TICLID
-
ticlopidine hydrochloride tablet, film coated
Genentech, Inc.
----------
WARNING:
TICLID can cause life-threatening hematological adverse reactions, including neutropenia/agranulocytosis, thrombotic thrombocytopenic purpura (TTP) and aplastic anemia.
Neutropenia/Agranulocytosis:
Among 2048 patients in clinical trials in stroke patients, there were 50 cases (2.4%) of neutropenia (less than 1200 neutrophils/mm3), and the neutrophil count was below 450/mm3 in 17 of these patients (0.8% of the total population).
TTP:
One case of thrombotic thrombocytopenic purpura was reported during clinical trials in stroke patients. Based on postmarketing data, US physicians reported about 100 cases between 1992 and 1997. Based on an estimated patient exposure of 2 million to 4 million, and assuming an event reporting rate of 10% (the true rate is not known), the incidence of ticlopidine-associated TTP may be as high as one case in every 2000 to 4000 patients exposed.
Aplastic Anemia:
Aplastic anemia was not seen during clinical trials in stroke patients, but US physicians reported about 50 cases between 1992 and 1998. Based on an estimated patient exposure of 2 million to 4 million, and assuming an event reporting rate of 10% (the true rate is not known), the incidence of ticlopidine-associated aplastic anemia may be as high as one case in every 4000 to 8000 patients exposed.
Monitoring of Clinical and Hematologic Status:
Severe hematological adverse reactions may occur within a few days of the start of therapy. The incidence of TTP peaks after about 3 to 4 weeks of therapy and neutropenia peaks at approximately 4 to 6 weeks. The incidence of aplastic anemia peaks after about 4 to 8 weeks of therapy. The incidence of the hematologic adverse reactions declines thereafter. Only a few cases of neutropenia, TTP, or aplastic anemia have arisen after more than 3 months of therapy.
Hematological adverse reactions cannot be reliably predicted by any identified demographic or clinical characteristics. During the first 3 months of treatment, patients receiving TICLID must, therefore, be hematologically and clinically monitored for evidence of neutropenia or TTP. If any such evidence is seen, TICLID should be immediately discontinued.
The detection and treatment of ticlopidine-associated hematological adverse reactions are further described under WARNINGS.
DESCRIPTION:
TICLID (ticlopidine hydrochloride) is a platelet aggregation inhibitor. Chemically it is 5-[(2-chlorophenyl)methyl]-4,5,6,7-tetrahydrothieno [3,2-c] pyridine hydrochloride. The structural formula is:
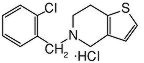
Ticlopidine hydrochloride is a white crystalline solid. It is freely soluble in water and self-buffers to a pH of 3.6. It also dissolves freely in methanol, is sparingly soluble in methylene chloride and ethanol, slightly soluble in acetone and insoluble in a buffer solution of pH 6.3. It has a molecular weight of 300.25.
TICLID tablets for oral administration are provided as white, oval, film-coated, blue-imprinted tablets containing 250 mg of ticlopidine hydrochloride. Each tablet also contains citric acid, magnesium stearate, microcrystalline cellulose, povidone, starch and stearic acid as inactive ingredients. The white film-coating contains hydroxypropylmethyl cellulose, polyethylene glycol and titanium dioxide. Each tablet is printed with blue ink, which includes FD&C Blue #1 aluminum lake as the colorant. The tablets are identified with Ticlid on one side and 250 on the reverse side.
CLINICAL PHARMACOLOGY:
Mechanism of Action:
When taken orally, ticlopidine hydrochloride causes a time- and dose-dependent inhibition of both platelet aggregation and release of platelet granule constituents, as well as a prolongation of bleeding time. The intact drug has no significant in vitro activity at the concentrations attained in vivo; and, although analysis of urine and plasma indicates at least 20 metabolites, no metabolite which accounts for the activity of ticlopidine has been isolated.
