ADBRY- tralokinumab-ldrm injection, solution
LEO Pharma Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ADBRY safely and effectively. See full prescribing information for ADBRY.
ADBRY™ (tralokinumab-ldrm) injection, for subcutaneous use Initial U.S. Approval: 2021 INDICATIONS AND USAGEADBRY is an interleukin-13 antagonist indicated for the treatment of moderate-to-severe atopic dermatitis in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. ADBRY can be used with or without topical corticosteroids. (1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSInjection: 150 mg/mL solution in a single-dose prefilled syringe with needle guard. (3) CONTRAINDICATIONSKnown hypersensitivity to tralokinumab-ldrm or any excipients in ADBRY. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence ≥ 1%) are upper respiratory tract infections, conjunctivitis, injection site reactions, and eosinophilia. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact LEO Pharma Inc. at 1-877-494-4536 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 7/2022 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
ADBRY is indicated for the treatment of moderate-to-severe atopic dermatitis in adult patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. ADBRY can be used with or without topical corticosteroids.
2 DOSAGE AND ADMINISTRATION
2.1 Vaccination Prior to Treatment
Complete all age-appropriate vaccinations as recommended by current immunization guidelines prior to initiating treatment with ADBRY [see Warnings and Precautions (5.4)].
2.2 Recommended Dosage
The recommended dosage of ADBRY is:
- An initial dose of 600 mg (four 150 mg injections), followed by 300 mg (two 150 mg injections) administered every other week.
- After 16 weeks of treatment, for patients with body weight below 100 kg who achieve clear or almost clear skin, a dosage of 300 mg every 4 weeks may be considered.
ADBRY is administered by subcutaneous injection [see Dosage and Administration (2.5, 2.6)].
2.3 Concomitant Topical Therapies
ADBRY can be used with or without topical corticosteroids. Topical calcineurin inhibitors may be used, but should be reserved for problem areas only, such as the face, neck, intertriginous and genital areas.
2.4 Missed Doses
If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
2.5 Preparation for Use
- Before injection, remove ADBRY prefilled syringes from the refrigerator and allow to reach room temperature (30 minutes for the 150 mg/mL prefilled syringes) without removing the needle cap.
- After removal from the refrigerator, prefilled syringes may be kept at room temperature up to 30°C (86°F) and must be used within 14 days or discarded.
- Inspect ADBRY visually for particulate matter and discoloration prior to administration. ADBRY injection is a clear to opalescent, colorless to pale yellow solution. Do not use if the liquid contains visible particulate matter, is discolored or cloudy (other than clear to opalescent, colorless to pale yellow).
- ADBRY does not contain preservatives; therefore, discard any unused product.
2.6 Important Administration Instructions
- ADBRY is intended for use under the guidance of a healthcare provider. A patient may self-inject ADBRY after training in subcutaneous injection technique. Provide proper training to patients and/or caregivers on the preparation and administration according to the "Instructions for Use" [see Instructions for Use].
- For the initial 600 mg dose, administer each of the four ADBRY 150 mg injections at different injection sites within the same body area.
- For the subsequent 300 mg doses, administer the two ADBRY 150 mg injections at different injection sites within the same body area.
- Administer subcutaneous injection into the thigh or abdomen, except for the 2 inches (5 cm) around the navel. The upper arm can also be used if a caregiver administers the injection.
- Rotate the body area with each subsequent set of injections. DO NOT inject ADBRY into skin that is tender, damaged, bruised, or scarred.
3 DOSAGE FORMS AND STRENGTHS
Injection: 150 mg/mL clear to opalescent, colorless to pale yellow solution in a single-dose prefilled syringe with needle guard
4 CONTRAINDICATIONS
ADBRY is contraindicated in patients who have known hypersensitivity to tralokinumab-ldrm or any excipients in ADBRY [see Warnings and Precautions (5.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Hypersensitivity reactions including anaphylaxis and angioedema, have been reported with use of ADBRY.
If a serious hypersensitivity reaction occurs, discontinue ADBRY immediately and initiate appropriate therapy.
5.2 Conjunctivitis and Keratitis
Conjunctivitis and keratitis occurred more frequently in atopic dermatitis subjects who received ADBRY. Conjunctivitis was the most frequently reported eye disorder. Most subjects with conjunctivitis or keratitis recovered or were recovering during the treatment period [see Adverse Reactions (6.1)].
Advise patients to report new onset or worsening eye symptoms to their healthcare provider.
5.3 Parasitic (Helminth) Infections
Patients with known helminth infections were excluded from participation in clinical studies. It is unknown if ADBRY will influence the immune response against helminth infections by inhibiting IL-13 signaling.
Treat patients with pre-existing helminth infections before initiating treatment with ADBRY. If patients become infected while receiving ADBRY and do not respond to antihelminth treatment, discontinue treatment with ADBRY until the infection resolves.
5.4 Risk of Infection with Live Vaccines
ADBRY may alter a patient's immunity and increase the risk of infection following administration of live vaccines. Prior to initiating therapy with ADBRY, complete all age appropriate vaccinations according to current immunization guidelines. Avoid use of live vaccines in patients treated with ADBRY. Limited data are available regarding coadministration of ADBRY with non-live vaccines [see Clinical Pharmacology (12.2)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail elsewhere in the labeling:
- Hypersensitivity [see Warnings and Precautions (5.1)]
- Conjunctivitis and Keratitis [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ADBRY was evaluated in a pool of 5 randomized, double-blind, placebo-controlled trials in subjects with moderate-to-severe atopic dermatitis including three phase 3 Eczema Tralokinumab trials (ECZTRA 1, ECZTRA 2, and ECZTRA 3), a dose-finding trial, and a vaccine response trial. The safety population had a mean age of 37 years; 43% of subjects were female, 67% were White, 21% were Asian, and 9% were Black. In terms of co-morbid conditions, 39% of the subjects had asthma, 49% had hay fever, 36% had food allergy, and 21% had allergic conjunctivitis at baseline.
