Label: DANTROLENE injection, powder, for solution



- NDC Code(s): 0143-9297-01
- Packager: Hikma Pharmaceuticals USA Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated February 9, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
Dantrolene Sodium for Injection, USP is a sterile, non-pyrogenic, lyophilized formulation. Dantrolene Sodium for Injection, USP is supplied in 100 mL vials containing 20 mg dantrolene sodium, 3,000 mg mannitol, and sufficient sodium hydroxide to yield a pH of approximately 9.5 when reconstituted with 60 mL sterile water for injection USP (without a bacteriostatic agent).
Dantrolene sodium is classified as a direct-acting skeletal muscle relaxant. Chemically, dantrolene sodium is hydrated 1-[[[5-(4-nitrophenyl)-2furanyl]methylene]amino]-2,4-imidazolidinedione sodium salt. The structural formula for the hydrated salt is:
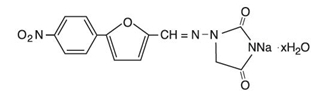
The hydrated salt contains approximately 15% water (3.5 moles) and has a molecular weight of 399. The anhydrous salt (dantrolene) has a molecular weight of 336.
-
CLINICAL PHARMACOLOGY
In isolated nerve-muscle preparation, dantrolene sodium has been shown to produce relaxation by affecting the contractile response of the muscle at a site beyond the myoneural junction. In skeletal muscle, dantrolene sodium dissociates the excitation-contraction coupling, probably by interfering with the release of Ca++ from the sarcoplasmic reticulum. The administration of intravenous dantrolene sodium to human volunteers is associated with loss of grip strength and weakness in the legs, as well as subjective CNS complaints (see also PRECAUTIONS, Information for Patients). Information concerning the passage of dantrolene sodium across the blood-brain barrier is not available.
In the anesthetic-induced malignant hyperthermia syndrome, evidence points to an intrinsic abnormality of skeletal muscle tissue. In affected humans, it has been postulated that "triggering agents" (e.g., general anesthetics and depolarizing neuromuscular blocking agents) produce a change within the cell which results in an elevated myoplasmic calcium. This elevated myoplasmic calcium activates acute cellular catabolic processes that cascade to the malignant hyperthermia crisis.
It is hypothesized that addition of dantrolene sodium to the "triggered" malignant hyperthermic muscle cell reestablishes a normal level of ionized calcium in the myoplasm. Inhibition of calcium release from the sarcoplasmic reticulum by dantrolene sodium reestablishes the myoplasmic calcium equilibrium, increasing the percentage of bound calcium. In this way, physiologic, metabolic, and biochemical changes associated with the malignant hyperthermia crisis may be reversed or attenuated. Experimental results in malignant hyperthermia susceptible swine show that prophylactic administration of intravenous or oral dantrolene prevents or attenuates the development of vital sign and blood gas changes characteristic of malignant hyperthermia in a dose related manner. The efficacy of intravenous dantrolene in the treatment of human and porcine malignant hyperthermia crisis, when considered along with prophylactic experiments in malignant hyperthermia susceptible swine, lends support to prophylactic use of oral or intravenous dantrolene in malignant hyperthermia susceptible humans. When prophylactic intravenous dantrolene is administered as directed, whole blood concentrations remain at a near steady state level for 3 or more hours after the infusion is completed. Clinical experience has shown that early vital sign and/or blood gas changes characteristic of malignant hyperthermia may appear during or after anesthesia and surgery despite the prophylactic use of dantrolene and adherence to currently accepted patient management practices. These signs are compatible with attenuated malignant hyperthermia and respond to the administration of additional intravenous dantrolene (see DOSAGE AND ADMINISTRATION). The administration of the recommended prophylactic dose of intravenous dantrolene to healthy volunteers was not associated with clinically significant cardiorespiratory changes.
Specific metabolic pathways for the degradation and elimination of dantrolene sodium in humans have been established. Dantrolene is found in measurable amounts in blood and urine. Its major metabolites in body fluids are 5-hydroxy dantrolene and an acetylamino metabolite of dantrolene. Another metabolite with an unknown structure appears related to the latter. Dantrolene sodium may also undergo hydrolysis and subsequent oxidation forming nitrophenylfuroic acid.
