ROSUVASTATIN CALCIUM- rosuvastatin calcium tablet, film coated
Denton Pharma, Inc. dba Northwind Pharmaceuticals
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ROSUVASTATIN TABLETS safely and effectively. See full prescribing information for ROSUVASTATIN TABLETS.
ROSUVASTATIN tablets, for oral use Initial U.S. Approval: 2003 RECENT MAJOR CHANGESINDICATIONS AND USAGERosuvastatin tablets are an HMG Co‑A reductase inhibitor indicated for:
Limitations of use (1.8): Rosuvastatin tablets have not been studied in Fredrickson Type I and V dyslipidemias. DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHSTablets: 5 mg, 10 mg, 20 mg, and 40 mg (3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost frequent adverse reactions (rate > 2%) are headache, myalgia, abdominal pain, asthenia, and nausea. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Accord Healthcare Inc. at 1-866-941-7875 or www.accord-healthcare.us or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
Pediatric use information for patients 7 to 17 years of age is approved for AstraZeneca’s CRESTOR (rosuvastatin calcium) tablets. However, due to AstraZeneca’s marketing exclusivity rights, this drug product is not labeled with that pediatric information. See 17 for PATIENT COUNSELING INFORMATION. Revised: 11/2020 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Pediatric use information for patients 7 to 17 years of age is approved for AstraZeneca’s CRESTOR (rosuvastatin calcium) tablets. However, due to AstraZeneca’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
1.3 Hypertriglyceridemia
Rosuvastatin tablets are indicated as adjunctive therapy to diet for the treatment of adult patients with hypertriglyceridemia.
1.4 Primary Dysbetalipoproteinemia (Type III Hyperlipoproteinemia)
Rosuvastatin tablets are indicated as an adjunct to diet for the treatment of adult patients with primary dysbetalipoproteinemia (Type III Hyperlipoproteinemia).
1.5 Adult Patients with Homozygous Familial Hypercholesterolemia
Rosuvastatin tablets are indicated as adjunctive therapy to other lipid-lowering treatments (e.g., LDL apheresis) or alone if such treatments are unavailable to reduce LDL-C, Total-C, and ApoB in adult patients with homozygous familial hypercholesterolemia.
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
The dose range for rosuvastatin tablet in adults is 5 to 40 mg orally once daily. The usual starting dose is 10 to 20 mg once daily. The usual starting dose in adult patients with homozygous familial hypercholesterolemia is 20 mg once daily.
The maximum rosuvastatin tablet dose of 40 mg should be used only for those patients who have not achieved their LDL-C goal utilizing the 20 mg dose [ see Warnings and Precautions (5.1)].
Rosuvastatin tablets can be administered as a single dose at any time of day, with or without food. The tablet should be swallowed whole.
When initiating rosuvastatin therapy or switching from another HMG‑CoA reductase inhibitor therapy, the appropriate rosuvastatin tablets starting dose should first be utilized, and only then titrated according to the patient’s response and individualized goal of therapy.
After initiation or upon titration of rosuvastatin, lipid levels should be analyzed within 2 to 4 weeks and the dosage adjusted accordingly.
Pediatric use information for patients 7 to 17 years of age is approved for AstraZeneca’s CRESTOR (rosuvastatin calcium) tablets. However, due to AstraZeneca’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
2.3 Dosing in Asian Patients
In Asian patients, consider initiation of rosuvastatin therapy with 5 mg once daily due to increased rosuvastatin plasma concentrations. The increased systemic exposure should be taken into consideration when treating Asian patients not adequately controlled at doses up to 20 mg/day [ see Use in Specific Populations (8.8) and Clinical Pharmacology (12.3)].
2.4 Use with Concomitant Therapy
Patients taking cyclosporine and darolutamide
The dose of rosuvastatin tablets should not exceed 5 mg once daily [
see Warnings and Precautions (5.1),
Drug Interactions (7.1),
Drug Interactions (7.4) and
Clinical Pharmacology (12.3)].
Patients taking gemfibrozil
Avoid concomitant use of rosuvastatin with gemfibrozil. If concomitant use cannot be avoided, initiate rosuvastatin tablets at 5 mg once daily. The dose of rosuvastatin tablets should not exceed 10 mg once daily. [
see Warnings and Precautions (5.1), Drug Interactions (7.2) and
Clinical Pharmacology (12.3)].
Patients taking regorafenib
Concomitant use of rosuvastatin tablets and regorafenib, the dose of rosuvastatin tablets should not exceed 10 mg once daily. [
see Warnings and Precautions (5.1), Drug Interactions (7.5) and
Clinical Pharmacology (12.3)].
Patients taking atazanavir and ritonavir, lopinavir and ritonavir, simeprevir or combination of dasabuvir/ombitasvir/paritaprevir/ritonavir, elbasvir/grazoprevir, sofosbuvir/velpatasvir and glecaprevir/pibrentasvir
Initiate rosuvastatin therapy with 5 mg once daily. The dose of rosuvastatin tablets should not exceed 10 mg once daily [
see Warnings and Precautions (5.1),
Drug Interactions (7.3), and
Clinical Pharmacology (12.3)].
2.5 Dosing in Patients with Severe Renal Impairment
For patients with severe renal impairment (CL cr <30 mL/min/1.73 m 2) not on hemodialysis, dosing of rosuvastatin tablets should be started at 5 mg once daily and not exceed 10 mg once daily [ see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Rosuvastatin tablets, USP 5 mg: Yellow, round, biconvex, film-coated tablet, debossed “5” on one side and “FI” on other side.
Rosuvastatin tablets, USP 10 mg: Pink, round, biconvex, film-coated tablet, debossed “10” on one side and “R” on other side.
Rosuvastatin tablets, USP 20 mg: Pink, round, biconvex, film-coated tablet, debossed “20” on one side and “R” on other side.
Rosuvastatin tablets, USP 40 mg: Pink, oval, biconvex, film-coated tablet, debossed “40” on one side and “R” on other side.
4 CONTRAINDICATIONS
Rosuvastatin tablets are contraindicated in the following conditions:
- Patients with a known hypersensitivity to any component of this product. Hypersensitivity reactions including rash, pruritus, urticaria, and angioedema have been reported with rosuvastatin [ see Adverse Reactions (6.1)].