In these 5 atopic dermatitis trials, 1964 subjects were treated with subcutaneous injections of ADBRY, with or without concomitant topical corticosteroids (TCS). A total of 807 subjects were treated with ADBRY for at least 1 year.
ECZTRA 1 and ECZTRA 2 compared the safety of ADBRY monotherapy to placebo through Week 52. ECZTRA 3 compared the safety of ADBRY + TCS to placebo + TCS through Week 32.
Weeks 0 to 16 (ECZTRA 1, ECZTRA 2, and ECZTRA 3):
Table 1 summarizes the adverse reactions identified in the pool of 3 trials (ECZTRA 1, ECZTRA 2, and ECZTRA 3) and that occurred at a rate of at least 1% in the ADBRY 300 mg every other week monotherapy group, and in the ADBRY 300 mg every other week + TCS study, all at a higher rate than placebo during the first 16 weeks of treatment.
| Adverse Reaction | ADBRY Monotherapy* | ADBRY + TCS† | ||
|---|---|---|---|---|
| ADBRY 300 mg Q2W‡
N=1180 n (%) | PLACEBO N=388 n (%) | ADBRY 300 mg Q2W‡ + TCS N=243 n (%) | PLACEBO + TCS N=123 n (%) |
|
|
||||
| Upper respiratory tract infections§ | 281 (23.8) | 79 (20.4) | 73 (30.0) | 19 (15.4) |
| Conjunctivitis¶ | 88 (7.5) | 12 (3.1) | 33 (13.6) | 6 (4.9) |
| Injection site reactions# | 87 (7.4) | 16 (4.1) | 27 (11.1) | 1 (0.8) |
| EosinophiliaÞ | 17 (1.4) | 2 (0.5) | 3 (1.2) | 0 |
In the monotherapy trials (ECZTRA 1 and ECZTRA 2) through Week 16, the proportion of subjects who discontinued treatment due to adverse reactions was 0.7% in the ADBRY 300 mg every other week group and 0% of the placebo group. In the concomitant TCS trial (ECZTRA 3) through Week 16, the proportion of subjects who discontinued treatment due to adverse reactions was 0.8% in the ADBRY 300 mg every other week +TCS group and 0% of the placebo + TCS group. The most common adverse reactions leading to discontinuation in the ADBRY group compared to the placebo group were injection site reaction (0.3% v. 0) and eosinophilia (0.3% v. 0) in ECZTRA 1 and ECZTRA 2; and injection site reaction (0.4% v. 0) and conjunctivitis (0.4% v. 0) in ECZTRA 3.
Safety Weeks 16-52 (ECZTRA 1 and ECZTRA 2) and Weeks 16-32 (ECZTRA 3):
The safety profile of ADBRY 300 mg every other week with or without TCS during maintenance treatment was consistent with that in the initial 16-week treatment period. In addition, the frequency of adverse reactions with ADBRY 300 mg every other week and every 4 weeks in ECZTRA 1 and ECZTRA 2 was 44% and 34%, respectively, and 43% and 26% with ADBRY 300 mg + TCS every other week and every 4 weeks in ECZTRA 3, respectively.
Specific Adverse Reactions
Conjunctivitis and Keratitis
Conjunctivitis, including allergic conjunctivitis, was reported in 7.5% of subjects treated with ADBRY 300 mg every other week (29 events per 100 subject-years of exposure) and in 3.1% of subjects treated with placebo (12 events per 100 subject-years of exposure) in the initial treatment period of up to 16 weeks in the pool of 5 trials. In the ADBRY group, 126 subjects reported 145 events of conjunctivitis, with 114 events resolved at the end of initial treatment period. Conjunctivitis led to discontinuation of treatment in 2 subjects.
During the maintenance treatment period of the monotherapy trials (ECZTRA 1 and ECZTRA 2) from 16 to 52 weeks, conjunctivitis was reported in 8.9% of subjects treated with ADBRY 300 mg every other week (20 events per 100 subject-years of exposure) and in 6.3% of subjects treated with ADBRY 300 mg every 4 weeks (14 events per 100 subject-years of exposure) compared to 7.7% of subjects treated with ADBRY 300 mg every other week in the initial treatment period (30 events per 100 subject-years of exposure). Conjunctivitis (including no serious events, 1 severe event, and 1 event that led to discontinuation) was reported in 24 subjects in the combined (every other week and every 4 weeks) ADBRY groups. A similar pattern was seen during the continuation treatment period of an additional 16 weeks in the ADBRY combination ECZTRA 3.
Keratitis (including keratoconjunctivitis) was reported in 0.5% of subjects treated with ADBRY and 0% treated with placebo during the initial treatment period of up to 16 weeks in the pool of 5 trials. Keratitis (including 1 ulcerative keratitis) was reported in 0.2% of subjects treated with ADBRY (0.9 events per 100 subject-years of exposure) and 0.2% of subjects treated with placebo (0.6 events per 100 subject-years of exposure). Keratoconjunctivitis (including 1 atopic keratoconjunctivitis) was reported in 0.3% of subjects treated with ADBRY (1.2 events per 100 subject-years of exposure), and in no subjects treated with placebo. In the ADBRY group, 9 subjects reported 10 events of keratitis or keratoconjunctivitis, with 5 events resolved during the trial following the initial treatment period. None of the events were serious or led to treatment discontinuation.
During the maintenance treatment period of the monotherapy trials (ECZTRA 1 and ECZTRA 2) from 16 to 52 weeks in the ADBRY 300 mg every other week group, keratitis was reported in 1 (0.6%) subject (ulcerative, severe, resolved after discontinuation) at an exposure-adjusted event rate of 1.2 per 100 subject-years, and keratoconjunctivitis (not serious or severe, resolved, not led to discontinuation) was reported in 3 (1.9%) subjects (3.6 events per 100 subject-years of exposure). No events of keratitis or keratoconjunctivitis was reported in ADBRY every 4 weeks or placebo groups, compared to keratitis event rate of 2 per 100 subject-years for ADBRY 300 mg every other week in the initial treatment period.