The mean biologic half-life of dantrolene sodium after intravenous administration is variable, between 4 to 8 hours under most experimental conditions. Based on assays of whole blood and plasma, slightly greater amounts of dantrolene are associated with red blood cells than with the plasma fraction of blood. Significant amounts of dantrolene are bound to plasma proteins, mostly albumin, and this binding is readily reversible.
Cardiopulmonary depression has not been observed in malignant hyperthermia susceptible swine following the administration of up to 7.5 mg/kg intravenous dantrolene. This is twice the amount needed to maximally diminish twitch response to single supramaximal peripheral nerve stimulation (95% inhibition). A transient, inconsistent, depressant effect on gastrointestinal smooth muscles has been observed at high doses.
-
INDICATIONS AND USAGE
Dantrolene Sodium for Injection is indicated, along with appropriate supportive measures, for the management of the fulminant hypermetabolism of skeletal muscle characteristic of malignant hyperthermia crises in patients of all ages. Dantrolene Sodium for Injection should be administered by continuous rapid intravenous push as soon as the malignant hyperthermia reaction is recognized (i.e., tachycardia, tachypnea, central venous desaturation, hypercarbia, metabolic acidosis, skeletal muscle rigidity, increased utilization of anesthesia circuit carbon dioxide absorber, cyanosis and mottling of the skin, and, in many cases, fever).
Dantrolene Sodium for Injection is also indicated preoperatively, and sometimes postoperatively, to prevent or attenuate the development of clinical and laboratory signs of malignant hyperthermia in individuals judged to be malignant hyperthermia susceptible.
- CONTRAINDICATIONS
-
WARNINGS
The use of Dantrolene Sodium for Injection in the management of malignant hyperthermia crisis is not a substitute for previously known supportive measures. These measures must be individualized, but it will usually be necessary to discontinue the suspect triggering agents, attend to increased oxygen requirements, manage the metabolic acidosis, institute cooling when necessary, monitor urinary output, and monitor for electrolyte imbalance.
Since the effect of disease state and other drugs on dantrolene sodium related skeletal muscle weakness, including possible respiratory depression, cannot be predicted, patients who receive intravenous dantrolene sodium preoperatively should have vital signs monitored.
If patients judged malignant hyperthermia susceptible are administered intravenous or oral dantrolene sodium preoperatively, anesthetic preparation must still follow a standard malignant hyperthermia susceptible regimen, including the avoidance of known triggering agents. Monitoring for early clinical and metabolic signs of malignant hyperthermia is indicated because attenuation of malignant hyperthermia, rather than prevention, is possible. These signs usually call for the administration of additional intravenous dantrolene.
-
PRECAUTIONS
General
Care must be taken to prevent extravasation of dantrolene sodium solution into the surrounding tissues due to the high pH of the intravenous formulation and potential for tissue necrosis.
When mannitol is used for prevention or treatment of late renal complications of malignant hyperthermia, the 3 g of mannitol needed to dissolve each 20 mg vial of intravenous dantrolene sodium should be taken into consideration.
Information for Patients
Based upon data in human volunteers, perioperatively, it is appropriate to tell patients who receive Dantrolene Sodium for Injection that symptoms of muscle weakness should be expected postoperatively (i.e. decrease in grip strength and weakness of leg muscles, especially walking down stairs). In addition, symptoms such as "lightheadedness" may be noted. Since some of these symptoms may persist for up to 48 hours, patients must not operate an automobile or engage in other hazardous activity during this time. Caution is also indicated at meals on the day of administration because difficulty swallowing and choking has been reported. Caution should be exercised in the concomitant administration of tranquilizing agents.
Hepatotoxicity Seen with Dantrolene Sodium Capsules
Dantrolene sodium has a potential for hepatotoxicity, and should not be used in conditions other than those recommended. Symptomatic hepatitis (fatal and non-fatal) has been reported at various dose levels of the drug. The incidence reported in patients taking up to 400 mg/day is much lower than in those taking doses of 800 mg or more per day. Even sporadic short courses of these higher dose levels within a treatment regimen markedly increased the risk of serious hepatic injury. Liver dysfunction as evidenced by blood chemical abnormalities alone (liver enzyme elevations) has been observed in patients exposed to dantrolene sodium for varying periods of time. Overt hepatitis has occurred at varying intervals after initiation of therapy, but has been most frequently observed between the third and twelfth month of therapy. The risk of hepatic injury appears to be greater in females, in patients over 35 years of age, and in patients taking other medication(s) in addition to dantrolene sodium. Dantrolene sodium should be used only in conjunction with appropriate monitoring of hepatic function including frequent determination of SGOT or SGPT.