- Patients with active liver disease, which may include unexplained persistent elevations of hepatic transaminase levels [ see Warnings and Precautions (5.2)].
- Pregnancy [ see Use in Specific Populations (8.1, 8.3)].
- Lactation. Limited data indicate that rosuvastatin is present in human milk. Because statins have the potential for serious adverse reactions in nursing infants, women who require rosuvastatin treatment should not breastfeed their infants [ see Use in Specific Populations (8.2)].
5 WARNINGS AND PRECAUTIONS
5.1 Skeletal Muscle Effects
Cases of myopathy and rhabdomyolysis with acute renal failure secondary to myoglobinuria have been reported with HMG-CoA reductase inhibitors, including rosuvastatin. These risks can occur at any dose level, but are increased at the highest dose (40 mg).
Rosuvastatin tablets should be prescribed with caution in patients with predisposing factors for myopathy (e.g., age ≥ 65 years, inadequately treated hypothyroidism, renal impairment). Rosuvastatin tablets should be prescribed with caution in patients with predisposing factors for myopathy (e.g., age ≥ 65 years, inadequately treated hypothyroidism, renal impairment).
The risk of myopathy during treatment with rosuvastatin may be increased with concurrent administration of gemfibrozil, some other lipid-lowering therapies (other fibrates or niacin), cyclosporine, darolutamide, regorafenib, atazanavir/ritonavir, lopinavir/ritonavir, simeprevir or combination of sofosbuvir/velpatasvir/voxilaprevir, dasabuvir/ombitasvir/paritaprevir/ritonavir, elbasvir/grazoprevir, sofosbuvir/velpatasvir, glecaprevir/pibrentasvir, all combinations with ledipasvir (including ledipasvir/sofosbuvir) [ see Dosage and Administration (2) and Drug Interactions (7)]. Cases of myopathy, including rhabdomyolysis, have been reported with HMG-CoA reductase inhibitors, including rosuvastatin, coadministered with colchicine, and caution should be exercised when prescribing rosuvastatin tablets with colchicine [see Drug Interactions (7.9)].
Rosuvastatin therapy should be discontinued if markedly elevated creatine kinase levels occur or myopathy is diagnosed or suspected. Rosuvastatin therapy should also be temporarily withheld in any patient with an acute, serious condition suggestive of myopathy or predisposing to the development of renal failure secondary to rhabdomyolysis (e.g., sepsis, hypotension, dehydration, major surgery, trauma, severe metabolic, endocrine, and electrolyte disorders, or uncontrolled seizures).Rosuvastatin therapy should be discontinued if markedly elevated creatine kinase levels occur or myopathy is diagnosed or suspected. Rosuvastatin therapy should also be temporarily withheld in any patient with an acute, serious condition suggestive of myopathy or predisposing to the development of renal failure secondary to rhabdomyolysis (e.g., sepsis, hypotension, dehydration, major surgery, trauma, severe metabolic, endocrine, and electrolyte disorders, or uncontrolled seizures).
There have been rare reports of immune-mediated necrotizing myopathy (IMNM), an autoimmune myopathy, associated with statin use. IMNM is characterized by: proximal muscle weakness and elevated serum creatine kinase, which persist despite discontinuation of statin treatment; muscle biopsy showing necrotizing myopathy without significant inflammation; improvement with immunosuppressive agents. There have been rare reports of immune-mediated necrotizing myopathy (IMNM), an autoimmune myopathy, associated with statin use. IMNM is characterized by: proximal muscle weakness and elevated serum creatine kinase, which persist despite discontinuation of statin treatment; muscle biopsy showing necrotizing myopathy without significant inflammation; improvement with immunosuppressive agents.
All patients should be advised to promptly report to their physician unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever or if muscle signs and symptoms persist after discontinuing rosuvastatin tablets. All patients should be advised to promptly report to their physician unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever or if muscle signs and symptoms persist after discontinuing rosuvastatin tablets.
5.2 Liver Enzyme Abnormalities
It is recommended that liver enzyme tests be performed before the initiation of rosuvastatin tablets, and if signs or symptoms of liver injury occur.
Increases in serum transaminases [AST (SGOT) or ALT (SGPT)] have been reported with HMG‑CoA reductase inhibitors, including rosuvastatin. In most cases, the elevations were transient and resolved or improved on continued therapy or after a brief interruption in therapy. There were two cases of jaundice, for which a relationship to rosuvastatin therapy could not be determined, which resolved after discontinuation of therapy. There were no cases of liver failure or irreversible liver disease in these trials.
In a pooled analysis of placebo-controlled trials, increases in serum transaminases to >3 times the upper limit of normal occurred in 1.1% of patients taking rosuvastatin versus 0.5% of patients treated with placebo.
There have been rare postmarketing reports of fatal and non-fatal hepatic failure in patients taking statins, including rosuvastatin. If serious liver injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs during treatment with rosuvastatin, promptly interrupt therapy. If an alternate etiology is not found, do not restart rosuvastatin tablets.
Rosuvastatin tablets should be used with caution in patients who consume substantial quantities of alcohol and/or have a history of chronic liver disease [ see Clinical Pharmacology (12.3)]. Active liver disease, which may include unexplained persistent transaminase elevations, is a contraindication to the use of rosuvastatin [ see Contraindications (4)].
5.3 Concomitant Coumarin Anticoagulants
Caution should be exercised when anticoagulants are given in conjunction with rosuvastatin because of its potentiation of the effect of coumarin-type anticoagulants in prolonging the prothrombin time/INR. In patients taking coumarin anticoagulants and rosuvastatin concomitantly, INR should be determined before starting rosuvastatin and frequently enough during early therapy to ensure that no significant alteration of INR occurs [ see Drug Interactions (7.6)].
5.4 Proteinuria and Hematuria
In the rosuvastatin clinical trial program, dipstick-positive proteinuria and microscopic hematuria were observed among rosuvastatin treated patients. These findings were more frequent in patients taking rosuvastatin tablets 40 mg, when compared to lower doses of rosuvastatin tablets or comparator HMG‑CoA reductase inhibitors, though it was generally transient and was not associated with worsening renal function. Although the clinical significance of this finding is unknown, a dose reduction should be considered for patients on rosuvastatin therapy with unexplained persistent proteinuria and/or hematuria during routine urinalysis testing.
