HIVID- zalcitabine tablet, film coated
Genentech, Inc.
----------
WARNING
THE USE OF HIVID HAS BEEN ASSOCIATED WITH SIGNIFICANT CLINICAL ADVERSE REACTIONS, SOME OF WHICH ARE POTENTIALLY FATAL. HIVID CAN CAUSE SEVERE PERIPHERAL NEUROPATHY AND BECAUSE OF THIS SHOULD BE USED WITH EXTREME CAUTION IN PATIENTS WITH PREEXISTING NEUROPATHY. HIVID MAY ALSO RARELY CAUSE PANCREATITIS AND PATIENTS WHO DEVELOP ANY SYMPTOMS SUGGESTIVE OF PANCREATITIS WHILE USING HIVID SHOULD HAVE THERAPY SUSPENDED IMMEDIATELY UNTIL THIS DIAGNOSIS IS EXCLUDED.
LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS, INCLUDING FATAL CASES, HAVE BEEN REPORTED WITH THE USE OF ANTIRETROVIRAL NUCLEOSIDE ANALOGUES ALONE OR IN COMBINATION, INCLUDING HIVID (SEE WARNINGS).
IN ADDITION, RARE CASES OF HEPATIC FAILURE AND DEATH CONSIDERED POSSIBLY RELATED TO UNDERLYING HEPATITIS B AND HIVID HAVE BEEN REPORTED (SEE WARNINGS ANDPRECAUTIONS).
DESCRIPTION
HIVID is the Hoffmann-La Roche brand of zalcitabine [formerly called 2',3'-dideoxycytidine (ddC)], a synthetic pyrimidine nucleoside analogue active against the human immunodeficiency virus (HIV). HIVID is available as film-coated tablets for oral administration in strengths of 0.375 mg and 0.750 mg. Each tablet also contains the inactive ingredients lactose, microcrystalline cellulose, croscarmellose sodium, magnesium stearate, hydroxypropyl methylcellulose, polyethylene glycol, and polysorbate 80 along with the following colorant system: 0.375 mg tablet — synthetic brown, black, red and yellow iron oxides, and titanium dioxide; 0.750 mg tablet — synthetic black iron oxide and titanium dioxide. The chemical name for zalcitabine is 4-amino-1-beta-D-2', 3'-dideoxyribofuranosyl-2-(1H)-pyrimidone or 2',3'-dideoxycytidine with the molecular formula C9H13N3O3 and a molecular weight of 211.22. Zalcitabine has the following structural formula:
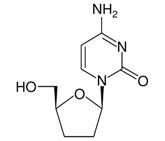
Zalcitabine is a white to off-white crystalline powder with an aqueous solubility of 76.4 mg/mL at 25°C.
MICROBIOLOGY
Mechanism of Action
Zalcitabine is a synthetic nucleoside analogue of the naturally occurring nucleoside deoxycytidine, in which the 3'-hydroxyl group is replaced by hydrogen. Within cells, zalcitabine is converted to the active metabolite, dideoxycytidine 5'-triphosphate (ddCTP), by the sequential action of cellular enzymes. Dideoxycytidine 5'-triphosphate inhibits the activity of the HIV-reverse transcriptase both by competing for utilization of the natural substrate, deoxycytidine 5'-triphosphate (dCTP), and by its incorporation into viral DNA. The lack of a 3'-OH group in the incorporated nucleoside analogue prevents the formation of the 5' to 3' phosphodiester linkage essential for DNA chain elongation and, therefore, the viral DNA growth is terminated. The active metabolite, ddCTP, is also an inhibitor of cellular DNA polymerase-beta and mitochondrial DNA polymerase-gamma and has been reported to be incorporated into the DNA of cells in culture.
In Vitro HIV Susceptibility
The in vitro anti-HIV activity of zalcitabine was assessed by infecting cell lines of lymphoblastic and monocytic origin and peripheral blood lymphocytes with laboratory and clinical isolates of HIV. The IC50 and IC95 values (50% and 95% inhibitory concentration) were in the range of 30 to 500 nM and 100 to 1000 nM, respectively (1 nM = 0.21 ng/mL). Zalcitabine showed antiviral activity in all acute infections; however, activity was substantially less in chronically infected cells. In drug combination studies with zidovudine (ZDV) or saquinavir, zalcitabine showed additive to synergistic activity in cell culture. The relationship between the in vitro susceptibility of HIV to reverse-transcriptase inhibitors and the inhibition of HIV replication in humans has not been established.
Drug Resistance
HIV isolates with a reduction in sensitivity to zalcitabine (ddC) have been isolated from a small number of patients treated with HIVID by 1 year of therapy. Genetic analysis of these isolates showed point mutations (Lys 65 Arg or Asn, Thr 69 Asp, Leu 74 Val, Val 75 Thr or Ala, Met 184 Val or Tyr 215 Cys) in the pol gene that encodes for the reverse transcriptase. Combination therapy with HIVID and ZDV does not appear to prevent the emergence of zidovudine-resistant isolates.
Cross-resistance
The potential for cross-resistance between HIV-reverse transcriptase inhibitors and HIV-protease inhibitors is low because of the different enzyme targets involved. The point mutation at position 69 appears to be specific to ddC in its selection and effect. Additionally, the point mutations at positions 65, 74, 75, and 184 are associated with resistance to didanosine (ddI), that at position 75 with resistance to stavudine (d4T), and those at positions 65 (Lys to Arg), and 184 (Met to Val) with resistance to lamivudine (3TC). HIV isolates with multidrug resistance to ZDV, ddI, ddC, d4T, and 3TC were recovered from a small number of patients treated for 1 year with the combination of ZDV, ddI or ddC. The pattern of resistance mutations in the combination therapy was different (Ala 62 Val, Val 75 Ile, Phe 77 Leu, Phe 116 Tyr and Gln 151 Met) from monotherapy with mutation 151 being most significant for multidrug resistance.
CLINICAL PHARMACOLOGY
Pharmacokinetics
The pharmacokinetics of zalcitabine has been evaluated in studies in HIV-infected patients following 0.01 mg/kg, 0.03 mg/kg, and 1.5 mg oral doses, and a 1.5 mg intravenous dose administered as a 1-hour infusion.