In the continuation treatment period of ECZTRA 3 (from 16 to 32 weeks), there were no additional events of keratitis reported for subjects randomized to ADBRY 300 mg + TCS.
Eosinophil Counts
ADBRY-treated subjects had a greater mean initial increase from baseline in eosinophil count compared to subjects treated with placebo. The mean and median increases in blood eosinophils from baseline to Week 4 were 190 and 100 cells/mcL, respectively. The increase in the ADBRY-treated subjects declined to baseline level with continued treatment. Eosinophilia (> 5000 cells/mcL) in the initial treatment period of up to 16 weeks was reported in 1.2% in the ADBRY-treated subjects and 0.3% in the placebo-treated subjects. The safety profile for subjects with eosinophilia was comparable to the safety profile for all subjects included in the pool of 5 atopic dermatitis trials.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity with ADBRY. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other tralokinumab products may be misleading.
In ECZTRA 1, ECZTRA 2, and ECZTRA 3, and the vaccine-response trial, the incidence of Anti-Drug-Antibodies (ADA) during the initial 16-week treatment period was 1.4% for subjects treated with ADBRY 300 mg every other week and in 1.3% for subjects treated with placebo; neutralizing antibodies were seen in 0.1% of subjects treated with ADBRY and 0.2% of subjects treated with placebo.
Across all trial periods, the ADA incidence for subjects who received ADBRY was 4.6%; 0.9% had persistent ADA and 1.0% had neutralizing antibodies.
No clinically meaningful differences in the pharmacokinetics, safety, or efficacy of tralokinumab-ldrm were observed in patients who tested positive for anti-tralokinumab-ldrm antibody (including neutralizing antibodies).
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are limited data from the use of ADBRY in pregnant women to inform a drug-associated risk of adverse developmental outcomes. Human IgG antibodies are known to cross the placental barrier; therefore, ADBRY may be transmitted from the mother to the developing fetus.
In an enhanced pre-and post-natal developmental study, no adverse developmental effects were observed in offspring born to pregnant monkeys after intravenous administration of tralokinumab-ldrm during organogenesis through parturition at doses up to 10 times the maximum recommended human dose (MRHD).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In a pre- and post-natal development study, intravenous doses up to 100 mg/kg tralokinumab-ldrm were administered to pregnant cynomolgus monkeys once every week from gestation day 20 to parturition. No maternal or developmental toxicity was observed at doses up to 100 mg/kg/week (10 times the MRHD on a mg/kg basis of 10 mg/kg/week).
In an enhanced pre- and post-natal development study, intravenous doses up to 100 mg/kg tralokinumab-ldrm (10 times the MRHD on a mg/kg basis of 10 mg/kg/week) were administered to pregnant cynomolgus monkeys once every week from the beginning of organogenesis to parturition. No treatment-related adverse effects on embryofetal toxicity or malformations, or on morphological, functional, or immunological development were observed in the infants from birth through 6 months of age.
8.2 Lactation
Risk Summary
There are no data on the presence of tralokinumab-ldrm in human milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is present in breast milk. The effects of local gastrointestinal exposure and limited systemic exposure to ADBRY on the breastfed infant are unknown. The development and health benefits of breastfeeding should be considered along with the mother's clinical need for ADBRY and any potential adverse effects on the breastfed child from ADBRY or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of ADBRY have not been established in pediatric patients.
8.5 Geriatric Use
Of the 1605 subjects exposed to ADBRY in 5 atopic dermatitis trials in the initial treatment period of up to 16 weeks, 77 subjects were 65 years or older. Clinical studies did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no specific treatment for ADBRY overdose. In the event of overdosage, contact Poison Control (1-800-222-1222) for latest recommendations and monitor the patient for any signs or symptoms of adverse reactions and institute appropriate symptomatic treatment immediately.
11 DESCRIPTION
Tralokinumab-ldrm, an interleukin-13 antagonist, is a human IgG4 monoclonal antibody. Tralokinumab-ldrm is produced in mouse myeloma cells by recombinant DNA technology, consists of 1326 amino acids, and has a molecular weight of approximately 147 kilodaltons.
ADBRY (tralokinumab-ldrm) injection is a sterile, preservative-free, clear to opalescent, colorless to pale yellow solution for subcutaneous use supplied as a single-dose prefilled syringe with needle guard in a siliconized Type-1 clear glass syringe. None of the components of the prefilled syringe or the needle guard are made with natural rubber latex.
Each prefilled syringe delivers 150 mg tralokinumab-ldrm in 1 mL and the inactive ingredients: acetic acid (0.3 mg), polysorbate 80 (0.1 mg), sodium acetate trihydrate (6 mg), sodium chloride (5 mg), and Water for Injection, at an approximate pH of 5.5.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tralokinumab-ldrm is a human IgG4 monoclonal antibody that specifically binds to human interleukin-13 (IL-13) and inhibits its interaction with the IL-13 receptor α1 and α2 subunits (IL-13Rα1 and IL-13Rα2). IL-13 is a naturally occurring cytokine of the Type 2 immune response. Tralokinumab-ldrm inhibits the bioactivity of IL-13 by blocking IL-13 interaction with IL-13Rα1/IL-4Rα receptor complex. Tralokinumab-ldrm inhibits IL-13-induced responses including the release of proinflammatory cytokines, chemokines and IgE.