5.5 Endocrine Effects
Increases in HbA1c and fasting serum glucose levels have been reported with HMG‑CoA reductase inhibitors, including rosuvastatin. Based on clinical trial data with rosuvastatin, in some instances these increases may exceed the threshold for the diagnosis of diabetes mellitus [ see Adverse Reactions (6.1)].
Although clinical studies have shown that rosuvastatin alone does not reduce basal plasma cortisol concentration or impair adrenal reserve, caution should be exercised if rosuvastatin is administered concomitantly with drugs that may decrease the levels or activity of endogenous steroid hormones such as ketoconazole, spironolactone, and cimetidine.
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Rhabdomyolysis with myoglobinuria and acute renal failure and myopathy (including myositis) [ see Warnings and Precautions (5.1)]
- Liver enzyme abnormalities [ see Warnings and Precautions (5.2)]
6.1 Clinical Studies Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice.
In the rosuvastatin controlled clinical trials database (placebo or active-controlled) of 5394 patients with a mean treatment duration of 15 weeks, 1.4% of patients discontinued due to adverse reactions. The most common adverse reactions that led to treatment discontinuation were:
- myalgia
- abdominal pain
- nausea
The most commonly reported adverse reactions (incidence ≥ 2%) in the rosuvastatin controlled clinical trial database of 5394 patients were:
- headache
- myalgia
- abdominal pain
- asthenia
- nausea
Adverse reactions reported in ≥ 2% of patients in placebo-controlled clinical studies and at a rate greater than placebo are shown in Table 1. These studies had a treatment duration of up to 12 weeks.
|
Adverse Reactions | Rosuvastatin 5 mg
N=291 | Rosuvastatin
10 mg
N=283 | Rosuvastatin
20 mg
N=64 | Rosuvastatin
40 mg
N=106 | Total Rosuvastatin
5 mg to 40 mg N=744 | Placebo
N=382 |
|
Headache |
5.5 |
4.9 |
3.1 |
8.5 |
5.5 |
5.0 |
|
Nausea |
3.8 |
3.5 |
6.3 |
0 |
3.4 |
3.1 |
|
Myalgia |
3.1 |
2.1 |
6.3 |
1.9 |
2.8 |
1.3 |
|
Asthenia |
2.4 |
3.2 |
4.7 |
0.9 |
2.7 |
2.6 |
|
Constipation |
2.1 |
2.1 |
4.7 |
2.8 |
2.4 |
2.4 |
1Adverse reactions by COSTART preferred term.
Other adverse reactions reported in clinical studies were abdominal pain, dizziness, hypersensitivity (including rash, pruritus, urticaria, and angioedema) and pancreatitis. The following laboratory abnormalities have also been reported: dipstick-positive proteinuria and microscopic hematuria [ see Warnings and Precautions (5.4)]; elevated creatine phosphokinase, transaminases, glucose, glutamyl transpeptidase, alkaline phosphatase, and bilirubin; and thyroid function abnormalities.
In a clinical trial, involving 981 participants treated with rosuvastatin 40 mg (n=700) or placebo (n=281) with a mean treatment duration of 1.7 years, 5.6% of subjects treated with rosuvastatin versus 2.8% of placebo-treated subjects discontinued due to adverse reactions. The most common adverse reactions that led to treatment discontinuation were: myalgia, hepatic enzyme increased, headache, and nausea.
Adverse reactions reported in ≥ 2% of patients and at a rate greater than placebo are shown in Table 2.
| Adverse Reactions | Rosuvastatin 40 mg
N=700 | Placebo
N=281 |
|---|---|---|
|
Myalgia |
12.7 |
12.1 |
|
Arthralgia |
10.1 |
7.1 |
|
Headache |
6.4 |
5.3 |
|
Dizziness |
4.0 |
2.8 |
|
Increased CPK |
2.6 |
0.7 |
|
Abdominal pain |
2.4 |
1.8 |
|
ALT >3x ULN 2 |
2.2 |
0.7 |
1 Adverse reactions by MedDRA preferred term.
2 Frequency recorded as abnormal laboratory value.
In a clinical trial, 17,802 participants were treated with rosuvastatin 20 mg (n=8901) or placebo (n=8901) for a mean duration of 2 years. A higher percentage of rosuvastatin- treated patients versus placebo-treated patients, 6.6% and 6.2%, respectively, discontinued study medication due to an adverse event, irrespective of treatment causality. Myalgia was the most common adverse reaction that led to treatment discontinuation.
There was a significantly higher frequency of diabetes mellitus reported in patients taking rosuvastatin (2.8%) versus patients taking placebo (2.3%). Mean HbA1c was significantly increased by 0.1% in rosuvastatin-treated patients compared to placebo-treated patients. The number of patients with a HbA1c > 6.5% at the end of the trial was significantly higher in rosuvastatin-treated versus placebo-treated patients [ see Warnings and Precautions (5.5].
Adverse reactions reported in ≥ 2% of patients and at a rate greater than placebo are shown in Table 3.
|
Adverse Reactions |
Rosuvastatin 20 mg N=8901 |
Placebo N=8901 |
|
Myalgia |
7.6 |
6.6 |
|
Arthralgia |
3.8 |
3.2 |
|
Constipation |
3.3 |
3.0 |
|
Diabetes mellitus |
2.8 |
2.3 |
|
Nausea |
2.4 |
2.3 |
1 Treatment-emergent adverse reactions by MedDRA preferred term.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of rosuvastatin: arthralgia, fatal and non-fatal hepatic failure, hepatitis, jaundice, thrombocytopenia, depression, sleep disorders (including insomnia and nightmares), peripheral neuropathy, interstitial lung disease and gynecomastia. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
There have been rare reports of immune-mediated necrotizing myopathy associated with statin use [ see Warnings and Precautions (5.1)].
There have been rare postmarketing reports of cognitive impairment (e.g., memory loss, forgetfulness, amnesia, memory impairment and confusion) associated with statin use. These cognitive issues have been reported for all statins. The reports are generally nonserious, and reversible upon statin discontinuation, with variable times to symptom onset (1 day to years) and symptom resolution (median of 3 weeks).
7. DRUG INTERACTIONS
7.1 Cyclosporine
Cyclosporine increased rosuvastatin exposure and may result in increased risk of myopathy. Therefore, in patients taking cyclosporine, the dose of rosuvastatin should not exceed 5 mg once daily [ see Dosage and Administration (2.4), Warnings and Precautions (5.1), and Clinical Pharmacology (12.3)].
