RILUTEK- riluzole tablet, film coated
sanofi-aventis U.S. LLC
----------
RILUTEK®
(riluzole) Tablets
DESCRIPTION
RILUTEK® (riluzole) is a member of the benzothiazole class. Chemically, riluzole is 2-amino-6-(trifluoromethoxy)benzothiazole. Its molecular formula is C8H5F3N2OS and its molecular weight is 234.2. Its structural formula is as follows:
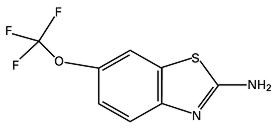
Riluzole is a white to slightly yellow powder that is very soluble in dimethylformamide, dimethylsulfoxide and methanol, freely soluble in dichloromethane, sparingly soluble in 0.1 N HCl and very slightly soluble in water and in 0.1 N NaOH. RILUTEK is available as a capsule-shaped, white, film-coated tablet for oral administration containing 50 mg of riluzole. Each tablet is engraved with "RPR 202" on one side.
CLINICAL PHARMACOLOGY
Mechanism of Action
The etiology and pathogenesis of amyotrophic lateral sclerosis (ALS) are not known, although a number of hypotheses have been advanced. One hypothesis is that motor neurons, made vulnerable through either genetic predisposition or environmental factors, are injured by glutamate. In some cases of familial ALS the enzyme superoxide dismutase has been found to be defective.
The mode of action of RILUTEK is unknown. Its pharmacological properties include the following, some of which may be related to its effect: 1) an inhibitory effect on glutamate release, 2) inactivation of voltage-dependent sodium channels, and 3) ability to interfere with intracellular events that follow transmitter binding at excitatory amino acid receptors.
Riluzole has also been shown, in a single study, to delay median time to death in a transgenic mouse model of ALS. These mice express human superoxide dismutase bearing one of the mutations found in one of the familial forms of human ALS.
It is also neuroprotective in various in vivo experimental models of neuronal injury involving excitotoxic mechanisms. In in vitro tests, riluzole protected cultured rat motor neurons from the excitotoxic effects of glutamic acid and prevented the death of cortical neurons induced by anoxia.
Due to its blockade of glutamatergic neurotransmission, riluzole also exhibits myorelaxant and sedative properties in animal models at doses of 30 mg/kg (about 20 times the recommended human daily dose) and anticonvulsant properties at a dose of 2.5 mg/kg (about 2 times the recommended human daily dose).
Pharmacokinetics
Riluzole is well-absorbed (approximately 90%), with average absolute oral bioavailability of about 60% (CV=30%). Pharmacokinetics are linear over a dose range of 25 to 100 mg given every 12 hours. A high fat meal decreases absorption, reducing AUC by about 20% and peak blood levels by about 45%. The mean elimination half-life of riluzole is 12 hours (CV=35%) after repeated doses. With multiple-dose administration, riluzole accumulates in plasma by about twofold and steady-state is reached in less than 5 days. Riluzole is 96% bound to plasma proteins, mainly to albumin and lipoproteins over the clinical concentration range.
The 50 mg market tablet was equivalent, with respect to AUC, to the tablet used in the dose ranging clinical trials, while the Cmax was approximately 30% higher. Both tablets have been used in clinical trials. However, if doses greater than those recommended are given, it is likely that higher plasma levels will be achieved, the safety of which has not been established (see DOSAGE AND ADMINISTRATION).
Metabolism and Elimination
Riluzole is extensively metabolized to six major and a number of minor metabolites, not all of which have been identified. Some metabolites appear pharmacologically active in in vitro assays. The metabolism of riluzole is mostly hepatic and consists of cytochrome P450-dependent hydroxylation and glucuronidation.
There is marked interindividual variability in the clearance of riluzole, probably attributable to variability of CYP 1A2 activity, the principal isozyme involved in N-hydroxylation.
In vitro studies using liver microsomes show that hydroxylation of the primary amine group producing N-hydroxyriluzole is the main metabolic pathway in human, monkey, dog and rabbit. In humans, cytochrome P450 1A2 is the principal isozyme involved in N-hydroxylation. In vitro studies predict that CYP 2D6, CYP 2C19, CYP 3A4 and CYP 2E1 are unlikely to contribute significantly to riluzole metabolism in humans. Whereas direct glucuroconjugation of riluzole (involving the glucurotransferase isoform UGT-HP4) is very slow in human liver microsomes, N-hydroxyriluzole is readily conjugated at the hydroxylamine group resulting in the formation of O- (>90%) and N-glucuronides.
