MERIDIA- sibutramine hydrochloride capsule
AbbVie Inc
----------
MERIDIA®
(sibutramine hydrochloride monohydrate) Capsule
CS-IV
DESCRIPTION
MERIDIA® (sibutramine hydrochloride monohydrate) is an orally administered agent for the treatment of obesity. Chemically, the active ingredient is a racemic mixture of the (+) and (-) enantiomers of cyclobutanemethanamine, 1-(4-chlorophenyl)-N,N-dimethyl-α-(2-methylpropyl)-, hydrochloride, monohydrate, and has an empirical formula of C17H29Cl2NO. Its molecular weight is 334.33.
The structural formula is shown below:
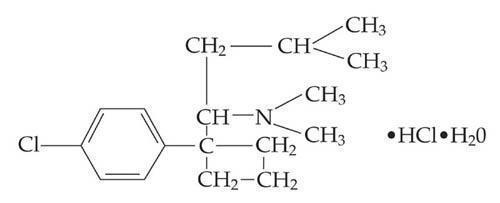
Sibutramine hydrochloride monohydrate is a white to cream crystalline powder with a solubility of 2.9 mg/mL in pH 5.2 water. Its octanol: water partition coefficient is 30.9 at pH 5.0.
Each MERIDIA capsule contains 5 mg, 10 mg, and 15 mg of sibutramine hydrochloride monohydrate. It also contains as inactive ingredients: lactose monohydrate, NF; microcrystalline cellulose, NF; colloidal silicon dioxide, NF; and magnesium stearate, NF in a hard-gelatin capsule [which contains titanium dioxide, USP; gelatin; FD&C Blue No. 2 (5- and 10-mg capsules only); D&C Yellow No. 10 (5- and 15-mg capsules only), and other inactive ingredients].
CLINICAL PHARMACOLOGY
Mode of Action
Sibutramine produces its therapeutic effects by norepinephrine, serotonin and dopamine reuptake inhibition. Sibutramine and its major pharmacologically active metabolites (M1 and M2) do not act via release of monoamines.
Pharmacodynamics
Sibutramine exerts its pharmacological actions predominantly via its secondary (M1) and primary (M2) amine metabolites. The parent compound, sibutramine, is a potent inhibitor of serotonin (5-hydroxytryptamine, 5-HT) and norepinephrine reuptake in vivo, but not in vitro. However, metabolites M1 and M2 inhibit the reuptake of these neurotransmitters both in vitro and in vivo.
In human brain tissue, M1 and M2 also inhibit dopamine reuptake in vitro, but with ~3-fold lower potency than for the reuptake inhibition of serotonin or norepinephrine.
| Serotonin | Norepinephrine | Dopamine | |
| Sibutramine | 298 | 5451 | 943 |
| M1 | 15 | 20 | 49 |
| M2 | 20 | 15 | 45 |
A study using plasma samples taken from sibutramine-treated volunteers showed monoamine reuptake inhibition of norepinephrine > serotonin > dopamine; maximum inhibitions were norepinephrine = 73%, serotonin = 54% and dopamine = 16%.
Sibutramine and its metabolites (M1 and M2) are not serotonin, norepinephrine or dopamine releasing agents. Following chronic administration of sibutramine to rats, no depletion of brain monoamines has been observed.
Sibutramine, M1 and M2 exhibit no evidence of anticholinergic or antihistaminergic actions. In addition, receptor binding profiles show that sibutramine, M1 and M2 have low affinity for serotonin (5-HT1, 5-HT1A, 5-HT1B, 5-HT2A, 5-HT2C), norepinephrine (β, β1, β3, α1 and α2), dopamine (D1 and D2), benzodiazepine, and glutamate (NMDA) receptors. These compounds also lack monoamine oxidase inhibitory activity in vitro and in vivo.
Pharmacokinetics
Absorption
Sibutramine is rapidly absorbed from the GI tract (Tmax of 1.2 hours) following oral administration and undergoes extensive first-pass metabolism in the liver (oral clearance of 1750 L/h and half-life of 1.1 h) to form the pharmacologically active mono- and di-desmethyl metabolites M1 and M2. Peak plasma concentrations of M1 and M2 are reached within 3 to 4 hours. On the basis of mass balance studies, on average, at least 77% of a single oral dose of sibutramine is absorbed. The absolute bioavailability of sibutramine has not been determined.
Distribution
Radiolabeled studies in animals indicated rapid and extensive distribution into tissues: highest concentrations of radiolabeled material were found in the eliminating organs, liver and kidney. In vitro, sibutramine, M1 and M2 are extensively bound (97%, 94% and 94%, respectively) to human plasma proteins at plasma concentrations seen following therapeutic doses.
Metabolism
Sibutramine is metabolized in the liver principally by the cytochrome P450 (3A4) isoenzyme, to desmethyl metabolites, M1 and M2. These active metabolites are further metabolized by hydroxylation and conjugation to pharmacologically inactive metabolites, M5 and M6. Following oral administration of radiolabeled sibutramine, essentially all of the peak radiolabeled material in plasma was accounted for by unchanged sibutramine (3%), M1 (6%), M2 (12%), M5 (52%), and M6 (27%).
M1 and M2 plasma concentrations reached steady-state within four days of dosing and were approximately two-fold higher than following a single dose. The elimination half-lives of M1 and M2, 14 and 16 hours, respectively, were unchanged following repeated dosing.
Excretion
Approximately 85% (range 68-95%) of a single orally administered radiolabeled dose was excreted in urine and feces over a 15-day collection period with the majority of the dose (77%) excreted in the urine. Major metabolites in urine were M5 and M6; unchanged sibutramine, M1, and M2 were not detected. The primary route of excretion for M1 and M2 is hepatic metabolism and for M5 and M6 is renal excretion.
| Mean (% CV) and 95% Confidence Intervals of Pharmacokinetic Parameters (Dose = 15 mg) | ||||
|
† Calculated only up to 24 hr for M1. |
||||
| Study Population | Cmax
(ng/mL) | Tmax
(h) | AUC†
(ng*h/mL) | T½ (h) |
| Metabolite M1 | ||||
| Target Population: | ||||
| Obese Subjects (n = 18) | 4.0 (42) 3.2 - 4.8 | 3.6 (28) 3.1 - 4.1 | 25.5 (63) 18.1 - 32.9 | – – |
| Special Population: | ||||
| Moderate Hepatic Impairment (n = 12) | 2.2 (36) 1.8 - 2.7 | 3.3 (33) 2.7 - 3.9 | 18.7 (65) 11.9 - 25.5 | – – |
| Metabolite M2 | ||||
| Target Population: | ||||
| Obese Subjects (n = 18) | 6.4 (28) 5.6 - 7.2 | 3.5 (17) 3.2 - 3.8 | 92.1 (26) 81.2 - 103 | 17.2 (58) 12.5 - 21.8 |
| Special Population: | ||||
| Moderate Hepatic Impairment (n = 12) | 4.3 (37) 3.4 - 5.2 | 3.8 (34) 3.1 - 4.5 | 90.5 (27) 76.9 - 104 | 22.7 (30) 18.9 - 26.5 |
Effect of Food
Administration of a single 20 mg dose of sibutramine with a standard breakfast resulted in reduced peak M1 and M2 concentrations (by 27% and 32%, respectively) and delayed the time to peak by approximately three hours. However, the AUCs of M1 and M2 were not significantly altered.
