Label: BRIDION- sugammadex injection, solution

- NDC Code(s): 71872-7187-1
- Packager: Medical Purchasing Solutions, LLC
- This is a repackaged label.
- Source NDC Code(s): 0006-5423
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated May 10, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BRIDION safely and effectively. See full prescribing information for BRIDION.
BRIDION ® (sugammadex) Injection, for intravenous use
Initial U.S. Approval: 2015INDICATIONS AND USAGE
BRIDION is indicated for the reversal of neuromuscular blockade induced by rocuronium bromide and vecuronium bromide in adults undergoing surgery. ( 1)
DOSAGE AND ADMINISTRATION
- Monitor for twitch responses to determine the timing and dose for BRIDION administration. ( 2)
- Administer as a single bolus injection. (
2)
For rocuronium and vecuronium: - 4 mg/kg is recommended if spontaneous recovery of the twitch response has reached 1 to 2 post-tetanic counts (PTC) and there are no twitch responses to train-of-four (TOF) stimulation. ( 2.1)
- 2 mg/kg is recommended if spontaneous recovery has reached the reappearance of the second twitch in response to TOF stimulation. (
2.1)
For rocuronium only: - 16 mg/kg is recommended if there is a clinical need to reverse neuromuscular blockade soon (approximately 3 minutes) after administration of a single dose of 1.2 mg/kg of rocuronium. ( 2.1)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Known hypersensitivity to sugammadex or any of its components. ( 4)
WARNINGS AND PRECAUTIONS
- Anaphylaxis: Anaphylaxis has occurred in 0.3% of healthy volunteers. Observe patients for an appropriate period of time after administration. ( 5.1)
- Marked Bradycardia: Cases of marked bradycardia, some of which have resulted in cardiac arrest, have been observed within minutes after administration. Monitor for hemodynamic changes and administer anticholinergic agents such as atropine if clinically significant bradycardia is observed. ( 5.2)
- Respiratory Function Monitoring: Ventilatory support is mandatory for patients until adequate spontaneous respiration is restored and the ability to maintain a patent airway is assured. Should neuromuscular blockade persist after BRIDION or recur following extubation, take appropriate steps to provide adequate ventilation. ( 5.3, 5.4)
- Waiting Times for Re-Administration of Neuromuscular Blocking Agent: If re-administration of a neuromuscular blocking agent is required after reversal with BRIDION, waiting times should be based on the dose of BRIDION, and the renal function of the patient. Consider use of a nonsteroidal neuromuscular blocking agent. ( 5.5)
ADVERSE REACTIONS
Most common adverse reactions (reported in ≥10% of patients at a 2, 4, or 16 mg/kg BRIDION dose and higher than the placebo rate): vomiting, pain, nausea, hypotension, and headache. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Drug Compatibility
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Hypersensitivity
5.2 Marked Bradycardia
5.3 Respiratory Function Monitoring During Recovery
5.4 Risk of Prolonged Neuromuscular Blockade
5.5 Waiting Times for Re-Administration of Neuromuscular Blocking Agents for Intubation Following Reversal with BRIDION
5.6 Interactions Potentially Affecting the Efficacy of Other Drugs
5.7 Risk of Recurrence of Neuromuscular Blockade Due to Displacement Interactions
5.8 Risk of Recurrence of Neuromuscular Blockade with Lower Than Recommended Dosing
5.9 Risk of Recurrence of Neuromuscular Blockade Due to the Administration of Drugs that Potentiate Neuromuscular Blockade
5.10 Risk of Coagulopathy and Bleeding
5.11 Renal Impairment
5.12 Light Anesthesia
5.13 Reversal after Rocuronium or Vecuronium Administration in the ICU
5.14 Reversal of Neuromuscular Blocking Agents Other Than Rocuronium or Vecuronium
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Summary
7.2 Interactions Potentially Affecting the Efficacy of BRIDION
7.3 Interaction Potentially Affecting the Efficacy of Hormonal Contraceptives
7.4 Interference with Laboratory Tests
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Cardiac Patients
8.9 Pulmonary Patients
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Controlled Clinical Studies
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
BRIDION (sugammadex) injection, for intravenous use, should be administered by trained healthcare providers familiar with the use, actions, characteristics, and complications of neuromuscular blocking agents (NMBA) and neuromuscular block reversal agents.
Doses and timing of BRIDION administration should be based on monitoring for twitch responses and the extent of spontaneous recovery that has occurred.
Administer BRIDION intravenously as a single bolus injection. The bolus injection may be given over 10 seconds, into an existing intravenous line. BRIDION has only been administered as a single bolus injection in clinical trials.
From the time of BRIDION administration until complete recovery of neuromuscular function, monitor the patient to assure adequate ventilation and maintenance of a patent airway. Satisfactory recovery should be determined through assessment of skeletal muscle tone and respiratory measurements in addition to the response to peripheral nerve stimulation.
The recommended dose of BRIDION does not depend on the anesthetic regimen.
2.1 Recommended Dosing
BRIDION can be used to reverse different levels of rocuronium- or vecuronium-induced neuromuscular blockade.
For rocuronium and vecuronium:
- A dose of 4 mg/kg BRIDION is recommended if spontaneous recovery of the twitch response has reached 1 to 2 post-tetanic counts (PTC) and there are no twitch responses to train-of-four (TOF) stimulation following rocuronium- or vecuronium-induced neuromuscular blockade [see Warnings and Precautions (5.8)].
- A dose of 2 mg/kg BRIDION is recommended if spontaneous recovery has reached the reappearance of the second twitch (T 2) in response to TOF stimulation following rocuronium- or vecuronium-induced neuromuscular blockade [see Warnings and Precautions (5.8)].
For rocuronium only:
- A dose of 16 mg/kg BRIDION is recommended if there is a clinical need to reverse neuromuscular blockade soon (approximately 3 minutes) after administration of a single dose of 1.2 mg/kg of rocuronium. The efficacy of the 16 mg/kg dose of BRIDION following administration of vecuronium has not been studied [see Clinical Studies (14.1)].
BRIDION dosing is based on actual body weight.
2.2 Drug Compatibility
May inject BRIDION into the intravenous line of a running infusion with the following intravenous solutions:
- 0.9% sodium chloride
- 5% dextrose
- 0.45% sodium chloride and 2.5% dextrose
- 5% dextrose in 0.9% sodium chloride
- isolyte P with 5% dextrose
- Ringer's lactate solution
- Ringer's solution
Ensure the infusion line is adequately flushed (e.g., with 0.9% sodium chloride) between administration of BRIDION and other drugs.
Do not mix BRIDION with other products except those listed above.
BRIDION is physically incompatible with verapamil, ondansetron, and ranitidine.
