection at a dose of 15 mg/m2 by continuous intravenous infusion over 3 hours repeated every 8 hours for 3 days. Repeat cycles every 6 weeks upon hematologic recovery (ANC at least 1,000/mcL and platelets at least 50,000/mcL) for a minimum of 4 cycles. A complete or partial response may take longer than 4 cycles. Delay and reduce dose for hematologic toxicity [see Dosage and Administration (2.2)].
Five Day Regimen
Administer decitabine for Injection at a dose of 20 mg/m2 by continuous intravenous infusion over 1 hour daily for 5 days. Delay and reduce dose for hematologic toxicity [see Dosage and Administration (2.2)]. Repeat cycles every 4 weeks upon hematologic recovery (ANC at least 1,000/mcL and platelets at least 50,000/mcL) for a minimum of 4 cycles. A complete or partial response may take longer than 4 cycles.
Patients with Renal or Severe Hepatic Impairment
Treatment with decitabine for injection has not been studied in patients with pre-existing renal or hepatic impairment. For patients with pre-existing renal or hepatic impairment, consider the potential risks and benefits before initiating treatment with decitabine for injection.
2.2 Dosage Modifications for Adverse Reactions
Hematologic Toxicity
If hematologic recovery from a previous decitabine for Injection treatment cycle requires more than 6 weeks, delay the next cycle of decitabine for Injection therapy and reduce decitabine for Injection dose temporarily by following this algorithm:
- Recovery requiring more than 6, but less than 8 weeks: delay decitabine for Injection dosing for up to 2 weeks and reduce the dose temporarily to 11 mg/m2 every 8 hours (33 mg/m2/day, 99 mg/m2/cycle) upon restarting therapy.
- Recovery requiring more than 8, but less than 10 weeks: Perform bone marrow aspirate to assess for disease progression. In the absence of progression, delay the decitabine for Injection dosing for up to 2 more weeks and reduce the dose to 11 mg/m2 every 8 hours (33 mg/m2/day, 99 mg/m2/cycle) upon restarting therapy, then maintain or increase dose in subsequent cycles as clinically indicated.
Non-hematologic Toxicity
Delay subsequent decitabine for injection treatment for any the following nonhematologic toxicities and do not restart until toxicities resolve:
- Serum creatinine greater than or equal to 2 mg/dL
- Alanine transaminase (ALT), total bilirubin greater than or equal to 2 times upper limit of normal (ULN)
- Active or uncontrolled infection
2.3 Preparation and Administration
Decitabine is a cytotoxic drug. Follow special handling and disposal procedures.1
Aseptically reconstitute decitabine for injection with room temperature (20° to 25°C) 10 mL of Sterile Water for Injection, USP. Upon reconstitution, the final concentration of the reconstituted decitabine for injection solution is 5 mg per mL. You must dilute the reconstituted solution with 0.9% Sodium Chloride Injection or 5% Dextrose Injection prior to administration. Temperature of the diluent (0.9% Sodium Chloride Injection or 5% Dextrose Injection) depends on time of administration after preparation.
For Administration Within 15 Minutes of Preparation
If decitabine for injection is intended to be administered within 15 minutes from the time of preparation, dilute the reconstituted solution with room temperature (20° to 25°C) 0.9% Sodium Chloride Injection or 5% Dextrose Injection to a final concentration of 0.1 mg per mL to 1 mg per mL. Discard unused portion.
For Delayed Administration
If decitabine for injection is intended to be administered after 15 minutes of preparation, dilute the reconstituted solution with cold (2° to 8°C) 0.9% Sodium Chloride Injection or 5% Dextrose Injection to a final concentration of 0.1 mg per mL to 1 mg per mL. Store at 2° to 8°C for up to 4 hours. Diluted stored solution must be used within 4 hours from the time of preparation. Discard unused portion.
Use the diluted, refrigerated solution within 4 hours from the time of preparation or discard.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if there is evidence of particulate matter or discoloration.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Fatal and serious myelosuppression occurs in decitabine-treated patients. Myelosuppression (anemia, neutropenia, and thrombocytopenia) is the most frequent cause of decitabine dose reduction, delay, and discontinuation. Neutropenia of any grade occurred in 90% of decitabine-treated patients with grade 3 or 4 occurring in 87% of patients. Grade 3 or 4 febrile neutropenia occurred in 23% of patients. Thrombocytopenia of any grade occurred in 89% of patients with grade 3 or 4 occurring in 85% of patients. Anemia of any grade occurred in 82% of patients. Perform complete blood count with platelets at baseline, prior to each cycle, and as needed to monitor response and toxicity. Manage toxicity using dose-delay, dose-reduction, growth factors, and anti-infective therapies as needed [see Dosage and Administration (2.2)]. Myelosuppression and worsening neutropenia may occur more frequently in the first or second treatment cycles and may not necessarily indicate progression of underlying MDS.