7.2 Gemfibrozil
Gemfibrozil significantly increased rosuvastatin exposure. Due to an observed increased risk of myopathy/rhabdomyolysis, combination therapy with rosuvastatin and gemfibrozil should be avoided. If used together, the dose of rosuvastatin should not exceed 10 mg once daily [ see Clinical Pharmacology (12.3)].
7.3 Anti-viral Medications
Coadministration of rosuvastatin with certain anti-viral drugs has differing effects on rosuvastatin exposure and may increase risk of myopathy.
The combination of sofosbuvir/velpatasvir/voxilaprevir which are anti-Hepatitis C virus (anti-HCV) drugs, increases rosuvastatin exposure. Similarly, the combination of ledipasvir/sofosbuvir may significantly increase rosuvastatin exposure. For these combinations of anti-HCV drugs, concomitant use with rosuvastatin is not recommended.
Simeprevir and combinations of dasabuvir/ombitasvir/paritaprevir/ritonavir, elbasvir/grazoprevir, sofosbuvir/velpatasvir and glecaprevir/pibrentasvir which are anti-HCV drugs, increase rosuvastatin exposure. Combinations of atazanavir/ritonavir and lopinavir/ritonavir, which are anti-HIV-1 drugs, increase rosuvastatin exposure [ see Table 4 – Clinical Pharmacology (12.3)]. For these anti-viral drugs, the dose of rosuvastatin tablets should not exceed 10 mg once daily.
The combinations of fosamprenavir/ritonavir or tipranavir/ritonavir, which are anti-HIV-1 drugs, produce little or no change in rosuvastatin exposure. No dose adjustment is needed for concomitant use with these combinations [ see Dosage and Administration (2.4), Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
7.4 Darolutamide
Darolutamide increased rosuvastatin exposure more than 5 fold. Therefore, in patients taking darolutamide, the dose of rosuvastatin tablets should not exceed 5 mg once daily [ see Dosage and Administration (2.4), Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
7.5 Regorafenib
Regorafenib increased rosuvastatin exposure and may increase the risk of myopathy. If used together, the dose of rosuvastatin tablets should not exceed 10 mg once daily [ see Dosage and Administration (2.4), Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
7.6 Coumarin Anticoagulants
When rosuvastatin was coadministered with fenofibrate, no clinically significant increase in the AUC of rosuvastatin or fenofibrate was observed. Because it is known that the risk of myopathy during treatment with HMG-CoA reductase inhibitors is increased with concomitant use of fenofibrates, caution should be used when prescribing fenofibrates with rosuvastatin [ see Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
7.7 Niacin
The risk of skeletal muscle effects may be enhanced when rosuvastatin is used in combination with lipid-modifying doses (≥1 g/day) of niacin; caution should be used when prescribing with rosuvastatin [ see Warnings and Precautions (5.1)].
7.8 Fenofibrate
When rosuvastatin was coadministered with fenofibrate, no clinically significant increase in the AUC of rosuvastatin or fenofibrate was observed. Because it is known that the risk of myopathy during treatment with HMG-CoA reductase inhibitors is increased with concomitant use of fenofibrates, caution should be used when prescribing fenofibrates with rosuvastatin [ see Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
7.9 Colchicine
Cases of myopathy, including rhabdomyolysis, have been reported with HMG‑CoA reductase inhibitors, including rosuvastatin, coadministered with colchicine, and caution should be exercised when prescribing rosuvastatin with colchicine [ see Warnings and Precautions (5.1)].
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Rosuvastatin is contraindicated for use in pregnant women since safety in pregnant women has not been established and there is no apparent benefit to therapy with rosuvastatin during pregnancy. Because HMG-CoA reductase inhibitors decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol, rosuvastatin may cause fetal harm when administered to pregnant women. Rosuvastatin should be discontinued as soon as pregnancy is recognized [see Contraindications (4)] . Limited published data on the use of rosuvastatin are insufficient to determine a drug-associated risk of major congenital malformations or miscarriage. In animal reproduction studies, there were no adverse developmental effects with oral administration of rosuvastatin during organogenesis at systemic exposures equivalent to a maximum recommended human dose (MRHD) of 40 mg/day in rats or rabbits (based on AUC and body surface area, respectively). In rats and rabbits, decreased pup/fetal survival occurred at 12 times and equivalent, respectively, to the MRHD of 40 mg/day [see Data] .
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Human Data
Limited published data on rosuvastatin have not shown an increased risk of major congenital malformations or miscarriage. Rare reports of congenital anomalies have been received following intrauterine exposure to other statins. In a review of approximately 100 prospectively followed pregnancies in women exposed to simvastatin or lovastatin, the incidences of congenital anomalies, spontaneous abortions, and fetal deaths/stillbirths did not exceed what would be expected in the general population. The number of cases is adequate to exclude a ≥3 to 4-fold increase in congenital anomalies over the background incidence. In 89% of the prospectively followed pregnancies, drug treatment was initiated prior to pregnancy and was discontinued at some point in the first trimester when pregnancy was identified.
Animal Data
Rosuvastatin crosses the placenta in rats and rabbits and is found in fetal tissue and amniotic fluid at 3% and 20%, respectively, of the maternal plasma concentration following a single 25 mg/kg oral gavage dose on gestation day 16 in rats. A higher fetal tissue distribution (25% maternal plasma concentration) was observed in rabbits after a single oral gavage dose of 1 mg/kg on gestation day 18.
Rosuvastatin administration did not indicate a teratogenic effect in rats at ≤25 mg/kg/day or in rabbits ≤3 mg/kg/day (doses equivalent to the MRHD of 40 mg/day based on AUC and body surface area, respectively).
In female rats given 5, 15 and 50 mg/kg/day before mating and continuing through to gestation day 7 resulted in decreased fetal body weight (female pups) and delayed ossification at 50 mg/kg/day (10 times the human exposure at the MRHD dose of 40 mg/day based on AUC).
In pregnant rats given 2, 10 and 50 mg/kg/day of rosuvastatin from gestation day 7 through lactation day 21 (weaning), decreased pup survival occurred at 50 mg/kg/day (dose equivalent to 12 times the MRHD of 40 mg/day based body surface area).