12.2 Pharmacodynamics
ADBRY was associated with decreased concentrations of Th2 and Th22 immunity biomarkers in the blood, such as thymus and activation-regulated chemokine (TARC/CCL17), periostin, IL-22, lactate dehydrogenase (LDH) and serum IgE. ADBRY decreased expression of keratin 16 and Ki-67 in AD skin, and upregulated protein expression of loricrin. ADBRY suppressed expression of genes in the Th2 pathway, including CCL17, CCL18 and CCL26 as well as markers of Th17- and Th22-regulated genes in lesional skin. The clinical relevance of these biomarkers is not fully understood.
Immune Response to Non-live Vaccines during Treatment
Immune responses to non-live vaccines were assessed in a trial in which adult subjects with atopic dermatitis were treated with an initial dose of 600 mg (four 150 mg injections) followed by 300 mg every other week administered as subcutaneous injection. After 12 weeks of ADBRY administration, subjects were vaccinated with a combined tetanus, diphtheria, and acellular pertussis vaccine, and a meningococcal vaccine. Antibody responses were assessed 4 weeks later. Antibody response to tetanus, diphtheria, acellular pertussis, and a meningococcal vaccine was similar in tralokinumab-ldrm treated and placebo treated subjects. Immune responses to other vaccines were not assessed.
12.3 Pharmacokinetics
The mean (SD) steady-state trough concentration of tralokinumab-ldrm ranged from 98.0 (41.1) mcg/mL to 101.4 (42.7) mcg/mL following administration of ADBRY at 300 mg every other week. Tralokinumab-ldrm exposure increased proportionally over a dosage range up to 2100 mg for a 70 kg subject (30 mg/kg IV) (3.5 times the maximum approved recommended dosage). Steady-state tralokinumab-ldrm concentrations were achieved by week 16 following a 600 mg starting dose and 300 mg every other week.
Absorption
The absolute bioavailability of tralokinumab-ldrm was estimated to be 76%. The time to maximum tralokinumab-ldrm concentrations (tmax) was 5 to 8 days after administration.
Distribution
The volume of distribution of tralokinumab-ldrm was estimated to be approximately 4.2 L.
Elimination
The half-life of tralokinumab-ldrm was 3 weeks and systemic clearance was estimated to be 0.149 L/day.
Specific Populations
No clinically significant differences in the pharmacokinetics of tralokinumab-ldrm were observed based on age (ranged from 18 – 92 years), sex, mild to moderate renal impairment, or mild hepatic impairment. The effect of severe renal impairment or moderate to severe hepatic impairment on the pharmacokinetics of tralokinumab-ldrm is unknown.
Body Weight
The exposure of tralokinumab-ldrm decreases with increasing body weight. After 300 mg dose every 4 weeks, the median tralokinumab-ldrm exposure (AUC) of subjects with body weight of above 100 kg is expected to be 1.46-fold lower than that of subjects weighing below 100 kg [see Dosage and Administration (2.2)].
Drug Interaction Studies
Cytochrome P450 Substrates
The effects of tralokinumab on the pharmacokinetics of midazolam (CYP3A4 substrate), warfarin (CYP2C9 substrate), omeprazole (CYP2C19 substrate), metoprolol (CYP2D6 substrate), and caffeine (CYP1A2 substrate) were evaluated in a clinical trial in 18-20 subjects with moderate to severe atopic dermatitis receiving an initial SC dose of 600 mg followed by 300 mg SC every other week for 14 weeks. No clinically significant changes in the exposure (AUC or Cmax) of the CYP450 substrates after multiple tralokinumab administrations were observed compared to that when administered alone.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies have not been conducted to evaluate the carcinogenic or mutagenic potential for tralokinumab-ldrm.
No effects on fertility parameters such as reproductive organs, menstrual cycle and sperm analysis were observed in male or female sexually mature cynomolgus monkeys that were subcutaneously administered tralokinumab-ldrm at doses up to 350 mg/animal (10 times the MRHD on a mg/kg basis of 10 mg/kg/week) in females once a week for three consecutive menstrual cycles (maximum of 15 doses) or 600 mg/animal (10 times the MRHD on a mg/kg basis of 10 mg/kg/week) in males once a week for 13 weeks. The monkeys were not mated to evaluate fertility.
14 CLINICAL STUDIES
The efficacy of ADBRY was assessed in three randomized, double-blind, placebo-controlled trials [ECZTRA 1 (NCT03131648), ECZTRA 2 (NCT03160885), and ECZTRA 3 (NCT03363854)]. Efficacy was assessed in a total of 1934 subjects 18 years of age and older with moderate-to-severe atopic dermatitis (AD) not adequately controlled by topical medication(s). Disease severity was defined by an Investigator's Global Assessment (IGA) score ≥ 3 in the overall assessment of AD lesions on a severity scale of 0 to 4, an Eczema Area and Severity Index (EASI) score ≥16 on a scale of 0 to 72, and a minimum body surface area (BSA) involvement of ≥10%. At baseline, 58% of subjects were male, 69% of subjects were White, 50% of subjects had a baseline IGA score of 3 (moderate AD), and 50% of subjects had a baseline IGA score of 4 (severe AD). The baseline mean EASI score was 32 and the baseline weekly averaged Worst Daily Pruritus Numeric Rating Scale (NRS) was 8 on a scale of 0-10.
In all three trials, subjects received subcutaneous injections of ADBRY 600 mg or placebo on Day 0, followed by 300 mg every other week or placebo for 16 weeks. Responders were defined as achieving an IGA 0 or 1 ("clear" or "almost clear") or EASI-75 (improvement of at least 75% in EASI score from baseline) at Week 16.
To evaluate maintenance of response in the monotherapy trials (ECZTRA 1 and ECZTRA 2), subjects responding to initial treatment with ADBRY 300 mg every other week were re-randomized to ADBRY 300 mg every other week, ADBRY 300 mg every 4 weeks or placebo every other week for another 36 weeks following first dose administration. Subjects randomized to placebo in the initial treatment period who achieved a clinical response at Week 16 continued to receive placebo every other week for another 36 weeks. Non-responders at Week 16, and subjects who lost clinical response during the maintenance period were placed on open-label treatment with ADBRY 300 mg every other week and optional use of TCS.