Special Populations
Geriatric
Plasma concentrations of M1 and M2 were similar between elderly (ages 61 to 77 yr) and young (ages 19 to 30 yr) subjects following a single 15-mg oral sibutramine dose. Plasma concentrations of the inactive metabolites M5 and M6 were higher in the elderly; these differences are not likely to be of clinical significance. Sibutramine is contraindicated in patients over 65 years of age (see CONTRAINDICATIONS).
Pediatric
The safety and effectiveness of sibutramine in pediatric patients under 16 years old have not been established.
Gender
Pooled pharmacokinetic parameters from 54 young, healthy volunteers (37 males and 17 females) receiving a 15-mg oral dose of sibutramine showed the mean Cmax and AUC of M1 and M2 to be slightly (≤ 19% and ≤ 36%, respectively) higher in females than males. Somewhat higher steady-state trough plasma levels were observed in female obese patients from a large clinical efficacy trial. However, these differences are not likely to be of clinical significance. Dosage adjustment based upon the gender of a patient is not necessary (see DOSAGE AND ADMINISTRATION).
Race
The relationship between race and steady-state trough M1 and M2 plasma concentrations was examined in a clinical trial in obese patients. A trend towards higher concentrations in Black patients over Caucasian patients was noted for M1 and M2. However, these differences are not considered to be of clinical significance.
Renal Insufficiency
The disposition of sibutramine metabolites (M1, M2, M5 and M6) following a single oral dose of sibutramine was studied in patients with varying degrees of renal function. Sibutramine itself was not measurable.
In patients with moderate and severe renal impairment, the AUC values of the active metabolite M1 were 24 to 46% higher and the AUC values of M2 were similar as compared to healthy subjects. Cross- study comparison showed that the patients with end - stage renal disease on dialysis had similar AUC values of M1 but approximately half of the AUC values of M2 measured in healthy subjects (CLcr ≥ 80 mL/ min). The AUC values of inactive metabolites M5 and M6 increased 2 - 3 fold (range 1 - to 7 - fold) in patients with moderate impairment (30 mL/ min < CLcr = 60 mL/ min) and 8 - 11 fold (range 5 - to 15 - fold) in patients with severe impairment (CLcr ≤ 30 mL/ min) as compared to healthy subjects. Cross - study comparison showed that the AUC values of M5 and M6 increased 22 - 33 fold in patients with end - stage renal disease on dialysis as compared to healthy subjects. Approximately 1% of the oral dose was recovered in the dialysate as a combination of M5 and M6 during the hemodialysis process, while M1 and M2 were not measurable in the dialysate.
Sibutramine should not be used in patients with severe renal impairment, including those with end-stage renal disease on dialysis.
Hepatic Insufficiency
In 12 patients with moderate hepatic impairment receiving a single 15-mg oral dose of sibutramine, the combined AUCs of M1 and M2 were increased by 24% compared to healthy subjects while M5 and M6 plasma concentrations were unchanged. The observed differences in M1 and M2 concentrations do not warrant dosage adjustment in patients with mild to moderate hepatic impairment. Sibutramine should not be used in patients with severe hepatic dysfunction.
Drug-Drug Interactions
In vitro studies indicated that the cytochrome P450 (3A4)-mediated metabolism of sibutramine was inhibited by ketoconazole and to a lesser extent by erythromycin. Phase 1 clinical trials were conducted to assess the interactions of sibutramine with drugs that are substrates and/or inhibitors of various cytochrome P450 isozymes. The potential for studied interactions is described below.
Ketoconazole
Concomitant administration of 200 mg doses of ketoconazole twice daily and 20 mg sibutramine once daily for 7 days in 12 uncomplicated obese subjects resulted in moderate increases in AUC and Cmax of 58% and 36% for M1 and of 20% and 19% for M2, respectively.
Erythromycin
The steady-state pharmacokinetics of sibutramine and metabolites M1 and M2 were evaluated in 12 uncomplicated obese subjects following concomitant administration of 500 mg of erythromycin three times daily and 20 mg of sibutramine once daily for 7 days. Concomitant erythromycin resulted in small increases in the AUC (less than 14%) for M1 and M2. A small reduction in Cmax for M1 (11%) and a slight increase in Cmax for M2 (10%) were observed.
Cimetidine
Concomitant administration of cimetidine 400 mg twice daily and sibutramine 15 mg once daily for 7 days in 12 volunteers resulted in small increases in combined (M1 and M2) plasma Cmax (3.4%) and AUC (7.3%).
Simvastatin
Steady-state pharmacokinetics of sibutramine and metabolites M1 and M2 were evaluated in 27 healthy volunteers after the administration of simvastatin 20 mg once daily in the evening and sibutramine 15 mg once daily in the morning for 7 days. Simvastatin had no significant effect on plasma Cmax and AUC of M2 or M1 and M2 combined. The Cmax (16%) and AUC (12%) of M1 were slightly decreased. Simvastatin slightly decreased sibutramine Cmax (14%) and AUC (21%). Sibutramine increased the AUC (7%) of the pharmacologically active moiety, simvastatin acid and reduced the Cmax (25%) and AUC (15%) of inactive simvastatin.
Omeprazole
Steady-state pharmacokinetics of sibutramine and metabolites M1 and M2 were evaluated in 26 healthy volunteers after the co-administration of omeprazole 20 mg once daily and sibutramine 15 mg once daily for 7 days. Omeprazole slightly increased plasma Cmax and AUC of M1 and M2 combined (approximately 15%). M2 Cmax and AUC were not significantly affected whereas M1 Cmax (30%) and AUC (40%) were modestly increased. Plasma Cmax (57%) and AUC (67%) of unchanged sibutramine were moderately increased. Sibutramine had no significant effect on omeprazole pharmacokinetics.
Olanzapine
Steady-state pharmacokinetics of sibutramine and metabolites M1 and M2 were evaluated in 24 healthy volunteers after the co-administration of sibutramine 15 mg once daily with olanzapine 5 mg twice daily for 3 days and 10 mg once daily thereafter for 7 days. Olanzapine had no significant effect on plasma Cmax and AUC of M2 and M1 and M2 combined, or the AUC of M1. Olanzapine slightly increased M1 Cmax (19%), and moderately increased sibutramine Cmax (47%) and AUC (63%). Sibutramine had no significant effect on olanzapine pharmacokinetics.