Fatal and non-fatal liver disorders of an idiosyncratic or hypersensitivity type may occur with dantrolene sodium therapy.
Drug Interactions
Dantrolene sodium is metabolized by the liver, and it is theoretically possible that its metabolism may be enhanced by drugs known to induce hepatic microsomal enzymes. However, neither phenobarbital nor diazepam appears to affect dantrolene sodium metabolism. Binding to plasma protein is not significantly altered by diazepam, diphenylhydantoin, or phenylbutazone. Binding to plasma proteins is reduced by warfarin and clofibrate and increased by tolbutamide.
Cardiovascular collapse in association with marked hyperkalemia has been reported in patients receiving dantrolene in combination with calcium channel blockers. It is recommended that the combination of intravenous dantrolene sodium and calcium channel blockers, such as verapamil, not be used together during the management of malignant hyperthermia crisis.
Administration of dantrolene may potentiate vecuronium-induced neuromuscular block.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Sprague-Dawley female rats fed dantrolene sodium for 18 months at dosage levels of 15, 30, and 60 mg/kg/day showed an increased incidence of benign and malignant mammary tumors compared with concurrent controls. At the highest dose level (approximately the same as the maximum recommended daily dose on a mg/m2 basis), there was an increase in the incidence of benign hepatic lymphatic neoplasms. In a 30-month study in Sprague-Dawley rats fed dantrolene sodium, the highest dose level (approximately the same as the maximum recommended daily dose on a mg/m2 basis) produced a decrease in the time of onset of mammary neoplasms. Female rats at the highest dose level showed an increased incidence of hepatic lymphangiomas and hepatic angiosarcomas.
The only drug-related effect seen in a 30-month study in Fischer-344 rats was a dose-related reduction in the time of onset of mammary and testicular tumors. A 24-month study in HaM/ICR mice revealed no evidence of carcinogenic activity.
The significance of carcinogenicity data relative to use of dantrolene sodium in humans is unknown.
Dantrolene sodium has produced positive results in the Ames S. Typhimurium bacterial mutagenesis assay in the presence and absence of a liver activating system.
Dantrolene sodium administered to male and female rats at dose levels up to 45 mg/kg/day (approximately 1.4 times the maximum recommended daily dose on a mg/m2 basis) showed no adverse effects on fertility or general reproductive performance.
Pregnancy
Pregnancy Category C
Dantrolene sodium has been shown to be embryocidal in the rabbit and has been shown to decrease pup survival in the rat when given at doses seven times the human oral dose. There are no adequate and well-controlled studies in pregnant women. Dantrolene Sodium for Injection should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Labor and Delivery
In one uncontrolled study, 100 mg per day of prophylactic oral dantrolene sodium was administered to term pregnant patients awaiting labor and delivery. Dantrolene readily crossed the placenta, with maternal and fetal whole blood levels approximately equal at delivery; neonatal levels then fell approximately 50% per day for 2 days before declining sharply. No neonatal respiratory and neuromuscular side effects were detected at low dose. More data, at higher doses, are needed before more definitive conclusions can be made.
Nursing Mothers
Dantrolene has been detected in human milk at low concentrations (less than 2 micrograms per mL) during repeat intravenous administration over 3 days. Because of the potential for serious adverse reactions in nursing infants from dantrolene, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of Dantrolene Sodium for Injection did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
There have been occasional reports of death following malignant hyperthermia crisis even when treated with intravenous dantrolene; incidence figures are not available (the pre-dantrolene mortality of malignant hyperthermia crisis was approximately 50%). Most of these deaths can be accounted for by late recognition, delayed treatment, inadequate dosage, lack of supportive therapy, intercurrent disease and/or the development of delayed complications such as renal failure or disseminated intravascular coagulopathy. In some cases there are insufficient data to completely rule out therapeutic failure of dantrolene.
There are reports of fatality in malignant hyperthermia crisis, despite initial satisfactory response to intravenous dantrolene, which involve patients who could not be weaned from dantrolene after initial treatment.