Visually inspect parenteral drug products for particulate matter and discoloration prior to administration, whenever the solution and container permit.
-
3 DOSAGE FORMS AND STRENGTHS
BRIDION (sugammadex) injection is a sterile, clear, colorless to slightly yellow-brown, non-pyrogenic aqueous solution intended for intravenous infusion. BRIDION is available as follows:
- 200 mg/2 mL (100 mg/mL) in a single-dose vial for bolus injection
- 500 mg/5 mL (100 mg/mL), in a single-dose vial for bolus injection
-
4 CONTRAINDICATIONS
BRIDION is contraindicated in patients with known hypersensitivity to sugammadex or any of its components. Hypersensitivity reactions that occurred varied from isolated skin reactions to serious systemic reactions (i.e., anaphylaxis, anaphylactic shock) and have occurred in patients with no prior exposure to sugammadex [see Warnings and Precautions (5.1), Adverse Reactions (6)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Hypersensitivity
Clinicians should be prepared for the possibility of drug hypersensitivity reactions (including anaphylactic reactions) and take the necessary precautions [see Contraindications (4), Adverse Reactions (6.1)] .
Potentially serious hypersensitivity reactions, including anaphylaxis, have occurred in patients treated with BRIDION. The nature and frequency of anaphylaxis and hypersensitivity associated with BRIDION administration were evaluated in a randomized, double-blind, placebo-controlled, parallel-group, repeat-dose study in which 375 subjects were randomized to receive 3 doses of BRIDION IV with a 5 week washout period: 151 subjects received 4 mg/kg, 148 received 16 mg/kg and 76 received placebo. The frequency of anaphylaxis for the 299 healthy volunteers treated with intravenous BRIDION was 0.3% (n=1 in the BRIDION 16 mg/kg group on the first dose). Signs and symptoms included conjunctival edema, urticaria, erythema, swelling of the uvula and reduction in peak expiratory flow within 5 minutes of dose administration. The most common hypersensitivity adverse reactions reported were nausea, pruritus and urticaria and showed a dose response relationship, occurring more frequently in the 16 mg/kg group compared to the 4 mg/kg and placebo groups.
Anaphylaxis has also been reported in the post-marketing setting, including at doses less than 16 mg/kg. The most commonly described clinical features in reports of anaphylaxis were dermatologic symptoms (including urticaria, rash, erythema, flushing and skin eruption); and clinically important hypotension often requiring the use of vasopressors for circulatory support. In addition, prolonged hospitalization and/or the use of additional respiratory support until full recovery (re-intubation, prolonged intubation, manual or mechanical ventilation) have been noted in a number of the anaphylaxis reports.
5.2 Marked Bradycardia
Cases of marked bradycardia, some of which have resulted in cardiac arrest, have been observed within minutes after the administration of BRIDION [see Adverse Reactions (6.2)] . Patients should be closely monitored for hemodynamic changes during and after reversal of neuromuscular blockade. Treatment with anticholinergic agents, such as atropine, should be administered if clinically significant bradycardia is observed.
5.3 Respiratory Function Monitoring During Recovery
Ventilatory support is mandatory for patients until adequate spontaneous respiration is restored and the ability to maintain a patent airway is assured. Even if recovery from neuromuscular blockade is complete, other drugs used in the peri- and post-operative period could depress respiratory function and therefore ventilatory support might still be required.
Should neuromuscular blockade persist after BRIDION administration or recur following extubation, take appropriate steps to provide adequate ventilation.
5.4 Risk of Prolonged Neuromuscular Blockade
In clinical trials, a small number of patients experienced a delayed or minimal response to the administration of BRIDION [see Clinical Studies (14.1)] . Thus, it is important to monitor ventilation until recovery occurs.
5.5 Waiting Times for Re-Administration of Neuromuscular Blocking Agents for Intubation Following Reversal with BRIDION
A minimum waiting time is necessary before administration of a steroidal neuromuscular blocking agent after administration of BRIDION.
Table 1: Re-administration of Rocuronium or Vecuronium after Reversal (up to 4 mg/kg BRIDION) Minimum Waiting Time NMBA and Dose to be Administered 5 minutes 1.2 mg/kg rocuronium 4 hours 0.6 mg/kg rocuronium or
0.1 mg/kg vecuroniumWhen rocuronium 1.2 mg/kg is administered within 30 minutes after reversal with BRIDION, the onset of neuromuscular blockade may be delayed up to approximately 4 minutes and the duration of neuromuscular blockade may be shortened up to approximately 15 minutes.
The recommended waiting time in patients with mild or moderate renal impairment for re-use of 0.6 mg/kg rocuronium or 0.1 mg/kg vecuronium after reversal with up to 4 mg/kg BRIDION should be 24 hours. If a shorter waiting time is required, the rocuronium dose for a new neuromuscular blockade should be 1.2 mg/kg.
For re-administration of rocuronium or administration of vecuronium after reversal of rocuronium with 16 mg/kg BRIDION, a waiting time of 24 hours is suggested.
If neuromuscular blockade is required before the recommended waiting time has elapsed, use a nonsteroidal neuromuscular blocking agent. The onset of a depolarizing neuromuscular blocking agent might be slower than expected, because a substantial fraction of postjunctional nicotinic receptors can still be occupied by the neuromuscular blocking agent.
5.6 Interactions Potentially Affecting the Efficacy of Other Drugs
Due to the administration of BRIDION, certain drugs, including hormonal contraceptives, could become less effective due to a lowering of the (free) plasma concentrations. In this situation, consider the re-administration of the other drug, the administration of a therapeutically equivalent drug (preferably from a different chemical class), and/or non-pharmacological interventions as appropriate [see Drug Interactions (7.3)] .
5.7 Risk of Recurrence of Neuromuscular Blockade Due to Displacement Interactions
Recurrence of neuromuscular blockade may occur due to displacement of rocuronium or vecuronium from BRIDION by other drugs [see Drug Interactions (7.2)] . In this situation the patient may require mechanical ventilation. Administration of the drug which caused displacement should be stopped in case of an infusion. The risk of displacement reactions will be the highest in the time period equivalent to 3 times the half-life of BRIDION [see Clinical Pharmacology (12.3)] .
5.8 Risk of Recurrence of Neuromuscular Blockade with Lower Than Recommended Dosing
The use of lower than recommended doses of BRIDION may lead to an increased risk of recurrence of neuromuscular blockade after initial reversal and is not recommended [see Dosage and Administration (2.1), Adverse Reactions (6.1)] .