In pregnant rabbits given 0.3, 1, and 3 mg/kg/day of rosuvastatin from gestation day 6 to day 18, decreased fetal viability and maternal mortality was observed at 3 mg/kg/day (dose equivalent to the MRHD of 40 mg/day based on body surface area).
8.2 Lactation
Risk Summary
Rosuvastatin use is contraindicated during breastfeeding [see Contraindications (4)] . Limited data indicate that rosuvastatin is present in human milk. There is no available information on the effects of the drug on the breastfed infant or the effects of the drug on milk production. Because of the potential for serious adverse reactions in a breastfed infant, advise patients that breastfeeding is not recommended during treatment with rosuvastatin.
8.3 Females and Males of Reproductive Potential
Contraception
Rosuvastatin may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with rosuvastatin.
8.4 Pediatric Use
Pediatric use information for patients 7 to 17 years of age is approved for AstraZeneca’s CRESTOR (rosuvastatin calcium) tablets. However, due to AstraZeneca’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
8.5 Geriatric Use
Of the 10,275 patients in clinical studies with rosuvastatin, 3159 (31%) were 65 years and older, and 698 (6.8%) were 75 years and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Elderly patients are at higher risk of myopathy and rosuvastatin should be prescribed with caution in the elderly [ see Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Rosuvastatin exposure is not influenced by mild to moderate renal impairment (CL cr ≥ 30 mL/min/1.73 m2). Exposure to rosuvastatin is increased to a clinically significant extent in patients with severe renal impairment (CL cr < 30 mL/min/1.73 m 2) who are not receiving hemodialysis and dose adjustment is required [ see Dosage and Administration (2.5), Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Rosuvastatin is contraindicated in patients with active liver disease, which may include unexplained persistent elevations of hepatic transaminase levels. Chronic alcohol liver disease is known to increase rosuvastatin exposure; rosuvastatin tablets should be used with caution in these patients [ see Contraindications (4), Warning and Precautions (5.2) and Clinical Pharmacology (12.3)].
8.8 Asian Patients
Pharmacokinetic studies have demonstrated an approximate 2‑fold increase in median exposure to rosuvastatin in Asian subjects when compared with Caucasian controls. Rosuvastatin dosage should be adjusted in Asian patients [ see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no specific treatment in the event of overdose. In the event of overdose, the patient should be treated symptomatically and supportive measures instituted as required. Hemodialysis does not significantly enhance clearance of rosuvastatin.
11 DESCRIPTION
Rosuvastatin calcium is a synthetic lipid-lowering agent for oral administration.
The chemical name for rosuvastatin calcium is bis[(E)-7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl(methylsulfonyl)amino] pyrimidin-5-yl](3R,5S)-3,5-dihydroxyhept-6-enoic acid] calcium salt with the following structural formula:
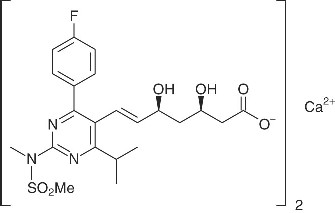
The empirical formula for rosuvastatin calcium is (C 22H 27FN 3O 6S) 2Ca and the molecular weight is 1001.14. Rosuvastatin calcium, USP is a white amorphous powder that is sparingly soluble in water and methanol, and slightly soluble in ethanol. Rosuvastatin calcium, USP is a hydrophilic compound with a partition coefficient (octanol/water) of 0.13 at pH of 7.0.
Rosuvastatin tablets, USP for oral administration contain 5, 10, 20, or 40 mg of rosuvastatin and the following inactive ingredients: For 5 mg: Each film coated tablet contains: microcrystalline cellulose, lactose monohydrate, anhydrous lactose, crospovidone, magnesium oxide, magnesium stearate, hypromellose, triacetin, titanium dioxide, and ferric oxide yellow.
For 10, 20 and 40 mg: Each film coated tablet contains: microcrystalline cellulose, lactose monohydrate, anhydrous lactose, crospovidone, magnesium oxide, magnesium stearate, hypromellose, triacetin, titanium dioxide, FD & C yellow No. 6, FD & C red No. 40, and FD & C blue No.1.
Rosuvastatin tablets, USP meet USP Dissolution Test 2.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Rosuvastatin is a selective and competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3‑hydroxy‑3‑methylglutaryl coenzyme A to mevalonate, a precursor of cholesterol. In vivo studies in animals, and in vitro studies in cultured animal and human cells have shown rosuvastatin to have a high uptake into, and selectivity for, action in the liver, the target organ for cholesterol lowering. In in vivo and in vitro studies, rosuvastatin produces its lipid-modifying effects in two ways. First, it increases the number of hepatic LDL receptors on the cell-surface to enhance uptake and catabolism of LDL. Second, rosuvastatin inhibits hepatic synthesis of VLDL, which reduces the total number of VLDL and LDL particles.
12.2 Pharmacodynamics
Rosuvastatin dose dependently reduces elevated LDL-cholesterol and reduces total cholesterol and triglycerides and increases HDL-cholesterol [see Clinical Studies (14)] . A therapeutic response to Rosuvastatin is evident within 1 week of commencing therapy and 90% of maximum response is usually achieved in 2 weeks. The maximum response is usually achieved by 4 weeks and is maintained after that. Individualization of drug dosage should be based on the therapeutic response [see Dosage and Administration (2)] .
12.3 Pharmacokinetics
Absorption
In clinical pharmacology studies in man, peak plasma concentrations of rosuvastatin were reached 3 to 5 hours following oral dosing. Both C max and AUC increased in approximate proportion to rosuvastatin dose. The absolute bioavailability of rosuvastatin is approximately 20%.
Administration of rosuvastatin tablet with food did not affect the AUC of rosuvastatin.
The AUC of rosuvastatin does not differ following evening or morning drug administration.
Distribution
Mean volume of distribution at steady-state of rosuvastatin is approximately 134 liters. Rosuvastatin is 88% bound to plasma proteins, mostly albumin. This binding is reversible and independent of plasma concentrations.
Elimination
Rosuvastatin is primarily eliminated by excretion in the feces. The elimination half-life of rosuvastatin is approximately 19 hours.
Metabolism
Rosuvastatin is not extensively metabolized; approximately 10% of a radiolabeled dose is recovered as metabolite. The major metabolite is N-desmethyl rosuvastatin, which is formed principally by cytochrome P450 \ 2C9, and in vitro studies have demonstrated that N-desmethyl rosuvastatin has approximately one-sixth to one-half the HMG-CoA reductase inhibitory activity of the parent compound. Overall, greater than 90% of active plasma HMG-CoA reductase inhibitory activity is accounted for by the parent compound.