Ticlopidine hydrochloride, after oral ingestion, interferes with platelet membrane function by inhibiting ADP-induced platelet-fibrinogen binding and subsequent platelet-platelet interactions. The effect on platelet function is irreversible for the life of the platelet, as shown both by persistent inhibition of fibrinogen binding after washing platelets ex vivo and by inhibition of platelet aggregation after resuspension of platelets in buffered medium.
Pharmacokinetics and Metabolism:
After oral administration of a single 250-mg dose, ticlopidine hydrochloride is rapidly absorbed with peak plasma levels occurring at approximately 2 hours after dosing and is extensively metabolized. Absorption is greater than 80%. Administration after meals results in a 20% increase in the AUC of ticlopidine.
Ticlopidine hydrochloride displays nonlinear pharmacokinetics and clearance decreases markedly on repeated dosing. In older volunteers the apparent half-life of ticlopidine after a single 250-mg dose is about 12.6 hours; with repeat dosing at 250 mg bid, the terminal elimination half-life rises to 4 to 5 days and steady-state levels of ticlopidine hydrochloride in plasma are obtained after approximately 14 to 21 days.
Ticlopidine hydrochloride binds reversibly (98%) to plasma proteins, mainly to serum albumin and lipoproteins. The binding to albumin and lipoproteins is nonsaturable over a wide concentration range. Ticlopidine also binds to alpha-1 acid glycoprotein. At concentrations attained with the recommended dose, only 15% or less ticlopidine in plasma is bound to this protein.
Ticlopidine hydrochloride is metabolized extensively by the liver; only trace amounts of intact drug are detected in the urine. Following an oral dose of radioactive ticlopidine hydrochloride administered in solution, 60% of the radioactivity is recovered in the urine and 23% in the feces. Approximately 1/3 of the dose excreted in the feces is intact ticlopidine hydrochloride, possibly excreted in the bile. Ticlopidine hydrochloride is a minor component in plasma (5%) after a single dose, but at steady-state is the major component (15%). Approximately 40% to 50% of the radioactive metabolites circulating in plasma are covalently bound to plasma proteins, probably by acylation.
Clearance of ticlopidine decreases with age. Steady-state trough values in elderly patients (mean age 70 years) are about twice those in younger volunteer populations.
Hepatically Impaired Patients:
The effect of decreased hepatic function on the pharmacokinetics of TICLID was studied in 17 patients with advanced cirrhosis. The average plasma concentration of ticlopidine in these subjects was slightly higher than that seen in older subjects in a separate trial (see CONTRAINDICATIONS).
Renally Impaired Patients:
Patients with mildly (Ccr 50 to 80 mL/min) or moderately (Ccr 20 to 50 mL/min) impaired renal function were compared to normal subjects (Ccr 80 to 150 mL/min) in a study of the pharmacokinetic and platelet pharmacodynamic effects of TICLID (250 mg bid) for 11 days. Concentrations of unchanged TICLID were measured after a single 250-mg dose and after the final 250-mg dose on Day 11.
AUC values of ticlopidine increased by 28% and 60% in mild and moderately impaired patients, respectively, and plasma clearance decreased by 37% and 52%, respectively, but there were no statistically significant differences in ADP-induced platelet aggregation. In this small study (26 patients), bleeding times showed significant prolongation only in the moderately impaired patients.
Pharmacodynamics:
In healthy volunteers over the age of 50, substantial inhibition (over 50%) of ADP-induced platelet aggregation is detected within 4 days after administration of ticlopidine hydrochloride 250 mg bid, and maximum platelet aggregation inhibition (60% to 70%) is achieved after 8 to 11 days. Lower doses cause less, and more delayed, platelet aggregation inhibition, while doses above 250 mg bid give little additional effect on platelet aggregation but an increased rate of adverse effects. The dose of 250 mg bid is the only dose that has been evaluated in controlled clinical trials.
After discontinuation of ticlopidine hydrochloride, bleeding time and other platelet function tests return to normal within 2 weeks, in the majority of patients.
At the recommended therapeutic dose (250 mg bid), ticlopidine hydrochloride has no known significant pharmacological actions in man other than inhibition of platelet function and prolongation of the bleeding time.