5.9 Risk of Recurrence of Neuromuscular Blockade Due to the Administration of Drugs that Potentiate Neuromuscular Blockade
When drugs which potentiate neuromuscular blockade are used in the post-operative phase, special attention should be paid to the possibility of recurrence of neuromuscular blockade. Refer to the package insert for rocuronium or vecuronium for a list of the specific drugs which potentiate neuromuscular blockade. In case recurrence of neuromuscular blockade is observed, the patient may require mechanical ventilation.
5.10 Risk of Coagulopathy and Bleeding
BRIDION doses up to 16 mg/kg were associated with increases in the coagulation parameters activated partial thromboplastin time (aPTT) and prothrombin time/international normalized ratio [PT(INR)] of up to 25% for up to 1 hour in healthy volunteers.
In patients undergoing major orthopedic surgery of the lower extremity who were concomitantly treated with heparin or low molecular weight heparin for thromboprophylaxis, increases in aPTT and PT(INR) of 5.5% and 3.0%, respectively, were observed in the hour following BRIDION 4 mg/kg administration. This clinical trial did not demonstrate an increased blood loss or anemia incidence with BRIDION compared with usual treatment. The rate of adjudicated bleeding events within 24 hours was 2.9% for sugammadex and 4.1% for usual care. The rate of post-operative anemia was 21% for sugammadex and 22% for usual care. The mean 24-hour drainage volume was 0.46 L for sugammadex and 0.48 L for usual care. The need for any post-operative transfusion was 37% for sugammadex and 39% for usual care.
In vitro experiments demonstrated additional aPTT and PT(INR) prolongations for sugammadex in combination with vitamin K antagonists, unfractionated heparin, low molecular weight heparinoids, rivaroxaban, and dabigatran up to ~25% and ~50% at C max levels of sugammadex corresponding to 4 mg/kg and 16 mg/kg doses, respectively.
Since bleeding risk has been studied systematically with only heparin and low molecular weight heparin thromboprophylaxis and 4 mg/kg doses of sugammadex coagulation parameters should be carefully monitored in patients with known coagulopathies, being treated with therapeutic anticoagulation, receiving thromboprophylaxis drugs other than heparin and low molecular weight heparin, or receiving thromboprophylaxis drugs and who then receive a dose of 16 mg/kg sugammadex.
5.11 Renal Impairment
BRIDION is not recommended for use in patients with severe renal impairment, including those requiring dialysis [see Use in Specific Populations (8.6)] . With regard to the recommended waiting time for re-administration in patients with mild or moderate renal impairment, see Waiting Times for Re-Administration of Neuromuscular Blocking Agents for Intubation Following Reversal with BRIDION [see Warnings and Precautions (5.5)] .
5.12 Light Anesthesia
When neuromuscular blockade was reversed intentionally in the middle of anesthesia in clinical trials, e.g., when investigating urgent reversal, signs of light anesthesia were noted occasionally (movement, coughing, grimacing and suckling of the tracheal tube).
5.13 Reversal after Rocuronium or Vecuronium Administration in the ICU
BRIDION has not been studied for reversal following rocuronium or vecuronium administration in the ICU.
5.14 Reversal of Neuromuscular Blocking Agents Other Than Rocuronium or Vecuronium
Do not use BRIDION to reverse blockade induced by nonsteroidal neuromuscular blocking agents such as succinylcholine or benzylisoquinolinium compounds.
Do not use BRIDION to reverse neuromuscular blockade induced by steroidal neuromuscular blocking agents other than rocuronium or vecuronium.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Anaphylaxis and Hypersensitivity [see Contraindications (4), Warnings and Precautions (5.1)]
- Marked Bradycardia [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect 2914 subjects exposed to 2, 4, or 16 mg/kg BRIDION and 544 to placebo in pooled Phase 1-3 studies. The population was 18 to 92 years old, 47% male and 53% female, 34% ASA (American Society of Anesthesiologists) Class 1, 51% ASA Class 2, and 14% ASA Class 3, and 82% Caucasian. Most subjects received a single dose of BRIDION 2 mg/kg or 4 mg/kg.
Adverse reactions reported in ≥10% of patients at a 2, 4, or 16 mg/kg BRIDION dose with a rate higher than the placebo rate are: vomiting, pain, nausea, hypotension, and headache.
All adverse reactions occurring in ≥2% of subjects treated with BRIDION and more often than placebo for adult subjects who received anesthesia and/or neuromuscular blocking agent in pooled Phase 1 to 3 studies are presented in Table 2.
Table 2: Percent of Subject Exposures in Pooled Phase 1 to 3 Studies with Adverse Reactions Incidence ≥ 2% Sugammadex Placebo Body System
Preferred Term2 mg/kg
(N=895)
n (%)4 mg/kg
(N=1921)
n (%)16 mg/kg
(N=98)
n (%)(N=544)
n (%)- *
- Combinations of preferred terms are as follows: Nausea includes preferred terms nausea and procedural nausea Vomiting includes preferred terms vomiting and procedural vomiting Abdominal pain includes preferred terms abdominal pain, abdominal pain upper, abdominal discomfort, abdominal pain lower, and epigastric discomfort Pain includes preferred terms pain and procedural pain Red blood cell count decreased includes preferred terms red blood cell count decreased, hemoglobin decreased, and hematocrit decreased Electrocardiogram QT interval abnormal includes preferred terms electrocardiogram QT interval abnormal and electrocardiogram QT interval prolonged Hypertension includes preferred terms hypertension, procedural hypertension, and blood pressure increased Hypotension includes preferred terms hypotension, procedural hypotension, and blood pressure decreased Tachycardia includes preferred terms tachycardia and heart rate increased Bradycardia includes preferred terms bradycardia and heart rate decreased