Following a single 150 mg dose of 14C-riluzole to 6 healthy males, 90% and 5% of the radioactivity was recovered in the urine and feces respectively over a period of 7 days. Glucuronides accounted for more than 85% of the metabolites in urine. Only 2% of a riluzole dose was recovered in the urine as unchanged drug.
Special Populations
Hepatic Impairment
The area-under-the-curve (AUC) of riluzole, after a single 50 mg oral dose, increases by about 1.7-fold in patients with mild chronic liver insufficiency (n=6; Child-Pugh's score A) and by about 3-fold in patients with moderate chronic liver insufficiency (n=6; Child-Pugh's score B) compared to healthy volunteers (n=12) (see WARNINGS and PRECAUTIONS). The pharmacokinetics of riluzole have not been studied in patients with severe hepatic impairment.
Renal Impairment
There is no significant difference in pharmacokinetic parameters between patients with moderate (n=5; creatinine clearance 30–50 ml.min-1) and severe (n=7; creatinine clearance <30 ml.min-1) renal insufficiency and healthy volunteers (n=12) after a single oral dose of 50 mg riluzole. The pharmacokinetics of riluzole have not been studied in patients undergoing hemodialysis.
Age
The pharmacokinetic parameters of riluzole after multiple dose administration (4.5 days of treatment at 50 mg riluzole b.i.d.) are not affected in the elderly (≥ 70 years).
Gender
No gender effect on riluzole pharmacokinetics has been found in young or elderly healthy subjects. However, in one placebo-controlled clinical trial with population pharmacokinetics, riluzole mean clearance was found to be 30% lower in female patients (corresponding to an approximate increase in AUC of 45%) as compared to male patients. No favorable or adverse effects of riluzole in relation to gender were seen in controlled trials, however.
Smoking
Patients who smoke cigarettes eliminate riluzole 20% faster than non-smoking patients, based on a population pharmacokinetic analysis on data from 128 ALS patients, of whom 19 were smokers. However, there is no need for dosage adjustment in these patients.
Race
A clinical study conducted to evaluate the pharmacokinetics of riluzole and its metabolite following repeated oral administration twice daily in healthy Japanese and Caucasian adult males showed that there were no significant racial differences in pharmacokinetic parameters between the Japanese and Caucasian subjects.
Clinical Trials
The efficacy of RILUTEK as a treatment of ALS was established in two adequate and well-controlled trials in which the time to tracheostomy or death was longer for patients randomized to RILUTEK than for those randomized to placebo.
These studies admitted patients with either familial or sporadic ALS, a disease duration of less than 5 years, and a baseline forced vital capacity greater than or equal to 60%.
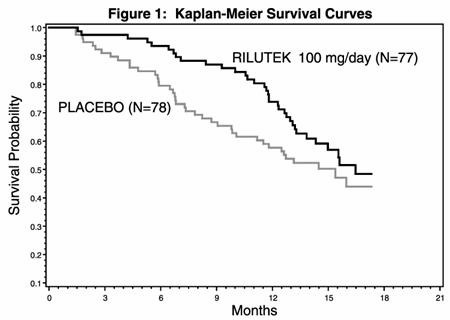
In one study, performed in France and Belgium, 155 ALS patients were followed for at least 13 months (maximum duration 18 months) after being randomized to either 100 mg/day (given 50 mg BID) of RILUTEK or placebo.
Figure 1, which follows, displays the survival curves for time to death or tracheostomy. The vertical axis represents the proportion of individuals alive without tracheostomy at various times following treatment initiation (horizontal axis). Although these survival curves were not statistically significantly different when evaluated by the analysis specified in the study protocol (Logrank test p=0.12), the difference was found to be significant by another appropriate analysis (Wilcoxon test p=0.05). As seen, the study showed an early increase in survival in patients given riluzole. Among the patients in whom treatment failed during the study (tracheostomy or death) there was a difference between the treatment groups in median survival of approximately 90 days. There was no statistically significant difference in mortality at the end of the study.