Absorption and Bioavailability in Adults
Following oral administration to HIV-infected patients, the mean absolute bioavailability of zalcitabine was >80% (30% CV, range 23% to 124%, n=19). The absorption rate of a 1.5 mg oral dose of zalcitabine (n=20) was reduced when administered with food. This resulted in a 39% decrease in mean maximum plasma concentrations (Cmax) from 25.2 ng/mL (35% CV, range 11.6 to 37.5 ng/mL) to 15.5 ng/mL (24% CV, range 9.1 to 23.7 ng/mL), and a twofold increase in time to achieve maximum plasma concentrations from a mean of 0.8 hours under fasting conditions to 1.6 hours when the drug was given with food. The extent of absorption (as reflected by AUC) was decreased by 14%, from 72 ng∙hr/mL (28% CV, range 43 to 119 ng∙hr/mL) to 62 ng∙hr/mL (23% CV, range 42 to 91 ng∙hr/mL). The clinical relevance of these decreases is unknown. Absorption of zalcitabine does not appear to be reduced in patients with diarrhea not caused by an identified pathogen.
Distribution in Adults
The steady-state volume of distribution following intravenous administration of a 1.5 mg dose of zalcitabine averaged 0.534 (± 0.127) L/kg (24% CV, range 0.304 to 0.734 L/kg, n=20). Cerebrospinal fluid obtained from 9 patients at 2 to 3.5 hours following 0.06 mg/kg or 0.09 mg/kg intravenous infusion showed measurable concentrations of zalcitabine. The CSF:plasma concentration ratio ranged from 9% to 37% (mean 20%), demonstrating penetration of the drug through the blood-brain barrier. The clinical relevance of these ratios has not been evaluated.
Metabolism and Elimination in Adults
Zalcitabine is phosphorylated intracellularly to zalcitabine triphosphate, the active substrate for HIV-reverse transcriptase. Concentrations of zalcitabine triphosphate are too low for quantitation following administration of therapeutic doses to humans.
Zalcitabine does not undergo a significant degree of metabolism by the liver. The primary metabolite of zalcitabine that has been identified is dideoxyuridine (ddU), which accounts for less than 15% of an oral dose in both urine and feces (n=4). Approximately 10% of an orally administered radiolabeled dose of zalcitabine appears in the feces (n=10), comprised primarily of unchanged drug and ddU. Renal excretion of unchanged drug appears to be the primary route of elimination, accounting for approximately 80% of an intravenous dose and 60% of an orally administered dose within 24 hours after dosing (n=19). The mean elimination half-life is 2 hours and generally ranges from 1 to 3 hours in individual patients. Total clearance following an intravenous dose averaged 285 mL/min (29% CV, range 165 to 447 mL/min, n=20). Renal clearance averaged approximately 235 mL/min or about 80% of total clearance (30% CV, range 129 to 348 mL/min, n=20). Renal clearance exceeds glomerular filtration rate suggesting renal tubular secretion contributes to the elimination of zalcitabine by the kidneys.
In patients with impaired kidney function, prolonged elimination of zalcitabine may be expected. Preliminary results from 7 patients with renal impairment (estimated creatinine clearance <55 mL/min) indicate that the half-life was prolonged (up to 8.5 hours) in these patients compared to those with normal renal function. Maximum plasma concentrations were higher in some patients after a single dose (see PRECAUTIONS).
In patients with normal renal function, the pharmacokinetics of zalcitabine was not altered during 3 times daily multiple dosing (n=9). Accumulation of drug in plasma during this regimen was negligible. The drug was <4% bound to plasma proteins, indicating that drug interactions involving binding-site displacement are unlikely (see Drug Interactions).
Drug Interactions
Zidovudine
There was no significant pharmacokinetic interaction between zidovudine and zalcitabine when single doses of zalcitabine (1.5 mg) and zidovudine (200 mg) were coadministered to 12 HIV-positive patients.
Probenecid
Following administration of a single oral 1.5 mg dose of zalcitabine alone during probenecid treatment (500 mg at 8 and 2 hours before and 4 hours after zalcitabine dosing) to 12 HIV-positive patients, mean renal clearance decreased from 310 mL/min (28% CV) to 180 mL/min (22% CV) and AUC increased from 59 ng∙hr/mL (27% CV) to 91 ng∙hr/mL (22% CV), indicating an increase in exposure of approximately 50% to zalcitabine. Mean half-life of zalcitabine increased from 1.7 to 2.5 hours (see PRECAUTIONS).
Cimetidine
Administration of a single dose of 1.5 mg zalcitabine with a single dose of 800 mg cimetidine to 12 HIV-positive patients resulted in a decrease in renal clearance from 224 mL/min (27% CV) to 171 mL/min (39% CV) and an increase in AUC from 75 ng∙hr/mL (29% CV) to 102 ng∙hr/mL (35% CV) (see PRECAUTIONS) indicating an increase in exposure of approximately 36% to zalcitabine.
Maalox
Concomitant administration of Maalox® TC (30 mL) with single dose of 1.5 mg zalcitabine to 12 HIV-positive patients resulted in a decrease in mean Cmax from 25.2 ng/mL (28% CV) to 18.4 ng/mL (34% CV) and AUC from 75 ng∙hr/mL (29% CV, n=10) to 58 ng∙hr/mL (36% CV, n=10) indicating a decrease in bioavailability of approximately 25% to zalcitabine (see PRECAUTIONS).
Metoclopramide
Administration of a single dose of 1.5 mg zalcitabine with 20 mg metoclopramide (10 mg 1 hour before and 10 mg 4 hours after zalcitabine dose) to 12 HIV-positive patients resulted in a decrease in AUC from 69 ng∙hr/mL (16% CV) to 62 ng∙hr/mL (21% CV) indicating a decrease in bioavailability of approximately 10% (see PRECAUTIONS).
Loperamide
Administration of a single dose of 1.5 mg zalcitabine during loperamide treatment (4 mg 16 hours before zalcitabine, 2 mg at 10 hours and 4 hours before zalcitabine, and 2 mg 2 hours after the zalcitabine dose) to 12 HIV-positive patients with diarrhea resulted in no significant pharmacokinetic interaction between zalcitabine and loperamide.
Pharmacokinetics in Pediatric Patients
For pharmacokinetic properties in pediatric patients, see PRECAUTIONS: Pediatric Use. Limited pharmacokinetic data have been reported for 5 HIV-positive pediatric patients using doses of 0.03 and 0.04 mg/kg HIVID administered orally every 6 hours.1 The mean bioavailability of zalcitabine in these pediatric patients was 54% and mean apparent systemic clearance was 150 mL/min/m2. Due to the small number of subjects and different analytical techniques, it is difficult to make comparisons between pediatric and adult data.
INDICATIONS AND USAGE
HIVID is indicated in combination with antiretroviral agents for the treatment of HIV infection. This indication is based on study results showing a reduction in the rate of disease progression (AIDS-defining events or death) in patients with limited prior antiretroviral therapy who were treated with the combination of HIVID and zidovudine (see Description of Clinical Studies). This indication is also based on a study showing a reduction in both mortality and AIDS-defining clinical events for patients who received INVIRASE® (saquinavir mesylate) in combination with HIVID compared to patients who received either HIVID or INVIRASE alone.