Injury, poisoning and procedural complications Incision site pain 58 (6) 106 (6) 4 (4) 6 (1) Procedural complication 13 (1) 27 (1) 8 (8) 3 (1) Airway complication of anesthesia 11 (1) 13 (1) 9 (9) 0 Anesthetic complication 8 (1) 14 (1) 9 (9) 1 (<1) Wound hemorrhage 5 (1) 38 (2) 0 8 (1) Recurrence of neuromuscular blockade 0 1 (<1) 2 (2) 0 Gastrointestinal disorders Nausea * 208 (23) 503 (26) 23 (23) 127 (23) Vomiting * 98 (11) 236 (12) 15 (15) 57 (10) Abdominal pain * 48 (5) 68 (4) 6 (6) 17 (3) Flatulence 17 (2) 51 (3) 1 (1) 10 (2) Dry mouth 9 (1) 5 (<1) 2 (2) 0 General disorders and administration site conditions Pain * 434 (48) 993 (52) 35 (36) 207 (38) Pyrexia 77 (9) 109 (6) 5 (5) 17 (3) Chills 30 (3) 61 (3) 7 (7) 27 (5) Nervous system disorders Headache 61 (7) 99 (5) 10 (10) 42 (8) Dizziness 44 (5) 67 (3) 6 (6) 13 (2) Hypoesthesia 12 (1) 24 (1) 3 (3) 9 (2) Respiratory, thoracic and mediastinal disorders Oropharyngeal pain 42 (5) 66 (3) 5 (5) 27 (5) Cough 13 (1) 49 (3) 8 (8) 11 (2) Musculoskeletal and connective tissue disorders Pain in extremity 13 (1) 35 (2) 6 (6) 15 (3) Musculoskeletal pain 16 (2) 33 (2) 1 (1) 6 (1) Myalgia 5 (1) 17 (1) 2 (2) 3 (1) Psychiatric disorders Insomnia 20 (2) 103 (5) 5 (5) 22 (4) Anxiety 14 (2) 19 (1) 3 (3) 1 (<1) Restlessness 3 (<1) 17 (1) 2 (2) 2 (<1) Depression 2 (<1) 5 (<1) 2 (2) 0 Investigations Red blood cell count decreased * 13 (1) 34 (2) 1 (1) 2 (<1) Electrocardiogram QT interval abnormal * 13 (1) 7 (<1) 6 (6) 4 (1) Blood creatine phosphokinase increased 9 (1) 14 (1) 2 (2) 1 (<1) Vascular disorders Hypertension * 48 (5) 96 (5) 9 (9) 38 (7) Hypotension * 33 (4) 102 (5) 13 (13) 20 (4) Skin and subcutaneous tissue disorders Pruritus 17 (2) 50 (3) 2 (2) 9 (2) Erythema 5 (1) 31 (2) 0 6 (1) Metabolism and nutrition disorders Hypocalcemia 15 (2) 12 (1) 0 4 (1) Cardiac disorders Tachycardia * 17 (2) 29 (2) 5 (5) 4 (1) Bradycardia * 9 (1) 21 (1) 5 (5) 6 (1) Surgical and medical procedures Hysterectomy 0 0 2 (2) 0 Anaphylaxis and Hypersensitivity
Hypersensitivity reactions, including anaphylaxis, have occurred in both premarketing clinical trials and in post-marketing spontaneous reports. In a dedicated hypersensitivity study in healthy volunteers, the frequency of anaphylaxis was 0.3% [see Warnings and Precautions (5.1)] . These reactions varied from isolated skin reactions to serious systemic reactions (i.e., anaphylaxis, anaphylactic shock) and have occurred in patients with no prior exposure to sugammadex.
Symptoms associated with these reactions can include: flushing, urticaria, erythematous rash, (severe) hypotension, tachycardia, swelling of tongue, swelling of pharynx, bronchospasm and pulmonary obstructive events. Severe hypersensitivity reactions can be fatal.
A randomized, double-blind study examined the incidence of drug hypersensitivity reactions in healthy volunteers given up to 3 doses of placebo (N=76), sugammadex 4 mg/kg (N=151) or sugammadex 16 mg/kg (N=148). Reports of suspected hypersensitivity were adjudicated by a blinded committee. The incidence of adjudicated hypersensitivity was 1%, 7% and 9% in the placebo, sugammadex 4 mg/kg and sugammadex 16 mg/kg groups, respectively. There were no reports of anaphylaxis after placebo or sugammadex 4 mg/kg. There was a single case of adjudicated anaphylaxis after the first dose of sugammadex 16 mg/kg. The frequency of anaphylaxis for the 299 healthy volunteers treated with intravenous sugammadex was 0.3%. There was no evidence of increased frequency or severity of hypersensitivity with repeat dosing.
In a previous study of similar design, there were three adjudicated cases of anaphylaxis, all after sugammadex 16 mg/kg (incidence 1% in the 298 healthy volunteers treated with sugammadex).
Recurrence of Neuromuscular Blockade
In clinical studies with subjects treated with rocuronium or vecuronium, where BRIDION was administered using a dose labeled for the depth of neuromuscular blockade (N=2022), an incidence of <1% was observed for recurrence of neuromuscular blockade as based on neuromuscular monitoring or clinical evidence [see Warnings and Precautions (5.8)] .
Bronchospasm
In one dedicated clinical trial and in post-marketing data, in patients with a history of pulmonary complications [see Use in Specific Populations (8.9)] , bronchospasm was reported as a possibly related adverse event.
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of BRIDION. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac Disorders: Cases of marked bradycardia and bradycardia with cardiac arrest have been observed within minutes after administration of sugammadex [see Warnings and Precautions (5.2)] . Other cardiac rhythm abnormalities have included atrial fibrillation, atrioventricular block, cardiac/cardiorespiratory arrest, ST segment changes, supraventricular tachycardia/extrasystoles, tachycardia, ventricular fibrillation, and ventricular tachycardia.
General Disorders and Administration Site Conditions: Cases of BRIDION not having the intended effect.
Immune System Disorders: Hypersensitivity events including anaphylactic shock, anaphylactic reaction, anaphylactoid reaction, and Type 1 hypersensitivity have been reported [see Warnings and Precautions (5.1)] .
-
7 DRUG INTERACTIONS
7.1 Summary
The information reported in sections 7.2 – 7.4 is based on binding affinity between BRIDION and other drugs, preclinical experiments, clinical studies and simulations of a pharmacokinetic-pharmacodynamic (PK-PD) model. Based on these considerations, no clinically significant pharmacodynamic interactions with other drugs are expected, with the exception of toremifene and hormonal contraceptives.
7.2 Interactions Potentially Affecting the Efficacy of BRIDION
Toremifene
For toremifene, which has a relatively high binding affinity for sugammadex and for which relatively high plasma concentrations might be present, some displacement of vecuronium or rocuronium from the complex with BRIDION could occur. The recovery to TOF ratio to 0.9 could therefore be delayed in patients who have received toremifene on the same day of surgery.
7.3 Interaction Potentially Affecting the Efficacy of Hormonal Contraceptives
In vitro binding studies indicate that BRIDION may bind to progestogen, thereby decreasing progestogen exposure. Therefore, the administration of a bolus dose of BRIDION is considered to be equivalent to missing dose(s) of oral contraceptives containing an estrogen or progestogen. If an oral contraceptive is taken on the same day that BRIDION is administered, the patient must use an additional, non-hormonal contraceptive method or back-up method of contraception (such as condoms and spermicides) for the next 7 days.
