Label: GEMFIBROZIL tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 55700-493-30, 55700-493-60, 55700-493-90 - Packager: Lake Erie Medical DBA Quality Care Products LLC
- This is a repackaged label.
- Source NDC Code(s): 69097-821
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated November 3, 2020
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Gemfibrozil, USP is a lipid regulating agent. It is available as tablets for oral administration. Each tablet contains 600 mg gemfibrozil. Each tablet also contains the following inactive ingredients: colloidal silicon dioxide, NF; croscarmellose sodium, NF; calcium stearate, NF; microcrystalline cellulose, NF; methylcellulose, USP and opadry white. The chemical name is 5-(2,5-dimethylphenoxy)-2,2-dimethylpentanoic acid, with the following structural formula:
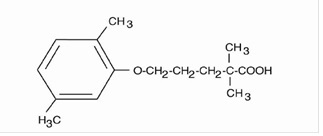
The empirical formula is C15H22O3 and the molecular weight is 250.35; the solubility in water and acid is 0.0019% and in dilute base it is greater than 1%. The melting point is 58° to 61°C. Gemfibrozil is a white solid which is stable under ordinary conditions.
-
CLINICAL PHARMACOLOGY
Gemfibrozil is a lipid regulating agent which decreases serum triglycerides and very low density lipoprotein (VLDL) cholesterol, and increases high density lipoprotein (HDL) cholesterol. While modest decreases in total and low density lipoprotein (LDL) cholesterol may be observed with gemfibrozil therapy, treatment of patients with elevated triglycerides due to Type IV hyperlipoproteinemia often results in a rise in LDL-cholesterol. LDL-cholesterol levels in Type IIb patients with elevations of both serum LDL-cholesterol and triglycerides are, in general, minimally affected by gemfibrozil treatment; however, gemfibrozil usually raises HDL-cholesterol significantly in this group. Gemfibrozil increases levels of high density lipoprotein (HDL) subfractions HDL2 and HDL3, as well as apolipoproteins AI and AII. Epidemiological studies have shown that both low HDL-cholesterol and high LDL-cholesterol are independent risk factors for coronary heart disease.
In the primary prevention component of the Helsinki Heart Study, in which 4081 male patients between the ages of 40 and 55 were studied in a randomized, double-blind, placebo-controlled fashion, gemfibrozil therapy was associated with significant reductions in total plasma triglycerides and a significant increase in high density lipoprotein cholesterol. Moderate reductions in total plasma cholesterol and low density lipoprotein cholesterol were observed for the gemfibrozil treatment group as a whole, but the lipid response was heterogeneous, especially among different Fredrickson types. The study involved subjects with serum non-HDL-cholesterol of over 200 mg/dL and no previous history of coronary heart disease. Over the five-year study period, the gemfibrozil group experienced a 1.4% absolute (34% relative) reduction in the rate of serious coronary events (sudden cardiac deaths plus fatal and nonfatal myocardial infarctions) compared to placebo, p=0.04 (see Table I).There was a 37% relative reduction in the rate of nonfatal myocardial infarction compared to placebo, equivalent to a treatment-related difference of 13.1 events per thousand persons. Deaths from any cause during the double-blind portion of the study totaled 44 (2.2%) in the gemfibrozil randomization group and 43 (2.1 %) in the placebo group.
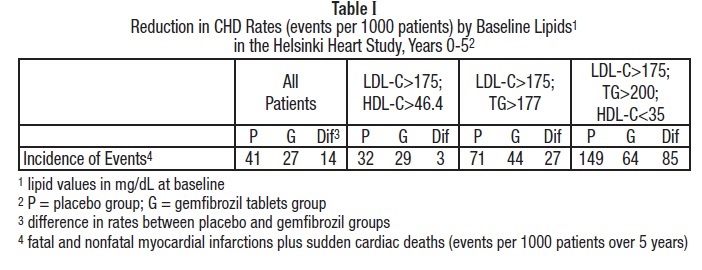
Among Fredrickson types, during the 5-year double-blind portion of the primary prevention component of the Helsinki Heart Study, the greatest reduction in the incidence of serious coronary events occurred in Type IIb patients who had elevations of both LDL-cholesterol and total plasma triglycerides. This subgroup of Type IIb gemfibrozil group patients had a lower mean HDL-cholesterol level at baseline than the Type IIa subgroup that had elevations of LDL-cholesterol and normal plasma triglycerides. The mean increase in HDL-cholesterol among the Type IIb patients in this study was 12.6% compared to placebo. The mean change in LDL-cholesterol among Type IIb patients was -4.1% with gemfibrozil compared to a rise of 3.9% in the placebo subgroup. The Type IIb subjects in the Helsinki Heart Study had 26 fewer coronary events per thousand persons over five years in the gemfibrozil group compared to placebo. The difference in coronary events was substantially greater between gemfibrozil and placebo for that subgroup of patients with the triad of LDL-cholesterol >175 mg/dL (>4.5 mmol), triglycerides >200 mg/dL (>2.2 mmol), and HDL-cholesterol <35 mg/dL (<0.90 mmol) (see Table I).