Lorazepam
Steady-state pharmacokinetics of sibutramine and metabolites M1 and M2 after sibutramine 15 mg once daily for 11 days were compared in 25 healthy volunteers in the presence or absence of lorazepam 2 mg twice daily for 3 days plus one morning dose. Lorazepam had no significant effect on the pharmacokinetics of sibutramine metabolites M1 and M2. Sibutramine had no significant effect on lorazepam pharmacokinetics.
Drugs Highly Bound to Plasma Proteins
Although sibutramine and its active metabolites M1 and M2 are extensively bound to plasma proteins (≥94%), the low therapeutic concentrations and basic characteristics of these compounds make them unlikely to result in clinically significant protein binding interactions with other highly protein bound drugs such as warfarin and phenytoin. In vitro protein binding interaction studies have not been conducted.
CLINICAL STUDIES
Observational epidemiologic studies have established a relationship between obesity and the risks for cardiovascular disease, non-insulin dependent diabetes mellitus (NIDDM), certain forms of cancer, gallstones, certain respiratory disorders, and an increase in overall mortality. These studies suggest that weight loss, if maintained, may produce health benefits for some patients with chronic obesity who may also be at risk for other diseases.
The long-term effects of sibutramine on the morbidity and mortality associated with obesity have not been established. Weight loss was examined in 11 double-blind, placebo-controlled obesity trials (BMI range across all studies 27-43) with study durations of 12 to 52 weeks and doses ranging from 1 to 30 mg once daily. Weight was significantly reduced in a dose-related manner in sibutramine-treated patients compared to placebo over the dose range of 5 to 20 mg once daily. In two 12-month studies, maximal weight loss was achieved by 6 months and statistically significant weight loss was maintained over 12 months. The amount of placebo-subtracted weight loss achieved on sibutramine was consistent across studies.
Analysis of the data in three long-term (≥ 6 months) obesity trials indicates that patients who lose at least 4 pounds in the first 4 weeks of therapy with a given dose of sibutramine are most likely to achieve significant long-term weight loss on that dose of sibutramine. Approximately 60% of such patients went on to achieve a placebo-subtracted weight loss of ≥ 5% of their initial body weight by month 6. Conversely, of those patients on a given dose of sibutramine who did not lose at least 4 pounds in the first 4 weeks of therapy, approximately 80% did not go on to achieve a placebo-subtracted weight loss of ≥ 5% of their initial body weight on that dose by month 6.
Significant dose-related reductions in waist circumference, an indicator of intra-abdominal fat, have also been observed over 6 and 12 months in placebo-controlled clinical trials. In a 12-week placebo-controlled study of non-insulin dependent diabetes mellitus patients randomized to placebo or 15 mg per day of sibutramine, Dual Energy X-Ray Absorptiometry (DEXA) assessment of changes in body composition showed that total body fat mass decreased by 1.8 kg in the sibutramine group versus 0.2 kg in the placebo group (p < 0.001). Similarly, truncal (android) fat mass decreased by 0.6 kg in the sibutramine group versus 0.1 kg in the placebo group (p < 0.01). The changes in lean mass, fasting blood sugar, and HbA1 were not statistically significantly different between the two groups.
Eleven double-blind, placebo-controlled obesity trials with study durations of 12 to 52 weeks have provided evidence that sibutramine does not adversely affect glycemia, serum lipid profiles, or serum uric acid in obese patients. Treatment with sibutramine (5 to 20 mg once daily) is associated with mean increases in blood pressure of 1 to 3 mm Hg and with mean increases in pulse rate of 4 to 5 beats per minute relative to placebo. These findings are similar in normotensives and in patients with hypertension controlled with medication. Those patients who lose significant (≥ 5% weight loss) amounts of weight on sibutramine tend to have smaller increases in blood pressure and pulse rate (see WARNINGS).
In Study 1, a 6-month, double-blind, placebo-controlled study in obese patients, Study 2, a 1-year, double-blind, placebo-controlled study in obese patients, and Study 3, a 1-year, double-blind, placebo-controlled study in obese patients who lost at least 6 kg on a 4-week very low calorie diet (VLCD), sibutramine produced significant reductions in weight, as shown below. In the two 1-year studies, maximal weight loss was achieved by 6 months and statistically significant weight loss was maintained over 12 months.
| Sibutramine (mg) | |||||
|
* Data for all patients who received study drug and who had any post-baseline measurement (last observation carried forward analysis). ** Data for patients who completed the entire 6-month (Study 1) or one-year period of dosing and have data recorded for the month 6 (Study 1) or month 12 visit. *** Data for patients who lost at least 4 lbs in the first 4 weeks of treatment and completed the study. **** Weight loss data shown describe changes in weight from the pre-VLCD; mean weight loss during the 4-week VLCD was 16.9 lbs for sibutramine and 16.3 lbs for placebo. |
|||||
| Study/Patient Group | Placebo (n) | 5 (n) | 10 (n) | 15 (n) | 20 (n) |
| Study 1 | |||||
| All patients* | 2.0 (142) | 6.6 (148) | 9.7 (148) | 12.1 (150) | 13.6 (145) |
| Completers** | 2.9 (84) | 8.1 (103) | 12.1 (95) | 15.4 (94) | 18.0 (89) |
| Early responders*** | 8.5 (17) | 13.0 (60) | 16.0 (64) | 18.2 (73) | 20.1 (76) |
| Study 2 | |||||
| All patients* | 3.5 (157) | 9.8 (154) | 14.0 (152) | ||
| Completers** | 4.8 (76) | 13.6 (80) | 15.2 (93) | ||
| Early responders*** | 10.7 (24) | 18.2 (57) | 18.8 (76) | ||
| Study 3**** | |||||
| All patients* | 15.2 (78) | 28.4 (81) | |||
| Completers** | 16.7 (48) | 29.7 (60) | |||
| Early responders*** | 21.5 (22) | 33.0 (46) | |||
Maintenance of weight loss with sibutramine was examined in a 2-year, double-blind, placebo-controlled trial. After a 6-month run-in phase in which all patients received sibutramine 10 mg (mean weight loss, 26 lbs.), patients were randomized to sibutramine (10 to 20 mg, 352 patients) or placebo (115 patients). The mean weight loss from initial body weight to endpoint was 21 lbs. and 12 lbs. for sibutramine and placebo patients, respectively. A statistically significantly (p < 0.001) greater proportion of sibutramine treated patients, 75%, 62%, and 43%, maintained at least 80% of their initial weight loss at 12, 18, and 24 months, respectively, compared with the placebo group (38%, 23%, and 16%). Also 67%, 37%, 17%, and 9% of sibutramine treated patients compared with 49%, 19%, 5%, and 3% of placebo patients lost ≥ 5%, ≥ 10%, ≥ 15%, and ≥ 20%, respectively, of their initial body weight at endpoint. From endpoint to the post-study follow-up visit (about 1 month), weight regain was approximately 4 lbs for the sibutramine patients and approximately 2 lbs for the placebo patients.