5.2 Embryo-Fetal Toxicity
Based on findings from human data, animal studies and its mechanism of action, decitabine can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)]. In preclinical studies in mice and rats, decitabine caused adverse developmental outcomes including embryo-fetal lethality and malformations. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception while receiving decitabine, and for 6 months following the last dose. Advise males with female partners of reproductive potential to use effective contraception while receiving treatment with decitabine and for 3 months following the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Myelosuppression [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of decitabine was studied in 3 single-arm studies (N = 66, N = 98, N = 99) and 1 controlled supportive care study (N = 83 Decitabine, N = 81 supportive care). The data described below reflect exposure to decitabine in 83 patients in the MDS trial. In the trial, patients received 15 mg/m2 intravenously every 8 hours for 3 days every 6 weeks. The median number of decitabine cycles was 3 (range 0 to 9).
Most Common Adverse Reactions: neutropenia, thrombocytopenia, anemia, fatigue, pyrexia, nausea, cough, petechiae, constipation, diarrhea, and hyperglycemia.
Adverse Reactions Most Frequently (≥ 1%) Resulting in Clinical Intervention and or Dose Modification in the Controlled Supportive Care Study in the Decitabine Arm:
- Discontinuation: thrombocytopenia, neutropenia, pneumonia, Mycobacterium avium complex infection, cardio-respiratory arrest, increased blood bilirubin, intracranial hemorrhage, abnormal liver function tests.
- Dose Delayed: neutropenia, pulmonary edema, atrial fibrillation, central line infection, febrile neutropenia.
- Dose Reduced: neutropenia, thrombocytopenia, anemia, lethargy, edema, tachycardia, depression, pharyngitis.
Table 1 presents all adverse reactions occurring in at least 5% of patients in the decitabine group and at a rate greater than supportive care.
Table 1 Adverse Reactions Reported in ≥ 5% of Patients in the Decitabine Group and at a Rate Greater than Supportive Care in the Controlled Trial in MDS Decitabine
N = 83 (%)Supportive Care
N = 81 (%)Blood and lymphatic system disorders Neutropenia 75 (90) 58 (72) Thrombocytopenia 74 (89) 64 (79) Anemia NOS 68 (82) 60 (74) Febrile neutropenia 24 (29) 5 (6) Leukopenia NOS 23 (28) 11 (14) Lymphadenopathy 10 (12) 6 (7) Thrombocythemia 4 (5) 1 (1) Cardiac disorders Pulmonary edema NOS 5 (6) 0 (0) Eye disorders Vision blurred 5 (6) 0 (0) Gastrointestinal disorders Nausea 35 (42) 13 (16) Constipation 29 (35) 11 (14) Diarrhea NOS 28 (34) 13 (16) Vomiting NOS 21 (25) 7 (9) Abdominal pain NOS 12 (14) 5 (6) Oral mucosal petechiae 11 (13) 4 (5) Stomatitis 10 (12) 5 (6) Dyspepsia 10 (12) 1 (1) Ascites 8 (10) 2 (2) Gingival bleeding 7 (8) 5 (6) Hemorrhoids 7 (8) 3 (4) Loose stools 6 (7) 3 (4) Tongue ulceration 6 (7) 2 (2) Dysphagia 5 (6) 2 (2) Oral soft tissue disorder NOS 5 (6) 1 (1) Lip ulceration 4 (5) 3 (4) Abdominal distension 4 (5) 1 (1) Abdominal pain upper 4 (5) 1 (1) Gastro-esophageal reflux disease 4 (5) 0 (0) Glossodynia 4 (5) 0 (0) General disorders and administrative site disorders Pyrexia 44 (53) 23 (28) Edema peripheral 21 (25) 13 (16) Rigors 18 (22) 14 (17) Edema NOS 15 (18) 5 (6) Pain NOS 11 (13) 5 (6) Lethargy 10 (12) 3 (4) Tenderness NOS 9 (11) 0 (0) Fall 7 (8) 3 (4) Chest discomfort 6 (7) 3 (4) Intermittent pyrexia 5 (6) 3 (4) Malaise 4 (5) 1 (1) Crepitations NOS 4 (5) 1 (1) Catheter site erythema 4 (5) 1 (1) Catheter site pain 4 (5) 0 (0) Injection site swelling 4 (5) 0 (0) Hepatobiliary disorders Hyperbilirubinemia 12 (14) 4 (5) Infections and infestations Pneumonia NOS 18 (22) 11 (14) Cellulitis 10 (12) 6 (7) Candidal infection NOS 8 (10) 1 (1) Catheter related infection 7 (8) 0 (0) Urinary tract infection NOS 6 (7) 1 (1) Staphylococcal infection 6 (7) 0 (0) Oral candidiasis 5 (6) 2 (2) Sinusitis NOS 4 (5) 2 (2) Bacteremia 4 (5) 0 (0) Injury, poisoning and procedural complications Transfusion reaction 6 (7) 3 (4) Abrasion NOS 4 (5) 1 (1) Investigations Cardiac murmur NOS 13 (16) 9 (11) Blood alkaline phosphatase NOS increased 9 (11) 7 (9) Aspartate aminotransferase increased 8 (10) 7 (9) Blood urea increased 8 (10) 1 (1) Blood lactate dehydrogenase increased 7 (8) 5 (6) Blood albumin decreased 6 (7) 0 (0) Blood bicarbonate increased 5 (6) 1 (1) Blood chloride decreased 5 (6) 1 (1) Protein total decreased 4 (5) 3 (4) Blood bicarbonate decreased 4 (5) 1 (1) Blood bilirubin decreased 4 (5) 1 (1) Metabolism and nutrition disorders Hyperglycemia NOS 27 (33) 16 (20) Hypoalbuminemia 20 (24) 14 (17) Hypomagnesemia 20 (24) 6 (7) Hypokalemia 18 (22) 10 (12) Hyponatremia 16 (19) 13 (16) Appetite decreased NOS 13 (16) 12 (15) Anorexia 13 (16) 8 (10) Hyperkalemia 11 (13) 3 (4) Dehydration 5 (6) 4 (5) Musculoskeletal and connective tissue disorders Arthralgia 17 (20) 8 (10) Pain in limb 16 (19) 8 (10) Back pain 14 (17) 5 (6) Chest wall pain 6 (7) 1 (1) Musculoskeletal discomfort 5 (6) 0 (0) Myalgia 4 (5) 1 (1) Nervous system disorders Headache 23 (28) 11 (14) Dizziness 15 (18) 10 (12) Hypoesthesia 9 (11) 1 (1) Psychiatric disorders Insomnia 23 (28) 11 (14) Confusional state 10 (12) 3 (4) Anxiety 9 (11) 8 (10) Renal and urinary disorders Dysuria 5 (6) 3 (4) Urinary frequency 4 (5) 1 (1) Respiratory, thoracic and Mediastinal disorders Cough 33 (40) 25 (31) Pharyngitis 13 (16) 6 (7) Crackles lung 12 (14) 1 (1) Breath sounds decreased 8 (10) 7 (9) Hypoxia 8 (10) 4 (5) Rales 7 (8) 2 (2) Postnasal drip 4 (5) 2 (2) Skin and subcutaneous tissue disorders Ecchymosis 18 (22) 12 (15) Rash NOS 16 (19) 7 (9) Erythema 12 (14) 5 (6) Skin lesion NOS 9 (11) 3 (4) Pruritis 9 (11) 2 (2) Alopecia 7 (8) 1 (1) Urticaria NOS 5 (6) 1 (1) Swelling face 5 (6) 0 (0) Vascular disorders Petechiae 32 (39) 13 (16) Pallor 19 (23) 10 (12) Hypotension NOS 5 (6) 4 (5) Hematoma NOS 4 (5) 3 (4) In a single-arm MDS study (N=99), decitabine was dosed at 20 mg/m2 intravenously, infused over one hour daily, for 5 consecutive days of a 4-week cycle. Table 2 presents all adverse reactions occurring in at least 5% of patients.
Table 2 Adverse Reactions Reported in ≥ 5% of Patients in a Single-arm Study* Decitabine N = 99 (%) * In this single arm study, investigators reported adverse events based on clinical signs and symptoms rather than predefined laboratory abnormalities. Thus, not all laboratory abnormalities were recorded as adverse events.