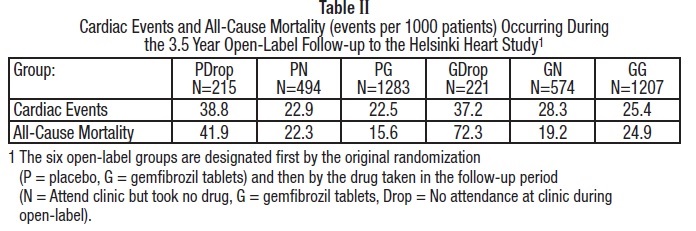
Further information is available from a 3.5 year (8.5 year cumulative) follow-up of all subjects who had participated in the Helsinki Heart Study. At the completion of the Helsinki Heart Study, subjects could choose to start, stop, or continue to receive gemfibrozil; without knowledge of their own lipid values or double-blind treatment, 60% of patients originally randomized to placebo began therapy with gemfibrozil and 60% of patients originally randomized to gemfibrozil continued medication. After approximately 6.5 years following randomization, all patients were informed of their original treatment group and lipid values during the five years of the double-blind treatment. After further elective changes in gemfibrozil treatment status, 61% of patients in the group originally randomized to gemfibrozil were taking drug; in the group originally randomized to placebo, 65% were taking gemfibrozil. The event rate per 1000 occurring during the open-label follow-up period is detailed in Table II.
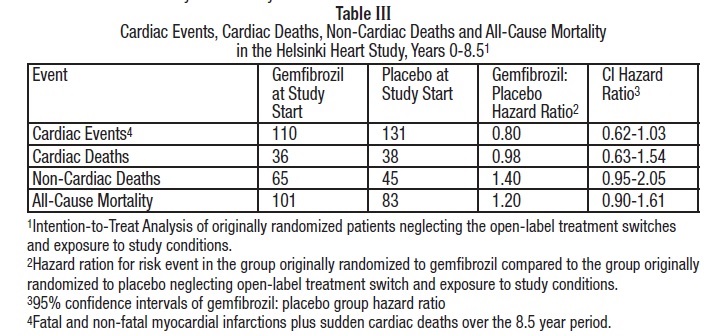
Cumulative mortality through 8.5 years showed a 20% relative excess of deaths in the group originally randomized to gemfibrozil versus the originally randomized placebo group and a 20% relative decrease in cardiac events in the group originally randomized to gemfibrozil versus the originally randomized placebo group (see Table III).This analysis of the originally randomized "intent-to-treat" population neglects the possible complicating effects of treatment switching during the open-label phase. Adjustment of hazard ratios, taking into account open-label treatment status from years 6.5 to 8.5, could change the reported hazard ratios for mortality toward unity.
It is not clear to what extent the findings of the primary prevention component of the Helsinki Heart Study can be extrapolated to other segments of the dyslipidemic population not studied (such as women, younger or older males, or those with lipid abnormalities limited solely to HDL-cholesterol) or to other lipid-altering drugs.
The secondary prevention component of the Helsinki Heart Study was conducted over five years in parallel and at the same centers in Finland in 628 middle-aged males excluded from the primary prevention component of the Helsinki Heart Study because of a history of angina, myocardial infarction or unexplained ECG changes. The primary efficacy endpoint of the study was cardiac events (the sum of fatal and non-fatal myocardial infarctions and sudden cardiac deaths). The hazard ratio (gemfibrozil: placebo) for cardiac events was 1.47 (95% confidence limits 0.88-2.48, p=0.14). Of the 35 patients in the gemfibrozil group who experienced cardiac events, 12 patients suffered events after discontinuation from the study. Of the 24 patients in the placebo group with cardiac events, 4 patients suffered events after discontinuation from the study. There were 17 cardiac deaths in the gemfibrozil group and 8 in the placebo group (hazard ratio 2.18; 95% confidence limits 0.94-5.05, p=0.06). Ten of these deaths in the gemfibrozil group and 3 in the placebo group occurred after discontinuation from therapy. In this study of patients with known or suspected coronary heart disease, no benefit from gemfibrozil treatment was observed in reducing cardiac events or cardiac deaths. Thus, gemfibrozil has shown benefit only in selected dyslipidemic patients without suspected or established coronary heart disease. Even in patients with coronary heart disease and the triad of elevated LDL-cholesterol, elevated triglycerides, plus low HDL-cholesterol, the possible effect of gemfibrozil on coronary events has not been adequately studied.
No efficacy in the patients with established coronary heart disease was observed during the Coronary Drug Project with the chemically and pharmacologically related drug, clofibrate. The Coronary Drug Project was a 6-year randomized, double-blind study involving 1000 clofibrate, 1000 nicotinic acid, and 3000 placebo patients with known coronary heart disease. A clinically and statistically significant reduction in myocardial infarctions was seen in the concurrent nicotinic acid group compared to placebo; no reduction was seen with clofibrate.