Sibutramine induced weight loss has been accompanied by beneficial changes in serum lipids that are similar to those seen with nonpharmacologically-mediated weight loss. A combined, weighted analysis of the changes in serum lipids in 11 placebo-controlled obesity studies ranging in length from 12 to 52 weeks is shown below for the last observation carried forward (LOCF) analysis.
| Category | TG
% (n) | CHOL
% (n) | LDL-C
% (n) | HDL-C
% (n) |
|
Baseline mean values: HDL-C: High Density Lipoprotein-Cholesterol |
||||
| All Placebo | 0.53 (475) | -1.53 (475) | -0.09 (233) | -0.56 (248) |
| < 5% Weight Loss | 4.52 (382) | -0.42 (382) | -0.70 (205) | -0.71 (217) |
| ≥ 5% Weight Loss | -15.30 (92) | -6.23 (92) | -6.19 (27) | 0.94 (30) |
| All Sibutramine | -8.75 (1164) | -2.21 (1165) | -1.85 (642) | 4.13 (664) |
| < 5% Weight Loss | -0.54 (547) | 0.17 (548) | -0.37 (320) | 3.19 (331) |
| ≥ 5% Weight Loss | -16.59 (612) | -4.87 (612) | -4.56 (317) | 4.68 (328) |
Sibutramine induced weight loss has been accompanied by reductions in serum uric acid. Certain centrally-acting weight loss agents that cause release of serotonin from nerve terminals have been associated with cardiac valve dysfunction. The possible occurrence of cardiac valve disease was specifically investigated in two studies. In one study 2-D and color Doppler echocardiography were performed on 210 patients (mean age, 54 years) receiving sibutramine 15 mg or placebo daily for periods of 2 weeks to 16 months (mean duration of treatment, 7.6 months). In patients without a prior history of valvular heart disease, the incidence of valvular heart disease was 3/132 (2.3%) in the sibutramine treatment group (all three cases were mild aortic insufficiency) and 2/77 (2.6%) in the placebo treatment group (one case of mild aortic insufficiency and one case of severe aortic insufficiency). In another study, 25 patients underwent 2-D and color Doppler echocardiography before treatment with sibutramine and again after treatment with sibutramine 5 to 30 mg daily for three months; there were no cases of valvular heart disease.
The effect of sibutramine 15 mg once daily on measures of 24-hour blood pressure was evaluated in a 12-week placebo-controlled study. Twenty-six male and female, primarily Caucasian individuals with an average BMI of 34 kg/m2 and an average age of 39 years underwent 24-hour ambulatory blood pressure monitoring (ABPM). The mean changes from baseline to Week 12 in various measures of ABPM are shown in the following table.
| Parameter | Systolic | Diastolic | ||||
| mm Hg | Placebo | Sibutramine | Placebo | Sibutramine | ||
| n=12 | 15 mg n=14 | 20 mg n=16 | 15 mg n=12 | 20 mg n=16 |
||
| Daytime | 0.2 | 3.9 | 4.4 | 0.5 | 5.0 | 5.7 |
| Nighttime | -0.3 | 4.1 | 6.4 | -1.0 | 4.3 | 5.4 |
| Early am | -0.9 | 9.4 | 5.3 | -3.0 | 6.7 | 5.8 |
| 24-hour mean | -0.1 | 4.0 | 4.7 | 0.1 | 5.0 | 5.6 |
Normal diurnal variation of blood pressure was maintained.
INDICATIONS AND USAGE
MERIDIA is indicated for the management of obesity, including weight loss and maintenance of weight loss, and should be used in conjunction with a reduced calorie diet. MERIDIA is recommended for obese patients with an initial body mass index ≥ 30 kg/m2, or ≥ 27 kg/m2 in the presence of other risk factors (e.g., diabetes, dyslipidemia, controlled hypertension).
Below is a chart of Body Mass Index (BMI) based on various heights and weights.
BMI is calculated by taking the patient's weight, in kg, and dividing by the patient's height, in meters, squared. Metric conversions are as follows: pounds ÷ 2.2 = kg; inches × 0.0254 = meters.

CONTRAINDICATIONS
MERIDIA is contraindicated in patients:
-
with a history of coronary artery disease (e.g., angina, history of myocardial infarction), congestive heart failure, tachycardia, peripheral arterial occlusive disease, arrhythmia or cerebrovascular disease (stroke or transient ischemic attack (TIA)) (see WARNINGS).
-
with inadequately controlled hypertension > 145/90 mm Hg (see WARNINGS).
-
over 65 years of age.
-
receiving monoamine oxidase inhibitors (MAOIs) (see WARNINGS).
-
with hypersensitivity to sibutramine or any of the inactive ingredients of MERIDIA.
-
who have a major eating disorder (anorexia nervosa or bulimia nervosa).
-
taking other centrally acting weight loss drugs.
WARNINGS
Concomitant Cardiovascular Disease
Due to an increased risk of heart attack and stroke in patients with cardiovascular disease, MERIDIA should not be used in patients with a history of coronary artery disease, congestive heart failure, arrhythmias, or stroke.
Blood Pressure and Pulse
MERIDIA SUBSTANTIALLY INCREASES BLOOD PRESSURE AND/OR PULSE RATE IN SOME PATIENTS. REGULAR MONITORING OF BLOOD PRESSURE AND PULSE RATE IS REQUIRED WHEN PRESCRIBING MERIDIA.
In placebo-controlled obesity studies, sibutramine 5 to 20 mg once daily was associated with mean increases in systolic and diastolic blood pressure of approximately 1 to 3 mm Hg relative to placebo, and with mean increases in pulse rate relative to placebo of approximately 4 to 5 beats per minute. Larger increases were seen in some patients, particularly when therapy with sibutramine was initiated at the higher doses (see table below). In premarketing placebo-controlled obesity studies, 0.4% of patients treated with sibutramine were discontinued for hypertension (SBP ≥160 mm Hg or DBP ≥ 95 mm Hg), compared with 0.4% in the placebo group, and 0.4% of patients treated with sibutramine were discontinued for tachycardia (pulse rate ≥ 100 bpm), compared with 0.1% in the placebo group. Blood pressure and pulse should be measured prior to starting therapy with MERIDIA and should be monitored at regular intervals thereafter. For patients who experience a sustained increase in blood pressure or pulse rate while receiving MERIDIA, either dose reduction or discontinuation should be considered. MERIDIA should be given with caution to those patients with a history of hypertension (see DOSAGE AND ADMINISTRATION), and should not be given to patients with uncontrolled or poorly controlled hypertension.
| Dose (mg) | % Outliers* | ||
|
* Outlier defined as increase from baseline of ≥ 15 mm Hg for three consecutive visits (SBP), ≥ 10 mm Hg for three consecutive visits (DBP), or pulse ≥ 10 bpm for three consecutive visits. |
|||
| SBP | DBP | Pulse | |
| Placebo | 9 | 7 | 12 |
| 5 | 6 | 20 | 16 |
| 10 | 12 | 15 | 28 |
| 15 | 13 | 17 | 24 |
| 20 | 14 | 22 | 37 |
Potential Interaction With Monoamine Oxidase Inhibitors
MERIDIA is a norepinephrine, serotonin and dopamine reuptake inhibitor and should not be used concomitantly with MAOIs (see PRECAUTIONS, Drug Interactions subsection). There should be at least a 2-week interval after stopping MAOIs before commencing treatment with MERIDIA. Similarly, there should be at least a 2-week interval after stopping MERIDIA before starting treatment with MAOIs.