In the case of non-oral hormonal contraceptives, the patient must use an additional, non-hormonal contraceptive method or back-up method of contraception (such as condoms and spermicides) for the next 7 days.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data on BRIDION use in pregnant women to inform any drug-associated risks. In animal reproduction studies, there was no evidence of teratogenicity following daily intravenous administration of sugammadex to rats and rabbits during organogenesis at exposures of up to 6 and 8 times, respectively, the maximum recommended human dose (MRHD) of 16 mg/kg. However, there was an increase in the incidence of incomplete ossification of the sternebra and reduced fetal body weights in the rabbit study at 8 times the MRHD, which is a dose level in which maternal toxicity was also observed. In a pre- and postnatal development study, sugammadex treatment resulted in an increase in early postnatal loss, which correlated with maternal behavior (increased incidence of pup cannibalism), at exposures equivalent to the MRHD and higher [see Data] . The background risk of major birth defects and miscarriage for the indicated population are unknown. However, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.
Data
Animal Data
In an embryofetal development study in rats, pregnant animals received daily intravenous administration of sugammadex at 0, 20, 100, and 500 mg/kg (0.2, 1, and 6-times the MRHD of 16 mg/kg/day, respectively, based on AUC comparison) during organogenesis (Gestational Days 6 - 17). No treatment-related maternal and embryofetal changes were observed.
In another embryofetal development study, pregnant New Zealand white rabbits received daily intravenous administration of sugammadex at 0, 20, 65, 200 mg/kg (0.6, 2, and 8 times the MRHD, respectively, based on AUC comparison) during organogenesis (Gestational Days 6-18). Fetal body weight decreases (10 and 14%, respectively) were observed in the offspring at maternal doses of 65 mg/kg and 200 mg/kg. In addition, incomplete ossification of sternebra, and unossified 1st metacarpal were noted at a maternal dose of 200 mg/kg/day. Maternal toxicity was also observed at 200 mg/kg. Considering the observed effects of sugammadex on bone [see Nonclinical Toxicology (13.2)] , it is possible that these findings may be attributable to drug. There was no evidence of teratogenicity at any dose.
In a prenatal and postnatal development study, pregnant rats were administered sugammadex intravenously at 0, 30, 120, and 500 mg/kg (0.3, 1, and 6 times the MRHD, respectively, based on AUC comparison) from Gestational Day (GD) 6 to Postnatal Day (PND) 21 (corresponding to the beginning of organogenesis through parturition and subsequent pup weaning). Postnatal loss during PND 1-4 was noted across control litters and treated litters from dams receiving sugammadex as a result of pup cannibalization by dams. Overall incidence of affected litters was 2, 1, 4, and 3 litters, respectively, at 0, 30, 120, or 500 mg/kg/day. The reason for the increased cannibalization is not known. An effect of sugammadex on steroidal hormones and/or pheromones cannot be ruled out. In addition, there were no drug-related effects on parturition in rats during evaluations for prenatal or postnatal development.
8.2 Lactation
Risk Summary
No data are available regarding the presence of sugammadex in human milk, the effects of sugammadex on the breast fed infant, or the effects of sugammadex on milk production. However, sugammadex is present in rat milk [see Data] . The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for BRIDION and any potential adverse effects on the breastfed infant from BRIDION or from the underlying maternal condition.
Data
In a milk excretion study in rat dams following single intravenous dose of 20 mg/kg sugammadex on Postnatal Day 9, the maximum drug level was achieved at about 30 minutes after dosing with a ratio of milk to plasma level approximately 1:1. The oral exposure via milk did not induce effects on survival, body weight and physical or the behavioral developmental parameters monitored in rats in the prenatal and postnatal development studies [see Use in Specific Populations (8.1)] .
8.3 Females and Males of Reproductive Potential
Contraception
Upon administration of BRIDION, the efficacy of hormonal contraceptives may be reduced for up to 7 days. Advise female patients of reproductive potential using hormonal contraceptives to use an additional, non-hormonal contraceptive for the next 7 days following BRIDION administration [see Drug Interactions (7.3)] .
8.4 Pediatric Use
The safety and efficacy of BRIDION in pediatric patients have not been established.
Juvenile Animal Studies
In a bone deposition study, sugammadex concentrations were significantly higher in juvenile rats compared to adult rats (13% vs. 3% of the administered dose, respectively) following a single intravenous (IV) dose at 30 mg/kg (0.3 times the MRHD based on adult AUC comparison).
In a juvenile animal bone toxicity study, 7-day old rats were dosed intravenously once daily for 28 days with 0, 30, 120, and 500 mg/kg sugammadex (approximately 0.1, 0.6, and 3 times the MRHD, respectively, by adult AUC comparison). Sugammadex at 120 and 500 mg/kg decreased ulna and femur bone lengths by approximately 3%, which did not recover after an 8-week treatment-free period. Reversible whitish discoloration and disturbance of enamel formation were also observed in the incisors at these dose levels. In molars, this effect was only observed at 500 mg/kg. The no-observed-effect-level (NOEL) was 30 mg/kg.
In a second juvenile animal bone toxicity study, 7-day old rats were dosed once weekly for 8 weeks with 0, 7.5, 30, and 120 mg/kg (up to 1.2 times the MRHD of 16 mg/kg based on adult AUC comparison). No adverse effects on bone or teeth were noted.
8.5 Geriatric Use
BRIDION has been administered in a dedicated clinical study to a total 102 geriatric patients that compared the time to recovery from neuromuscular blockade induced by rocuronium (0.6 mg/kg) following administration of 2 mg/kg BRIDION given at the reappearance of T 2 in 65-74 year-olds (N=62) and ≥75 year-olds (N=40) compared with 18-64 year-olds (N=48). The median time to recovery of the TOF (T 4/T 1) ratio to 0.9 in 18-64 year-olds was 2.2 minutes; in 65-74 year-olds it was 2.5 minutes, and in ≥75 year-olds it was 3.6 minutes. For time to recovery from neuromuscular blockade induced by rocuronium following administration of 4 mg/kg BRIDION given at 1-2 PTCs, results across clinical trials revealed a median recovery of 2.5 minutes for geriatric patients (≥65 years, N=63) versus 2.0 minutes, for adults aged 18-64 years (N=359). Hence no dose adjustment is necessary in geriatric patients with normal organ function [see Dosage and Administration (2.1)] .
This drug is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)] .
8.6 Renal Impairment
This drug is known to be substantially excreted by the kidney. Effect of mild or moderate renal impairment (creatinine clearance ≥30 and ≤80 mL/min) on sugammadex PK and PD was obtained from a study in elderly patients [see Use in Specific Populations (8.5)] . Although clearance of drug decreased in elderly subjects with mild and moderate renal impairment, there was no significant difference in the ability of sugammadex to reverse the pharmacodynamic effect of rocuronium. Hence, no dosage adjustment is necessary for mild and moderate renal impairment. BRIDION is not recommended for use in patients with severe renal impairment (creatinine clearance <30 mL/min) due to insufficient safety information combined with the prolonged and increased overall exposure in these patients [see Warnings and Precautions (5.11), Clinical Pharmacology (12.3)] .