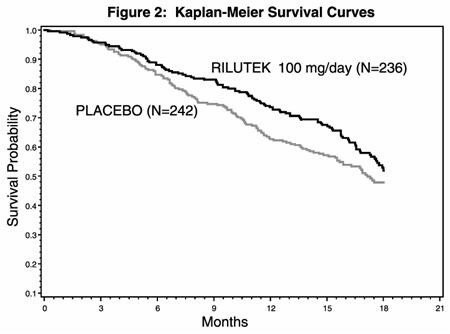
In the second study, performed in both Europe and North America, 959 ALS patients were followed for at least 1 year (North American centers) and up to 18 months (European centers) after being randomized to either 50, 100, 200 mg/day of RILUTEK or placebo.
Figure 2, which follows, displays the survival curves for time to death or tracheostomy for patients randomized to either 100 mg/day of RILUTEK or placebo. Although these survival curves were not statistically significantly different when evaluated by the analysis specified in the study protocol (Logrank test p = 0.076), the difference was found to be significant by another appropriate analysis (Wilcoxon test p = 0.05). Not displayed in Figure 2 are the results of 50 mg/day of RILUTEK which could not be statistically distinguished from placebo and the results of 200 mg/day which are essentially identical to 100 mg/day. As seen, the study showed an early increase in survival in patients given riluzole. Among the patients in whom treatment failed during the study (tracheostomy or death) there was a difference between the treatment groups in median survival of approximately 60 days. There was no statistically significant difference in mortality at the end of the study.
Although riluzole improved early survival in both studies, measures of muscle strength and neurological function did not show a benefit.
INDICATIONS AND USAGE
RILUTEK is indicated for the treatment of patients with amyotrophic lateral sclerosis (ALS). Riluzole extends survival and/or time to tracheostomy.
CONTRAINDICATIONS
RILUTEK is contraindicated in patients who have a history of severe hypersensitivity reactions to riluzole or any of the tablet components.
WARNINGS
Liver Injury / Monitoring Liver Chemistries
RILUTEK should be prescribed with care in patients with current evidence or history of abnormal liver function indicated by significant abnormalities in serum transaminase (ALT/SGPT; AST/SGOT), bilirubin, and/or gamma-glutamate transferase (GGT) levels (see PRECAUTIONS and DOSAGE AND ADMINISTRATION sections). Baseline elevations of several LFTs (especially elevated bilirubin) should preclude the use of RILUTEK.
RILUTEK, even in patients without a prior history of liver disease, causes serum aminotransferase elevations. Treatment should be discontinued if ALT levels are ≥ 5 × ULN or if clinical jaundice develops.
Experience in almost 800 ALS patients indicates that about 50% of riluzole-treated patients will experience at least one ALT/SGPT level above the upper limit of normal, about 8% will have elevations > 3 × ULN, and about 2% of patients will have elevations > 5 × ULN. A single non-ALS patient with epilepsy treated with concomitant carbamazepine and phenobarbital experienced marked, rapid elevations of liver enzymes with jaundice (ALT 26 × ULN, AST 17 × ULN, and bilirubin 11 × ULN) four months after starting RILUTEK; these returned to normal 7 weeks after treatment discontinuation.
Maximum increases in serum ALT usually occurred within 3 months after the start of riluzole therapy and were usually transient when < 5 times ULN. In trials, if ALT levels were < 5 times ULN, treatment continued and ALT levels usually returned to below 2 times ULN within 2 to 6 months. Treatment in studies was discontinued, however, if ALT levels exceeded 5 × ULN, so that there is no experience with continued treatment of ALS patients once ALT values exceed 5 times ULN. There were rare instances of jaundice. There is limited experience with rechallenge of patients who have had RILUTEK discontinued for ALT > 5 × ULN, but there is the possibility of increased ALT values reoccurring (see PRECAUTIONS: Laboratory Tests). Therefore, rechallenge is not recommended.
In postmarketing experience, cases of clinical hepatitis associated with riluzole have been reported, including with fatal outcome.
Neutropenia
Among approximately 4000 patients given riluzole for ALS, there were three cases of marked neutropenia (absolute neutrophil count less than 500/mm3), all seen within the first 2 months of riluzole treatment. In one case, neutrophil counts rose on continued treatment. In a second case, counts rose after therapy was stopped. A third case was more complex, with marked anemia as well as neutropenia and the etiology of both is uncertain. Patients should be warned to report any febrile illness to their physicians. The report of a febrile illness should prompt treating physicians to check white blood cell counts.