Blood and lymphatic system disorders Anemia 31 (31) Febrile neutropenia 20 (20) Leukopenia 6 (6) Neutropenia 38 (38) Pancytopenia 5 (5) Thrombocythemia 5 (5) Thrombocytopenia 27 (27) Cardiac disorders Cardiac failure congestive 5 (5) Tachycardia 8 (8) Ear and labyrinth disorders Ear pain 6 (6) Gastrointestinal disorders Abdominal pain 14 (14) Abdominal pain upper 6 (6) Constipation 30 (30) Diarrhea 28 (28) Dyspepsia 10 (10) Dysphagia 5 (5) Gastro-esophageal reflux disease 5 (5) Nausea 40 (40) Oral pain 5 (5) Stomatitis 11 (11) Toothache 6 (6) Vomiting 16 (16) General disorders and administration site conditions Asthenia 15 (15) Chest pain 6 (6) Chills 16 (16) Fatigue 46 (46) Mucosal inflammation 9 (9) Edema 5 (5) Edema peripheral 27 (27) Pain 5 (5) Pyrexia 36 (36) Infections and infestations Cellulitis 9 (9) Oral candidiasis 6 (6) Pneumonia 20 (20) Sinusitis 6 (6) Staphylococcal bacteremia 8 (8) Tooth abscess 5 (5) Upper respiratory tract infection 10 (10) Urinary tract infection 7 (7) Injury, poisoning and procedural complications Contusion 9 (9) Investigations Blood bilirubin increased 6 (6) Breath sounds abnormal 5 (5) Weight decreased 9 (9) Metabolism and nutrition disorders Anorexia 23 (23) Decreased appetite 8 (8) Dehydration 8 (8) Hyperglycemia 6 (6) Hypokalemia 12 (12) Hypomagnesemia 5 (5) Musculoskeletal and connective tissue disorders Arthralgia 17 (17) Back pain 18 (18) Bone pain 6 (6) Muscle spasms 7 (7) Muscular weakness 5 (5) Musculoskeletal pain 5 (5) Myalgia 9 (9) Pain in extremity 18 (18) Nervous system disorders Dizziness 21 (21) Headache 23 (23) Psychiatric disorders Anxiety 9 (9) Confusional state 8 (8) Depression 9 (9) Insomnia 14 (14) Respiratory, thoracic and mediastinal disorders Cough 27 (27) Dyspnea 29 (29) Epistaxis 13 (13) Pharyngolaryngeal pain 8 (8) Pleural effusion 5 (5) Sinus congestion 5 (5) Skin and subcutaneous tissue disorders Dry skin 8 (8) Ecchymosis 9 (9) Erythema 5 (5) Night sweats 5 (5) Petechiae 12 (12) Pruritus 9 (9) Rash 11 (11) Skin lesion 5 (5) Vascular disorders Hypertension 6 (6) Hypotension 11 (11) No overall difference in safety was detected between patients > 65 years of age and younger patients in these MDS trials. No significant differences in safety were detected between males and females. Patients with renal or hepatic dysfunction were not studied. Insufficient numbers of non-White patients were available to draw conclusions in these clinical trials.
Serious adverse reactions that occurred in patients receiving decitabine, not previously reported in Tables 1 and 2 include:
- Allergic Reaction: hypersensitivity (anaphylactic reaction).
- Blood and Lymphatic System Disorders: myelosuppression, splenomegaly.
- Cardiac Disorders: myocardial infarction, cardio-respiratory arrest, cardiomyopathy, atrial fibrillation, supraventricular tachycardia.
- Gastrointestinal Disorders: gingival pain, upper gastrointestinal hemorrhage.
- General Disorders and Administrative Site Conditions: chest pain, catheter site hemorrhage.
- Hepatobiliary Disorders: cholecystitis.
- Infections and Infestations: fungal infection, sepsis, bronchopulmonary aspergillosis, peridiverticular abscess, respiratory tract infection, pseudomonal lung infection, Mycobacterium avium complex infection.
- Injury, Poisoning and Procedural Complications: post procedural pain, post procedural hemorrhage.
- Nervous System Disorders: intracranial hemorrhage.
- Psychiatric Disorders: mental status changes.
- Renal and Urinary Disorders: renal failure, urethral hemorrhage.
- Respiratory, Thoracic and Mediastinal Disorders: hemoptysis, lung infiltration, pulmonary embolism, respiratory arrest, pulmonary mass.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of decitabine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Sweet's syndrome (acute febrile neutrophilic dermatosis)
- Differentiation syndrome
- Interstitial lung disease
-
7 DRUG INTERACTIONS
Drug interaction studies with decitabine have not been conducted. In vitro studies in human liver microsomes suggest that decitabine is unlikely to inhibit or induce cytochrome P450 enzymes. In vitro metabolism studies have suggested that decitabine is not a substrate for human liver cytochrome P450 enzymes. As plasma protein binding of decitabine is negligible (< 1%), interactions due to displacement of more highly protein bound drugs from plasma proteins are not expected.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from human data, animal studies, and the mechanism of action, decitabine can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)]. Limited published data on decitabine use throughout the first trimester during pregnancy describe adverse developmental outcomes including major birth defects (structural abnormalities). In animal reproduction studies, administration of decitabine to pregnant mice and rats during organogenesis caused adverse developmental outcomes including malformations and embryo-fetal lethality starting at doses approximately 7% of the recommended human dose on a mg/m2 basis (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage in the U.S. general population is 2% to 4% and 15% to 20% of clinically recognized pregnancies, respectively.