8.7 Hepatic Impairment
BRIDION is not metabolized nor excreted by the liver; therefore, dedicated trials in patients with hepatic impairment have not been conducted. Exercise caution when administering BRIDION to patients with hepatic impairment accompanied by coagulopathy or severe edema [see Warnings and Precautions (5.10, 5.14)] .
8.8 Cardiac Patients
One trial of 76 patients who were diagnosed with or have a history of cardiac disease (e.g., patients with ischemic heart disease, chronic heart failure, or arrhythmia) of primarily NYHA (New York Heart Association) Class II investigated time to recovery from neuromuscular blockade induced by rocuronium 0.6 mg/kg following administration of 2 mg/kg or 4 mg/kg BRIDION given at the reappearance of T 2. The trial showed that the median time to recovery of the T 4/T 1 ratio to 0.9 was 1.7 minutes and 1.3 minutes, respectively, in the 2 mg/kg and 4 mg/kg BRIDION dose groups. This is similar to the median values observed in the other trials; therefore, no dosage adjustment is necessary [see Dosage and Administration (2.1)] .
8.9 Pulmonary Patients
One trial of 77 patients who were diagnosed with or have a history of pulmonary complications investigated the time to recovery from neuromuscular blockade induced by rocuronium (0.6 mg/kg) following administration of 2 mg/kg or 4 mg/kg BRIDION given at the first signs of recovery (reappearance of T 2). The trial showed that for these patients the median time to recovery of the T 4/T 1 ratio to 0.9 was 2.1 minutes after a dose of 2 mg/kg BRIDION and 1.9 minutes after a dose of 4 mg/kg BRIDION. This is similar to the median values observed in the other trials; therefore, no dosage adjustment is necessary. [See Dosage and Administration (2.1), Adverse Reactions (6.1).]
-
10 OVERDOSAGE
In premarketing clinical trials, one case of accidental overdose with 40 mg/kg BRIDION was reported without significant effects.
BRIDION can be removed using hemodialysis with a high-flux filter, but not with a low-flux filter. Based upon clinical studies, BRIDION concentrations in plasma are reduced with a high-flux filter by about 70% after a 3- to 6-hour dialysis session.
-
11 DESCRIPTION
BRIDION (sugammadex) injection, for intravenous use, contains sugammadex sodium, a modified gamma cyclodextrin chemically designated as 6 A,6 B,6 C,6 D,6 E,6 F,6 G,6 H-Octakis-S-(2-carboxyethyl)-6 A,6 B,6 C,6 D,6 E,6 F,6 G,6 H-octathio- γ-cyclodextrin sodium salt (1:8) with a molecular weight of 2178.01. The structural formula is:
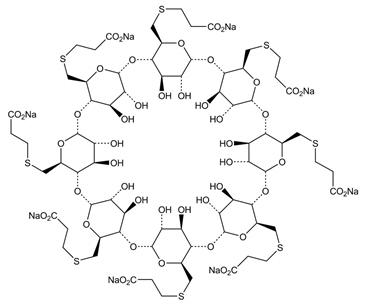
BRIDION is supplied as a sterile, non-pyrogenic aqueous solution that is clear, colorless to slightly yellow-brown for intravenous injection only. Each mL contains 100 mg sugammadex, which is equivalent to 108.8 mg sugammadex sodium. The aqueous solution is adjusted to a pH of between 7 and 8 with hydrochloric acid and/or sodium hydroxide. The osmolality of the product is between 300 and 500 mOsmol/kg.
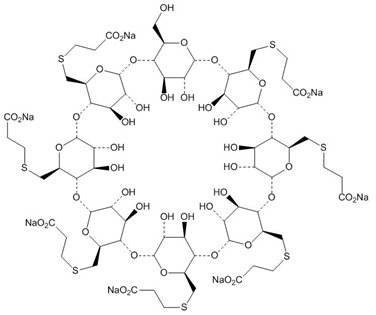
BRIDION may contain up to 7 mg/mL of the mono OH-derivative of sugammadex [see Clinical Pharmacology (12.2)] . This derivative is chemically designated as 6 A,6 B,6 C,6 D,6 E,6 F,6 G-Heptakis-S-(2-carboxyethyl)-6 A,6 B,6 C,6 D,6 E,6 F,6 G-heptathio- γ-cyclodextrin sodium salt (1:7) with a molecular weight of 2067.90. The structural formula is:
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
BRIDION is a modified gamma cyclodextrin. It forms a complex with the neuromuscular blocking agents rocuronium and vecuronium, and it reduces the amount of neuromuscular blocking agent available to bind to nicotinic cholinergic receptors in the neuromuscular junction. This results in the reversal of neuromuscular blockade induced by rocuronium and vecuronium.
12.2 Pharmacodynamics
BRIDION has been administered in doses ranging from 0.5 mg/kg to 16 mg/kg in dose response trials of rocuronium-induced blockade (0.6, 0.9, 1 and 1.2 mg/kg with and without maintenance doses) and vecuronium-induced blockade (0.1 mg/kg with or without maintenance doses) at different time points/depths of block. In these trials a clear dose-response relationship was observed.
BRIDION may contain up to 7% of the mono OH-derivative of sugammadex. In preclinical pharmacology studies, the mono OH-derivative was demonstrated to have ~50% of the affinity as sugammadex for rocuronium and vecuronium and that product with up to 7% of the mono OH-derivative has nearly similar efficacy in reversing rocuronium- or vecuronium-induced blockade.
Although sugammadex has greatest affinity for aminosteroid neuromuscular blocking agents such as rocuronium and vecuronium, plasma levels of endogenous or exogenous compounds with a similar steroidal structure, such as some hormones, hormonal contraceptives, and pheromones may also be reduced following administration of sugammadex [see Drug Interactions (7.3)] .
12.3 Pharmacokinetics
The sugammadex pharmacokinetic parameters were calculated from the total sum of non-complex-bound and complex-bound concentrations of sugammadex. Pharmacokinetic parameters as clearance and volume of distribution are assumed to be the same for non-complex-bound and complex-bound sugammadex in anesthetized patients.
Distribution
The observed steady-state volume of distribution of sugammadex is approximately 11 to 14 liters in adult patients with normal renal function (based on conventional, non-compartmental pharmacokinetic analysis). Neither sugammadex nor the complex of sugammadex and rocuronium binds to plasma proteins or erythrocytes, as was shown in vitro using male human plasma and whole blood. Sugammadex exhibits linear kinetics in the dosage range of 1 to 16 mg/kg when administered as an IV bolus dose.
In nonclinical drug distribution studies, sugammadex is retained in sites of active mineralization, such as bone and teeth, with a mean half-life of 172 and 8 days, respectively [see Use in Specific Populations (8.4), Nonclinical Toxicology (13.2)] .