Description of Clinical Studies
The use of HIVID in combination with zidovudine is based on the clinical results from study ACTG 175. ACTG 175 was a randomized, double-blind, controlled trial that compared zidovudine 200 mg three times daily; didanosine 200 mg twice daily; zidovudine+didanosine; and zidovudine+HIVID 0.750 mg three times daily. A total of 2467 HIV-infected adults (mean baseline CD4 count = 352 cells/mm3) with no prior AIDS-defining event enrolled with the following demographics: male (82%), Caucasian (70%), mean age of 35 years, asymptomatic HIV infection (81%) and prior antiretroviral use (57%, mean duration = 89.5 weeks). The overall mean duration of study treatment was 99 weeks. The incidence of AIDS-defining events or death is shown in Table 1.
| Antiretroviral Experience | Event | Treatment | |||
|---|---|---|---|---|---|
| zidovudine | Zidovudine + didanosine | zidovudine + HIVID | didanosine | ||
| Overall | n | 619 | 613 | 615 | 620 |
| AIDS/Death | 96 (16%) | 65 (11%) | 76 (12%) | 71 (11%) | |
| Death Only | 54 (9%) | 31 (5%) | 40 (7%) | 29 (5%) | |
| Naive | n | 269 | 263 | 267 | 268 |
| AIDS/Death | 32 (12%) | 20 (8%) | 16 (6%) | 23 (9%) | |
| Death Only | 18 (7%) | 11 (4%) | 9 (3%) | 11 (4%) | |
| Experienced | n | 350 | 350 | 348 | 352 |
| AIDS/Death | 64 (18%) | 45 (13%) | 60 (17%) | 48 (14%) | |
| Death Only | 36 (10%) | 20 (6%) | 31 (9%) | 18 (5%) | |
Although no antiretroviral agent should be used as monotherapy, a description of CPCRA 002 is included here as it provides a comparison of the safety and efficacy of HIVID compared to ddI.
CPCRA 002 was a randomized, multicenter, open-label study in which HIVID was compared to ddI as treatment for patients with advanced HIV infection (median CD4 cell count = 37 cells/mm3) who were clinically intolerant to ZDV, or who had met criteria for having disease progression while receiving ZDV.2 Patients in this study had a mean of 17.5 months of prior ZDV use. The median duration of treatment for both HIVID and ddI was 34 weeks. The results demonstrate that HIVID was at least as efficacious as ddI in terms of time to an AIDS-defining event or death, while for survival alone the results favored HIVID. However, most of the patients (66%) in either group had disease progression over the median 16 months of follow-up. Overall rates of study drug intolerance, discontinuation and adverse events were similar for the two groups, although the types of events were different.
A clinical study (N3300/ACTG 114) has demonstrated ZDV to be superior to HIVID as monotherapy for advanced HIV disease (CD4 cell count ≤200 cells/mm3) in previously untreated patients.3,4 The final analysis of this study indicated that 134 patients (42%) in the HIVID group with a median follow-up of 85 weeks and 120 patients (38%) in the ZDV group with a median follow-up of 96 weeks died with a relative risk for mortality of ZDV to HIVID of 0.54.
CONTRAINDICATIONS
HIVID is contraindicated in patients with clinically significant hypersensitivity to zalcitabine or to any of the excipients contained in the tablets.
WARNINGS
SIGNIFICANT CLINICAL ADVERSE REACTIONS, SOME OF WHICH ARE POTENTIALLY FATAL, HAVE BEEN REPORTED WITH HIVID. PATIENTS WITH DECREASED CD4 CELL COUNTS APPEAR TO HAVE AN INCREASED INCIDENCE OF ADVERSE EVENTS.
Peripheral Neuropathy
THE MAJOR CLINICAL TOXICITY OF HIVID IS PERIPHERAL NEUROPATHY, WHICH MAY OCCUR IN UP TO 1/3 OF PATIENTS WITH ADVANCED DISEASE TREATED WITH HIVID. The incidence in patients with less-advanced disease is lower.
HIVID-related peripheral neuropathy is a sensorimotor neuropathy characterized initially by numbness and burning dysesthesia involving the distal extremities. These symptoms may be followed by sharp shooting pains or severe continuous burning pain if the drug is not withdrawn. The neuropathy may progress to severe pain requiring narcotic analgesics and is potentially irreversible. In some patients, symptoms of neuropathy may initially progress despite discontinuation of HIVID. With prompt discontinuation of HIVID, the neuropathy is usually slowly reversible.
There are no data regarding the use of HIVID in patients with preexisting peripheral neuropathy since these patients were excluded from clinical trials; therefore, HIVID should be used with extreme caution in these patients. Individuals with moderate or severe peripheral neuropathy, as evidenced by symptoms accompanied by objective findings, are advised to avoid HIVID.
HIVID should be used with caution in patients with a risk of developing peripheral neuropathy: patients with low CD4 cell counts (CD4<50 cells/mm3), diabetes, weight loss and/or patients receiving HIVID concomitantly with drugs that have the potential to cause peripheral neuropathy (see PRECAUTIONS: Drug Interactions). Careful monitoring is strongly recommended for these individuals.
HIVID should be stopped promptly if signs or symptoms of peripheral neuropathy occur, such as when moderate discomfort from numbness, tingling, burning or pain of the extremities progresses, or any related symptoms occur that are accompanied by an objective finding (see DOSAGE AND ADMINISTRATION).
Pancreatitis
PANCREATITIS, WHICH HAS BEEN FATAL IN SOME CASES, HAS BEEN OBSERVED WITH THE ADMINISTRATION OF HIVID. Pancreatitis is an uncommon complication of HIVID occurring in up to 1.1% of patients.
Patients with a history of pancreatitis or known risk factors for the development of pancreatitis should be followed more closely while on HIVID therapy. Of 528 HIVID-treated patients enrolled in an expanded-access safety study (N3544), who had a history of prior pancreatitis or increased amylase, 28 (5.3%) developed pancreatitis and an additional 23 (4.4%) developed asymptomatic elevated serum amylase.
Treatment with HIVID should be stopped immediately if clinical signs or symptoms (nausea, vomiting, abdominal pain) or if abnormalities in laboratory values (hyperamylasemia associated with dysglycemia, rising triglyceride level, decreasing serum calcium) suggestive of pancreatitis should occur. If clinical pancreatitis develops during HIVID administration, it is recommended that HIVID be permanently discontinued. Treatment with HIVID should also be interrupted if treatment with another drug known to cause pancreatitis (eg, intravenous pentamidine) is required (see Drug Interactions).