The mechanism of action of gemfibrozil has not been definitely established. In man, gemfibrozil has been shown to inhibit peripheral lipolysis and to decrease the hepatic extraction of free fatty acids, thus reducing hepatic triglyceride production. Gemfibrozil inhibits synthesis and increases clearance of VLDL carrier apolipoprotein B, leading to a decrease in VLDL production.
Animal studies suggest that gemfibrozil may, in addition to elevating HDL-cholesterol, reduce incorporation of long-chain fatty acids into newly formed triglycerides, accelerate turnover and removal of cholesterol from the liver, and increase excretion of cholesterol in the feces. Gemfibrozil is well absorbed from the gastrointestinal tract after oral administration. Peak plasma levels occur in 1 to 2 hours with a plasma half-life of 1.5 hours following multiple doses.
Gemfibrozil is completely absorbed after oral administration of gemfibrozil tablets, reaching peak plasma concentrations 1 to 2 hours after dosing. Gemfibrozil pharmacokinetics are affected by the timing of meals relative to time of dosing. In one study (ref.4), both the rate and extent of absorption of the drug were significantly increased when administered 0.5 hour before meals. Average AUC was reduced by 14-44% when gemfibrozil was administered after meals compared to 0.5 hour before meals. In a subsequent study, rate of absorption of gemfibrozil was maximum when administered 0.5 hour before meals with the Cmax 50 to 60% greater than when given either with meals or fasting. In this study, there were no significant effects on AUC of timing of dose relative to meals (see DOSAGE AND ADMINISTRATION).
Gemfibrozil mainly undergoes oxidation of a ring methyl group to successively form a hydroxymethyl and a carboxyl metabolite. Approximately seventy percent of the administered human dose is excreted in the urine, mostly as the glucuronide conjugate, with less than 2% excreted as unchanged gemfibrozil. Six percent of the dose is accounted for in the feces. Gemfibrozil is highly bound to plasma proteins and there is potential for displacement interactions with other drugs (see PRECAUTIONS).
-
INDICATIONS AND USAGE
Gemfibrozil Tablets, USP are indicated as adjunctive therapy to diet for:
1. Treatment of adult patients with very high elevations of serum triglyceride levels (Types IV and V hyperlipidemia) who present a risk of pancreatitis and who do not respond adequately to a determined dietary effort to control them. Patients who present such risk typically have serum
triglycerides over 2000 mg/dL and have elevations of VLDL-cholesterol as well as fasting chylomicrons (Type V hyperlipidemia). Subjects who consistently have total serum or plasma triglycerides below 1000 mg/dL are unlikely to present a risk of pancreatitis. Gemfibrozil therapy may be considered for those subjects with triglyceride elevations between 1000 and 2000 mg/dL who have a history of pancreatitis or of recurrent abdominal pain typical of pancreatitis. It is recognized that some Type IV patients with triglycerides under 1000 mg/dL may, through dietary or alcoholic indiscretion, convert to a Type V pattern with massive triglyceride elevations accompanying fasting chylomicronemia, but the influence of gemfibrozil therapy on the risk of pancreatitis in such situations has not been adequately studied. Drug therapy is not indicated for patients with Type I hyperlipoproteinemia, who have elevations of chylomicrons and plasma triglycerides, but who have normal levels of very low density lipoprotein (VLDL). Inspection of plasma refrigerated for 14 hours is helpful in distinguishing Types I, IV, and V hyperlipoproteinemia.
2. Reducing the risk of developing coronary heart disease only in Type IIb patients without history of or symptoms of existing coronary heart disease who have had an inadequate response to weight loss, dietary therapy, exercise, and other pharmacologic agents (such as bile acid sequestrants and nicotinic acid, known to reduce LDL- and raise HDL-cholesterol) and who have the following triad of lipid abnormalities: low HDL-cholesterol levels in addition to elevated LDL-cholesterol and elevated triglycerides (see WARNINGS, PRECAUTIONS, and CLINICAL PHARMACOLOGY). The National Cholesterol Education Program has defined a serum HDL-cholesterol value that is consistently below 35 mg/dL as constituting an independent risk factor for coronary heart disease. Patients with significantly elevated triglycerides should be closely observed when treated with gemfibrozil. In some patients with high triglyceride levels,
treatment with gemfibrozil is associated with a significant increase in LDL-cholesterol. BECAUSE OF POTENTIAL TOXICITY SUCH AS MALIGNANCY, GALLBLADDER DISEASE, ABDOMINAL PAIN LEADING TO APPENDECTOMY AND OTHER ABDOMINAL SURGERIES, AN INCREASED INCIDENCE IN NON-CORONARY MORTALITY, AND THE 44% RELATIVE INCREASE DURING THE TRIAL PERIOD IN AGE-ADJUSTED ALL-CAUSE MORTALITY SEEN WITH THE CHEMICALLY AND PHARMACOLOGICALLY RELATED DRUG, CLOFIBRATE, THE POTENTIAL BENEFIT OF GEMFIBROZIL IN TREATING TYPE IIA PATIENTS WITH ELEVATIONS OF LDL-CHOLESTEROL ONLY IS NOT LIKELY TO OUTWEIGH THE RISKS. GEMFIBROZIL IS ALSO NOT INDICATED FOR THE TREATMENT OF PATIENTS WITH LOW HDL-CHOLESTEROL AS THEIR ONLY LIPID ABNORMALITY.