Interstitial Lung Disease
Cases of interstitial lung disease (see ADVERSE REACTIONS) have been reported in patients treated with riluzole, some of them severe; upon further investigation, many of these cases were hypersensitivity pneumonitis. If respiratory symptoms develop such as dry cough and/or dyspnea, chest radiography should be performed, and in case of findings suggestive of interstitial lung disease or hypersensitivity pneumonitis (e.g., bilateral diffuse lung opacities), riluzole should be discontinued immediately. In the majority of the reported cases, symptoms resolved after drug discontinuation and symptomatic treatment.
PRECAUTIONS
Use in Patients with Concomitant Disease
RILUTEK should be used with caution in patients with concomitant liver insufficiency (see WARNINGS, CLINICAL PHARMACOLOGY). In particular, in cases of RILUTEK-induced hepatic injury manifested by elevated liver enzymes, the effect of the hepatic injury on RILUTEK metabolism is unknown.
Special Populations
Riluzole should be used with caution in elderly patients whose hepatic function may be compromised due to age. Also, female patients may possess a lower metabolic capacity to eliminate riluzole compared to males (see CLINICAL PHARMACOLOGY: Special Populations).
Information for the Patient
Patients should be advised to report any febrile illness to their physicians (see WARNINGS: Neutropenia).
Patients should be advised to report any cough or difficulties in breathing to their physicians (see WARNINGS: Interstitial Lung Disease).
Patients and caregivers should be advised that RILUTEK should be taken on a regular basis and at the same time of the day (e.g., in the morning and evening) each day. If a dose is missed, take the next tablet as originally planned (see DOSAGE AND ADMINISTRATION).
Patients should be warned about the potential for dizziness, vertigo, or somnolence and advised not to drive or operate machinery until they have gained sufficient experience on RILUTEK to gauge whether or not it affects their mental and/or motor performance adversely.
Whether alcohol increases the risk of serious hepatotoxicity with RILUTEK is unknown; therefore, patients being treated with RILUTEK should be discouraged from drinking excessive amounts of alcohol.
Patients should also be made aware that RILUTEK should be stored at temperatures between 20°–25°C (68°–77°F) and protected from bright light.
RILUTEK must be kept out of the reach of children.
Laboratory Tests
Serum aminotransferases including ALT levels should be measured before and during riluzole therapy. Serum ALT levels should be evaluated every month during the first 3 months of treatment, every 3 months during the remainder of the first year, and periodically thereafter. Serum ALT levels should be evaluated more frequently in patients who develop elevations (see WARNINGS).
As noted in the WARNINGS Section, there is no experience with continued treatment of patients once ALT exceeds 5 × ULN. Treatment should be discontinued if ALT levels are ≥ 5 × ULN or if clinical jaundice develops. There is limited experience with rechallenge of patients who have had RILUTEK discontinued for ALT > 5 × ULN, but there is the possibility of increased ALT values reoccurring. Therefore, rechallenge is not recommended.
In the two controlled trials in patients with ALS, the frequency with which values for hemoglobin, hematocrit, and erythrocyte counts fell below the lower limit of normal was greater in RILUTEK-treated patients than in placebo-treated patients; however, these changes were mild and transient. The proportions of patients observed with abnormally low values for these parameters showed a dose-response relationship. Only one patient was discontinued from treatment because of severe anemia. The significance of this finding is unknown.
Drug Interactions
There have been no clinical studies designed to evaluate the interaction of riluzole with other drugs.
As with all drugs, the potential for interaction by a variety of mechanisms is a possibility.
Hepatotoxic Drugs
The clinical trials in ALS excluded patients on concomitant medications which were potentially hepatotoxic, (e.g., allopurinol, methyldopa, sulfasalazine). Accordingly, there is no information about the safety of administering RILUTEK in conjunction with such medications. If the practitioner chooses to prescribe such a combination, caution should be exercised.