Lactic Acidosis/Severe Hepatomegaly With Steatosis and Hepatic Toxicity
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination, including HIVID and other antiretrovirals.5,6 A majority of these cases have been in women. Obesity and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering HIVID to any patient with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors. Treatment with HIVID should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
IN ADDITION, RARE CASES OF HEPATIC FAILURE AND DEATH CONSIDERED POSSIBLY RELATED TO UNDERLYING HEPATITIS B AND HIVID HAVE BEEN REPORTED. Treatment with HIVID in patients with preexisting liver disease, liver enzyme abnormalities, a history of ethanol abuse or hepatitis should be approached with caution. Treatment with HIVID should be suspended in any patient who develops clinical or laboratory findings suggestive of pronounced hepatotoxicity. In clinical trials, drug interruption was recommended if liver function tests exceeded >5 times the upper limit of normal.
Other Serious Toxicities
- a)
- Oral Ulcers: Severe oral ulcers occurred in up to 3% of patients receiving HIVID in CPCRA 002 and ACTG 175; less severe oral ulcerations have occurred at higher frequencies in other clinical trials.
- b)
- Esophageal Ulcers: Infrequent cases of esophageal ulcers have also been attributed to HIVID therapy. Interruption of HIVID should be considered in patients whodevelop esophageal ulcers that do not respond to specific treatment for opportunistic pathogens in order to assess a possible relationship to HIVID.
- c)
- Cardiomyopathy/Congestive Heart Failure: Cardiomyopathy and congestive heart failure in patients with AIDS have been associated with the use of nucleoside analogues. Infrequent cases have been reported in patients receiving HIVID. Treatment with HIVID in patients with baseline cardiomyopathy or history of congestive heart failure should be approached with caution.
- d)
- Anaphylactoid Reaction: An anaphylactoid reaction was reported in a patient receiving both HIVID and zidovudine. In addition, there have been several reports of hypersensitivity reactions (including anaphylactic reaction or urticaria without other signs of anaphylaxis).
PRECAUTIONS
General
- Renal Impairment: Patients with renal impairment (estimated creatinine clearance <55 mL/min) may be at a greater risk of toxicity from HIVID due to decreased drug clearance. Dosage adjustment is recommended in these patients (see DOSAGE AND ADMINISTRATION).
-
Lymphoma: High doses of zalcitabine, administered for 3 months to B6C3F1 mice (resulting in plasma concentrations over 1000 times those seen in patients taking the recommended doses of HIVID) induced an increased incidence of thymic lymphoma.7 Although the pathogenesis of the effect is uncertain, a predisposition to chemically induced thymic lymphoma and high rates of spontaneous lymphoreticular neoplasms have previously been noted in this strain of mice.8
The incidence of lymphomas was reviewed in 13 comparative studies conducted by Roche, the NIAID and the NCI, as well as 7 Roche expanded-access studies that included HIVID. In one study, ACTG 155, a statistically significant increased rate of lymphomas was seen in patients receiving HIVID or combination HIVID and zidovudine compared to zidovudine alone (rates of 0, 1.3, and 2.3 per 100 person years for zidovudine, HIVID, and combination HIVID and zidovudine, respectively; log rank p-value=0.01, pooling HIVID, and combination HIVID and zidovudine vs zidovudine, p-value=0.003). Based on review of the literature, the incidence of lymphomas in HIV-infected patients with advanced disease on zidovudine monotherapy would be expected to be approximately 1 to 2 per 100 person years of follow-up.
None of the other comparative studies evaluated showed a statistically significant difference in rates of lymphomas in patients receiving HIVID. In a large, controlled clinical trial (ACTG 175) HIVID in combination with zidovudine was not associated with an increase in the incidence of lymphoma over that seen with zidovudine monotherapy (6 of 615 and 9 of 619, respectively).
Lymphoma has been identified as a consequence of HIV infection. This most likely represents a consequence of prolonged immunosuppression; however, an association between the occurrence of lymphoma and antiviral therapy cannot be excluded.
- Fat Redistribution: Redistribution/accumulation of body fat including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and "cushingoid appearance" have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
Patients receiving HIVID or any other antiretroviral therapy may continue to develop opportunistic infections and other complications of HIV infections, and therefore should remain under close clinical observation by physicians experienced in the treatment of patients with associated HIV diseases.
The duration of clinical benefit from antiretroviral therapy may be limited. Alterations in antiretroviral therapy should be considered in cases of disease progression, either clinical or as demonstrated by viral rebound (increase in HIV RNA after initial decline).
Information for Patients
Patients should be informed that HIVID is not a cure for HIV infection and that they may continue to acquire illnesses associated with advanced HIV infection, including opportunistic infections.
Patients should be told that there is currently no data demonstrating that HIVID therapy can reduce the risk of transmitting HIV to others through sexual contact or blood contamination.
Patients should be advised to take HIVID every day as prescribed. Patients should not alter the dose or discontinue therapy without consulting with their physician. If a dose is missed, patients should take the dose as soon as possible and then return to their normal schedule. However, if a dose is skipped, the patient should not double the next dose.
Patients should be instructed that the major toxicity of HIVID is peripheral neuropathy. Pancreatitis and hepatic toxicity are other serious potentially life-threatening toxicities that have been reported in patients treated with HIVID. Patients should be advised of the early symptoms of these conditions and instructed to promptly report them to their physician. Since the development of peripheral neuropathy appears to be dose-related to HIVID, patients should be advised to follow their physicians' instructions regarding the prescribed dose.
Patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long-term health effects of these conditions are not known at this time.
Laboratory Tests
Complete blood counts and clinical chemistry tests should be performed prior to initiating HIVID therapy and at appropriate intervals thereafter. Baseline testing of serum amylase and triglyceride levels should be performed in individuals with a prior history of pancreatitis, increased amylase, those on parenteral nutrition or with a history of ethanol abuse.
Drug Interactions
Zidovudine
There is no significant pharmacokinetic interaction between ZDV and zalcitabine which has been confirmed clinically. Zalcitabine also has no significant effect on the intracellular phosphorylation of ZDV, as shown in vitro in peripheral blood mononuclear cells or in two other cell lines (U937 and Molt-4). In the same study it was shown that didanosine and stavudine had no significant effect on the intracellular phosphorylation of zalcitabine in peripheral blood mononuclear cells.