CLINICAL TRIALS:
Stroke Patients:
The effect of ticlopidine on the risk of stroke and cardiovascular events was studied in two multicenter, randomized, double-blind trials.
1. Study in Patients Experiencing Stroke Precursors:
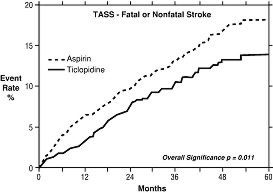
In a trial comparing ticlopidine and aspirin (The Ticlopidine Aspirin Stroke Study or TASS), 3069 patients (1987 men, 1082 women) who had experienced such stroke precursors as transient ischemic attack (TIA), transient monocular blindness (amaurosis fugax), reversible ischemic neurological deficit or minor stroke, were randomized to ticlopidine 250 mg bid or aspirin 650 mg bid. The study was designed to follow patients for at least 2 years and up to 5 years.
Over the duration of the study, TICLID significantly reduced the risk of fatal and nonfatal stroke by 24% (p = .011) from 18.1 to 13.8 per 100 patients followed for 5 years, compared to aspirin. During the first year, when the risk of stroke is greatest, the reduction in risk of stroke (fatal and nonfatal) compared to aspirin was 48%; the reduction was similar in men and women.
2. Study in Patients Who Had a Completed Atherothrombotic Stroke:
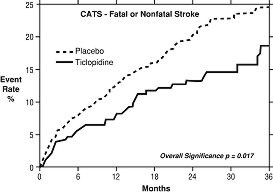
In a trial comparing ticlopidine with placebo (The Canadian American Ticlopidine Study or CATS) 1073 patients who had experienced a previous atherothrombotic stroke were treated with TICLID 250 mg bid or placebo for up to 3 years.
TICLID significantly reduced the overall risk of stroke by 24% (p = .017) from 24.6 to 18.6 per 100 patients followed for 3 years, compared to placebo. During the first year the reduction in risk of fatal and nonfatal stroke over placebo was 33%.
Stent Patients:
The ability of TICLID to reduce the rate of thrombotic events after the placement of coronary artery stents has been studied in five randomized trials, one of substantial size (Stent Anticoagulation Restenosis Study or STARS) described below, and four smaller studies. In these trials, ticlopidine 250 mg bid with ASA (dose range from 100 mg bid to 325 mg qd) was compared to aspirin alone or to anticoagulant therapy plus aspirin. The trials enrolled patients undergoing both planned (elective) and unplanned coronary stent placement. The types of stents used, the use of intravascular ultrasound, and the use of high-pressure stent deployment varied among the trials, although all patients in STARS received a Palmaz-Schatz stent. The primary efficacy endpoints of the trials were similar, and included death, myocardial infarction and the need for repeat coronary angioplasty or CABG. All trials followed patients for at least 30 days.
In STARS, patients were randomized to receive one of three regimens for 4 weeks: aspirin alone, aspirin plus coumadin, or aspirin plus ticlopidine. Therapy was initiated following successful coronary stent placement. The primary endpoint was the incidence of stent thrombosis, defined as death, Q-Wave MI, or angiographic thrombus within the stented vessel demonstrated at the time of documented ischemia requiring emergent revascularization. The incidence rates for the primary endpoint and its components at 30 days are shown in the table below.
| STARS | TICLID + Aspirin N=546 | Aspirin N=557 | Coumadin + Aspirin N=550 | Odds Ratio (95% C.I.)* | p-Value* |
|---|---|---|---|---|---|
|
|||||
| Primary Endpoint | 3 (0.5%) | 20 (3.6%) | 15 (2.7%) | 0.15 (0.03, 0.51) | <0.001 |
| Deaths | 0 (0%) | 1 (0.2%) | 0 (0%) | — | — |
| Q-Wave MI (Recurrent and Procedure Related) | 1 (0.2%) | 12 (2.2%) | 8 (1.5%) | 0.08 (0.002, 0.57) | 0.004 |
| Angiographically Evident Thrombosis | 3 (0.5%) | 16 (2.9%) | 15 (2.7%) | 0.19 (0.03, 0.66) | 0.005 |
The use of ticlopidine plus aspirin did not affect the rate of non-Q-wave MIs when compared with aspirin alone or aspirin plus anticoagulants in STARS.