In the combination therapy trial (ECZTRA 3), subjects received either ADBRY 300 mg every other week with TCS or placebo with TCS and as needed topical calcineurin inhibitors (TCI) until Week 16. Subjects in the ADBRY 300 mg with TCS group who achieved clinical response at Week 16 were re-randomized to ADBRY 300 mg every other week with TCS or ADBRY every 4 weeks with TCS for another 16 weeks following first dose administration. Subjects in the placebo with TCS group who achieved clinical response at Week 16 continued on placebo with TCS for another 16 weeks. Subjects who did not achieve clinical response at Week 16 received ADBRY 300 mg every other week for another 16 weeks. A mid-potency TCS (i.e., mometasone furoate 0.1% cream) was dispensed at each dosing visit. Subjects were instructed to apply a thin film of the dispensed TCS as needed once daily to active lesions from Week 0 to Week 32 and were to discontinue treatment with TCS when control was achieved. An additional, lower potency TCS or TCI could be used at the investigator's discretion on areas of the body where use of the supplied TCS was not advisable, such as areas of thin skin.
All three trials assessed the primary endpoints of the proportion of subjects with an IGA 0 or 1 at Week 16 and the proportion of subjects with EASI-75 at Week 16. Secondary endpoints included the reduction of Worst Daily Pruritus NRS (weekly average) of at least 4 points on the 11-point itch NRS from baseline to Week 16.
Clinical Response at Week 16 (ECZTRA 1, ECZTRA 2, and ECZTRA 3)
The results of the ADBRY monotherapy trials (ECZTRA 1 and ECZTRA 2) and the ADBRY with TCS trial (ECZTRA 3) are presented in Table 2.
| ECZTRA 1 | ECZTRA 2 | ECZTRA 3 | ||||
|---|---|---|---|---|---|---|
| ADBRY 300 mg every other week | Placebo | ADBRY 300 mg every other week | Placebo | ADBRY 300 mg every other week + TCS | Placebo + TCS | |
| Abbreviations: AD = Atopic Dermatitis CI = Confidence Interval. | ||||||
| Note: Difference and 95% CI are based on the CMH test stratified by region and baseline IGA score. | ||||||
| Number of subjects randomized and dosed (FAS)* | 601 | 197 | 577 | 193 | 243 | 123 |
| IGA 0 or 1†,‡ | 16% | 7% | 21% | 9% | 38% | 27% |
| Difference from Placebo (95% CI) | 9% (4%, 13%) | 12% (7%, 17%) | 11% (1%, 21%) |
|||
| EASI-75‡ | 25% | 13% | 33% | 10% | 56% | 37% |
| Difference from Placebo (95% CI) | 12% (6%, 18%) | 22% (17%, 28%) | 20% (9%, 30%) |
|||
| Number of subjects with baseline Worst Daily Pruritus NRS (weekly average) score ≥4 | 594 | 194 | 563 | 192 | 240 | 123 |
| Worst Daily Pruritus NRS (≥4 point reduction)‡ | 20% | 10% | 25% | 9% | 46% | 35% |
| Difference from Placebo (95% CI) | 10% (4%, 15%) | 16% (11%, 21%) | 11% (1%, 22%) |
|||
A higher proportion of subjects in the ADBRY 300 mg every other week arm achieved EASI-90 compared to placebo in the three pivotal trials.
Examination of age, gender, race, body weight, and previous treatment, including immunosuppressants, did not identify differences in response to ADBRY 300 mg every other week among these subgroups.
Monotherapy Trials (ECZTRA 1 and ECZTRA 2) – Maintenance Period (Week 16-52)
In ECZTRA 1, 179 ADBRY 300 mg every other week responders (IGA 0/1 or EASI-75) were re-randomized (and dosed) at Week 16 to ADBRY 300 mg every other week (68 subjects), ADBRY 300 mg every 4 weeks (76 subjects) or placebo (35 subjects). Among these subjects, 39 subjects in ADBRY 300 mg every other week arm, 36 subjects in ADBRY 300 mg every 4 weeks arm and 19 subjects in placebo arm were IGA 0/1 responders at Week 16. Maintenance of IGA 0/1 response at Week 52 was as follows: 20 subjects (51%) in the every other week arm, 14 subjects (39%) in the every 4 weeks arm and 9 subjects (47%) in the placebo arm. Among the re-randomized subjects, 47 subjects in ADBRY 300 mg every other week arm, 57 subjects in ADBRY 300 mg every 4 weeks arm and 30 subjects in placebo arm were EASI-75 responders at Week 16. Maintenance of EASI-75 response at Week 52 was as follows: 28 subjects (60%) in the every other week arm, 28 subjects (49%) in the every 4 weeks arm and 10 subjects (33%) in the placebo arm.
In ECZTRA 2, 218 ADBRY 300 mg every other week responders (IGA 0/1 or EASI-75) were re-randomized (and dosed) at Week 16 to ADBRY 300 mg every other week (90 subjects), ADBRY 300 mg every 4 weeks (84 subjects) or placebo (44 subjects). Among these subjects, 53 subjects in ADBRY 300 mg every other week arm, 44 subjects in ADBRY 300 mg every 4 weeks arm and 26 subjects in placebo arm were IGA 0/1 responders at Week 16. Maintenance of IGA 0/1 response at Week 52 was as follows: 32 subjects (60%) in the every other week arm, 22 subjects (50%) in the every 4 weeks arm and 6 subjects (23%) in the placebo arm. Among the re-randomized subjects, 76 subjects in ADBRY 300 mg every other week arm, 69 subjects in ADBRY 300 mg every 4 weeks arm and 40 subjects in placebo arm were EASI-75 responders at Week 16. Maintenance of EASI-75 response at Week 52 was as follows: 43 subjects (57%) in the every other week arm, 38 subjects (55%) in the every 4 weeks arm and 8 subjects (20%) in the placebo arm.