Lamivudine
In vitro studies in peripheral blood mononuclear cells, U937 and Molt-4 cells revealed that lamivudine significantly inhibited zalcitabine phosphorylation in a dose dependent manner. Effects were already seen with doses corresponding to relevant plasma levels in humans, and the intracellular phosphorylation of zalcitabine to its three metabolites (including the active zalcitabine triphosphate metabolite) was significantly inhibited. Zalcitabine inhibited lamivudine phosphorylation at high concentration ratios (10 and 100); however, it is considered to be unlikely that this decrease of phosphorylated lamivudine concentration is of clinical significance, as lamivudine is a more efficient substrate for deoxycytidine kinase than zalcitabine. These in vitro studies suggest that concomitant administration of zalcitabine and lamivudine in humans may result in sub-therapeutic concentrations of active phosphorylated zalcitabine, which may lead to a decreased antiretroviral effect of zalcitabine. It is unknown how the effect seen in these in vitro studies translates into clinical consequences. Concomitant use of zalcitabine and lamivudine is not recommended.
Saquinavir
The combination of HIVID, saquinavir, and ZDV has been studied (as triple combination) in adults. Pharmacokinetic data suggest that absorption, metabolism, and elimination of each of these drugs are unchanged when they are used together.
Drugs Associated With Peripheral Neuropathy
The concomitant use of HIVID with drugs that have the potential to cause peripheral neuropathy should be avoided where possible. Drugs that have been associated with peripheral neuropathy include antiretroviral nucleoside analogues, chloramphenicol, cisplatin, dapsone, disulfiram, ethionamide, glutethimide, gold, hydralazine, iodoquinol, isoniazid, metronidazole, nitrofurantoin, phenytoin, ribavirin, and vincristine. Concomitant use of HIVID with didanosine is not recommended.
Intravenous Pentamidine
Treatment with HIVID should be interrupted when the use of a drug that has the potential to cause pancreatitis is required. Death due to fulminant pancreatitis possibly related to intravenous pentamidine and HIVID has been reported. If intravenous pentamidine is required to treat Pneumocystis carinii pneumonia, treatment with HIVID should be interrupted (see WARNINGS).
Amphotericin, Foscarnet, and Aminoglycosides
Drugs such as amphotericin, foscarnet, and aminoglycosides may increase the risk of developing peripheral neuropathy (see WARNINGS: Peripheral Neuropathy) or other HIVID-associated adverse events by interfering with the renal clearance of zalcitabine (thereby raising systemic exposure). Patients who require the use of one of these drugs with HIVID should have frequent clinical and laboratory monitoring with dosage adjustment for any significant change in renal function.
Probenecid or Cimetidine
Concomitant administration of probenecid or cimetidine decreases the elimination of zalcitabine, most likely by inhibition of renal tubular secretion of zalcitabine. Patients receiving these drugs in combination with zalcitabine should be monitored for signs of toxicity and the dose of zalcitabine reduced if warranted.
Magnesium/Aluminum-containing Antacid Products
Absorption of zalcitabine is moderately reduced (approximately 25%) when coadministered with magnesium/aluminum-containing antacid products. The clinical significance of this reduction is not known, hence zalcitabine is not recommended to be ingested simultaneously with magnesium/aluminum-containing antacids.
Metoclopramide
Bioavailability is mildly reduced (approximately 10%) when zalcitabine and metoclopramide are coadministered (see CLINICAL PHARMACOLOGY: Drug Interactions).
Doxorubicin
Doxorubicin caused a decrease in zalcitabine phosphorylation (>50% inhibition of total phosphate formation) in U937/Molt 4 cells. Although there may be decreased zalcitabine activity because of lessened active metabolite formation, the clinical relevance of these in vitro results are not known.
Carcinogenesis, Mutagenesis and Impairment of Fertility
Carcinogenesis
Zalcitabine was administered orally by dietary admixture to CRL:CD-1® (ICR) Br mice at dosages of 3, 83, or 250 mg/kg/day for 2 years. Plasma exposures (as measured by AUC) at these doses were 6-fold to 704-fold greater than the systemic exposure in humans with the therapeutic dose. Zalcitabine was administered orally by dietary admixture to CDF® (F-344)/CrlBR/CdBR rats at dosages of 3, 28, 83, or 250 mg/kg/day. At the highest dose tested, the systemic exposure to zalcitabine was 833 times the systemic exposure in humans with the therapeutic dose.
A significant increase in thymic lymphoma in all zalcitabine dose groups and Harderian gland (a gland of the eye of rodents) adenoma in the two highest dose groups was observed in female CD-1 mice after 2 years of dosing. No increase in tumor incidence was observed in rats or male mice treated with zalcitabine. In an independent study, administration of zalcitabine to B6C3F1 mice at a dose of 1000 mg/kg/day for 3 months induced an increased incidence of thymic lymphoma. A high rate of spontaneous lymphoreticular neoplasms have previously been noted in this strain of mice.
Mutagenesis
Zalcitabine was positive in a cell transformationassay and induced chromosomal aberrations in vitro in human peripheral blood lymphocytes. Oral doses of zalcitabine at 2500 and 4500 mg/kg were clastogenic in the mouse micronucleus assay. Zalcitabine showed no evidence of mutagenicity in Ames tests, Chinese hamster lung cell assays and the mouse lymphoma assay. An unscheduled DNA synthesis assay in rat hepatocytes showed that zalcitabine had no effect on DNA repair.
Impairment of Fertility
Fertility and reproductive performance were assessed in rats at plasma concentrations up to 2142 times those achieved with the maximum recommended human dose (MRHD) based on AUC measurements. No adverse effects on rate of conception or general reproductive performance were observed. The highest dose was associated with embryolethality and evidence of teratogenicity. The next lower dose studied (plasma concentrations equivalent to 485 times the MRHD) was associated with a lower frequency of embryotoxicity but no teratogenicity. The fertility of F1 males was significantly reduced at a calculated dose of 2142 (but not 485) times the MRHD (based on AUC measurements) in a teratology study in which rat mothers were dosed on gestation days 7 to 15. No adverse effects were observed on the fertility of parents or F1 generation in the study of fertility and general reproductive performance or in the perinatal and postnatal reproduction study.
Pregnancy
Teratogenic Effects
Pregnancy Category C. Zalcitabine has been shown to be teratogenic in mice at calculated exposure levels of 1365 and 2730 times that of the MRHD (based on AUC measurements). In rats, zalcitabine was teratogenic at a calculated exposure level of 2142 times the MRHD but not at an exposure level of 485 times the MRHD. In a perinatal and postnatal study in the rat, a high incidence of hydrocephalus was observed in the F1 offspring derived from litters of dams treated with 1071 (but not 485) times the MRHD (based on AUC measurements). There are no adequate and well-controlled studies of zalcitabine in pregnant women. HIVID should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Fertile women should not receive HIVID unless they are using effective contraception during therapy. If pregnancy occurs, physicians are encouraged to report such cases by calling (800) 526-6367.