Excretion
Following oral administration, rosuvastatin and its metabolites are primarily excreted in the feces (90%). After an intravenous dose, approximately 28% of total body clearance was via the renal route, and 72% by the hepatic route.
Specific Populations
Racial or Ethnic Groups
A population pharmacokinetic analysis revealed no clinically relevant differences in pharmacokinetics among Caucasian, Hispanic, and Black or Afro-Caribbean groups. However, pharmacokinetic studies, including one conducted in the US, have demonstrated an approximate 2-fold elevation in median exposure (AUC and C max) in Asian subjects when compared with a Caucasian control group.
Male and Female Patients
There were no differences in plasma concentrations of rosuvastatin between men and women.
Pediatric use information for patients ages 8 to less than 10 years is approved for AstraZeneca’s CRESTOR (rosuvastatin calcium) tablets. However, due to AstraZeneca’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
Geriatric Patients
There were no differences in plasma concentrations of rosuvastatin between the nonelderly and elderly populations (age ≥ 65 years).
Patients with Renal Impairment
Mild to moderate renal impairment (CL cr ≥ 30 mL/min/1.73 m 2) had no influence on plasma concentrations of rosuvastatin. However, plasma concentrations of rosuvastatin increased to a clinically significant extent (about 3-fold) in patients with severe renal impairment (CLcr < 30 mL/min/1.73 m 2) not receiving hemodialysis compared with healthy subjects (CL cr > 80 mL/min/1.73 m 2).
Hemodialysis
Steady-state plasma concentrations of rosuvastatin in patients on chronic hemodialysis were approximately 50% greater compared with healthy volunteer subjects with normal renal function.
Patients with Hepatic Impairment
In patients with chronic alcohol liver disease, plasma concentrations of rosuvastatin were modestly increased.
In patients with Child-Pugh A disease, C max and AUC were increased by 60% and 5%, respectively, as compared with patients with normal liver function. In patients with Child-Pugh B disease, C max and AUC were increased 100% and 21%, respectively, compared with patients with normal liver function.
Drug Interactions Studies
Rosuvastatin clearance is not dependent on metabolism by cytochrome P450 3A4 to a clinically significant extent.
Rosuvastatin is a substrate for certain transporter proteins including the hepatic uptake transporter organic anion-transporting polyprotein 1B1 (OATP1B1) and efflux transporter breast cancer resistance protein (BCRP). Concomitant administration of rosuvastatin with medications that are inhibitors of these transporter proteins (e.g. cyclosporine, certain HIV protease inhibitors) may result in increased rosuvastatin plasma concentrations [ see Dosage and Administration (2.4) and Drug Interactions (7.1, 7.3)].
|
Coadministered drug and dosing regimen |
Rosuvastatin |
||
|
Mean Ratio (ratio with/without coadministered drug) No Effect = 1.0 |
|||
|
Dose (mg)1 |
Change in AUC |
Change in C max |
|
|
Sofosbuvir/velpatasvir/voxilaprevir (400 mg-100 mg-100 mg) + Voxilaprevir (100 mg) once daily for 15 days |
10 mg single dose |
7.39
2
|
18.88
2
|
|
Cyclosporine – stable dose required (75 mg to 200 mg BID) |
10 mg QD for 10 days |
7.1 2 |
11 2 |
|
Darolutamide 600 mg BID, 5 days |
5mg, single dose |
5.2 2 |
~5 2 |
|
Regorafenib 160mg OD, 14 days |
5mg, single dose |
3.8 2 |
4.6 2 |
|
Atazanavir/ritonavir combination 300 mg/100 mg QD for 8 days |
10 mg |
3.1 2 |
7 2 |
|
Simeprevir 150 mg QD, 7 days |
10 mg, single dose |
2.8
2
|
3.2
2
|
|
Velpatasvir 100mg once daily |
10 mg single dose |
2.69
2
|
2.61
2
|
|
Ombitasvir 25mg/paritaprevir 150mg/ ritonavir 100mg + dasabuvir 400mg BID |
5mg single dose |
2.59
2
|
7.13
2
|
|
Elbasvir 50mg/grazoprevir 200mg once daily |
10 mg single dose |
2.26
2
|
5.49
2
|
|
Glecaprevir 400mg/pibrentasvir 120mg once daily |
5mg once daily |
2.15
2
|
5.62
2
|
|
Lopinavir/ritonavir combination 400 mg/100 mg BID for 17 days |
20 mg QD for 7 days |
2.1
2
|
5
2
|
|
Gemfibrozil 600 mg BID for 7 days |
80 mg |
1.9
2
|
2.2
2
|
|
Eltrombopag 75 mg QD, 5 days |
10 mg |
1.6
|
2
|
|
Darunavir 600 mg/ritonavir 100 mg BID, 7 days |
10 mg QD for 7 days
|
1.5
|
2.4
|
|
Tipranavir/ritonavir combination 500 mg/200mg BID for 11 days |
10 mg |
1.4
|
2.2
|
|
Dronedarone 400 mg BID |
10 mg |
1.4 | |
|
Itraconazole 200 mg QD, 5 days |
10 mg or 80 mg |
1.4
1.3
|
1.4
1.2
|
|
Ezetimibe 10 mg QD, 14 days |
10 mg QD for 14 days |
1.2
|
1.2
|
|
Fosamprenavir/ritonavir 700 mg/100 mg BID for 7 days |
10 mg |
1.1 |
1.5 |
|
Fenofibrate 67 mg TID for 7 days |
10 mg |
↔ |
1.2
|
|
Rifampicin 450 mg QD, 7 days |
20 mg |
↔ | |
|
Aluminum & magnesium hydroxide
|
|
0.8
|
0.8
|
|
Ketoconazole 200 mg BID for 7 days |
80 mg |
1.0
|
1.0
|
|
Fluconazole 200 mg QD for 11 days |
80 mg |
1.1
|
1.1
|
|
Erythromycin 500 mg QID for 7 days |
80 mg |
0.8
|
0.7
|
QD= Once daily, BID= Twice daily, TID= Three times daily, QID= Four times daily
1Single dose unless otherwise noted
2Clinically significant [ see Dosage and Administration (2) and Warnings and Precautions (5)]