Concomitant TCS Trial (ECZTRA 3) – Maintenance Period (Week 16-32)
In ECZTRA 3, 131 ADBRY 300 mg every other week + TCS responders (IGA 0/1 or EASI-75) were re-randomized (and dosed) at Week 16 to ADBRY 300 mg every other week + TCS (65 subjects) or ADBRY 300 mg every 4 weeks + TCS (66 subjects). Among these subjects, 45 subjects in ADBRY 300 mg every other week + TCS arm and 46 subjects in ADBRY 300 mg every 4 weeks + TCS arm were IGA 0/1 responders at Week 16. Maintenance of IGA 0/1 response at Week 32 was as follows: 40 subjects (89%) in the every other week arm and 35 subjects (76%) every 4 weeks arm. Among the re-randomized subjects, 65 subjects in ADBRY 300 mg every other week arm and 62 subjects in ADBRY 300 mg every 4 weeks arm were EASI-75 responders at Week 16. Maintenance of EASI-75 response at Week 32 was as follows: 60 subjects (92%) in the every other week arm and 56 subjects (90%) in the every 4 weeks arm.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ADBRY (tralokinumab-ldrm) injection is a sterile, clear to opalescent, colorless to pale yellow solution, supplied in single-dose prefilled syringe with a 27-gauge, ½ inch needle and a needle guard.
Each prefilled syringe delivers 150 mg/mL of ADBRY.
ADBRY is available in pack sizes containing 2 or 4 prefilled syringes with needle guard.
| Pack Size | NDC # |
|---|---|
| Two cartons (multipack) containing 4 prefilled syringes | NDC 50222-346-04 |
| One carton containing 2 prefilled syringes | NDC 50222-346-02 |
Storage and Handling
ADBRY does not contain preservatives. Discard any unused product remaining in the prefilled syringe.
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light.
If necessary, prefilled syringes may be kept at room temperature up to 30°C (86°F) for a maximum of 14 days in the original carton. Do not store above 30°C (86°F). If the carton needs to be removed permanently from refrigerator, the date of removal may be recorded on the outer carton in the space provided. After removal from the refrigerator, ADBRY must be used within 14 days or discarded.
Do not expose the prefilled syringe to heat or direct sunlight.
Do not freeze. Do not shake.
17 PATIENT COUNSELING INFORMATION
Advise the patients to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Administration Instructions
Instruct patients or caregivers:
- to perform the first self-injection under the supervision and guidance of a qualified healthcare provider for proper training in subcutaneous injection technique.
- to inject the full dose of ADBRY.
- to follow sharps disposal recommendations [see Instructions for Use].
Hypersensitivity
Advise patients to discontinue ADBRY and to seek immediate medical attention if they experience any symptoms of systemic hypersensitivity reactions [see Warnings and Precautions (5.1)].
Conjunctivitis and Keratitis
Advise patients to consult their healthcare provider if new onset or worsening eye symptoms develop [see Adverse Reactions (6.1)].
Risk of Infection with Live Vaccines
Advise patients that ADBRY may increase the risk of infection following administration of live vaccines and that vaccination with live vaccines is not recommended during ADBRY treatment. Instruct patients to inform the healthcare provider that they are taking ADBRY prior to a potential vaccination [see Warnings and Precautions (5.4)].
Manufactured by:
LEO Pharma A/S
Industriparken 55
Ballerup, Denmark DK-2750
U.S. License No. 2169
Distributed by:
LEO Pharma Inc.
Madison, NJ 07940, USA
ADBRY™ is a trademark of LEO Pharma A/S.
© 2022 LEO Pharma Inc. All rights reserved.
| PATIENT INFORMATION ADBRY™ [ad'-bree] (tralokinumab-ldrm) injection, for subcutaneous use |
||
|---|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Issued: 12/2021 | |
| What is ADBRY? | ||
|
||
| Do not use ADBRY if you are allergic to tralokinumab or to any of the ingredients in ADBRY. See the end of this leaflet for a complete list of ingredients in ADBRY. | ||
| Before using ADBRY, tell your healthcare provider about all your medical conditions, including if you: | ||
|
||
| Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | ||
| How should I use ADBRY? | ||
|
||
| What are the possible side effects of ADBRY? | ||
| ADBRY can cause serious side effects, including: | ||
|
||
|
|
|
|
||
| The most common side effects of ADBRY include: | ||
|
||
| These are not all of the possible side effects of ADBRY. | ||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||
| General information about the safe and effective use of ADBRY. | ||
| Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ADBRY for a condition for which it was not prescribed. Do not give ADBRY to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ADBRY that is written for health professionals. | ||
| What are the ingredients in ADBRY? | ||
| Active ingredient: tralokinumab-ldrm | ||
| Inactive ingredients: acetic acid, polysorbate 80, sodium acetate trihydrate, sodium chloride, and water for injection. | ||
| Manufactured by: LEO Pharma A/S Industriparken 55, DK-2750 Ballerup, Denmark Distributed by: LEO Pharma Inc., Madison, NJ 07940, USA U.S. License No. 2169 ADBRY™ is a trademark of LEO Pharma A/S. © 2021 LEO Pharma Inc. All rights reserved. For more information about ADBRY, go to www.ADBRY.com or call 1-844-MY-ADBRY (1-844-692-3279). |
||
INSTRUCTIONS FOR USE
ADBRY™ [ad'-bree]
(tralokinumab-ldrm)
injection, for subcutaneous use
This Instructions for Use contains information on how to inject ADBRY.
Read this Instructions for Use before you start using ADBRY prefilled syringes and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Keep this Instructions for Use and refer to it as needed.