Serotonin Syndrome or Neuroleptic Malignant Syndrome (NMS)-Like Reactions
The development of a potentially life-threatening serotonin syndrome, or Neuroleptic Malignant Syndrome (NMS)-like reactions, has been reported with SNRIs and SSRIs alone, including MERIDIA treatment, but particularly with concomitant use of serotonergic drugs (including triptans), with drugs which impair metabolism of serotonin (including MAOIs), or with antipsychotics or other dopamine antagonists. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination) and/or gastrointestinal symptoms [e.g., nausea, vomiting, diarrhea] (see PRECAUTIONS, Drug Interactions). Serotonin syndrome, in its most severe form, can resemble neuroleptic malignant syndrome, which includes hyperthermia, muscle rigidity, autonomic instability with possible rapid fluctuation of vital signs, and mental status changes. Patients should be monitored for the emergence of serotonin syndrome or NMS-like signs and symptoms.
PRECAUTIONS
Pulmonary Hypertension
Certain centrally-acting weight loss agents that cause release of serotonin from nerve terminals have been associated with pulmonary hypertension (PPH), a rare but lethal disease. In premarketing clinical studies, no cases of PPH have been reported with sibutramine capsules. Because of the low incidence of this disease in the underlying population, however, it is not known whether or not MERIDIA may cause this disease.
Seizures
During premarketing testing, seizures were reported in < 0.1% of sibutramine treated patients. MERIDIA should be used cautiously in patients with a history of seizures. It should be discontinued in any patient who develops seizures.
Bleeding
There have been reports of bleeding in patients taking sibutramine. While a causal relationship is unclear, caution is advised in patients predisposed to bleeding events and those taking concomitant medications known to affect hemostasis or platelet function.
Renal Impairment
MERIDIA should be used with caution in patients with mild to moderate renal impairment. MERIDIA should not be used in patients with severe renal impairment, including those with end stage renal disease on dialysis (see Pharmacokinetics-Special Populations-Renal Insufficiency).
Hepatic Dysfunction
Patients with severe hepatic dysfunction have not been systematically studied; MERIDIA should therefore not be used in such patients.
Interference With Cognitive and Motor Performance
Although sibutramine did not affect psychomotor or cognitive performance in healthy volunteers, any CNS active drug has the potential to impair judgment, thinking or motor skills.
Information For Patients
Physicians should instruct their patients to read the Medication Guide before starting therapy with MERIDIA and to reread it each time the prescription is renewed.
Physicians should also discuss with their patients any part of the package insert that is relevant to them. In particular, the importance of keeping appointments for follow-up visits should be emphasized.
Patients should be advised to notify their physician if they develop a rash, hives, or other allergic reactions.
Patients should be advised to inform their physicians if they are taking, or plan to take, any prescription or over-the-counter drugs, especially weight-reducing agents, decongestants, antidepressants, cough suppressants, lithium, dihydroergotamine, sumatriptan (Imitrex®), or tryptophan, since there is a potential for interactions.
Patients should be reminded of the importance of having their blood pressure and pulse monitored at regular intervals.
Drug Interactions
CNS Active Drugs:
The use of MERIDIA in combination with other CNS-active drugs, particularly serotonergic agents, has not been systematically evaluated. Consequently, caution is advised if the concomitant administration of MERIDIA with other centrally-acting drugs is indicated (see CONTRAINDICATIONS and WARNINGS).
In patients receiving monoamine oxidase inhibitors (MAOIs) (e.g., phenelzine, selegiline) in combination with serotonergic agents (e.g., fluoxetine, fluvoxamine, paroxetine, sertraline, venlafaxine), there have been reports of serious, sometimes fatal, reactions ("serotonin syndrome;" see below). Because sibutramine inhibits serotonin reuptake, MERIDIA should not be used concomitantly with a MAOI (see CONTRAINDICATIONS ). At least 2 weeks should elapse between discontinuation of a MAOI and initiation of treatment with MERIDIA. Similarly, at least 2 weeks should elapse between discontinuation of MERIDIA and initiation of treatment with a MAOI.
The rare, but serious, constellation of symptoms termed "serotonin syndrome" has also been reported with the concomitant use of selective serotonin reuptake inhibitors and agents for migraine therapy, such as Imitrex® (sumatriptan succinate) and dihydroergotamine, certain opioids, such as dextromethorphan, meperidine, pentazocine and fentanyl, lithium, or tryptophan. Serotonin syndrome has also been reported with the concomitant use of two serotonin reuptake inhibitors. The syndrome requires immediate medical attention and may include one or more of the following symptoms: excitement, hypomania, restlessness, loss of consciousness, confusion, disorientation, anxiety, agitation, motor weakness, myoclonus, tremor, hemiballismus, hyperreflexia, ataxia, dysarthria, incoordination, hyperthermia, shivering, pupillary dilation, diaphoresis, emesis, and tachycardia.
Because sibutramine inhibits serotonin reuptake, in general, it should not be administered with other serotonergic agents such as those listed above. However, if such a combination is clinically indicated, appropriate observation of the patient is warranted.
Drugs That May Raise Blood Pressure and/or Heart Rate
Concomitant use of MERIDIA and other agents that may raise blood pressure or heart rate have not been evaluated. These include certain decongestants, cough, cold, and allergy medications that contain agents such as ephedrine, or pseudoephedrine. Caution should be used when prescribing MERIDIA to patients who use these medications.
Alcohol
In a double-blind, placebo-controlled, crossover study in 19 volunteers, administration of a single dose of ethanol (0.5 mL/kg) together with 20 mg of sibutramine resulted in no psychomotor interactions of clinical significance between alcohol and sibutramine. However, the concomitant use of MERIDIA and excess alcohol is not recommended.
Oral Contraceptives
The suppression of ovulation by oral contraceptives was not inhibited by sibutramine. In a crossover study, 12 healthy female volunteers on oral steroid contraceptives received placebo in one period and 15 mg sibutramine in another period over the course of 8 weeks. No clinically significant systemic interaction was observed; therefore, no requirement for alternative contraceptive precautions are needed when patients taking oral contraceptives are concurrently prescribed sibutramine.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
Sibutramine was administered in the diet to mice (1.25, 5 or 20 mg/kg/day) and rats (1, 3, or 9 mg/kg/day) for two years generating combined maximum plasma AUC's of the two major active metabolites equivalent to 0.4 and 16 times, respectively, those following a daily human dose of 15 mg. There was no evidence of carcinogenicity in mice or in female rats. In male rats there was a higher incidence of benign tumors of the testicular interstitial cells; such tumors are commonly seen in rats and are hormonally mediated. The relevance of these tumors to humans is not known.