Data
Human Data
A single published case report of decitabine pregnancy exposure in a 39-year old woman with a hematologic malignancy described multiple structural abnormalities after 6 cycles of therapy in the 18th week of gestation. These abnormalities included holoprosencephaly, absence of nasal bone, mid-facial deformity, cleft lip and palate, polydactyly and rocker-bottom feet. The pregnancy was terminated.
Animal Data
In utero exposure to decitabine causes temporal related defects in the rat and/or mouse, which include growth suppression, exencephaly, defective skull bones, rib/sternabrae defects, phocomelia, digit defects, micrognathia, gastroschisis, micromelia. Decitabine inhibits proliferation and increases apoptosis of neural progenitor cells of the fetal CNS and induces palatal clefting in the developing murine fetus. Studies in mice have also shown that decitabine administration during osteoblastogenesis (day 10 of gestation) induces bone loss in offspring.
In mice exposed to single IP (intraperitoneal) injections (0, 0.9 and 3.0 mg/m2, approximately 2% and 7% of the recommended daily clinical dose, respectively) over gestation days 8, 9, 10 or 11, no maternal toxicity was observed but reduced fetal survival was observed after treatment at 3 mg/m2 and decreased fetal weight was observed at both dose levels. The 3 mg/m2 dose elicited characteristic fetal defects for each treatment day, including supernumerary ribs (both dose levels), fused vertebrae and ribs, cleft palate, vertebral defects, hind-limb defects and digital defects of fore- and hind-limbs.
In rats given a single IP injection of 2.4, 3.6 or 6 mg/m2 (approximately 5%, 8%, or 13% the daily recommended clinical dose, respectively) on gestation days 9 to 12, no maternal toxicity was observed. No live fetuses were seen at any dose when decitabine was injected on gestation day 9. A significant decrease in fetal survival and reduced fetal weight at doses greater than 3.6 mg/m2 was seen when decitabine was given on gestation day 10. Increased incidences of vertebral and rib anomalies were seen at all dose levels, and induction of exophthalmia, exencephaly, and cleft palate were observed at 6.0 mg/m2. Increased incidence of foredigit defects was seen in fetuses at doses greater than 3.6 mg/m2. Reduced size and ossification of long bones of the fore-limb and hind-limb were noted at 6.0 mg/m2.
The effect of decitabine on postnatal development and reproductive capacity was evaluated in mice administered a single 3 mg/m2 IP injection (approximately 7% the recommended daily clinical dose) on day 10 of gestation. Body weights of males and females exposed in utero to decitabine were significantly reduced relative to controls at all postnatal time points. No consistent effect on fertility was seen when female mice exposed in utero were mated to untreated males. Untreated females mated to males exposed in utero showed decreased fertility at 3 and 5 months of age (36% and 0% pregnancy rate, respectively). Follow up studies indicated that treatment of pregnant mice with decitabine on gestation day 10 was associated with a reduced pregnancy rate resulting from effects on sperm production in the F1-generation.
8.2 Lactation
Risk Summary
There are no data on the presence of decitabine or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions from decitabine in a breastfed child, advise women not to breastfeed while receiving decitabine and for at least 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Conduct pregnancy testing of females of reproductive potential prior to initiating decitabine.
Contraception
Females
Decitabine for injection can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception while receiving decitabine and for 6 months following the last dose.
Males
Advise males with female partners of reproductive potential to use effective contraception while receiving treatment with decitabine and for 3 months following the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings of decitabine in animals, male fertility may be compromised by treatment with Decitabine. The reversibility of the effect on fertility is unknown [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of decitabine in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients exposed to decitabine in the controlled clinical trial, 61 of 83 patients were age 65 years and over, while 21 of 83 patients were age 75 years and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
- 10 OVERDOSAGE
-
11 DESCRIPTION
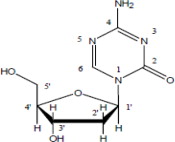
Decitabine is a nucleoside metabolic inhibitor. Decitabine is a fine, white to almost white powder with the molecular formula of C8H12N4O4 and a molecular weight of 228.21 gm/mol. Its chemical name is 4-amino-1-(2-deoxy-β-D-erythro-pentofuranosyl)-1,3,5-triazin-2(1H)-one and it has the following structural formula:
Decitabine is slightly soluble in ethanol/water (50/50), methanol/water (50/50) and methanol; sparingly soluble in water and soluble in dimethylsulfoxide (DMSO).