Each single-dose prefilled syringe contains 150 mg of ADBRY. The ADBRY prefilled syringes are for single-dose only.
Important Information You Need to Know Before Injecting ADBRY:
- Your healthcare provider should show you how to prepare and inject ADBRY using the prefilled syringes before you inject ADBRY for the first time.
- Do not inject yourself or someone else until you have been shown how to inject ADBRY the right way.
- Talk to your healthcare provider if you have any questions about how to inject ADBRY the right way.
-
To receive your full prescribed dose, you will need to give more than 1 ADBRY injection.
- To get your full prescribed initial dose of 600 mg, you will need to give 4 injections at different injection sites within the same body area.
- To get your full prescribed dose of 300 mg, you will need to give 2 injections at different injection sites within the same body area.
- It is recommended to rotate the body area with the next set of injections.
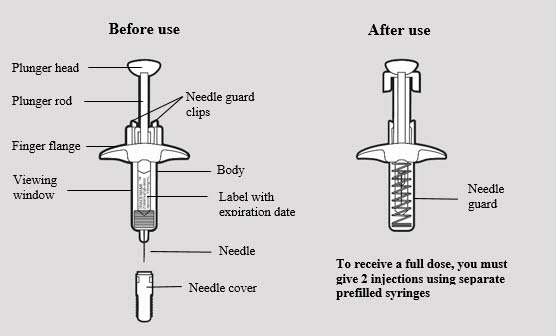
- The ADBRY prefilled syringes have a needle guard that will be activated to cover the needle after the injection is finished.
- For subcutaneous injection only (inject directly into fatty layer under the skin).
- Do not remove the needle cover until just before you give the injection.
- Do not share or reuse your ADBRY prefilled syringes.
- Do not inject through clothes.
ADBRY prefilled syringe parts (see Figure A):
Storing ADBRY
- Store ADBRY prefilled syringes in a refrigerator between 36°F to 46°F (2°C to 8°C).
- Store ADBRY prefilled syringes in the original carton and protect from light until you are ready to use them.
- ADBRY can be stored in the original carton at room temperature up to 86°F (30°C) for up to 14 days. Throw away (dispose of) syringes if left out of the refrigerator for more than 14 days.
- Do not freeze ADBRY prefilled syringes. Do not use if they have been frozen.
- Do not shake the ADBRY prefilled syringe.
- Do not heat the ADBRY prefilled syringe.
- Do not put the ADBRY prefilled syringe into direct sunlight.
- Keep ADBRY prefilled syringes and all medicines out of the reach of children.
Step 1: Setting up ADBRY injection
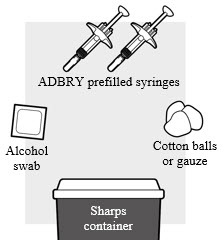
1a: Gather the supplies needed for your injection. For each ADBRY dose you will need (see Figure B):
- A clean flat, well-lit work surface, like a table
- 1 ADBRY carton that contains 2 ADBRY prefilled syringes
- An alcohol swab (not included in the carton)
- Clean gauze pads or cotton balls (not included in the carton)
- A puncture-resistant sharps disposal container (not included in the carton)
See Step 5 "Disposing of ADBRY" at the end of this Instructions for Use.
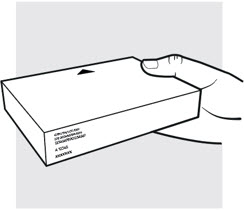
1b: Take the ADBRY prefilled syringe carton out of the refrigerator
- Check the expiration date (EXP) on the carton (see Figure C). Do not use if the expiration date on the carton has passed.
- Check to make sure the seal on the ADBRY carton is intact. Do not use the ADBRY prefilled syringes if the seal on the carton is broken.

1c: Let the ADBRY prefilled syringes warm up to room temperature (see Figure D)
Set the ADBRY carton on the flat surface and wait 30 minutes before you inject ADBRY to let the prefilled syringes warm up to room temperature 68°F to 86°F (20°C to 30°C). This will help to reduce discomfort and make it easier to inject ADBRY.
- Do not microwave the prefilled syringes, run hot water over them, or leave them in direct sunlight.
- Do not shake the syringes.
- Do not remove the needle cover on the prefilled syringes until you have reached Step 3 and are ready to inject.
- Do not put the syringes back in the refrigerator once they have reached room temperature.
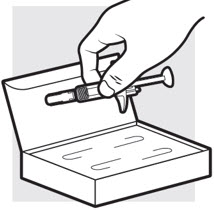
1d: Remove the ADBRY prefilled syringes from the carton
- Remove the 2 ADBRY prefilled syringes one by one from the carton by grasping the body (not the plunger rod) of the ADBRY prefilled syringes (see Figure E).
- Do not touch the needle guard clips to keep from activating the safety device (needle guard) too soon.
- Do not remove the needle cover on the prefilled syringes until you have reached Step 3 and are ready to inject.
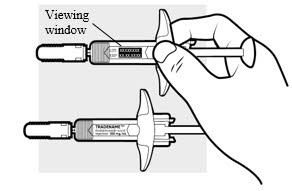
1e: Inspect the 2 ADBRY prefilled syringes (see Figure F)
- Make sure the ADBRY appears on the labels.
- Check the expiration date printed on the syringes.
- Check the medicine through the viewing windows. The medicine inside should be clear to slightly pearly and colorless to pale yellow.
- Do not use the ADBRY prefilled syringes, throw away and get new ones if:
- the expiration date printed on the syringes has passed
- the medicine is cloudy, discolored, or has particles in it
- they look damaged or have been dropped
- You may see small air bubbles in the liquid. This is normal. You do not need to do anything about it.