The use of ticlopidine plus aspirin was associated with a lower rate of recurrent cardiovascular events when compared with aspirin alone or aspirin plus anticoagulants in the other four randomized trials.
The rate of serious bleeding complications and neutropenia in STARS are shown in the table below. There were no cases of thrombotic thrombocytopenic purpura (TTP) or aplastic anemia reported in 1346 patients who received ticlopidine plus aspirin in the five randomized trials.
| STARS | TICLID + Aspirin N=546 | Aspirin N=557 | Coumadin + Aspirin N=550 |
|---|---|---|---|
| Hemorrhagic Complications | 30 (5.5%) | 10 (1.8%) | 34 (6.2%) |
| Cerebrovascular Accident | 0 (0%) | 2 (0.4%) | 1 (0.2%) |
| Neutropenia (≤1200/mm3) | 3 (0.5%) | 0 (0%) | 1 (0.2%) |
INDICATIONS AND USAGE:
TICLID is indicated:
- to reduce the risk of thrombotic stroke (fatal or nonfatal) in patients who have experienced stroke precursors, and in patients who have had a completed thrombotic stroke. Because TICLID is associated with a risk of life-threatening blood dyscrasias including thrombotic thrombocytopenic purpura (TTP), neutropenia/agranulocytosis and aplastic anemia (see BOXED WARNING and WARNINGS), TICLID should be reserved for patients who are intolerant or allergic to aspirin therapy or who have failed aspirin therapy.
- as adjunctive therapy with aspirin to reduce the incidence of subacute stent thrombosis in patients undergoing successful coronary stent implantation (see CLINICAL TRIALS).
CONTRAINDICATIONS:
The use of TICLID is contraindicated in the following conditions:
- Hypersensitivity to the drug
- Presence of hematopoietic disorders such as neutropenia and thrombocytopenia or a past history of either TTP or aplastic anemia
- Presence of a hemostatic disorder or active pathological bleeding (such as bleeding peptic ulcer or intracranial bleeding)
- Patients with severe liver impairment
WARNINGS:
Hematological Adverse Reactions:
Neutropenia:
Neutropenia may occur suddenly. Bone-marrow examination typically shows a reduction in white blood cell precursors. After withdrawal of ticlopidine, the neutrophil count usually rises to >1200/mm3 within 1 to 3 weeks.
Thrombotic Thrombocytopenic Purpura (TTP):
TTP is characterized by thrombocytopenia, microangiopathic hemolytic anemia (schistocytes [fragmented RBCs] seen on peripheral smear), neurological findings, renal dysfunction, and fever. The signs and symptoms can occur in any order, in particular, clinical symptoms may precede laboratory findings by hours or days. With prompt treatment (often including plasmapheresis), 70% to 80% of patients will survive with minimal or no sequelae. Because platelet transfusions may accelerate thrombosis in patients with TTP on ticlopidine, they should, if possible, be avoided.
Aplastic Anemia:
Aplastic anemia is characterized by anemia, thrombocytopenia and neutropenia together with a bone marrow examination that shows decreases in the precursor cells for red blood cells, white blood cells, and platelets. Patients may present with signs or symptoms suggestive of infection, in association with low white blood cell and platelet counts. Prompt treatment, which may include the use of drugs to stimulate the bone marrow, can minimize the mortality associated with aplastic anemia.