Metabolism
In clinical studies, no metabolites of sugammadex have been observed and only renal excretion of the unchanged product was observed as the route of elimination.
Elimination
In adult anesthetized patients with normal renal function, the elimination half-life (t 1/2) of sugammadex is about 2 hours and the estimated plasma clearance is about 88 mL/min (based on compartmental pharmacokinetic analysis). A mass balance study demonstrated that >90% of the dose was excreted within 24 hours. Ninety-six percent (96%) of the dose was excreted in urine, of which at least 95% could be attributed to unchanged sugammadex. Excretion via feces or expired air was less than 0.02% of the dose. Administration of BRIDION to healthy volunteers resulted in increased renal elimination of rocuronium in complex.
Patients with Renal Impairment
Sugammadex is known to be substantially excreted by the kidney. The half-life of sugammadex in patients with mild, moderate and severe renal impairment is 4, 6, and 19 hours, respectively.
In one study, exposure to sugammadex was prolonged, leading to 17-fold higher overall exposure in patients with severe renal impairment. Low concentrations of sugammadex are detectable for at least 48 hours post-dose in patients with severe renal impairment.
In a second study comparing subjects with moderate or severe renal impairment to subjects with normal renal function, sugammadex clearance progressively decreased and t 1/2 was progressively prolonged with declining renal function. Exposure was 2-fold and 5-fold higher in subjects with moderate and severe renal impairment, respectively. Sugammadex concentrations were no longer detectable beyond 7 days post-dose in subjects with severe renal impairment.
Age: Geriatric Population
Geriatric patients may have mild or moderate renal impairment. Population pharmacokinetic analysis indicated that, beyond the effects of a decreased creatinine clearance, increased age has limited impact on sugammadex PK parameters [see Use in Specific Populations (8.5, 8.6)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term animal studies to evaluate the carcinogenic potential of sugammadex have not been conducted.
Mutagenesis
Sugammadex and its mono OH-derivative tested negatively in in vitro bacterial reverse mutation assays (Ames test), in vitro chromosomal aberration assays in human peripheral blood lymphocytes, and in vivo micronucleus assays in mice and rats.
Impairment of Fertility
A fertility and early embryonic development study in Sprague-Dawley rats in which male rats were treated daily for 29 days prior to mating and through the mating period and female rats were treated daily for 14 days prior to mating to Day 5 post-coitum via intravenous administration of sugammadex at 20, 100, and 500 mg/kg (0.2, 1, and 6 times the MRHD of 16 mg/kg, respectively, based on AUC comparison) did not show adverse effects on fertility.
13.2 Animal Toxicology and/or Pharmacology
Bone and teeth retention of sugammadex occurred in rats after intravenous injection, with mean half-lives of 172 and 8 days, respectively. Sugammadex bound to hydroxyapatite in an in vitro study and distributed in the bone formation area where hydroxyapatite is present for mineralization in vivo.
In adult rat bone toxicity studies, a single dose of sugammadex at 2000 mg/kg (approximately 24 times the maximum recommended human dose (MRHD) of 16 mg/kg by AUC comparison) administered to adult rats caused a slight increase in bone resorption, but had no effect on teeth color. No adverse bone effects were seen following a single dose of sugammadex at 500 mg/kg (4 times the MRHD dose of 16 mg/kg based on plasma AUC comparison).
In a bone repair study, adult rats were treated with intravenous sugammadex weekly for 6 weeks at 0, 30, 120, and 500 mg/kg (approximately 0.4, 1, and 6 times the MRHD, respectively, by AUC comparison). Based on histological data, high dose animals with post-fracture treatment, showed a statistically significant increase in callus formation and decrease in bone formation, suggesting a potential for a slight delay in the bone healing process. However, there were no statistically significant effects on bone volume or bone mineral density.
In juvenile animal studies, bone and teeth deposition was significantly higher in juvenile rats compared to adults. In addition, sugammadex administered to juvenile rats daily for 4 weeks caused slight bone length decrease (approximately 3%), which did not recover after an 8-week treatment-free period, and reversible whitish discoloration of the teeth at a dose approximately 0.6 times the MRHD, while weekly administration for 8 weeks did not produce similar changes in bone and teeth at doses up to 1.2 times the MRHD [see Use in Specific Populations (8.4)] .
-
14 CLINICAL STUDIES
14.1 Controlled Clinical Studies
Comparative Study of BRIDION versus Neostigmine as a Reversal Agent for Neuromuscular Blockade Induced by Rocuronium or Vecuronium at Reappearance of T 2 (Moderate Blockade)
A multicenter, randomized, parallel-group, active-controlled, safety-assessor blinded study comparing BRIDION and neostigmine enrolled 189 patients (87 women and 102 men, 95% were ASA class 1 and 2 and 99% were Caucasian, median weights were 72 kg and 76 kg and median ages were 50 years and 51 years in the rocuronium and vecuronium groups, respectively). Patients were randomly assigned to the rocuronium or vecuronium group and underwent elective surgical procedures under general anesthesia that required endotracheal intubation and maintenance of neuromuscular blockade. The surgical procedures were mainly endocrine, ocular, ENT, abdominal (gynecological, colorectal, urological), orthopedic, vascular, or dermatological. At the reappearance of T 2, after the last dose of rocuronium or vecuronium, 2 mg/kg BRIDION or 50 mcg/kg neostigmine was administered in a randomized order as a single bolus injection. The time from start of administration of BRIDION or neostigmine to recovery of the TOF (T 4/T 1) ratio to 0.9 was assessed. Generally, a T 4/T 1 ratio ≥0.9 correlates with recovery from neuromuscular blockade.
Return of the T 4/T 1 ratio to 0.9 after the reappearance of T 2 was overall faster with BRIDION 2 mg/kg as compared to neostigmine 50 mcg/kg in the setting of rocuronium or vecuronium-induced neuromuscular blockade (Figures 1 and 2).