In a subgroup analysis of patients in the Helsinki Heart Study with above-median HDL-cholesterol values at baseline (greater than 46.4 mg/dL), the incidence of serious coronary events was similar for gemfibrozil and placebo subgroups (see Table I).
The initial treatment for dyslipidemia is dietary therapy specific for the type of lipoprotein abnormality. Excess body weight and excess alcohol intake may be important factors in hypertriglyceridemia and should be managed prior to any drug therapy. Physical exercise can be an important ancillary measure, and has been associated with rises in HDL-cholesterol. Diseases contributory to hyperlipidemia such as hypothyroidism or diabetes mellitus should be looked for and adequately treated. Estrogen therapy is sometimes associated with massive rises in plasma triglycerides, especially in subjects with familial hypertriglyceridemia. In such cases, discontinuation of estrogen therapy may obviate the need for specific drug therapy of hypertriglyceridemia. The use of drugs should be considered only when reasonable attempts have been made to obtain satisfactory results with nondrug methods. If the decision is made to use drugs, the patient should be instructed that this does not reduce the importance of adhering to diet.
-
CONTRAINDICATIONS
- Hepatic or severe renal dysfunction, including primary biliary cirrhosis.
- Preexisting gallbladder disease (see WARNINGS).
- Hypersensitivity to gemfibrozil.
- Combination therapy of gemfibrozil with simvastatin (see WARNINGS and PRECAUTIONS).
- Combination therapy of gemfibrozil with repaglinide (see PRECAUTIONS).
- Combination therapy of gemfibrozil with dasabuvir (see PRECAUTIONS)
-
WARNINGS
1. Because of chemical, pharmacological, and clinical similarities between gemfibrozil and clofibrate, the adverse findings with clofibrate in two large clinical studies may also apply to gemfibrozil. In the first of those studies, the Coronary Drug Project, 1000 subjects with previous myocardial infarction were treated for five years with clofibrate. There was no difference in mortality between the clofibrate-treated subjects and 3000 placebo-treated subjects, but twice as many clofibrate-treated subjects developed cholelithiasis and cholecystitis requiring surgery. In the other study, conducted by the World Health Organization (WHO), 5000 subjects without known coronary heart disease were treated with clofibrate for five years and followed one year beyond. There was a statistically significant (44%) higher age-adjusted total mortality in the clofibrate-treated group than in a comparable placebo-treated control group during the trial period. The excess mortality was due to a 33% increase in non-cardiovascular causes, including malignancy, post-cholecystectomy complications, and pancreatitis. The higher risk of clofibrate- treated subjects for gallbladder disease was confirmed.
Because of the more limited size of the Helsinki Heart Study, the observed difference in mortality from any cause between the gemfibrozil and placebo groups is not statistically significantly different from the 29% excess mortality reported in the clofibrate group in the separate WHO study at the nine year follow-up (see CLINICAL PHARMACOLOGY). Noncoronary heart disease related mortality showed an excess in the group originally randomized to gemfibrozil primarily due to cancer deaths observed during the open-label extension.
During the five year primary prevention component of the Helsinki Heart Study, mortality from any cause was 44 (2.2%) in the gemfibrozil group and 43 (2.1%) in the placebo group; including the 3.5 year follow-up period since the trial was completed, cumulative mortality from any cause was 101 (4.9%) in the gemfibrozil group and 83 (4.1%) in the group originally randomized to placebo (hazard ratio 1:20 in favor of placebo). Because of the more limited size of the Helsinki Heart Study, the observed difference in mortality from any cause between the gemfibrozil and placebo groups at Year-5 or at Year-8.5 is not statistically significantly different from the 29% excess mortality reported in the clofibrate group in the separate WHO study at the nine year follow-up. Noncoronary heart disease related mortality showed an excess in the group originally randomized to gemfibrozil at the 8.5 year follow-up (65 gemfibrozil versus 45 placebo noncoronary deaths).
The incidence of cancer (excluding basal cell carcinoma) discovered during the trial and in the 3.5 years after the trial was completed was 51 (2.5%) in both originally randomized groups. In addition, there were 16 basal cell carcinomas in the group originally randomized to gemfibrozil and 9 in the group randomized to placebo (p=0.22). There were 30 (1.5%) deaths attributed to cancer in the group originally randomized to gemfibrozil and 18 (0.9%) in the group originally randomized to placebo (p=0.11). Adverse outcomes, including coronary events, were higher in gemfibrozil patients in a corresponding study in men with a history of known or suspected coronary heart disease in the secondary prevention component of the Helsinki Heart Study (see CLINICAL PHARMACOLOGY).