Decitabine for Injection, for intravenous use is a sterile, white to almost white lyophilized powder supplied in a clear colorless glass single-dose vial. Each 20 mL vial contains 50 mg decitabine, 68 mg monobasic potassium phosphate (potassium dihydrogen phosphate) and 11.6 mg sodium hydroxide. Sodium hydroxide and/or hydrochloric acid are used for pH adjustment.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Decitabine is believed to exert its antineoplastic effects after phosphorylation and direct incorporation into DNA and inhibition of DNA methyltransferase, causing hypomethylation of DNA and cellular differentiation or apoptosis. Decitabine inhibits DNA methylation in vitro, which is achieved at concentrations that do not cause major suppression of DNA synthesis. Decitabine-induced hypomethylation in neoplastic cells may restore normal function to genes that are critical for the control of cellular differentiation and proliferation. In rapidly dividing cells, the cytotoxicity of decitabine may also be attributed to the formation of covalent adducts between DNA methyltransferase and decitabine incorporated into DNA. Non-proliferating cells are relatively insensitive to decitabine.
12.2 Pharmacodynamics
Decitabine has been shown to induce hypomethylation both in vitro and in vivo. However, there have been no studies of decitabine-induced hypomethylation and pharmacokinetic parameters.
12.3 Pharmacokinetics
Pharmacokinetic (PK) parameters were evaluated in patients. Eleven patients received 20 mg/m2 infused over 1 hour intravenously (treatment Option 2). Fourteen patients received 15 mg/m2 infused over 3 hours intravenously (treatment Option 1). PK parameters are shown in Table 3. Plasma concentration-time profiles after discontinuation of infusion showed a biexponential decline. The clearance (CL) of decitabine was higher following treatment Option 2. Upon repeat doses, there was no systemic accumulation of decitabine or any changes in PK parameters. Population PK analysis (N=35) showed that the cumulative AUC per cycle for treatment Option 2 was 2.3-fold lower than the cumulative AUC per cycle following treatment Option 1.
Table 3 Mean (CV% or 95% CI) Pharmacokinetic Parameters of Decitabine * N=14, **N=11, ***N=35 Cumulative AUC per cycle
Dose Cmax (ng/mL) AUC0-INF
(ng·h/mL)T½ (h) CL (L/h/m2) AUCCumulative*** (ng·h/mL) 15 mg/m2 3-hr infusion every 8 hours for 3 days (Option 1)* 73.8 (66) 163
(62)0.62 (49) 125
(53)1332
(1010 to 1730)20 mg/m2 1-hr infusion daily for 5 days (Option 2)** 147
(49)115
(43)0.54 (43) 210
(47)570
(470 to 700)The exact route of elimination and metabolic fate of decitabine is not known in humans. One of the pathways of elimination of decitabine appears to be deamination by cytidine deaminase found principally in the liver but also in granulocytes, intestinal epithelium and whole blood.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis and Impairment of Fertility
Carcinogenicity studies with decitabine have not been conducted.
The mutagenic potential of decitabine was tested in several in vitro and in vivo systems. Decitabine increased mutation frequency in L5178Y mouse lymphoma cells, and mutations were produced in an Escherichia coli lac-I transgene in colonic DNA of decitabine-treated mice. Decitabine caused chromosomal rearrangements in larvae of fruit flies.
In male mice given IP injections of 0.15, 0.3 or 0.45 mg/m2 decitabine (approximately 0.3% to 1% the recommended clinical dose) 3 times a week for 7 weeks, decitabine did not affect survival, body weight gain or hematological measures (hemoglobin and white blood cell counts). Testes weights were reduced, abnormal histology was observed and significant decreases in sperm number were found at doses ≥ 0.3 mg/m2. In females mated to males dosed with ≥ 0.3 mg/m2 decitabine, pregnancy rate was reduced, and preimplantation loss was significantly increased.