Mutagenicity
Sibutramine was not mutagenic in the Ames test, in vitro Chinese hamster V79 cell mutation assay, in vitro clastogenicity assay in human lymphocytes or micronucleus assay in mice. Its two major active metabolites were found to have equivocal bacterial mutagenic activity in the Ames test. However, both metabolites gave consistently negative results in the in vitro Chinese hamster V79 cell mutation assay, in vitro clastogenicity assay in human lymphocytes, in vitro DNA-repair assay in HeLa cells, micronucleus assay in mice and in vivo unscheduled DNA-synthesis assay in rat hepatocytes.
Impairment of Fertility
In rats, there were no effects on fertility at doses generating combined plasma AUC's of the two major active metabolites up to 32 times those following a human dose of 15 mg. At 13 times the human combined AUC, there was maternal toxicity, and the dams' nest-building behavior was impaired, leading to a higher incidence of perinatal mortality; there was no effect at approximately 4 times the human combined AUC.
Pregnancy
Teratogenic Effects
Pregnancy Category C
Radiolabeled studies in animals indicated that tissue distribution was unaffected by pregnancy, with relatively low transfer to the fetus. In rats, there was no evidence of teratogenicity at doses of 1, 3, or 10 mg/kg/day generating combined plasma AUC's of the two major active metabolites up to approximately 32 times those following the human dose of 15 mg. In rabbits dosed at 3, 15, or 75 mg/kg/day, plasma AUC's greater than approximately 5 times those following the human dose of 15 mg caused maternal toxicity. At markedly toxic doses, Dutch Belted rabbits had a slightly higher than control incidence of pups with a broad short snout, short rounded pinnae, short tail and, in some, shorter thickened long bones in the limbs; at comparably high doses in New Zealand White rabbits, one study showed a slightly higher than control incidence of pups with cardiovascular anomalies while a second study showed a lower incidence than in the control group.
No adequate and well controlled studies with sibutramine have been conducted in pregnant women. The use of MERIDIA during pregnancy is not recommended. Women of childbearing potential should employ adequate contraception while taking MERIDIA. Patients should be advised to notify their physician if they become pregnant or intend to become pregnant while taking MERIDIA.
Nursing Mothers
It is not known whether sibutramine or its metabolites are excreted in human milk. MERIDIA is not recommended for use in nursing mothers. Patients should be advised to notify their physician if they are breast-feeding.
Pediatric Use
The efficacy of sibutramine in adolescents who are obese has not been adequately studied.
Sibutramine's mechanism of action inhibiting the reuptake of serotonin and norepinephrine is similar to the mechanism of action of some antidepressants. Pooled analyses of short-term placebo-controlled trials of antidepressants in children and adolescents with major depressive disorder (MDD), obsessive compulsive disorder (OCD), and other psychiatric disorders have revealed a greater risk of adverse events representing suicidal behavior or thinking during the first few months of treatment in those receiving antidepressants. The average risk of such events in patients receiving antidepressants was 4%, twice the placebo risk of 2%.
No placebo-controlled trials of sibutramine have been conducted in children or adolescents with MDD, OCD, or other psychiatric disorders. In a study of adolescents with obesity in which 368 patients were treated with sibutramine and 130 patients with placebo, one patient in the sibutramine group and one patient in the placebo group attempted suicide. Suicidal ideation was reported by 2 sibutramine-treated patients and none of the placebo patients. It is unknown if sibutramine increases the risk of suicidal behavior or thinking in pediatric patients.
The data are inadequate to recommend the use of sibutramine for the treatment of obesity in pediatric patients.
ADVERSE REACTIONS
In placebo-controlled studies, 9% of patients treated with sibutramine (n = 2068) and 7% of patients treated with placebo (n = 884) withdrew for adverse events.
In placebo-controlled studies, the most common events were dry mouth, anorexia, insomnia, constipation and headache. Adverse events in these studies occurring in ≥ 1% of sibutramine treated patients and more frequently than in the placebo group are shown in the following table.
| BODY SYSTEM Adverse Event | Sibutramine (n = 2068) | Placebo (n = 884) |
| % Incidence | % Incidence | |
| BODY AS A WHOLE: | ||
| Headache | 30.3 | 18.6 |
| Back pain | 8.2 | 5.5 |
| Flu syndrome | 8.2 | 5.8 |
| Injury accident | 5.9 | 4.1 |
| Asthenia | 5.9 | 5.3 |
| Abdominal pain | 4.5 | 3.6 |
| Chest pain | 1.8 | 1.2 |
| Neck pain | 1.6 | 1.1 |
| Allergic reaction | 1.5 | 0.8 |
| CARDIOVASCULAR SYSTEM | ||
| Tachycardia | 2.6 | 0.6 |
| Vasodilation | 2.4 | 0.9 |
| Migraine | 2.4 | 2.0 |
| Hypertension/increased blood pressure | 2.1 | 0.9 |
| Palpitation | 2.0 | 0.8 |
| DIGESTIVE SYSTEM | ||
| Anorexia | 13.0 | 3.5 |
| Constipation | 11.5 | 6.0 |
| Increased appetite | 8.7 | 2.7 |
| Nausea | 5.9 | 2.8 |
| Dyspepsia | 5.0 | 2.6 |
| Gastritis | 1.7 | 1.2 |
| Vomiting | 1.5 | 1.4 |
| Rectal disorder | 1.2 | 0.5 |
| METABOLIC & NUTRITIONAL | ||
| Thirst | 1.7 | 0.9 |
| Generalized edema | 1.2 | 0.8 |
| MUSCULOSKELETAL SYSTEM | ||
| Arthralgia | 5.9 | 5.0 |
| Myalgia | 1.9 | 1.1 |
| Tenosynovitis | 1.2 | 0.5 |
| Joint disorder | 1.1 | 0.6 |
| NERVOUS SYSTEM | ||
| Dry mouth | 17.2 | 4.2 |
| Insomnia | 10.7 | 4.5 |
| Dizziness | 7.0 | 3.4 |
| Nervousness | 5.2 | 2.9 |
| Anxiety | 4.5 | 3.4 |
| Depression | 4.3 | 2.5 |
| Paresthesia | 2.0 | 0.5 |
| Somnolence | 1.7 | 0.9 |
| CNS stimulation | 1.5 | 0.5 |
| Emotional lability | 1.3 | 0.6 |
| RESPIRATORY SYSTEM | ||
| Rhinitis | 10.2 | 7.1 |
| Pharyngitis | 10.0 | 8.4 |
| Sinusitis | 5.0 | 2.6 |
| Cough increase | 3.8 | 3.3 |
| Laryngitis | 1.3 | 0.9 |
| SKIN & APPENDAGES | ||
| Rash | 3.8 | 2.5 |
| Sweating | 2.5 | 0.9 |
| Herpes simplex | 1.3 | 1.0 |
| Acne | 1.0 | 0.8 |
| SPECIAL SENSES | ||
| Taste perversion | 2.2 | 0.8 |
| Ear disorder | 1.7 | 0.9 |
| Ear pain | 1.1 | 0.7 |
| UROGENITAL SYSTEM | ||
| Dysmenorrhea | 3.5 | 1.4 |
| Urinary tract infection | 2.3 | 2.0 |
| Vaginal monilia | 1.2 | 0.5 |
| Metrorrhagia | 1.0 | 0.8 |
The following additional adverse events were reported in ≥ 1% of all patients who received sibutramine in controlled and uncontrolled premarketing studies.