3Mean ratio with 90% CI (with/without coadministered drug, e.g., 1= no change, 0.7 = 30% decrease, 11=11 fold increase in exposure)
| Rosuvastatin Dosage Regimen | Coadministered Drug | ||
|---|---|---|---|
| Mean Ratio (ratio with/without coadministered drug) No Effect = 1.0 | |||
| Name and Dose | Change in
AUC | Change in
C max |
|
|
40 mg QD for 10 days |
Warfarin 1 25 mg single dose |
R- Warfarin
S- Warfarin
|
R- Warfarin
S- Warfarin
|
|
40 mg QD for 12 days |
Digoxin 0.5 mg single dose |
1.0
|
1.0
|
|
40 mg QD for 28 days |
Oral Contraceptive (ethinyl estradiol 0.035 mg & norgestrel 0.180, 0.215 and 0.250 mg) QD for 21 Days |
NG 1.3
|
NG 1.2
|
EE = ethinyl estradiol, NG = norgestrel, QD= Once daily
1. Clinically significant pharmacodynamic effects [ see Warnings and Precautions (5.3)]
2. Mean ratio with 90% CI (with/without coadministered drug, e.g., 1= no change, 0.7=30% decrease, 11=11-fold increase in exposure)
12.5 Pharmacogenomics
Disposition of HMG-CoA reductase inhibitors, including rosuvastatin, involves OATP1B1 and other transporter proteins. Higher plasma concentrations of rosuvastatin have been reported in very small groups of patients (n=3 to 5) who have two reduced function alleles of the gene that encodes OATP1B1 ( SLCO1B1 521T>C). The frequency of this genotype (i.e., SLCO1B1 521 C/C) is generally lower than 5% in most racial/ethnic groups. The impact of this polymorphism on efficacy and/or safety of rosuvastatin has not been clearly established.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 104-week carcinogenicity study in rats at dose levels of 2, 20, 60, or 80 mg/kg/day by oral gavage, the incidence of uterine stromal polyps was significantly increased in females at 80 mg/kg/day at systemic exposure 20 times the human exposure at 40 mg/day based on AUC. Increased incidence of polyps was not seen at lower doses.
In a 107-week carcinogenicity study in mice given 10, 60, or 200 mg/kg/day by oral gavage, an increased incidence of hepatocellular adenoma/carcinoma was observed at 200 mg/kg/day at systemic exposures 20 times the human exposure at 40 mg/day based on AUC. An increased incidence of hepatocellular tumors was not seen at lower doses.
Rosuvastatin was not mutagenic or clastogenic with or without metabolic activation in the Ames test with Salmonella typhimurium and Escherichia coli, the mouse lymphoma assay, and the chromosomal aberration assay in Chinese hamster lung cells. Rosuvastatin was negative in the in vivo mouse micronucleus test.
In rat fertility studies with oral gavage doses of 5, 15, 50 mg/kg/day, males were treated for 9 weeks prior to and throughout mating and females were treated 2 weeks prior to mating and throughout mating until gestation day 7. No adverse effect on fertility was observed at 50 mg/kg/day (systemic exposures up to 10 times the human exposure at 40 mg/day based on AUC). In testicles of dogs treated with rosuvastatin at 30 mg/kg/day for one month, spermatidic giant cells were seen. Spermatidic giant cells were observed in monkeys after 6‑month treatment at 30 mg/kg/day in addition to vacuolation of seminiferous tubular epithelium. Exposures in the dog were 20 times and in the monkey 10 times the human exposure at 40 mg/day based on body surface area. Similar findings have been seen with other drugs in this class.
13.2 Animal Toxicology and/or Pharmacology
Central Nervous System Toxicity
CNS vascular lesions, characterized by perivascular hemorrhages, edema, and mononuclear cell infiltration of perivascular spaces, have been observed in dogs treated with several other members of this drug class. A chemically similar drug in this class produced dose-dependent optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in dogs, at a dose that produced plasma drug levels about 30 times higher than the mean drug level in humans taking the highest recommended dose. Edema, hemorrhage, and partial necrosis in the interstitium of the choroid plexus was observed in a female dog sacrificed moribund at day 24 at 90 mg/kg/day by oral gavage (systemic exposures 100 times the human exposure at 40 mg/day based on AUC). Corneal opacity was seen in dogs treated for 52 weeks at 6 mg/kg/day by oral gavage (systemic exposures 20 times the human exposure at 40 mg/day based on AUC). Cataracts were seen in dogs treated for 12 weeks by oral gavage at 30 mg/kg/day (systemic exposures 60 times the human exposure at 40 mg/day based on AUC). Retinal dysplasia and retinal loss were seen in dogs treated for 4 weeks by oral gavage at 90 mg/kg/day (systemic exposures 100 times the human exposure at 40 mg/day based on AUC). Doses ≤30 mg/kg/day (systemic exposures ≤60 times the human exposure at 40 mg/day based on AUC) did not reveal retinal findings during treatment for up to one year.
Juvenile Toxicology Study
In a juvenile study, rats were dosed by oral gavage with 10 or 50 mg/kg/day from weaning for 9 weeks prior to pairing, throughout pairing and up to the day before necropsy for males or up to gestation day 7 for females. No effects on sexual development, testicular and epididymal appearance or fertility were observed at either dose level.