-
14 CLINICAL STUDIES
14.1 Controlled Trial in Myelodysplastic Syndrome
A randomized open-label, multicenter, controlled trial evaluated 170 adult patients with myelodysplastic syndromes (MDS) meeting French-American-British (FAB) classification criteria and International Prognostic Scoring System (IPSS) High-Risk, Intermediate-2 and Intermediate-1 prognostic scores. Eighty-nine patients were randomized to decitabine therapy plus supportive care (only 83 received decitabine), and 81 to Supportive Care (SC) alone. Patients with Acute Myeloid Leukemia (AML) were not intended to be included. Of the 170 patients included in the study, independent review (adjudicated diagnosis) found that 12 patients (9 in the decitabine arm and 3 in the SC arm) had the diagnosis of AML at baseline. Baseline demographics and other patient characteristics in the Intent-to-Treat (ITT) population were similar between the 2 groups, as shown in Table 4.
Table 4 Baseline Demographics and Other Patient Characteristics (ITT) Demographic or Other Patient Characteristic Decitabine
N = 89Supportive Care
N = 81Age (years) Mean (±SD) 69±10 67±10 Median (IQR) 70 (65 to 76) 70 (62 to 74) (Range: min-max) (31 to 85) (30 to 82) Sex n (%) Male 59 (66) 57 (70) Female 30 (34) 24 (30) Race n (%) White 83 (93) 76 (94) Black 4 (4) 2 (2) Other 2 (2) 3 (4) Weeks Since MDS Diagnosis Mean (±SD) 86±131 77±119 Median (IQR) 29 (10 to 87) 35 (7 to 98) (Range: min-max) (2 to 667) (2 to 865) Previous MDS Therapy n (%) Yes 27 (30) 19 (23) No 62 (70) 62 (77) RBC Transfusion Status n (%) Independent 23 (26) 27 (33) Dependent 66 (74) 54 (67) Platelet Transfusion Status n (%) Independent 69 (78) 62 (77) Dependent 20 (22) 19 (23) IPSS Classification n (%) Intermediate–1 28 (31) 24 (30) Intermediate–2 38 (43) 36 (44) High Risk 23 (26) 21 (26) FAB Classification n (%) RA 12 (13) 12 (15) RARS 7 (8) 4 (5) RAEB 47 (53) 43 (53) RAEB-t 17 (19) 14 (17) CMML 6 (7) 8 (10) Patients randomized to the decitabine arm received decitabine intravenously infused at a dose of 15 mg/m2 over a 3-hour period, every 8 hours, for 3 consecutive days. This cycle was repeated every 6 weeks, depending on the patient's clinical response and toxicity. Supportive care consisted of blood and blood product transfusions, prophylactic antibiotics, and hematopoietic growth factors. The study endpoints were overall response rate (complete response + partial response) and time to AML or death. Responses were classified using the MDS International Working Group (IWG) criteria; patients were required to be RBC and platelet transfusion independent during the time of response. Response criteria are given in Table 5.
Table 5 Response Criteria for the Controlled Trial in MDS* * Cheson BD, Bennett JM, et al. Report of an International Working Group to Standardize Response Criteria for MDS. Blood. 2000; 96:3671 to 3674.
Complete Response (CR) ≥ 8 weeks Bone Marrow On repeat aspirates:
• < 5% myeloblasts
• No dysplastic changesPeripheral Blood In all samples during response:
• Hgb> 11 g/dL (no transfusions or erythropoietin
• ANC ≥ 1500/mcL (no growth factor)
• Platelets ≥ 100,000/mcL (no thrombopoietic agent)
• No blasts and no dysplasiaPartial Response (PR) ≥ 8 weeks Bone Marrow On repeat aspirates:
• ≥ 50% decrease in blasts over pretreatment values
OR
• Improvement to a less advanced MDS FAB classificationPeripheral Blood Same as for CR The overall response rate (CR+PR) in the ITT population was 17% in decitabine-treated patients and 0% in the SC group (p<0.001) (see Table 6). The overall response rate was 21% (12/56) in decitabine-treated patients considered evaluable for response (i.e., those patients with pathologically confirmed MDS at baseline who received at least 2 cycles of treatment). The median duration of response (range) for patients who responded to decitabine was 288 days (116 to 388) and median time to response (range) was 93 days (55 to 272). All but one of the decitabine-treated patients who responded did so by the fourth cycle. Benefit was seen in an additional 13% of decitabine-treated patients who had hematologic improvement, defined as a response less than PR lasting at least 8 weeks, compared to 7% of SC patients. Decitabine treatment did not significantly delay the median time to AML or death versus supportive care.
Table 6 Analysis of Response (ITT) Parameter Decitabine
N=89Supportive Care
N=81**p-value <0.001 from two-sided Fisher's Exact Test comparing decitabine vs. Supportive Care.
†In the statistical analysis plan, a p-value of ≤ 0.024 was required to achieve statistical significance.