Monitoring for Hematologic Adverse Reactions:
Starting just before initiating treatment and continuing through the third month of therapy, patients receiving TICLID must be monitored every 2 weeks. Because of ticlopidine's long plasma half-life, patients who discontinue ticlopidine during this 3-month period should continue to be monitored for 2 weeks after discontinuation. More frequent monitoring, and monitoring after the first 3 months of therapy, is necessary only in patients with clinical signs (eg, signs or symptoms suggestive of infection) or laboratory signs (eg, neutrophil count less than 70% of the baseline count, decrease in hematocrit or platelet count) that suggest incipient hematological adverse reactions.
Clinically, fever might suggest neutropenia, TTP, or aplastic anemia; TTP might also be suggested by weakness, pallor, petechiae or purpura, dark urine (due to blood, bile pigments, or hemoglobin) or jaundice, or neurological changes. Patients should be told to discontinue TICLID and to contact the physician immediately upon the occurrence of any of these findings.
Laboratory monitoring should include a complete blood count, with special attention to the absolute neutrophil count (WBC x % neutrophils), platelet count, and the appearance of the peripheral smear. Ticlopidine is occasionally associated with thrombocytopenia unrelated to TTP or aplastic anemia. Any acute, unexplained reduction in hemoglobin or platelet count should prompt further investigation for a diagnosis of TTP, and the appearance of schistocytes (fragmented RBCs) on the smear should be treated as presumptive evidence of TTP. A simultaneous decrease in platelet count and WBC count should prompt further investigation for a diagnosis of aplastic anemia. If there are laboratory signs of TTP or aplastic anemia, or if the neutrophil count is confirmed to be <1200/mm3, then TICLID should be discontinued immediately.
Other Hematological Effects:
Rare cases of agranulocytosis, pancytopenia, or leukemia have been reported in postmarketing experience, some of which have been fatal. All forms of hematological adverse reactions are potentially fatal.
Cholesterol Elevation:
TICLID therapy causes increased serum cholesterol and triglycerides. Serum total cholesterol levels are increased 8% to 10% within 1 month of therapy and persist at that level. The ratios of the lipoprotein subfractions are unchanged.
Anticoagulant Drugs:
The tolerance and long-term safety of coadministration of TICLID with heparin, oral anticoagulants or fibrinolytic agents have not been established. In trials for cardiac stenting, patients received heparin and TICLID concomitantly for approximately 12 hours. If a patient is switched from an anticoagulant or fibrinolytic drug to TICLID, the former drug should be discontinued prior to TICLID administration.
PRECAUTIONS:
General:
TICLID should be used with caution in patients who may be at risk of increased bleeding from trauma, surgery or pathological conditions. If it is desired to eliminate the antiplatelet effects of TICLID prior to elective surgery, the drug should be discontinued 10 to 14 days prior to surgery. Several controlled clinical studies have found increased surgical blood loss in patients undergoing surgery during treatment with ticlopidine. In TASS and CATS it was recommended that patients have ticlopidine discontinued prior to elective surgery. Several hundred patients underwent surgery during the trials, and no excessive surgical bleeding was reported.
Prolonged bleeding time is normalized within 2 hours after administration of 20 mg methylprednisolone IV. Platelet transfusions may also be used to reverse the effect of TICLID on bleeding. Because platelet transfusions may accelerate thrombosis in patients with TTP on ticlopidine, they should, if possible, be avoided.
GI Bleeding:
TICLID prolongs template bleeding time. The drug should be used with caution in patients who have lesions with a propensity to bleed (such as ulcers). Drugs that might induce such lesions should be used with caution in patients on TICLID (see CONTRAINDICATIONS).
Use in Hepatically Impaired Patients:
Since ticlopidine is metabolized by the liver, dosing of TICLID or other drugs metabolized in the liver may require adjustment upon starting or stopping concomitant therapy. Because of limited experience in patients with severe hepatic disease, who may have bleeding diatheses, the use of TICLID is not recommended in this population (see CLINICAL PHARMACOLOGY and CONTRAINDICATIONS).