Nonteratogenic Effects
Increased embryolethality was observed in pregnant mice at doses 2730 times the MRHD and in pregnant rats above 485 (but not 98) times the MRHD (based on AUC measurements). Average fetal body weight was significantly decreased in mice at doses of 1365 times the MRHD and in rats at 2142 times the MRHD (based on AUC measurements). In a perinatal and postnatal study, the learning and memory of a significant number of F1 offspring were impaired, and they tended to stay hyperactive for a longer period of time. These effects, observed at a calculated exposure level of 1071 (but not 485) times the MRHD (based on AUC measurements), were considered to result from extensive damage to or gross underdevelopment of the brain of these F1 offspring consistent with the finding of hydrocephalus.
Antiretroviral Pregnancy Registry
To monitor maternal-fetal outcomes of pregnant women exposed to HIVID, an Antiretroviral Pregnancy Registry has been established. Physicians are encouraged to register patients by calling 1-800-258-4263.
Nursing Mothers
The Centers for Disease Control and Prevention recommend HIV-infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV. It is not known whether zalcitabine is excreted in human milk. Because of both the potential for HIV transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breast-feed if they are receiving antiretroviral medications, including HIVID.
Pediatric Use
Pharmacokinetics in Pediatric Patients
Limited pharmacokinetic data have been reported for 5 HIV-positive pediatric patients using doses of 0.03 and 0.04 mg/kg HIVID administered orally every 6 hours.1 The mean bioavailability of zalcitabine in these pediatric patients was 54% and mean apparent systemic clearance was 150 mL/min/m2. Due to the small number of subjects and different analytical techniques, it is difficult to make comparisons between pediatric and adult data.
Safety and effectiveness of HIVID in HIV-infected pediatric patients younger than 13 years of age have not been established.
Geriatric Use
Clinical studies of HIVID did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. HIVID is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection. In addition, renal function should be monitored and dosage adjustments should be made accordingly (see PRECAUTIONS: General: Renal Impairment and DOSAGE AND ADMINISTRATION).
ADVERSE REACTIONS
(SEE WARNINGS.)
Table 2 and Table 3 summarize the clinical adverse events and laboratory abnormalities, respectively, that occurred in≥1% of patients in the comparative monotherapy trial (CPCRA 002) of HIVID vs didanosine (ddI), and the comparative combination trial (ACTG 175) of zidovudine (ZDV) monotherapy vs HIVID and zidovudine combination therapy, respectively. Other studies have found a higher or lower incidence of adverse experiences depending upon disease status, generally being lower in patients with less advanced disease.
| CPCRA 002*
ZDV Intolerant or Failure | ACTG 175‡
ZDV Naive/Experienced |
|||
|---|---|---|---|---|
| HIVID 0.750 mg q8h | ddI 250 mg q12h | ZDV 200 mg q8h | HIVID+ZDV 0.750 mg q8h +200 mg q8h |
|
| n=237 | n=230 | n=619 | n=615 | |
| Body System/Adverse Event | ||||
|
||||
| Systemic | ||||
| Fatigue | 3.8 | 2.6 | 2.7 | 2.3 |
| Headache | 2.1 | 1.3 | 2.4 | 2.6 |
| Fever | 1.7 | 0.4 | 2.7 | 2.9 |
| Gastrointestinal | ||||
| Abdominal Pain | 3.0 | 7.0 | 2.3 | 1.8 |
| Oral Lesions/Stomatitis§ | 3.0 | 0.0 | 0.6 | 1.5 |
| Vomiting/Nausea§ | 3.4 | 7.0 | 4.9 | 2.1 |
| Diarrhea/Constipation§ | 2.5 | 17.4 | 2.9 | 1.0 |
| Hepatic | ||||
| Abnormal Hepatic Function | 8.9 | 7.0 | ¶ | ¶ |
| Neurological | ||||
| Convulsions | 1.3 | 2.2 | ||
| Peripheral Neuropathy# | 28.3 | 13.0 | 3.1 | 3.3 |
| Skin | ||||
| Rash/Pruritus/Urticaria | 3.4 | 3.9 | 1.8 | 1.6 |
| Metabolic and Nutrition | ||||
| Pancreatitis | 0.0 | 1.7 | 0.2 | 0.5 |
| Psychological | ||||
| Depression | 0.4 | 0.0 | 1.1 | 1.8 |
| Musculoskeletal | ||||
| Painful/Swollen Joints | 0.4 | 0.0 | 0.3 | 1.0 |
| CPCRA 002*
ZDV Intolerant or Failure | ACTG 175 ZDV Naive/Experienced |
|||
|---|---|---|---|---|
| HIVID 0.750 mg q8h n=237 | ddI 250 mg q12h n=230 | ZDV 200 mg q8h n=619 | HIVID+ZDV 0.750 mg q8h +200 mg q8h n=615 |
|
| Laboratory Abnormality | ||||
| Anemia (<7.5 gm/dL) | 8.4 | 7.4 | 1.8 | 3.1 |
| Leukopenia (<1500 cells/mm3) | 13.1 | 9.6 | N/A† | N/A† |
| Eosinophilia (>1000 cells/mm3 or 25%) | 2.5 | 1.7 | N/A† | N/A† |
| Neutropenia (<750 cells/mm3) | 16.9 | 11.7 | 1.9 | 4.2 |
| Thrombocytopenia (<50,000 cells/mm3) | 1.3 | 4.8 | 1.1 | 1.8 |
| CPK Elevation* (>4× ULN) | 0.8 | 0.0 | 5.8 | 5.7 |
| ALT (SGPT) (>5 × ULN) | N/A† | N/A† | 3.6 | 5.0 |
| AST (SGOT) (>5 × ULN) | 7.6 | 5.7 | 2.9 | 4.1 |
| Bilirubin (>2.5 × ULN) | 0.8 | 0.9 | 0.5 | 1.0 |
| GGT (>5 × ULN) | N/A† | N/A† | 0.5 | 1.0 |
| Amylase (>2 × ULN) | 5.1 | 3.9 | 1.0 | 1.5 |
| Hyperglycemia* (>250 mg/dL) | 0.0 | 1.7 | 0.8 | 2.0 |
Additional clinical adverse experiences associated with HIVID that occurred in <1% of patients in CPCRA 002 (at least possibly related, Grade 3 or higher), ACTG 175 (any relationship, Grade 3/4) or in other clinical studies are listed below by body system. Several of these events occurred in slightly higher rates in other studies. The incidence of adverse experiences varied in different studies, generally being lower in patients with less-advanced disease.