Drugs Highly Bound To Plasma Proteins
Riluzole is highly bound (96%) to plasma proteins, binding mainly to serum albumin and to lipoproteins. The effect of riluzole (up to 5 mcg/mL) on warfarin (5 mcg/mL) binding did not show any displacement of warfarin. Conversely, riluzole binding was unaffected by the addition of warfarin, digoxin, imipramine and quinine at high therapeutic concentrations.
Effect of Other Drugs On Riluzole Metabolism
In vitro studies using human liver microsomal preparations suggest that CYP 1A2 is the principal isozyme involved in the initial oxidative metabolism of riluzole and, therefore, potential interactions may occur when riluzole is given concurrently with agents that affect CYP 1A2 activity. Potential inhibitors of CYP 1A2 (e.g., caffeine, phenacetin, theophylline, amitriptyline, and quinolones) could decrease the rate of riluzole elimination, while inducers of CYP 1A2 (e.g., cigarette smoke, charcoal-broiled food, rifampicin, and omeprazole) could increase the rate of riluzole elimination.
Effect of Riluzole On the Metabolism of Other Drugs
CYP 1A2 is the principal isoenzyme involved in the initial oxidative metabolism of riluzole; potential interactions may occur when riluzole is given concurrently with other agents which are also metabolized primarily by CYP 1A2 (e.g., theophylline, caffeine, and tacrine). Currently, it is not known whether riluzole has any potential for enzyme induction in humans.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Riluzole was not carcinogenic in mice or rats when administered for 2 years at daily oral doses up to 20 mg/kg and 10 mg/kg, respectively, which are approximately equivalent to the maximum human dose on a mg/m2 basis.
The genotoxic potential of riluzole was evaluated in the bacterial mutagenicity (Ames) test, the mouse lymphoma mutation assay in L5178Y cells, the in vitro chromosomal aberration assay in human lymphocytes and the in vivo rat cytogenetic assay and in vivo mouse micronucleus assay in bone marrow. There was no evidence of mutagenic or clastogenic potential in the Ames test, the mouse lymphoma assay, or the in vivo assays in the mouse and rat. There was an equivocal clastogenic response in the in vitro human lymphocyte chromosomal aberration assay, which was not reproduced in a second assay performed at equal or higher concentrations; riluzole was therefore considered non-clastogenic in the human lymphocyte assay.
N-hydroxyriluzole, the major active metabolite of riluzole, caused chromosomal damage in the in vitro mammalian mouse lymphoma assay and in the in vitro micronucleus assay that used the same mouse lymphoma cell line, L5178Y. N-hydroxyriluzole was not mutagenic in this cell line when tested in the HPRT gene mutation assay, and was negative in the Ames bacterial gene mutation assay (with and without rat or hamster S9), the in vitro UDS assay in rat hepatocytes, the chromosomal aberration test in human lymphocytes, and the in vivo mouse bone marrow micronucleus test.
Riluzole impaired fertility when administered to male and female rats prior to and during mating at an oral dose of 15 mg/kg or 1.5 times the maximum daily dose on a mg/m2 basis (see PRECAUTIONS: "Pregnancy" for effects on fertility).
Pregnancy
Pregnancy category C
Oral administration of riluzole to pregnant animals during the period of organogenesis caused embryotoxicity in rats and rabbits at doses of 27 mg/kg and 60 mg/kg, respectively, or 2.6 and 11.5 times, respectively, the recommended maximum human daily dose on a mg/m2 basis. Evidence of maternal toxicity was also observed at these doses.
When administered to rats prior to and during mating (males and females) and throughout gestation and lactation (females), riluzole produced adverse effects on pregnancy (decreased implantations, increased intrauterine death) and offspring viability and growth at an oral dose of 15 mg/kg or 1.5 times the maximum daily dose on a mg/m2 basis.
There are no adequate and well-controlled studies in pregnant women. Riluzole should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Women
In rat studies, 14C-riluzole was detected in maternal milk. It is not known whether riluzole is excreted in human breast milk. Because many drugs are excreted in human milk, and because the potential for serious adverse reactions in nursing infants from RILUTEK® is unknown, women should be advised not to breast-feed during treatment with RILUTEK.
Geriatric Use
Age-related compromised renal and hepatic function may cause a decrease in clearance of riluzole (see CLINICAL PHARMACOLOGY: Special Populations). In controlled clinical trials, about 30% of patients were over 65. There were no differences in adverse effects between younger and older patients.