Other Adverse Events
Clinical Studies
Seizures
Convulsions were reported as an adverse event in three of 2068 (0.1%) sibutramine treated patients and in none of 884 placebo-treated patients in placebo-controlled premarketing obesity studies. Two of the three patients with seizures had potentially predisposing factors (one had a prior history of epilepsy; one had a subsequent diagnosis of brain tumor). The incidence in all subjects who received sibutramine (three of 4,588 subjects) was less than 0.1%.
Ecchymosis/Bleeding Disorders
Ecchymosis (bruising) was observed in 0.7% of sibutramine treated patients and in 0.2% of placebo-treated patients in premarketing placebo-controlled obesity studies. One patient had prolonged bleeding of a small amount which occurred during minor facial surgery. Sibutramine may have an effect on platelet function due to its effect on serotonin uptake.
Interstitial Nephritis
Acute interstitial nephritis (confirmed by biopsy) was reported in one obese patient receiving sibutramine during premarketing studies. After discontinuation of the medication, dialysis and oral corticosteroids were administered; renal function normalized. The patient made a full recovery.
Altered Laboratory Findings
Abnormal liver function tests, including increases in AST, ALT, GGT, LDH, alkaline phosphatase and bilirubin, were reported as adverse events in 1.6% of sibutramine-treated obese patients in placebo-controlled trials compared with 0.8% of placebo patients. In these studies, potentially clinically significant values (total bilirubin ≥ 2 mg/dL; ALT, AST, GGT, LDH, or alkaline phosphatase ≥ 3 × upper limit of normal) occurred in 0% (alkaline phosphatase) to 0.6% (ALT) of the sibutramine treated patients and in none of the placebo-treated patients. Abnormal values tended to be sporadic, often diminished with continued treatment, and did not show a clear dose-response relationship.
Postmarketing Reports
Voluntary reports of adverse events temporally associated with the use of sibutramine are listed below. It is important to emphasize that although these events occurred during treatment with sibutramine, they may have no causal relationship with the drug. Obesity itself, concurrent disease states/risk factors, or weight reduction may be associated with an increased risk for some of these events.
Psychiatric
Cases of depression, psychosis, mania, suicidal ideation and suicide have been reported rarely in patients on sibutramine treatment. However, a relationship has not been established between these events and the use of sibutramine. If any of these events should occur during treatment with sibutramine, discontinuation should be considered.
Hypersensitivity
Allergic hypersensitivity reactions ranging from mild skin eruptions and urticaria to angioedema and anaphylaxis have been reported (see CONTRAINDICATIONS and PRECAUTIONS-Information For Patients, and other reports of allergic reactions listed below).
Other Postmarketing Reported Events:
Body as a Whole
anaphylactic shock, anaphylactoid reaction, chest pressure, chest tightness, facial edema, limb pain, sudden unexplained death.
Cardiovascular System
angina pectoris, atrial fibrillation, congestive heart failure, heart arrest, heart rate decreased, myocardial infarction, supraventricular tachycardia, syncope, torsade de pointes, vascular headache, ventricular tachycardia, ventricular extrasystoles, ventricular fibrillation.
Digestive System
cholecystitis, cholelithiasis, duodenal ulcer, eructation, gastrointestinal hemorrhage, increased salivation, intestinal obstruction, mouth ulcer, stomach ulcer, tongue edema.
Nervous System
abnormal dreams, abnormal gait, amnesia, anger, cerebrovascular accident, concentration impaired, confusion, depression aggravated, Gilles de la Tourette’s syndrome, hypesthesia, libido decreased, libido increased, mood changes, nightmares, short term memory loss, speech disorder, transient ischemic attack, tremor, twitch, vertigo.
OVERDOSAGE
Overdose Management
There is limited experience of overdose with sibutramine. The most frequently noted adverse events associated with overdose are tachycardia, hypertension, headache and dizziness. Treatment should consist of general measures employed in the management of overdosage: an airway should be established as needed; cardiac and vital sign monitoring is recommended; general symptomatic and supportive measures should be instituted. Cautious use of β-blockers may be indicated to control elevated blood pressure or tachycardia. The results from a study in patients with end-stage renal disease on dialysis showed that sibutramine metabolites were not eliminated to a significant degree with hemodialysis. (see Pharmacokinetics-Special Populations-Renal Insufficiency).
DOSAGE AND ADMINISTRATION
The recommended starting dose of MERIDIA is 10 mg administered once daily with or without food. If there is inadequate weight loss, the dose may be titrated after four weeks to a total of 15 mg once daily. The 5 mg dose should be reserved for patients who do not tolerate the 10 mg dose. Blood pressure and heart rate changes should be taken into account when making decisions regarding dose titration (see WARNINGS and PRECAUTIONS).
Doses above 15 mg daily are not recommended. In most of the clinical trials, MERIDIA was given in the morning.
Analysis of numerous variables has indicated that approximately 60% of patients who lose at least 4 pounds in the first 4 weeks of treatment with a given dose of MERIDIA in combination with a reduced-calorie diet lose at least 5% (placebo-subtracted) of their initial body weight by the end of 6 months to 1 year of treatment on that dose of MERIDIA. Conversely, approximately 80% of patients who do not lose at least 4 pounds in the first 4 weeks of treatment with a given dose of MERIDIA do not lose at least 5% (placebo-subtracted) of their initial body weight by the end of 6 months to 1 year of treatment on that dose. If a patient has not lost at least 4 pounds in the first 4 weeks of treatment, the physician should consider reevaluation of therapy which may include increasing the dose or discontinuation of MERIDIA.
The safety and effectiveness of MERIDIA, as demonstrated in double-blind, placebo-controlled trials, have not been determined beyond 2 years at this time.
HOW SUPPLIED
MERIDIA® (sibutramine hydrochloride monohydrate) Capsules contain 5 mg, 10 mg, or 15 mg sibutramine hydrochloride monohydrate and are supplied as follows:
5 mg, NDC 0074-2456-11, blue/yellow capsules imprinted with "MERIDIA” on the cap and "-5-" on the body, in bottles of 30 capsules.
10 mg, NDC 0074-2457-11, blue/white capsules imprinted with "MERIDIA” on the cap and "-10-” on the body, in bottles of 30 capsules.
15 mg, NDC 0074-2458-11, yellow/white capsules imprinted with "MERIDIA” on the cap and "-15-” on the body, in bottles of 30 capsules.