The administration of intravenous Dantrolene Sodium for Injection to human volunteers is associated with loss of grip strength and weakness in the legs, as well as drowsiness and dizziness.
The following adverse reactions are in approximate order of severity:
There are rare reports of pulmonary edema developing during the treatment of malignant hyperthermia crisis in which the diluent volume and mannitol needed to deliver intravenous dantrolene possibly contributed.
There have been reports of thrombophlebitis following administration of intravenous dantrolene; actual incidence figures are not available. Tissue necrosis secondary to extravasation has been reported.
There have been rare reports of urticaria and erythema possibly associated with the administration of intravenous dantrolene sodium. There has been one case of anaphylaxis.
Injection site reactions (pain, erythema, swelling), commonly due to extravasation, have been reported.
None of the serious reactions occasionally reported with long-term oral dantrolene sodium use, such as hepatitis, seizures, and pleural effusion with pericarditis, have been reasonably associated with short-term Dantrolene Sodium for Injection therapy.
The following events have been reported in patients receiving oral dantrolene: aplastic anemia, leukopenia, lymphocytic lymphoma, and heart failure. (See package insert for dantrolene sodium capsules for a complete listing of adverse reactions.)
The published literature has included some reports of dantrolene sodium use in patients with Neuroleptic Malignant Syndrome (NMS). Dantrolene Sodium for Injection is not indicated for the treatment of NMS and patients may expire despite treatment with Dantrolene Sodium for Injection.
For medical advice about adverse reactions contact your medical professional. To report SUSPECTED ADVERSE REACTIONS, contact Hikma Pharmaceuticals USA Inc. at 1-877-233-2001 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
OVERDOSAGE
Because Dantrolene Sodium for Injection must be administered at a low concentration in a large volume of fluid, acute toxicity of dantrolene sodium could not be assessed in animals. In 14-day (subacute) studies, the intravenous formulation of dantrolene sodium was relatively non-toxic to rats at doses of 10 mg/kg/day and 20 mg/kg/day. While 10 mg/kg/day in dogs for 14 days evoked little toxicity, 20 mg/kg/day for 14 days caused hepatic changes of questionable biologic significance.
Symptoms which may occur in case of overdose include, but are not limited to, muscular weakness and alterations in the state of consciousness (e.g., lethargy, coma), vomiting, diarrhea, and crystalluria.
For acute overdosage, general supportive measures should be employed.
Intravenous fluids should be administered in fairly large quantities to avert the possibility of crystalluria. An adequate airway should be maintained and artificial resuscitation equipment should be at hand. Electrocardiographic monitoring should be instituted, and the patient carefully observed. The value of dialysis in dantrolene sodium overdose is not known.
-
DOSAGE AND ADMINISTRATION
As soon as the malignant hyperthermia reaction is recognized, all anesthetic agents should be discontinued; the administration of 100% oxygen is recommended. Dantrolene Sodium for Injection should be administered by continuous rapid intravenous push beginning at a minimum dose of 1 mg/kg, and continuing until symptoms subside or the maximum cumulative dose of 10 mg/kg has been reached.
If the physiologic and metabolic abnormalities reappear, the regimen may be repeated. It is important to note that administration of Dantrolene Sodium for Injection should be continuous until symptoms subside. The effective dose to reverse the crisis is directly dependent upon the individual's degree of susceptibility to malignant hyperthermia, the amount and time of exposure to the triggering agent, and the time elapsed between onset of the crisis and initiation of treatment.
Pediatric Dose
Experience to date indicates that the dose of Dantrolene Sodium for Injection for pediatric patients is the same as for adults.
Preoperatively
Dantrolene Sodium for Injection and/or Dantrolene Sodium Capsules may be administered preoperatively to patients judged malignant hyperthermia susceptible as part of the overall patient management to prevent or attenuate the development of clinical and laboratory signs of malignant hyperthermia.
The recommended prophylactic dose of Dantrolene Sodium for Injection is 2.5 mg/kg, starting approximately 1-1/4 hours before anticipated anesthesia and infused over approximately 1 hour. This dose should prevent or attenuate the development of clinical and laboratory signs of malignant hyperthermia provided that the usual precautions, such as avoidance of established malignant hyperthermia triggering agents, are followed.