ADVERSE REACTIONS
The most commonly observed AEs associated with the use of RILUTEK more frequently than placebo treated patients were: asthenia, nausea, dizziness, decreased lung function, diarrhea, abdominal pain, pneumonia, vomiting, vertigo, circumoral paresthesia, anorexia, and somnolence. Asthenia, nausea, dizziness, diarrhea, anorexia, vertigo, somnolence, and circumoral paresthesia were dose related.
Approximately 14% (n = 141) of the 982 individuals with ALS who received RILUTEK in pre-marketing clinical trials discontinued treatment because of an adverse experience. Of those patients who discontinued due to adverse events, the most commonly reported were: nausea, abdominal pain, constipation, and ALT elevations. In a dose response study in ALS patients, the rates of discontinuation of RILUTEK for asthenia, nausea, abdominal pain, and ALT elevation were dose related.
Incidence in Controlled ALS Clinical Studies
Table 1 lists treatment-emergent signs and symptoms that occurred in at least 2% of patients with ALS treated with RILUTEK (n=794) participating in placebo-controlled trials and were numerically greater in the patients treated with RILUTEK 100 mg/day than with placebo or for which a dose response relationship is suggested.
The prescriber should be aware that these figures cannot be used to predict the frequency of adverse experiences in the course of usual medical practice where patient characteristics and other factors may differ from those prevailing during clinical studies. Inspection of these frequencies, however, does provide the prescriber with one basis to estimate the relative contribution of drug and non-drug factors to the AE incidences in the population studied.
| Body System / Adverse Event* | Riluzole 50 mg/day (N=237) | Riluzole 100 mg/day (N=313) | Riluzole 200 mg/day (N=244) | Placebo (N=320) |
|---|---|---|---|---|
|
||||
| Body as a Whole | ||||
| Asthenia | 14.8 | 19.2 | 20.1 | 12.2 |
| Headache | 8.0 | 7.3 | 7.0 | 6.6 |
| Abdominal pain | 6.8 | 5.1 | 7.8 | 3.8 |
| Back pain | 1.7 | 3.2 | 4.1 | 2.5 |
| Aggravation reaction | 0.4 | 1.3 | 2.0 | 0.9 |
| Malaise | 0.4 | 0.6 | 1.2 | 0.0 |
| Digestive | ||||
| Nausea | 12.2 | 16.3 | 20.5 | 10.6 |
| Vomiting | 4.2 | 4.2 | 4.5 | 1.6 |
| Dyspepsia | 2.5 | 3.8 | 6.1 | 5.0 |
| Anorexia | 3.8 | 3.2 | 8.6 | 3.8 |
| Diarrhea | 5.5 | 2.9 | 9.0 | 3.1 |
| Flatulence | 2.5 | 2.6 | 2.0 | 1.9 |
| Stomatitis | 0.8 | 1.0 | 1.2 | 0.0 |
| Tooth disorder | 0.0 | 1.0 | 1.2 | 0.3 |
| Oral Moniliasis | 0.4 | 0.6 | 1.2 | 0.3 |
| Nervous | ||||
| Hypertonia | 5.9 | 6.1 | 5.3 | 5.9 |
| Depression | 4.2 | 4.5 | 6.1 | 5.0 |
| Dizziness | 5.1 | 3.8 | 12.7 | 2.5 |
| Dry mouth | 3.0 | 3.5 | 2.0 | 3.4 |
| Insomnia | 2.1 | 3.5 | 2.9 | 3.4 |
| Somnolence | 0.8 | 1.9 | 4.1 | 1.3 |
| Vertigo | 2.5 | 1.9 | 4.5 | 0.9 |
| Circumoral paresthesia | 1.3 | 1.6 | 3.3 | 0.0 |
| Skin and Appendages | ||||
| Pruritus | 3.8 | 3.8 | 2.5 | 3.1 |
| Eczema | 0.8 | 1.6 | 1.6 | 0.6 |
| Alopecia | 0.0 | 1.0 | 1.2 | 0.6 |
| Exfoliative dermatitis | 0.0 | 0.6 | 1.2 | 0.0 |
| Respiratory | ||||
| Decreased lung function | 13.1 | 10.2 | 16.0 | 9.4 |
| Rhinitis | 8.9 | 6.4 | 7.8 | 6.3 |
| Increased cough | 2.1 | 2.6 | 3.7 | 1.6 |
| Sinusitis | 0.4 | 1.0 | 1.6 | 0.9 |
| Cardiovascular | ||||
| Hypertension | 6.8 | 5.1 | 3.3 | 4.1 |
| Tachycardia | 1.3 | 2.6 | 2.0 | 1.3 |
| Phlebitis | 0.4 | 1.0 | 0.8 | 0.3 |
| Palpitation | 0.4 | 0.6 | 1.2 | 0.9 |
| Postural hypotension | 0.8 | 0.0 | 1.6 | 0.6 |
| Metabolic and Nutritional Disorders | ||||
| Weight loss | 4.6 | 4.8 | 3.7 | 4.7 |
| Peripheral edema | 4.2 | 2.9 | 3.3 | 2.2 |
| Musculoskeletal System | ||||