A comparative carcinogenicity study was also done in rats comparing three drugs in this class: fenofibrate (10 and 60 mg/kg; 0.3 and 1.6 times the human dose, respectively), clofibrate (400 mg/kg; 1.6 times the human dose), and gemfibrozil (250 mg/kg; 1.7 times the human dose). Pancreatic acinar adenomas were increased in males and females on fenofibrate; hepatocellular carcinoma and pancreatic acinar adenomas were increased in males and hepatic neoplastic nodules in females treated with clofibrate; hepatic neoplastic nodules were increased in males and females treated with clofibrate; hepatic neoplastic nodules were increased in males and females treated with gemfibrozil while testicular interstitial cell (Leydig cell) tumors were increased in males on all three drugs.
2. A gallstone prevalence substudy of 450 Helsinki Heart Study participants showed a trend toward a greater prevalence of gallstones during the study within the gemfibrozil treatment group (7.5% versus 4.9% for the placebo group, a 55% excess for the gemfibrozil group). A trend toward a greater incidence of gallbladder surgery was observed for the gemfibrozil group (17 versus 11 subjects, a 54% excess). This result did not differ statistically from the increased incidence of cholecystectomy observed in the WHO study in the group treated with clofibrate. Both clofibrate and gemfibrozil may increase cholesterol excretion into the bile, leading to cholelithiasis. If cholelithiasis is suspected, gallbladder studies are indicated. Gemfibrozil therapy should be discontinued if gallstones are found. Cases of cholelithiasis have been reported with gemfibrozil therapy.
3. Since a reduction of mortality from coronary heart disease has not been demonstrated and because liver and interstitial cell testicular tumors were increased in rats, gemfibrozil should be administered only to those patients described in the INDICATIONS AND USAGE section. If a significant serum lipid response is not obtained, gemfibrozil should be discontinued.
4. Concomitant Anticoagulants-Caution should be exercised when warfarin is given in conjunction with gemfibrozil. The dosage of the warfarin should be reduced to maintain the prothrombin time at the desired level to prevent bleeding complications. Frequent prothrombin determinations are advisable until it has been definitely determined that the prothrombin level has stabilized.
5. The concomitant administration of gemfibrozil with simvastatin is contraindicated (see CONTRAINDICATIONSand PRECAUTIONS). Concomitant therapy with gemfibrozil and an HMG-CoA reductase inhibitor is associated with an increased risk of skeletal muscle toxicity manifested as rhabdomyolysis, markedly elevated creatine kinase (CPK) levels, and myoglobinuria, leading in a high proportion of cases to acute renal failure and death. IN PATIENTS WHO HAVE HAD AN UNSATISFACTORY LIPID RESPONSE TO EITHER DRUG ALONE, THE BENEFIT OF COMBINED THERAPY WITH GEMFIBROZIL an HMG-CoA REDUCTASE INHIBITOR DOES NOT OUTWEIGH THE RISKS OF SEVERE MYOPATHY, RHABDOMYOLYSIS, AND ACUTE RENAL FAILURE (see PRECAUTIONS, Drug Interactions). The use of fibrates alone, including gemfibrozil, may occasionally be associated with myositis. Patients receiving gemfibrozil and complaining of muscle pain, tenderness, or weakness should have prompt medical evaluation for myositis, including serum creatine-kinase level determination. If myositis is suspected or diagnosed, gemfibrozil therapy should be withdrawn.
6. Cataracts-Subcapsular bilateral cataracts occurred in 10%, and unilateral in 6.3%, of male rats treated with gemfibrozil at 10 times the human dose.
7. CYP2C8 substrates -Gemfibrozil, an inhibitor of CYP2C8, may increase exposure of CYP2C8 substrates when administered concomitantly (see PRECAUTIONS, Drug Interactions).
8. OATP1B1 substrates – Gemfibrozil is an inhibitor of organic anion-transporter polyprotein (OATP) 1B1 and may increase exposure of drugs that are substrates of OATP1B1 (e.g., atrasentan, atorvastatin, bosentan, ezetimibe, fluvastatin, glyburide, SN38 [active metabolite of irinotecan], rosuvastatin, pitavastatin, pravastatin, rifampin, valsartan, olmesartan). Therefore, dosing reductions of drugs that are substrates of OATP1B1 may be required when gemfibrozil is used concomitantly (see PRECAUTIONS, Drug Interactions). Combination therapy of gemfibrozil with simvastatin or with repaglinide, which are OATP1B1 substrates, is contraindicated (see CONTRAINDICATIONS).