Additional Dantrolene Sodium for Injection may be indicated during anesthesia and surgery because of the appearance of early clinical and/or blood gas signs of malignant hyperthermia or because of prolonged surgery (see also CLINICAL PHARMACOLOGY, WARNINGS, and PRECAUTIONS). Additional doses must be individualized.
Oral Administration of Dantrolene Oral Capsules: Administer 4 to 8 mg/kg/day of oral Dantrolene Sodium in three or four divided doses for 1 or 2 days prior to surgery, with the last dose being given with a minimum of water approximately 3 to 4 hours before scheduled surgery. Adjustment can usually be made within the recommended dosage range to avoid incapacitation (weakness, drowsiness, etc.) or excessive gastrointestinal irritation (nausea and/or vomiting).
See also the package insert for Dantrolene Sodium Capsules, USP.
Post Crisis Follow-Up
Dantrolene Sodium Capsules, 4 to 8 mg/kg/day, in four divided doses should be administered 1 to 3 days following a malignant hyperthermia crisis to prevent recurrence of the manifestations of malignant hyperthermia.
Intravenous dantrolene sodium may be used postoperatively to prevent or attenuate the recurrence of signs of malignant hyperthermia when oral dantrolene sodium administration is not practical. The intravenous dose of dantrolene sodium in the postoperative period must be individualized, starting with 1 mg/kg or more as the clinical situation dictates.
Preparation
Each vial of Dantrolene Sodium for Injection should be reconstituted by adding 60 mL of sterile water for injection USP (without a bacteriostatic agent), and the vial shaken until the solution is clear. 5% Dextrose Injection USP, 0.9% Sodium Chloride Injection USP, and other acidic solutions are not compatible with Dantrolene Sodium for Injection and should not be used. The contents of the vial must be protected from direct light and used within 6 hours after reconstitution. Store reconstituted solutions at controlled room temperature (59°F to 86°F or 15°C to 30°C).
Reconstituted Dantrolene Sodium for Injection should not be transferred to large glass bottles for prophylactic infusion due to precipitate formation observed with the use of some glass bottles as reservoirs.
For prophylactic infusion, the required number of individual vials of Dantrolene Sodium for Injection should be reconstituted as outlined above. The contents of individual vials are then transferred to a larger volume sterile intravenous plastic bag. Stability data on file indicate commercially available sterile plastic bags are acceptable drug delivery devices. However, it is recommended that the prepared infusion be inspected carefully for cloudiness and/or precipitation prior to dispensing and administration. Such solutions should not be used. While stable for 6 hours, it is recommended that the infusion be prepared immediately prior to the anticipated dosage administration time.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.
-
HOW SUPPLIED
Dantrolene Sodium for Injection, USP (NDC 0143-9297-01) is available in vials containing a sterile lyophilized mixture of 20 mg dantrolene sodium, 3,000 mg mannitol, and sufficient sodium hydroxide to yield a pH of approximately 9.5 when reconstituted with 60 mL sterile water for injection USP (without a bacteriostatic agent).
Store reconstituted solution at 20° to 25°C (68º to 77°F) [See USP Controlled Room Temperature].
Protect from direct light.
Store unreconstituted product at 20° to 25°C (68º to 77°F) [See USP Controlled Room Temperature]. Avoid prolonged exposure to light.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL
NDC 0143-9297-01 Rx only
Dantrolene
Sodium for
Injection, USP
20 mg per vial
For treatment of malignant hyperthermia
For Intravenous use ONLY
Single Dose Vial

NDC 0143-9297-01 Rx only
Dantrolene
Sodium for Injection, USP
20 mg per vial
For treatment of malignant hyperthermia
For Intravenous use ONLY
1 Single Dose Vial, 20 mg in each vial

- SERIALIZATION IMAGE
-
INGREDIENTS AND APPEARANCE
DANTROLENE
dantrolene injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0143-9297 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DANTROLENE SODIUM (UNII: 287M0347EV) (DANTROLENE - UNII:F64QU97QCR) DANTROLENE SODIUM 20 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 3000 mg SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0143-9297-01 1 in 1 VIAL; Type 0: Not a Combination Product 06/19/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA204762 06/19/2017 Labeler - Hikma Pharmaceuticals USA Inc. (001230762)