| Arthralgia | 5.1 | 3.5 | 1.6 | 3.4 |
| Urogenital System | ||||
| Urinary tract infection | 2.5 | 2.6 | 4.5 | 2.2 |
| Dysuria | 0.0 | 1.0 | 1.2 | 0.3 |
Other Adverse Events Observed
Other events which occurred in more than 2% of patients treated with RILUTEK 100 mg/day but equally or more frequently in the placebo group included: accidental injury, apnea, bronchitis, constipation, death, dysphagia, dyspnea, flu syndrome, heart arrest, increased sputum, pneumonia, and respiratory disorder.
The overall adverse event profile for RILUTEK was similar between females and males, and was independent of age. Because the largest non-white racial subgroup was only 2% of patients exposed to RILUTEK (18/794) in placebo-controlled trials, there are insufficient data to support a statement regarding the distribution of adverse experience reports by race. In ALS studies, dizziness did occur more commonly in females (11%) than in males (4%). There was not a difference between females and males in the rates of discontinuation of RILUTEK for individual adverse experiences.
Other Adverse Events Observed During All Clinical Trials
RILUTEK has been administered to 1713 individuals during all clinical trials, some of which were placebo-controlled. During these trials, all adverse events were recorded by the clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals having adverse events, similar types of events were grouped into a smaller number of standardized categories using modified COSTART dictionary terminology. The frequencies presented represent the proportion of the 1713 individuals exposed to RILUTEK who experienced an event of the type cited on at least one occasion while receiving RILUTEK. All reported events are included except those already listed in the previous table, those too general to be informative, and those not reasonably associated with the use of the drug.
Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring in at least 1/100 patients; infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare adverse events are those occurring in fewer than 1/1000 patients.
Body as a Whole: Frequent: Hostility1. Infrequent: Abscess1, sepsis1, photosensitivity reaction1, cellulitis, face edema1, hernia, peritonitis, attempted suicide, injection site reaction, chills1, flu syndrome, intentional injury, enlarged abdomen, neoplasm. Rare: Acrodynia, hypothermia, moniliasis1, rheumatoid arthritis.
Digestive System: Infrequent: Increased appetite, intestinal obstruction1, fecal impaction, gastrointestinal hemorrhage, gastrointestinal ulceration, gastritis1, fecal incontinence, jaundice, hepatitis, glossitis, gum hemorrhage1, pancreatitis, tenesmus, esophageal stenosis. Rare: Cheilitis1, cholecystitis, hematemesis, melena1, biliary pain, proctitis, pseudomembranous enterocolitis, enlarged salivary gland, tongue discoloration, tooth caries.
Immune System Disorders: Infrequent: Anaphylactoid reaction and anaphylaxis.
Nervous System: Frequent: Agitation1, tremor. Infrequent: Hallucinations, personality disorder1, abnormal thinking1, coma, paranoid reaction1, manic reaction, ataxia, extrapyramidal syndrome, hypokinesia, urinary retention, emotional lability, delusions, apathy, hypesthesia, incoordination, confusion1, convulsion, leg cramps, amnesia, dysarthria, increased libido, stupor, subdural hematoma, abnormal gait, delirium, depersonalization, facial paralysis, hemiplegia, decreased libido, myoclonus. Rare: Abnormal dreams, acute brain syndrome, CNS depression, dementia, cerebral embolism, euphoria1, hypotonia, ileus1, peripheral neuritis, psychosis1, psychotic depression, schizophrenic reaction, trismus, wristdrop.