-
PRECAUTIONS
1. Initial Therapy-Laboratory studies should be done to ascertain that the lipid levels are consistently abnormal. Before instituting gemfibrozil therapy, every attempt should be made to control serum lipids with appropriate diet, exercise, weight loss in obese patients, and control of any medical problems such as diabetes mellitus and hypothyroidism that are contributing to the lipid abnormalities.
2. Continued Therapy-Periodic determination of serum lipids should be obtained, and the drug withdrawn if lipid response is inadequate after three months of therapy.
3. Drug Interactions-(A) HMG-CoA reductase inhibitors: The concomitant administration of gemfibrozil with simvastatin is contraindicated (see CONTRAINDICATIONSand WARNINGS). The risk of myopathy and rhabdomyolysis is increased with combined gemfibrozil and HMG-CoA reductase inhibitor therapy. Myopathy or rhabdomyolysis with or without acute renal failure have been reported as early as three weeks after initiation of combined therapy or after several months (see WARNINGS). There is no assurance that periodic monitoring of creatine kinase will prevent the occurrence of severe myopathy and kidney damage.
(B)Anticoagulants: CAUTION SHOULD BE EXERCISED WHEN WARFARIN IS GIVEN IN CONJUNCTION WITH GEMFIBROZIL. THE DOSAGE OF THE WARFARIN SHOULD BE REDUCED TO MAINTAIN THE PROTHROMBIN TIME AT THE DESIRED LEVEL TO PREVENT BLEEDING COMPLICATIONS. FREQUENT PROTHROMBIN DETERMINATIONS ARE ADVISABLE UNTIL IT HAS BEEN DEFINITELY DETERMINED THAT THE PROTHROMBIN LEVEL HAS STABILIZED.
(C) CYP2C8 Substrates: Gemfibrozil is an inhibitor of CYP2C8 and may increase exposure of drugs mainly metabolized by CYP2C8 (e.g., dabrafenib, loperamide, montelukast, paclitaxel, pioglitazone, rosiglitazone). Therefore, dosing reduction of drugs that are mainly metabolized by CYP2C8 enzyme may be required when gemfibrozil is used concomitantly (see WARNINGS).
Repaglinide: In healthy volunteers, co-administration with gemfibrozil (600 mg twice daily for 3 days) resulted in an 8.1-fold (range 5.5-to 15.0-fold) higher repaglinide AUC and a 28.6-fold (range 18.5-to 80.1-fold) higher repaglinide plasma concentration 7 hours after the dose. In the same study, gemfibrozil (600 mg twice daily for 3 days) + itraconazole (200 mg in the morning and 100 mg in the evening at Day 1, then 100 mg twice daily at Day 2-3) resulted in a 19.4-(range 12.9-to 24.7-fold) higher repaglinide AUC and a 70.4-fold (range 42.9-to 119.2-fold) higher repaglinide plasma concentration 7 hours after the dose. In addition, gemfibrozil alone or gemfibrozil + itraconazole prolonged the hypoglycemic effects of repaglinide. Co-administration of gemfibrozil and repaglinide increases the risk of severe hypoglycemia and is contraindicated (see CONTRAINDICATIONS).
Dasabuvir: Co-administration of gemfibrozil with dasabuvir increased dasabuvir AUC and Cmax (ratios: 11.3 and 2.01, respectively) due to CYP2C8 inhibition. Increased dasabuvir exposure may increase the risk of QT prolongation, therefore, coadministration of gemfibrozil with dasabuvir is contraindicated (see CONTRAINDICATIONS).
(D) OATP1B1 substrates: Gemfibrozil is an inhibitor of OATP1B1 transporter and may increase exposure of drugs that are substrates of OATP1B1 (e.g., atrasentan, atorvastatin, bosentan, ezetimibe, fluvastatin, glyburide, SN-38 [active metabolite of irinotecan], rosuvastatin, pitavastatin, pravastatin, rifampin, valsartan, olmesartan). Therefore, dosing reductions of drugs that are substrates of OATP1B1 may be required when gemfibrozil is used concomitantly (see WARNINGS). Combination therapy of gemfibrozil with simvastatin or with repaglinide, which are OATP1B1 substrates, is contraindicated (see CONTRAINDICATIONS).
(E) In vitro studies of CYP enzymes, UGTA enzymes and OATP1B1 transporter: In vitro studies have shown that gemfibrozil is an inhibitor of CYP1A2, CYP2C8, CYP2C9, CYP2C19, OATP1B1, and UDP-glucuronosyltransferase (UGT) 1A1 and 1A3 (see WARNINGS).
(F) Bile Acid-Binding Resins: Gemfibrozil AUC was reduced by 30% when gemfibrozil was given (600 mg) simultaneously with resin-granule drugs such as colestipol (5 g). Administration of the drugs two hours or more apart is recommended because gemfibrozil exposure was not significantly affected when it was administered two hours apart from colestipol.