Use in Renally Impaired Patients:
There is limited experience in patients with renal impairment. Decreased plasma clearance, increased AUC values and prolonged bleeding times can occur in renally impaired patients. In controlled clinical trials no unexpected problems have been encountered in patients having mild renal impairment, and there is no experience with dosage adjustment in patients with greater degrees of renal impairment. Nevertheless, for renally impaired patients, it may be necessary to reduce the dosage of ticlopidine or discontinue it altogether if hemorrhagic or hematopoietic problems are encountered (see CLINICAL PHARMACOLOGY).
Information for the Patient (see Patient Leaflet):
Patients should be told that a decrease in the number of white blood cells (neutropenia) or platelets (thrombocytopenia) can occur with TICLID, especially during the first 3 months of treatment and that neutropenia, if it is severe, can result in an increased risk of infection. They should be told it is critically important to obtain the scheduled blood tests to detect neutropenia or thrombocytopenia. Patients should also be reminded to contact their physicians if they experience any indication of infection such as fever, chills, or sore throat, any of which might be a consequence of neutropenia. Thrombocytopenia may be part of a syndrome called TTP. Symptoms and signs of TTP, such as fever, weakness, difficulty speaking, seizures, yellowing of skin or eyes, dark or bloody urine, pallor or petechiae (pinpoint hemorrhagic spots on the skin), should be reported immediately.
All patients should be told that it may take them longer than usual to stop bleeding when they take TICLID and that they should report any unusual bleeding to their physician. Patients should tell physicians and dentists that they are taking TICLID before any surgery is scheduled and before any new drug is prescribed.
Patients should be told to promptly report side effects of TICLID such as severe or persistent diarrhea, skin rashes or subcutaneous bleeding or any signs of cholestasis, such as yellow skin or sclera, dark urine, or light-colored stools.
Patients should be told to take TICLID with food or just after eating in order to minimize gastrointestinal discomfort.
Laboratory Tests:
Liver Function:
TICLID therapy has been associated with elevations of alkaline phosphatase, bilirubin, and transaminases, which generally occurred within 1 to 4 months of therapy initiation. In controlled clinical trials in stroke patients, the incidence of elevated alkaline phosphatase (greater than two times upper limit of normal) was 7.6% in ticlopidine patients, 6% in placebo patients and 2.5% in aspirin patients. The incidence of elevated AST (SGOT) (greater than two times upper limit of normal) was 3.1% in ticlopidine patients, 4% in placebo patients and 2.1% in aspirin patients. No progressive increases were observed in closely monitored clinical trials (eg, no transaminase greater than 10 times the upper limit of normal was seen), but most patients with these abnormalities had therapy discontinued. Occasionally patients had developed minor elevations in bilirubin.
Postmarketing experience includes rare individuals with elevations in their transaminases and bilirubin to >10X above the upper limits of normal. Based on postmarketing and clinical trial experience, liver function testing, including ALT, AST, and GGT, should be considered whenever liver dysfunction is suspected, particularly during the first 4 months of treatment.
Drug Interactions:
Therapeutic doses of TICLID caused a 30% increase in the plasma half-life of antipyrine and may cause analogous effects on similarly metabolized drugs. Therefore, the dose of drugs metabolized by hepatic microsomal enzymes with low therapeutic ratios or being given to patients with hepatic impairment may require adjustment to maintain optimal therapeutic blood levels when starting or stopping concomitant therapy with ticlopidine. Studies of specific drug interactions yielded the following results:
Aspirin and Other NSAIDs:
Ticlopidine potentiates the effect of aspirin or other NSAIDs on platelet aggregation. The safety of concomitant use of ticlopidine and NSAIDs has not been established. The safety of concomitant use of ticlopidine and aspirin beyond 30 days has not been established (see CLINICAL TRIALS: Stent Patients). Aspirin did not modify the ticlopidine-mediated inhibition of ADP-induced platelet aggregation, but ticlopidine potentiated the effect of aspirin on collagen-induced platelet aggregation. Caution should be exercised in patients who have lesions with a propensity to bleed, such as ulcers. Long-term concomitant use of aspirin and ticlopidine is not recommended (see PRECAUTIONS: GI Bleeding).
Antacids:
Administration of TICLID after antacids resulted in an 18% decrease in plasma levels of ticlopidine.