Body as a Whole: abnormal weight loss, asthenia, cachexia, chest tightness or pain, chills, cutaneous/allergic reaction, debilitation, difficulty moving, dry eyes/mouth, edema, facial pain or swelling, flank pain, flushing, increased sweating, lymphadenopathy, hypersensitivity reactions (see WARNINGS), malaise, night sweats, pain, pelvic/groin pain, rigors, redistribution/accumulation of body fat (see PRECAUTIONS: Fat Redistribution).
Cardiovascular: abnormal cardiac movement, arrhythmia, atrial fibrillation, cardiac failure, cardiac dysrhythmias, cardiomyopathy, heart racing, hypertension, palpitation, subarachnoid hemorrhage, syncope, tachycardia, ventricular ectopy.
Endocrine/Metabolic: abnormal triglycerides, abnormal lipase, altered serum glucose, decreased bicarbonate, diabetes mellitus, glycosuria, gout, hot flushes, hypercalcemia, hyperkalemia, hyperlipemia, hypernatremia, hyperuricemia, hypocalcemia, hypoglycemia, hypokalemia, hypomagnesemia, hyponatremia, hypophosphatemia, increased nonprotein nitrogen, lactic acidosis.
Gastrointestinal: abdominal bloating or cramps, acute pancreatitis, anal/rectal pain, anorexia, bleeding gums, bloody or black stools, colitis, dental abscess, dry mouth, dyspepsia, dysphagia, enlarged abdomen, epigastric pain, eructation, esophageal pain, esophageal ulcers, esophagitis, flatulence, gagging with pills, gastritis, gastrointestinal hemorrhage, gingivitis, glossitis, gum disorder, heartburn, hemorrhagic pancreatitis, hemorrhoids, increased saliva, left quadrant pain, melena, mouth lesion, odynophagia, painful sore gums, painful swallowing, pancreatitis, rectal hemorrhage, rectal mass, rectal ulcers, salivary gland enlargement, sore tongue, sore throat, tongue disorder, tongue ulcer, toothache, unformed/loose stools, vomiting.
Hematologic: absolute neutrophil count alteration, anemia, epistaxis, decreased hematocrit, granulocytosis, hemoglobinemia, leukopenia, neutrophilia, platelet alteration, purpura, thrombus, unspecified hematologic toxicity, white blood cell alteration.
Hepatic: abnormal lactate dehydrogenase, bilirubinemia, cholecystitis, decreased alkaline phosphatase, hepatitis, hepatocellular damage, hepatomegaly, increased alkaline phosphatase, jaundice.
Musculoskeletal: arthralgia, arthritis, arthropathy, arthrosis, back pain, backache, bone pains/aches, bursitis, cold extremities, extremity pain, joint inflammation, leg cramps, muscle aches, muscle weakness, muscle disorder, muscle stiffness, muscle cramps, myalgia, myopathy, myositis, neck pain, rib pain, stiff neck.
Neurological: abnormal coordination, aphasia, ataxia, Bell's palsy, confusion, decreased concentration, decreased neurological function, disequilibrium, dizziness, dysphonia, facial nerve palsy, focal motor seizures, grand mal seizure, hyperkinesia, hypertonia, hypokinesia, memory loss, migraine, neuralgia, neuritis, paralysis, seizures, speech disorder, status epilepticus, stupor, tremor, twitch, vertigo.
Psychological: acute psychotic disorder, acute stress reaction, agitation, amnesia, anxiety, confusion, decreased motivation, decreased sexual desire, depersonalization, emotional lability, euphoria, hallucination, impaired concentration, insomnia, manic reaction, mood swings, nervousness, paranoid state, somnolence, suicide attempt, dementia.
Respiratory: acute nasopharyngitis, chest congestion, coughing, cyanosis, difficulty breathing, dry nasal mucosa, dyspnea, flu-like symptoms, hemoptysis, nasal discharge, pharyngitis, rales/rhonchi, respiratory distress, sinus congestion, sinus pain, sinusitis, wheezing.
Skin: acne, alopecia, bullous eruptions, carbuncle/furuncle, cellulitis, cold sore, dermatitis, dry skin, dry rash desquamation, erythematous rash, exfoliative dermatitis, finger inflammation, follicular rash, impetigo, infection, itchy rash, lip blisters/lesions, macular/papular rash, maculopapular rash, moniliasis, mucocutaneous/skin disorder, nail disorder, photosensitivity reaction, pruritic disorder, pruritus, skin disorder, skin lesions, skin fissure, skin ulcer, urticaria.
Special Senses: abnormal vision, blurred vision, burning eyes, decreased taste, decreased vision, ear pain/problem, ear blockage, eye abnormality, eye inflammation, eye itching, eye pain, eye irritation, eye redness, eye hemorrhage, fluid in ears, hearing loss, increased tears, loss of taste, mucopurulent conjunctivitis, parosmia, photophobia, smell dysfunction, taste perversion, tinnitus, unequal-sized pupils, xerophthalmia, yellow sclera.
Urogenital: abnormal renal function, acute renal failure, albuminuria, bladder pain, dysuria, frequent urination, genital lesion/ulcer, increased blood urea nitrogen, increased creatinine, micturition frequency, nocturia, painful penis sore, pain on urination, penile edema, polyuria, renal cyst, renal calculus, testicular swelling, toxic nephropathy, urinary retention, vaginal itch, vaginal ulcer, vaginal pain, vaginal/cervix disorder, vaginal discharge.
OVERDOSAGE
Acute Overdosage
Inadvertent pediatric overdoses have occurred with doses up to 1.5 mg/kg HIVID. Pediatric patients had prompt gastric lavage and treatment with activated charcoal and had no sequelae. Mixed overdoses including HIVID and other drugs have led to drowsiness and vomiting (with HIVID or placebo, zidovudine and trimethoprim/sulfamethoxazole [TMP/SMX]), or increased GGT (with 18.75 mg HIVID with zidovudine and lormetazepam) or increased creatine phosphokinase (with HIVID or placebo, zidovudine, fluconazole, dapsone and wine). There is no experience with acute HIVID overdosage at higher doses and sequelae are unknown. There is no known antidote for HIVID overdosage. It is not known whether zalcitabine is dialyzable by peritoneal dialysis or hemodialysis.
Chronic Overdosage
In an initial dose-finding study in which zalcitabine was administered at doses 25 times (0.25 mg/kg every 8 hours) the currently recommended dose, one patient discontinued HIVID after 1½ weeks of treatment subsequent to the development of a rash and fever. In the early Phase 1 studies, all patients receiving zalcitabine at approximately 6 times the current total daily recommended dose experienced peripheral neuropathy by week 10. Eighty percent of patients who received approximately 2 times the current total daily recommended dose experienced peripheral neuropathy by week 12.