(G) Colchicine: Myopathy, including rhabdomyolysis, has been reported with chronic administration of colchicine at therapeutic doses. Concomitant use of gemfibrozil may potentiate the development of myopathy. Patients with renal dysfunction and elderly patients are at increased risk. Caution should be exercised when prescribing gemfibrozil with colchicine, especially in elderly patients or patients with renal dysfunction.
4. Carcinogenesis, Mutagenesis, Impairment of Fertility – Long-term studies have been conducted in rats at 0.2 and 1.3 times the human exposure (based on AUC). The incidence of benign liver nodules and liver carcinomas was significantly increased in high dose male rats. The incidence of liver carcinomas increased also in low dose males, but this increase was not statistically significant (p=0.1). Male rats had a dose-related and statistically significant increase of benign Leydig cell tumors. The higher dose female rats had a significant increase in the combined incidence of benign and malignant liver neoplasms.
Long-term studies have been conducted in mice at 0.1 and 0.7 times the human exposure (based on AUC). There were no statistically significant differences from controls in the incidence of liver tumors, but the doses tested were lower than those shown to be carcinogenic with other fibrates.
Electron microscopy studies have demonstrated a florid hepatic peroxisome proliferation following gemfibrozil administration to the male rat. An adequate study to test for peroxisome proliferation has not been done in humans but changes in peroxisome morphology have been observed. Peroxisome proliferation has been shown to occur in humans with either of two other drugs of the fibrate class when liver biopsies were compared before and after treatment in the same individual.
Administration of approximately 2 times the human dose (based on surface area) to male rats for 10 weeks resulted in a dose-related decrease of fertility. Subsequent studies demonstrated that this effect was reversed after a drug-free period of about eight weeks, and it was not transmitted to the offspring.
5.Pregnancy Category C-Gemfibrozil has been shown to produce adverse effects in rats and rabbits at doses between 0.5 and 3 times the human dose (based on surface area). There are no adequate and well-controlled studies in pregnant women. Gemfibrozil should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Administration of gemfibrozil to female rats at 2 times the human dose (based on surface area) before and throughout gestation caused a dose-related decrease in conception rate, an increase in stillborns, and a slight reduction in pup weight during lactation. There were also dose-related increased skeletal variations. Anophthalmia occurred, but rarely.
Administration of 0.6 and 2 times the human dose (based on surface area) of gemfibrozil to female rats from gestation day 15 through weaning caused dose-related decreases in birth weight and suppressions of pup growth during lactation.
Administration of 1 and 3 times the human dose (based on surface area) of gemfibrozil to female rabbits during organogenesis caused a dose-related decrease in litter size and, at the high dose, an increased incidence of parietal bone variations.
6.Nursing Mothers-It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for tumorigenicity shown for gemfibrozil in animal studies, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
7.Hematologic Changes-Mild hemoglobin, hematocrit and white blood cell decreases have been observed in occasional patients following initiation of gemfibrozil therapy. However, these levels stabilize during long-term administration. Rarely, severe anemia, leukopenia, thrombocytopenia, and bone marrow hypoplasia have been reported. Therefore, periodic blood counts are recommended during the first 12 months of gemfibrozil administration.
8.Liver Function–Abnormal liver function tests have been observed occasionally during gemfibrozil administration, including elevations of AST, ALT, LDH, bilirubin, and alkaline phosphatase. These are usually reversible when gemfibrozil is discontinued. Therefore, periodic liver function studies are recommended and gemfibrozil therapy should be terminated if abnormalities persist.
9.Kidney Function-There have been reports of worsening renal insufficiency upon the addition of gemfibrozil therapy in individuals with baseline plasma creatinine >2.0 mg/dL. In such patients, the use of alternative therapy should be considered against the risks and benefits of a lower dose of gemfibrozil.
10. Pediatric Use-Safety and efficacy in pediatric patients have not been established.
-
ADVERSE REACTIONS
In the double-blind controlled phase of the primary prevention component of the Helsinki Heart Study, 2046 patients received gemfibrozil for up to five years. In that study, the following adverse reactions were statistically more frequent in subjects in the gemfibrozil group:
GEMFIBROZIL
(N = 2046)
PLACEBO
(N = 2035)
Frequency in percent of subjects
Gastrointestinal reactions
34.2
23.8
Dyspepsia
19.6
11.9
Abdominal pain
9.8
5.6
Acute appendicitis
1.2
0.6
(histologically confirmed in most cases where data were available)
Atrial fibrillation
0.7
0.1
Adverse events reported by more than 1% of subjects, but without a significant difference between groups:
Diarrhea
7.2
6.5
Fatigue
3.8
3.5
Nausea/Vomiting
2.5
2.1
Eczema
1.9
1.2
Rash
1.7
1.3
Vertigo
1.5
1.3
Constipation
1.4
1.3
Headache
1.2
1.1
Gallbladder surgery was performed in 0.9% of gemfibrozil and 0.5% of placebo subjects in the primary prevention component, a 64% excess, which is not statistically different from the excess of gallbladder surgery observed in the clofibrate group compared to the placebo group of the WHO study. Gallbladder surgery was also performed more frequently in the gemfibrozil group compared to the placebo group (1.9% versus 0.3%, p=0.07) in the secondary prevention component. A statistically significant increase in appendectomy in the gemfibrozil group was seen also in the secondary prevention component (6 on gemfibrozil vs 0 on placebo, p=0.014).