Pediatric information is approved for AstraZeneca’s CRESTOR (rosuvastatin calcium) tablets. However, due to AstraZeneca’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
14 CLINICAL STUDIES
14.3 Hypertriglyceridemia
Dose-Response Study: In a double-blind, placebo-controlled dose-response study in patients with baseline TG levels from 273 to 817 mg/dL, rosuvastatin given as a single daily dose (5 to 40 mg) over 6 weeks significantly reduced serum TG levels (Table 9).
| Dose | Placebo
(n=26) | Rosuvastatin
5 mg (n=25) | Rosuvastatin 10 mg
(n=23) | Rosuvastatin 20 mg
(n=27) | Rosuvastatin
40 mg (n=25) |
|---|---|---|---|---|---|
|
Triglycerides |
1 (-40, 72) |
-21 (-58, 38) |
-37 (-65, 5) |
-37 (-72, 11) |
-43 (-80, -7) |
|
nonHDL-C |
2 (-13, 19) |
-29 (-43, -8) |
-49 (-59, -20) |
-43 (-74, 12) |
-51 (-62, -6) |
|
VLDL-C |
2 (-36, 53) |
-25 (-62, 49) |
-48 (-72, 14) |
-49 (-83, 20) |
-56 (-83, 10) |
|
Total-C |
1 (-13, 17) |
-24 (-40, -4) |
-40 (-51, -14) |
-34 (-61, -11) |
-40 (-51, -4) |
|
LDL-C |
5 (-30, 52) |
-28 (-71, 2) |
-45 (-59, 7) |
-31 (-66, 34) |
-43 (-61, -3) |
|
HDL-C |
-3 (-25, 18) |
3 (-38, 33) |
8 (-8, 24) |
22 (-5, 50) |
17 (-14, 63) |
14.4 Primary Dysbetalipoproteinemia (Type III Hyperlipoproteinemia)
In a randomized, multicenter, double-blind crossover study, 32 patients (27 with ε2/ε2 and 4 with apo E mutation [Arg145Cys] with primary dysbetalipoproteinemia (Type III Hyperlipoproteinemia) entered a 6-week dietary lead-in period on the NCEP Therapeutic Lifestyle Change (TLC) diet. Following dietary lead-in, patients were randomized to a sequence of treatments in conjunction with the TLC diet for 6 weeks each: rosuvastatin 10 mg followed by rosuvastatin 20 mg or rosuvastatin 20 mg followed by rosuvastatin 10 mg. Rosuvastatin reduced nonHDL-C (primary end point) and circulating remnant lipoprotein levels. Results are shown in the table below.
| Median at Baseline (mg/dL) | Median percent change from baseline
(95% CI) Rosuvastatin 10 mg | Median percent change from baseline
(95% CI) Rosuvastatin 20 mg |
|
|---|---|---|---|
|
Total-C |
342.5 |
-43.3 (-46.9, -37.5) |
-47.6 (-51.6, -42.8) |
|
Triglycerides |
503.5 |
-40.1 (-44.9, -33.6) |
-43.0 (-52.5, -33.1) |
|
NonHDL-C |
294.5 |
-48.2 (-56.7, -45.6) |
-56.4 (-61.4, -48.5) |
|
VLDL-C + IDL-C |
209.5 |
-46.8 (-53.7, -39.4) |
-56.2 (-67.7, -43.7) |
|
LDL-C |
112.5 |
-54.4 (-59.1, -47.3) |
-57.3 (-59.4, -52.1) |
|
HDL-C |
35.5 |
10.2 (1.9, 12.3) |
11.2 (8.3, 20.5) |
|
RLP-C |
82.0 |
-56.4 (-67.1, -49.0) |
-64.9 (-74.0, -56.6) |
|
Apo-E |
16.0 |
-42.9 (-46.3, -33.3) |
-42.5 (-47.1, -35.6) |
14.5 Homozygous Familial Hypercholesterolemia
Dose-Titration Study: In an open-label, forced-titration study, homozygous FH patients (n=40, 8‑63 years) were evaluated for their response to rosuvastatin 20 to 40 mg titrated at a 6‑week interval. In the overall population, the mean LDL‑C reduction from baseline was 22%. About one-third of the patients benefited from increasing their dose from 20 mg to 40 mg with further LDL lowering of greater than 6%. In the 27 patients with at least a 15% reduction in LDL‑C, the mean LDL-C reduction was 30% (median 28% reduction). Among 13 patients with an LDL‑C reduction of <15%, 3 had no change or an increase in LDL‑C. Reductions in LDL‑C of 15% or greater were observed in 3 of 5 patients with known receptor negative status.
Pediatric use information for patients 7 to 17 years of age is approved for AstraZeneca’s CRESTOR (rosuvastatin calcium) tablets. However, due to AstraZeneca’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
16 HOW SUPPLIED/STORAGE AND HANDLING
Rosuvastatin tablets, USP are supplied as:
- 10 mg. Pink, round, biconvex, film-coated tablet, debossed “10” on one side and “R” on other side;
Bottle of 30 tablets with child-resistant closure NDC 70934-275-30
Bottle of 90 tablets with child-resistant closure NDC 70934-275-90
Bottle of 180 tablets with child-resistant closure NDC 70934-275-96
Storage
Store at 20ºC to 25ºC (68ºF to 77ºF) [see USP Controlled Room Temperature]. Protect from moisture.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Patients should be instructed not to take 2 doses of rosuvastatin tablets within 12 hours of each other.
Skeletal Muscle Effects
Patients should be advised to report promptly unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever or if these muscle signs or symptoms persist after discontinuing rosuvastatin tablets.
Concomitant Use of Antacids
When taking rosuvastatin tablets with an aluminum and magnesium hydroxide combination antacid, the antacid should be taken at least 2 hours after rosuvastatin administration.
Embryofetal Toxicity
Advise females of reproductive potential of the risk to a fetus, to use effective contraception during treatment, and to inform their healthcare provider of a known or suspected pregnancy. [see Contraindications (4) and Use in Specific Populations (8.1, 8.3)] .
Lactation
Advise women not to breastfeed during treatment with rosuvastatin tablets [see Contraindications (4) and Use in Specific Populations (8.2)] .
Liver Enzymes
It is recommended that liver enzyme tests be performed before the initiation of rosuvastatin tablets and if signs or symptoms of liver injury occur. All patients treated with rosuvastatin tablets should be advised to promptly report any symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice.
Manufactured For:
Accord Healthcare, Inc.,
1009 Slater Road,
Suite 210-B,
Durham, NC 27703,
USA
Manufactured By:
Intas Pharmaceuticals Limited.
Plot No 5 to 14, Pharmez,
Sarkhej-Bavla, National Highway No 8-A, Near Village Matoda, Tal Sanand,
Ahmedabad - 382213, Gujarat,
India.
51 2359 4 724860
Issued September 2020

| ROSUVASTATIN CALCIUM
rosuvastatin calcium tablet, film coated |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| Labeler - Denton Pharma, Inc. dba Northwind Pharmaceuticals (080355546) |
| Registrant - Denton Pharma, Inc. dba Northwind Pharmaceuticals (080355546) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Denton Pharma, Inc. dba Northwind Pharmaceuticals | 080355546 | repack(70934-275) | |