Skin and Appendages: Infrequent: Skin ulceration, urticaria, psoriasis, seborrhea1, skin disorder, fungal dermatitis1. Rare: Angioedema, contact dermatitis, erythema multiforme, furunculosis1, skin moniliasis, skin granuloma, skin nodule.
Respiratory System: Infrequent: Hiccup, pleural disorder1, asthma, epistaxis, hemoptysis, yawn, hyperventilation1, lung edema1, hypoventilation1, lung carcinoma, hypoxia, laryngitis, pleural effusion, pneumothorax1, respiratory moniliasis, stridor, interstitial lung disease, hypersensitivity pneumonitis.
Cardiovascular System: Infrequent: Syncope1, hypotension, heart failure, migraine, peripheral vascular disease, angina pectoris1, myocardial infarction1, ventricular extrasystoles, cerebral hemorrhage, atrial fibrillation1, bundle branch block, congestive heart failure, pericarditis, lower extremity embolus, myocardial ischemia1, shock1. Rare: Bradycardia, cerebral ischemia, hemorrhage, mesenteric artery occlusion, subarachnoid hemorrhage, supraventricular tachycardia1, thrombosis, ventricular fibrillation, ventricular tachycardia.
Metabolic and Nutritional Disorders: Infrequent: Gout1, respiratory acidosis, edema, thirst1, hypokalemia, hyponatremia, weight gain1. Rare: Generalized edema, hypercalcemia, hypercholesteremia.
Endocrine System: Infrequent: Diabetes mellitus, thyroid neoplasia. Rare: Diabetes insipidus, parathyroid disorder.
Hemic and Lymphatic System: Infrequent: Anemia1, leukocytosis, leukopenia, ecchymosis. Rare: Neutropenia, aplastic anemia, cyanosis, hypochromic anemia, iron deficiency anemia, lymphadenopathy, petechiae1, purpura.
Musculoskeletal System: Infrequent: Arthrosis, myasthenia1, bone neoplasm. Rare: Bone necrosis, osteoporosis, tetany.
Special Senses: Infrequent: Amblyopia, ophthalmitis. Rare: Blepharitis, cataract, deafness, diplopia1, ear pain, glaucoma, hyperacusis, photophobia, taste loss, vestibular disorder.
Urogenital System: Infrequent: Urinary urgency, urine abnormality, urinary incontinence, kidney calculus, hematuria, impotence, prostate carcinoma, kidney pain, metrorrhagia, priapism. Rare: Amenorrhea, breast abscess, breast pain, nephritis1, nocturia, pyelonephritis, enlarged uterine fibroids, uterine hemorrhage, vaginal moniliasis.
Laboratory Tests: Infrequent: Increased gamma glutamyl transferase, abnormal liver function/tests, increased alkaline phosphatase, positive direct Coombs test, increased gamma globulins. Rare: increased lactic dehydrogenase.
- 1
- = AE frequency ≤ to placebo
OVERDOSAGE
No specific antidote or information on treatment of overdosage with RILUTEK is available. In the event of overdose, RILUTEK therapy should be discontinued immediately. Experience with riluzole overdose in humans is limited. Neurological and psychiatric symptoms, acute toxic encephalopathy with stupor, coma, and methemoglobinemia have been observed in isolated cases. Treatment should be supportive and directed toward alleviating symptoms.
Severe methemoglobinemia may be rapidly reversible after treatment with methylene blue.
The estimated oral median lethal dose is 94 mg/kg and 39 mg/kg for male mice and rats, respectively.
DOSAGE AND ADMINISTRATION
The recommended dose for RILUTEK is 50 mg every 12 hours. No increased benefit can be expected from higher daily doses, but adverse events are increased.
RILUTEK tablets should be taken at least an hour before, or two hours after, a meal to avoid a food-related decrease in bioavailability.
HOW SUPPLIED
RILUTEK 50 mg tablets are white, film-coated, capsule-shaped and engraved with "RPR 202" on one side. RILUTEK is supplied in bottles of 60 tablets, NDC 0075-7700-60.
| RILUTEK
riluzole tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (824676584) |