DOSAGE AND ADMINISTRATION
Patients should be advised that HIVID is recommended for use in combination with active antiretroviral therapy. Greater activity has been observed when new antiretroviral therapies are begun at the same time as HIVID. Concomitant therapy should be based on a patient's prior drug exposure. The recommended regimen is one 0.750 mg tablet of HIVID orally every 8 hours (2.25 mg HIVID total daily dose) in combination with other antiretroviral agents. Please refer to the complete product information for each of the other antiretroviral agents for the recommended doses of these agents. Based on preliminary data, the recommended HIVID dosage reduction for patients with impaired renal function is: creatinine clearance 10 to 40 mL/min: 0.750 mg of HIVID every 12 hours; creatinine clearance <10 mL/min: 0.750 mg of HIVID every 24 hours.
Monitoring of Patients
Complete blood counts and clinical chemistry tests should be performed prior to initiating HIVID therapy and at appropriate intervals thereafter. For comprehensive patient monitoring recommendations for other antiretroviral therapies, physicians should refer to the complete product information for these drugs. Serum amylase levels should be monitored in those individuals who have a history of elevated amylase, pancreatitis, ethanol abuse, who are on parenteral nutrition or who are otherwise at high risk of pancreatitis. Careful monitoring for signs or symptoms suggestive of peripheral neuropathy is recommended, particularly in individuals with a low CD4 cell count or who are at a greater risk of developing peripheral neuropathy while on therapy (see WARNINGS).
Dose Adjustment for HIVID
For toxicities that are likely to be associated with HIVID (eg, peripheral neuropathy, severe oral ulcers, pancreatitis, elevated liver function tests especially in patients with chronic Hepatitis B), HIVID should be interrupted or dose reduced. FOR SEVERE TOXICITIES OR THOSE PERSISTING AFTER DOSE REDUCTION, HIVID SHOULD BE INTERRUPTED. For recipients of combination therapy with HIVID and other antiretroviral agents, dose adjustments or interruption for each drug should be based on the known toxicity profile of the individual drugs. SEE INFORMATION FOR EACH DRUG USED IN COMBINATION FOR A DESCRIPTION OF KNOWN DRUG-ASSOCIATED ADVERSE REACTIONS.
Patients developing moderate discomfort with signs or symptoms of peripheral neuropathy should stop HIVID. HIVID-associated peripheral neuropathy may continue to worsen despite interruption of HIVID. HIVID should be reintroduced at 50% dose — 0.375 mg every 8 hours only if all findings related to peripheral neuropathy have improved to mild symptoms. HIVID should be permanently discontinued if patients experience severe discomfort related to peripheral neuropathy or moderate discomfort that progresses. If other moderate to severe clinical adverse reactions or laboratory abnormalities (such as increased liver function tests) occur, then HIVID and/or the other potential causative agent(s) should be interrupted until the adverse reaction abates. HIVID and/or the other potential causative agent(s) should then be carefully reintroduced at lower doses if appropriate. If adverse reactions recur at the reduced dose, therapy should be discontinued. The minimum effective dose of HIVID in combination with zidovudine for the treatment of adult patients with advanced HIV infection has not been established.
In patients with poor bone marrow reserve, particularly those patients with advanced symptomatic HIV disease, frequent monitoring of hematologic indices is recommended to detect serious anemia or granulocytopenia. Significant toxicities, such as anemia (hemoglobin of <7.5 gm/dL or reduction of >25% of baseline) and/or granulocytopenia (granulocyte count of <750 cells/mm3 or reduction of >50% from baseline), may require a treatment interruption of HIVID and zidovudine until evidence of marrow recovery is observed. For less severe anemia or granulocytopenia, a reduction in daily dose of zidovudine in those patients receiving combination therapy may be adequate. In patients who experience hematologic toxicity, reduction in hemoglobin may occur as early as 2 to 4 weeks after initiation of therapy, and granulocytopenia usually occurs after 6 to 8 weeks of therapy. In patients who develop significant anemia, dose modification does not necessarily eliminate the need for transfusion. If marrow recovery occurs following dose modification, gradual increases in dose may be appropriate depending on hematologic indices and patient tolerance. For more details, refer to the complete product information for zidovudine.
HOW SUPPLIED
HIVID 0.375 mg tablets are oval, beige, film-coated tablets with "HIVID 0.375" imprinted on one side and "ROCHE" on the other side — bottles of 100 (NDC 0004-0220-01). HIVID 0.750 mg tablets are oval, gray, film-coated tablets with "HIVID 0.750" imprinted on one side and "ROCHE" on the other side — bottles of 100 (NDC 0004-0221-01).
REFERENCES
- Pizzo PA, Butler K, Balis F, et al. Dideoxycytidine alone and in an alternating schedule with zidovudine in children with symptomatic human immunodeficiency virus infection. J Pediatr. 1990;117(5): 799-808.
- Abrams DI, Goldman AI, Launer C, et al. A comparative trial of didanosine or zalcitabine after treatment with zidovudine in patients with human immunodeficiency virus infection. N Engl J Med. 1994;330(10): 657-662.
- Follansbee S, Drew L, Olson R, et al. The efficacy of zalcitabine (ddC, HIVID) versus zidovudine (ZDV) as monotherapy in ZDV-naive patients with advanced HIV disease; a randomized, double-blind, comparative trial (ACTG 114; N3300). IXth International Conference on AIDS/IV STD World Congress, Berlin, Germany, June 7-11, 1993. Poster PO-B26-2113.
- Remick S, Follansbee S, Olson R, et al. Safety and tolerance of zalcitabine (ddC, HIVID) in a double-blind comparative trial (ACTG 114; N3300). IXth International Conference on AIDS/IV STD World Congress, Berlin, Germany, June 7-11, 1993. Poster PO-B26-2115.
- "Dear Doctor" letter, Burroughs Wellcome Co., June 1, 1993.
- Food and Drug Administration Antiviral Drugs Advisory Committee Meeting, "Mitochondrial Damage Associated with Nucleoside Analogues," Rockville, MD, September 21, 1993.
- Sanders VM, Elwell MR, Heath JE, et al. Induction of Thymic Lymphoma in Mice Administered the Dideoxynucleoside ddC. Fundamental and Applied Toxicology. 1995;27: 263-269.
- Irons RD, Le AT, Som DB, et al. 2'3'-Dideoxycytidine-induced Thymic Lymphoma Correlates with Species-specific Suppression of a Subpopulation of Primitive Hematopoietic Progenitor Cells in Mouse but Not Rat or Human Bone Marrow. J Clin Invest. 1995;95: 2777-2782.


| HIVID
zalcitabine tablet, film coated |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| HIVID
zalcitabine tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Genentech, Inc. (080129000) |