Nervous system and special senses adverse reactions were more common in the gemfibrozil group. These included hypesthesia, paresthesias, and taste perversion. Other adverse reactions that were more common among gemfibrozil treatment group subjects but where a causal relationship was not established include cataracts, peripheral vascular disease, and intracerebral hemorrhage.
From other studies it seems probable that gemfibrozil is causally related to the occurrence of MUSCULOSKELETAL SYMPTOMS (see WARNINGS), and to ABNORMAL LIVER FUNCTION TESTS and HEMATOLOGIC CHANGES (see PRECAUTIONS).
Reports of viral and bacterial infections (common cold, cough, urinary tract infections) were more common in gemfibrozil treated patients in other controlled clinical trials of 805 patients. Additional adverse reactions that have been reported for gemfibrozil are listed below by system. These are categorized according to whether a causal relationship to treatment with gemfibrozil is probable or not established:
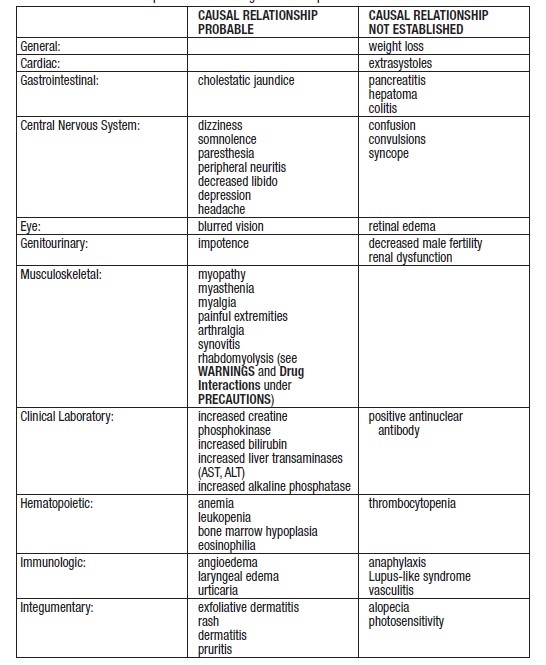
Additional adverse reactions that have been reported include cholecystitis and cholelithiasis (seeWARNINGS).
-
DOSAGE AND ADMINISTRATION
The recommended dose for adults is 1200 mg administered in two divided doses 30 minutes before the morning and evening meal (see CLINICAL PHARMACOLOGY).
-
OVERDOSAGE
There have been reported cases of overdosage with gemfibrozil. In one case, a 7-year-old child recovered after ingesting up to 9 grams of gemfibrozil. Symptoms reported with overdosage were abdominal cramps, abnormal liver function tests, diarrhea, increased CPK, joint and muscle pain, nausea and vomiting. Symptomatic supportive measures should be taken, should an overdose occur.
-
HOW SUPPLIED
Supplied as White film-coated, capsule shaped, biconvex tablets de-bossed with I on the left side of bisect and G on the right side of bisect on one side and 225 on the other.
55700-493-30
55700-493-60
Store at controlled room temperature 20° - 25°C (68° - 77°F) [see USP]. Protect from light and humidity. Preserve in tight containers.
Manufactured for:
Cipla USA Inc.,
9100 S. Dadeland Blvd., Suite 1500
Miami, FL 33156
Manufactured by:
InvaGen Pharmaceuticals, Inc.
(a subsidiary of Cipla Ltd.)
Hauppauge, NY 11788
Revised: 9/2016
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
GEMFIBROZIL
gemfibrozil tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55700-493(NDC:69097-821) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength GEMFIBROZIL (UNII: Q8X02027X3) (GEMFIBROZIL - UNII:Q8X02027X3) GEMFIBROZIL 600 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) CALCIUM STEARATE (UNII: 776XM7047L) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) METHYLCELLULOSE (15 CPS) (UNII: NPU9M2E6L8) HYPROMELLOSE 2910 (3 MPA.S) (UNII: 0VUT3PMY82) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYSORBATE 80 (UNII: 6OZP39ZG8H) Product Characteristics Color WHITE Score 2 pieces Shape OVAL Size 19mm Flavor Imprint Code 225;IG Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55700-493-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 02/10/2017 10/11/2019 2 NDC:55700-493-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 02/10/2017 02/28/2017 3 NDC:55700-493-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 02/06/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077836 10/13/2016 Labeler - Lake Erie Medical DBA Quality Care Products LLC (831276758) Establishment Name Address ID/FEI Business Operations Lake Erie Medical DBA Quality Care Products LLC 831276758 repack(55700-493)