All patients with a CR or PR were RBC and platelet transfusion independent in the absence of growth factors.
Responses occurred in patients with an adjudicated baseline diagnosis of AML.
Overall Response Rate (CR+PR)†
Complete Response (CR)
Partial Response (PR)15 (17%)*
8 (9%)
7 (8%)0 (0%)
0 (0%)
0 (0%)Duration of Response
Median time to (CR+PR) response - Days (range) Median Duration of (CR+PR) response - Days (range)
93 (55 to 272)
288 (116 to 388)
NA
NA14.2 Single-arm Studies in Myelodysplastic Syndrome
Three open-label, single-arm, multicenter studies were conducted to evaluate the safety and efficacy of decitabine in MDS patients with any of the FAB subtypes. In one study conducted in North America, 99 patients with IPSS Intermediate-1, Intermediate-2, or high-risk prognostic scores received decitabine 20 mg/m2 as an intravenous infusion over 1-hour daily, on days 1 to 5 of week 1 every 4 weeks (1 cycle). The results were consistent with the results of the controlled trial and are summarized in Table 8.
Table 7 Baseline Demographics and Other Patient Characteristics (ITT) Demographic or Other Patient Characteristic Decitabine
N = 99Age (years) Mean (±SD) 71±9 Median (Range: min-max) 72 (34 to 87) Sex n (%) Male 71 (72) Female 28 (28) Race n (%) White 86 (87) Black 6 (6) Asian 4 (4) Other 3 (3) Days From MDS Diagnosis to First Dose Mean (±SD)
Median (Range: min-max)444±626
154 (7 to 3079)Previous MDS Therapy n (%) Yes 27 (27) No 72 (73) RBC Transfusion Status n (%)
Independent
33 (33)Dependent 66 (67) Platelet Transfusion Status n (%) Independent 84 (85) Dependent 15 (15) IPSS Classification n (%) Low Risk
Intermediate–1
Intermediate–2
High Risk1 (1)
52 (53)
23 (23)
23 (23)FAB Classification n (%) RA 20 (20) RARS 17 (17) RAEB 45 (45) RAEB-t 6 (6) CMML 11 (11) Table 8 Analysis of Response (ITT)* Parameter Decitabine
N = 99* Cheson BD, Bennett JM, et al. Report of an International Working Group to Standardize Response Criteria for MDS. Blood. 2000; 96:3671 to 3674.
+ indicates censored observation
Overall Response Rate (CR+PR)
Complete Response (CR)
Partial Response (PR)16 (16%)
15 (15%)
1 (1%)Duration of Response
Median time to (CR+PR) response - Days (range)
Median Duration of (CR+PR) response - Days (range)
162 (50 to 267)
443 (72 to 722+) - 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Decitabine for Injection is a sterile white to almost white lyophilized powder for intravenous use supplied as follows:
NDC Decitabine for Injection Package Factor 25021-231-20 50 mg Single-Dose Vial 1 vial per carton Store vials at 20° to 25°C (68° to 77°F); excursions permitted between 15° to 30°C (59° to 86°F). [See USP Controlled Room Temperature.]
Discard unused portion.
Sterile, Nonpyrogenic, Preservative-free.
The container closure is not made with natural rubber latex. -
17 PATIENT COUNSELING INFORMATION
Myelosuppression
Advise patients of the risk of myelosuppression and to report any symptoms of infection, anemia, or bleeding to their healthcare provider as soon as possible. Advise patients for the need for laboratory monitoring [see Warnings and Precautions (5.1)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.2) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception while receiving decitabine for injection and for 6 months after last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception while receiving treatment with decitabine for injection and for 3 months after the last dose [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Lactation
Advise women to avoid breastfeeding while receiving decitabine for injection and for at least 2 weeks after the last dose [see Use in Specific Populations (8.2)].
SAGENT®
Mfd. for SAGENT Pharmaceuticals
Schaumburg, IL 60195 (USA)
Made in India
©2023 Sagent PharmaceuticalsJune 2023
SAGENT Pharmaceuticals®
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
DECITABINE
decitabine injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:25021-231 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength decitabine (UNII: 776B62CQ27) (decitabine - UNII:776B62CQ27) decitabine 50 mg in 10 mL Inactive Ingredients Ingredient Name Strength potassium phosphate, monobasic (UNII: 4J9FJ0HL51) sodium hydroxide (UNII: 55X04QC32I) water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:25021-231-20 1 in 1 CARTON 08/15/2018 1 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA205539 08/15/2018 Labeler - Sagent Pharmaceuticals (080579617)