Figure 1: Time (Minutes) from Administration of BRIDION or Neostigmine at the Reappearance of T 2 after Rocuronium to Recovery of the T 4/T 1 Ratio to 0.9 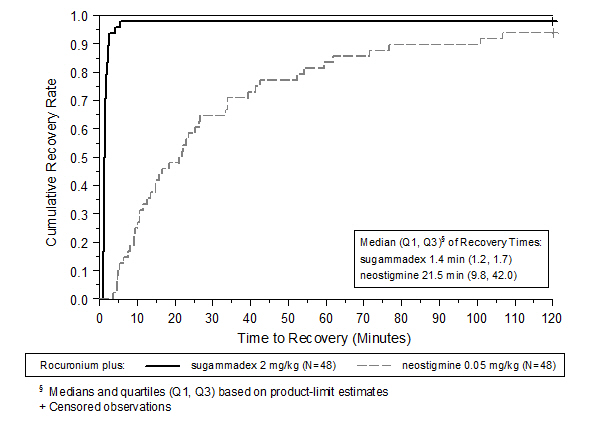
Figure 2: Time (Minutes) from Administration of BRIDION or Neostigmine at the Reappearance of T 2 after Vecuronium to Recovery of the T 4/T 1 Ratio to 0.9 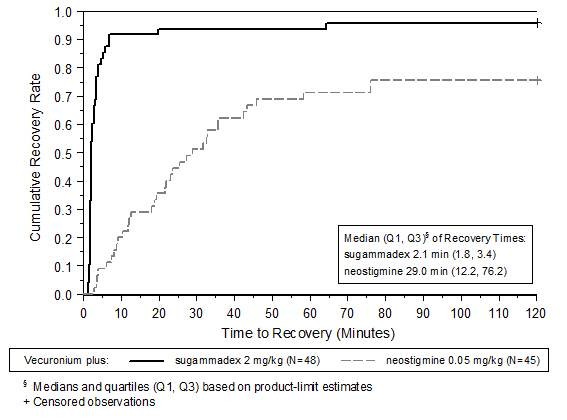
Comparative Study of BRIDION versus Neostigmine as a Reversal Agent for Neuromuscular Blockade Induced by Rocuronium or Vecuronium at 1 to 2 PTCs (Deep Blockade)
A multicenter, randomized, parallel-group, active-controlled, safety-assessor blinded study comparing BRIDION and neostigmine enrolled 157 patients (86 women and 71 men; 8% ASA class 1, 71% class 2, and 21% class 3; 79% Caucasian; median weights of 81 kg and 84 kg, and median ages of 54 years and 56 years in the rocuronium and vecuronium groups, respectively). Patients were randomly assigned to the rocuronium or vecuronium group and underwent elective surgical procedures under general anesthesia that required endotracheal intubation and maintenance of neuromuscular blockade. The surgical procedures were mainly abdominal (gynecological, colorectal, urological), orthopedic, reconstructive, or neurological. At 1 to 2 PTCs, after the last dose of rocuronium or vecuronium, 4 mg/kg BRIDION or 70 mcg/kg neostigmine was administered in a randomized order as a single bolus injection. The time from start of administration of BRIDION or neostigmine to recovery of the TOF (T 4/T 1) ratio to 0.9 was assessed, although neostigmine was not expected to reverse neuromuscular blockade at a depth of 1 to 2 PTCs. Generally, a T 4/T 1 ratio ≥0.9 correlates with recovery from neuromuscular blockade.
Return of the T 4/T 1 ratio to 0.9 in patients with 1 to 2 PTCs with BRIDION 4 mg/kg had a wider range of recovery times but the median time to recovery was comparable to the study of reversal at T 2 (2.7 minutes with 25 th and 75 th percentiles of 2.1 and 4.3 minutes for rocuronium [N=37], and 3.3 minutes with 25 th and 75 th percentiles of 2.3 and 6.6 minutes for vecuronium [N=47]). There were 7 and 6 censored observations in the rocuronium and vecuronium groups, respectively.
Reversal of Neuromuscular Blockade 3 Minutes after Rocuronium 1.2 mg/kg
Time to recovery from neuromuscular blockade induced by succinylcholine compared with recovery from neuromuscular blockade induced by rocuronium followed 3 minutes later with BRIDION was assessed in a multicenter, randomized, parallel-group, active-controlled, safety-assessor blinded study. The study was conducted in 110 patients (64 women and 46 men, ASA class 1 and 2, 78% were Caucasian, median weight was 70 kg, median age was 43 years). Patients underwent elective surgical procedures under general anesthesia that required endotracheal intubation and a short duration of neuromuscular relaxation. The laparoscopic or open surgical procedures were mainly gynecological, orthopedic, or reconstructive. Return of the first twitch in a TOF (T 1) to 10% of baseline was compared between BRIDION 16 mg/kg for reversal of rocuronium 1.2 mg/kg versus spontaneous recovery from succinylcholine 1 mg/kg.
Recovery to T 1 of 10% of baseline (relative to the time of administration of rocuronium or succinylcholine) was overall faster in the rocuronium/BRIDION group compared with succinylcholine alone (Table 3).
Table 3: Time (minutes) from Start of Administration of Rocuronium or Succinylcholine to Recovery of T 1 to 10% of Baseline Treatment Regimen Rocuronium (1.2 mg/kg) and BRIDION (16 mg/kg) Succinylcholine (1 mg/kg) N 55 55 Mean (SD) 4.4 (0.7) 7.1 (1.6) Median (Range) 4.2 (3.5 – 7.7) 7.1 (3.8 – 10.5) -
16 HOW SUPPLIED/STORAGE AND HANDLING
BRIDION (sugammadex) injection is a clear, colorless to slightly yellow-brown, non-pyrogenic aqueous solution intended for intravenous infusion. BRIDION is available in the following presentations:
- BRIDION 2-mL single-dose vials containing 200 mg sugammadex (100 mg/mL)
Box of 10 NDC 0006-5423-12 - BRIDION 5-mL single-dose vials containing 500 mg sugammadex (100 mg/mL)
Box of 10 NDC 0006-5423-15, NDC 0006-5425-15
The packaging of this product is not made with natural rubber latex.
- BRIDION 2-mL single-dose vials containing 200 mg sugammadex (100 mg/mL)
-
17 PATIENT COUNSELING INFORMATION
- Advise females of reproductive potential using hormonal contraceptives that BRIDION may reduce the contraceptive effect . Instruct females to use an additional, non-hormonal method of contraception for the next 7 days following BRIDION administration [see Drug Interactions (7.3)].
-
SPL UNCLASSIFIED SECTION
Distributed by: Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC., Whitehouse Station, NJ 08889, USAFor patent information: www.merck.com/product/patent/home.html
Copyright © 2015-2018 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved.uspi-mk8616-soi-1812r004
- PRINCIPAL DISPLAY PANEL - OUTER PACKAGE
-
INGREDIENTS AND APPEARANCE
BRIDION
sugammadex injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71872-7187(NDC:0006-5423) Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SUGAMMADEX SODIUM (UNII: ERJ6X2MXV7) (SUGAMMADEX - UNII:361LPM2T56) SUGAMMADEX 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71872-7187-1 1 in 1 BAG 12/16/2019 1 2 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022225 12/15/2015 Labeler - Medical Purchasing Solutions, LLC (601458529) Establishment Name Address ID/FEI Business Operations Medical Purchasing Solutions, LLC 601458529 repack(71872-7187)