Step 2: Choosing and preparing injection area
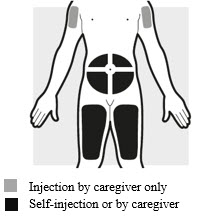
2a: Choose the area for your injections (see Figure G)
- You may inject into:
- your stomach area (abdomen)
- your thighs
- your upper arm. To inject into your upper arm, you will need a caregiver to give you the injections.
- Do not inject within 2 inches (5 cm) of your belly button (navel).
- Rotate the body area with each following set of injections. Do not use the same body area 2 times in a row.
- Do not inject where the skin is tender, damaged, bruised or scarred.
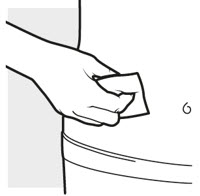
2b: Wash your hands and prepare your skin
- Wash your hands with soap and water.
- Clean the injection area for the 2 injections with an alcohol swab using a circular motion (see Figure H).
- Let the area dry completely.
- Do not blow or touch the cleaned area before injecting.
Step 3: Injecting ADBRY
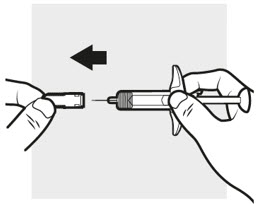
3a: Pull off the ADBRY needle cover
Hold the ADBRY prefilled syringe body with one hand, pull the needle cover straight off with your other hand (see Figure I) and throw it away in the sharps container.
- Do not try to recap the ADBRY prefilled syringes.
- Do not hold the plunger rod or plunger head while removing the needle cover.
- You may see a drop of liquid at the end of the needle. This is normal.
- Do not touch the needle, or let it touch any surface. If either of these occur, throw away the syringe and get a new one.
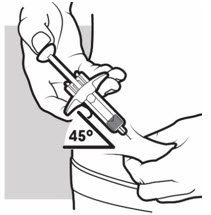
3b: Insert the needle
With one hand, gently pinch and hold a fold of skin where you cleaned the injection area. With the other hand, insert the needle completely at about a 45 degree angle into your skin (see Figure J).
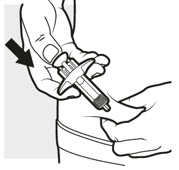
3c: Inject the medicine
Use your thumb to firmly push down the plunger head all the way down (see Figure K). All the medicine is injected when you cannot push the plunger head any further.
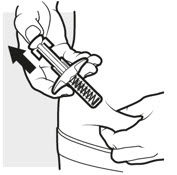
3d: Release and remove
Lift your thumb off the plunger head. The needle will automatically move back inside the syringe body and lock into place (see Figure L).
- Place a dry cotton ball or gauze pad over the injection site for a few seconds. Do not rub the injection site. If needed, cover the injection site with a small bandage.
- There may be a small amount of blood or liquid where you injected. This is normal.
Throw away the used ADBRY prefilled syringe in the sharps disposal container. See Step 5 "Disposing of ADBRY".
Step 4: Injecting the next syringe
To get your full prescribed dose, you will need to give more than 1 injection.
- To get your full prescribed initial dose of 600 mg, you will need to give 4 injections.
- To get your full prescribed dose of 300 mg, you will need to give 2 injections.
Get a new ADBRY prefilled syringe and repeat Steps 3 and 5 (see Figure M) for each injection you need to give for your full prescribed dose.
Note
Make sure each injection is at least 1 inch (3 cm) from the prior injection site and within the same body area.
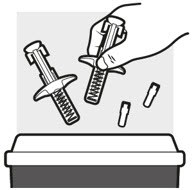
- Put the used ADBRY prefilled syringes in an FDA-cleared sharps disposal container right away after use (see Figure N). Do not throw away the ADBRY prefilled syringes in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not recycle your used sharps disposal container.
For more information go to www.ADBRY.com or call 1-844-692-3279. If you still have questions, call your healthcare provider.
Manufactured by: LEO Pharma A/S Industriparken 55, DK-2750 Ballerup, Denmark
Distributed by: LEO Pharma Inc., Madison, NJ 07940, USA
U.S. License No. 2169
ADBRY™ is a trademark of LEO Pharma A/S.
© 2021 LEO Pharma Inc. All rights reserved.
This "Instructions for Use" has been approved by the U.S. Food and Drug Administration.
Approved January 2022
PRINCIPAL DISPLAY PANEL - 150 mg/mL Syringe Carton - NDC 50222-346-02
Adbry™
(tralokinumab-ldrm)
Injection
150 mg/mL per syringe
x 2
NDC 50222-346-02
Rx only
For Subcutaneous Use Only
Two 1-mL single-dose prefilled syringes equal 300 mg
Carton contains:
- Two 1-mL single-dose prefilled syringes with needle guard
- Prescribing Information
- Instructions for Use and Patient Information
Store refrigerated at 36°F to 46°F (2°C to 8°C) in
the original carton to protect from light.
LEO®

PRINCIPAL DISPLAY PANEL - 150 mg/mL Syringe Carton - NDC 50222-346-04
Adbry™
(tralokinumab-ldrm)
Injection
150 mg/mL per syringe
x 4
NDC 50222-346-04
Rx only
For Subcutaneous Use Only
Four 1-mL single-dose prefilled syringes equal 600 mg
Multipack contains:
- Two cartons
- Each carton contains two 1-mL single-dose prefilled syringes
with needle guard which equal 300 mg
Store refrigerated at 36°F to 46°F (2°C to 8°C) in
the original carton to protect from light.
LEO®

| ADBRY
tralokinumab-ldrm injection, solution |
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||
| Labeler - LEO Pharma Inc. (832692615) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Catalent Indiana, LLC. | 172209277 | LABEL(50222-346) , PACK(50222-346) , MANUFACTURE(50222-346) , ANALYSIS(50222-346) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Astra Zeneca Pharmaceuticals LP | 968459953 | API MANUFACTURE(50222-346) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| LEO Pharma A/S | 306218108 | MANUFACTURE(50222-346) | |