MEDICATION GUIDE
MERIDIA® (mer-ID-dee-uh) (CIV)
(sibutramine hydrochloride monohydrate)
Capsules
Read this Medication Guide before you start taking MERIDIA and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment.
What is the most important information I should know about MERIDIA?
MERIDIA can cause serious side effects including a large increase in your blood pressure or heart rate (pulse). Do not take MERIDIA if your blood pressure is not well controlled. Call your doctor right away if you check your blood pressure and it is higher than normal for you, or if you have symptoms of high blood pressure such as headache, dizziness or blurred vision.
Before you start taking MERIDIA, your doctor should check your blood pressure and heart rate. Your doctor should continue checking your blood pressure regularly while you are taking MERIDIA. It is important that you have regular check-ups while you are taking MERIDIA.
What is MERIDIA?
MERIDIA is a prescription medicine used to help overweight or obese people lose weight and keep the weight off. MERIDIA should be used together with a low calorie diet.
MERIDIA contains sibutramine, a substance that people can become addicted to. Keep your MERIDIA in a safe place to protect it from theft. Never give your MERIDIA to anyone else, because it may cause death or harm them. Selling or giving away this medicine is against the law.
The use of MERIDIA for more than 2 years has not been studied.
It is not known if MERIDIA is safe and effective in children younger than 16 years old.
Who should not take MERIDIA?
Do not take MERIDIA if you:
- have or have had, heart problems, including:
- heart attack
- chest pain
- heart failure
- fast or irregular heart beat
- hardening of your arteries or other blood vessels
- poor circulation in your legs
- have or have ever had, a stroke or symptoms of a stroke
- uncontrolled high blood pressure (above 145/90)
- are over age 65
- are taking or have taken a type of medicine used to treat depression called a monoamine oxidase inhibitor (MAOI) in the past 2 weeks. Do not take MAOIs for at least 2 weeks before using MERIDIA. Do not take MAOIs for at least 2 weeks after stopping MERIDIA. Ask your doctor or pharmacist if you are not sure if any of your medicines are MAOIs.
- have an eating problem called anorexia nervosa or bulimia nervosa.
- are taking certain other weight loss medicines.
- are allergic to sibutramine hydrochloride monohydrate or any other ingredients in MERIDIA. See the end of this Medication Guide for a complete list of ingredients in MERIDIA.
Talk to your doctor before taking this medicine if you have any of these conditions.
What should I tell my doctor before taking MERIDIA?
Before you take MERIDIA, tell your doctor if you:
- have liver or kidney problems
- have glaucoma
- have or had seizures (convulsions, fits)
- have bleeding problems
- have or had gallstones
- are pregnant or plan to become pregnant. It is not known if MERIDIA will harm your unborn baby. Talk to your doctor if you are pregnant or plan to become pregnant. If you can become pregnant, you should use birth control while taking MERIDIA. Tell your doctor right away if you become pregnant while taking MERIDIA.
- are breastfeeding or plan to breastfeed. It is not known if MERIDIA passes into your breast milk. You and your doctor should decide if you will take MERIDIA or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Using MERIDIA with certain other medicines may affect how MERIDIA or the other medicines work. Using MERIDIA with other medicines can cause serious side effects.
Especially tell your doctor if you take:
- a monoamine oxidase inhibitors (MAOIs) medicine. See "Who should not take MERIDIA?"
- other weight loss medicines
- cough and cold medicines
- migraine headache medicines like sumatriptan (Imitrex, Imitrex Statdose) or dihydroergotamine (D.H.E 45, Migranal)
- medicines to treat depression
- narcotic pain medicines
- lithium (Lithobid)
- tryptophan
- medicines that thin the blood
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get new medicine.
How should I take MERIDIA?
- Take MERIDIA exactly as your doctor tells you to.
- Take MERIDIA 1 time a day.
- If you miss a dose of MERIDIA, just skip it. Do not take an extra dose to make up for missed doses.
- If you take too much MERIDIA, call your doctor or Poison Control Center right away, or go to the emergency room.
- Your doctor may change your dose if needed.
- Take MERIDIA with or without food.
- You should see your doctor regularly for check-ups.
What should I avoid while taking MERIDIA?
- Do not drive, operate heavy machinery or do other dangerous activities until you know how MERIDIA affects you.
- Do not have more than two standard alcoholic drinks per day while you take MERIDIA.
What are the possible side effects of MERIDIA?
MERIDIA may cause serious side effects, including:
- See “What is the most important information I should know about MERIDIA?”
- serotonin syndrome. Serotonin syndrome may happen when people take MERIDIA with certain other medicines that affect a brain chemical called serotonin. Do not take other medicines with MERIDIA unless your doctor has told you to. Get medical help right away if you have any of the following symptoms:
- feel weak, restless, confused, or anxious
- lose consciousness (faint)
- have a fever, vomiting, sweating, shivering or shaking
- have a fast heartbeat
- seizures (convulsions, fits)
- bleeding. Bleeding may happen if you have a condition that causes bleeding or if you take a blood thinning medicine.
Certain weight loss medicines have a rare but life-threatening problem that affects blood pressure in the lungs (pulmonary hypertension). It is not known if MERIDIA may cause this problem because pulmonary hypertension is so rare. Call your doctor right away if you have new or worsening shortness of breath.
The most common side effects of MERIDIA include:
- dry mouth
- loss of appetite
- trouble sleeping
- constipation
- headache
Tell your doctor if you get a rash or hives while taking MERIDIA. You may be having an allergic reaction.
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the side effects of MERIDIA. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store MERIDIA?
- Store MERIDIA between 59°F to 86° F (15°C to 30° C).
- Keep MERIDIA capsules dry and away from heat.
- Keep MERIDIA in a tightly closed container, and keep MERIDIA out of the light.
- Safely throw away medicine that is out of date or no longer needed.
Keep MERIDIA and all medicines out of reach of children.
General information about MERIDIA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use MERIDIA for a condition for which it was not prescribed. Do not give MERIDIA to other people, even if they have the same symptoms you have. It may harm them and it is against the law.
This Medication Guide summarizes the most important information about MERIDIA. If you would like more information, talk with your doctor. You can also ask your doctor or pharmacist for information about MERIDIA that is written for health professionals.
For more information, go to www.Meridia.net, or call 1-800-633-9110.
What are the ingredients in MERIDIA?
Active ingredient: sibutramine hydrochloride monohydrate
Inactive ingredients: lactose monohydrate, microcrystalline cellulose, colloidal silicon dioxide, and magnesium stearate.
Hard-gelatin capsule: titanium dioxide, gelatin, FD&C Blue No. 2 (5 mg and 10 mg capsules only), D&C Yellow No. 10 (5 mg and 15 mg capsules only), and other inactive ingredients.



| MERIDIA
sibutramine hydrochloride capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| MERIDIA
sibutramine hydrochloride capsule |
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
| MERIDIA
sibutramine hydrochloride capsule |
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
| Labeler - AbbVie Inc (078458370) |