Cimetidine:
Chronic administration of cimetidine reduced the clearance of a single dose of TICLID by 50%.
Digoxin:
Coadministration of TICLID with digoxin resulted in a slight decrease (approximately 15%) in digoxin plasma levels. Little or no change in therapeutic efficacy of digoxin would be expected.
Theophylline:
In normal volunteers, concomitant administration of TICLID resulted in a significant increase in the theophylline elimination half-life from 8.6 to 12.2 hours and a comparable reduction in total plasma clearance of theophylline.
Phenobarbital:
In 6 normal volunteers, the inhibitory effects of TICLID on platelet aggregation were not altered by chronic administration of phenobarbital.
Phenytoin:
In vitro studies demonstrated that ticlopidine does not alter the plasma protein binding of phenytoin. However, the protein binding interactions of ticlopidine and its metabolites have not been studied in vivo. Several cases of elevated phenytoin plasma levels with associated somnolence and lethargy have been reported following coadministration with TICLID. Caution should be exercised in coadministering this drug with TICLID, and it may be useful to remeasure phenytoin blood concentrations.
Propranolol:
In vitro studies demonstrated that ticlopidine does not alter the plasma protein binding of propranolol. However, the protein binding interactions of ticlopidine and its metabolites have not been studied in vivo. Caution should be exercised in coadministering this drug with TICLID.
Other Concomitant Therapy:
Although specific interaction studies were not performed, in clinical studies TICLID was used concomitantly with beta blockers, calcium channel blockers and diuretics without evidence of clinically significant adverse interactions (see PRECAUTIONS).
Food Interaction:
The oral bioavailability of ticlopidine is increased by 20% when taken after a meal. Administration of TICLID with food is recommended to maximize gastrointestinal tolerance. In controlled trials in stroke patients, TICLID was taken with meals.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
In a 2-year oral carcinogenicity study in rats, ticlopidine at daily doses of up to 100 mg/kg (610 mg/m2) was not tumorigenic. For a 70-kg person (1.73 m2 body surface area) the dose represents 14 times the recommended clinical dose on a mg/kg basis and two times the clinical dose on body surface area basis. In a 78-week oral carcinogenicity study in mice, ticlopidine at daily doses up to 275 mg/kg (1180 mg/m2) was not tumorigenic. The dose represents 40 times the recommended clinical dose on a mg/kg basis and four times the clinical dose on body surface area basis.
Ticlopidine was not mutagenic in vitro in the Ames test, the rat hepatocyte DNA-repair assay, or the Chinese-hamster fibroblast chromosomal aberration test; or in vivo in the mouse spermatozoid morphology test, the Chinese-hamster micronucleus test, or the Chinese-hamster bone-marrow-cell sister-chromatid exchange test. Ticlopidine was found to have no effect on fertility of male and female rats at oral doses up to 400 mg/kg/day.
Pregnancy: Teratogenic Effects: Pregnancy: Category B.
Teratology studies have been conducted in mice (doses up to 200 mg/kg/day), rats (doses up to 400 mg/kg/day) and rabbits (doses up to 200 mg/kg/day). Doses of 400 mg/kg in rats, 200 mg/kg/day in mice and 100 mg/kg in rabbits produced maternal toxicity, as well as fetal toxicity, but there was no evidence of a teratogenic potential of ticlopidine. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of a human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers:
Studies in rats have shown ticlopidine is excreted in the milk. It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from ticlopidine, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use:
Clearance of ticlopidine is somewhat lower in elderly patients and trough levels are increased. The major clinical trials with TICLID in stroke patients were conducted in an elderly population with an average age of 64 years. Of the total number of patients in the therapeutic trials, 45% of patients were over 65 years old and 12% were over 75 years old. No overall differences in effectiveness or safety were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
ADVERSE REACTIONS:
Adverse reactions in stroke patients were relatively frequent with over 50% of patients reporting at least one. Most (30% to 40%) involved the gastrointestinal tract. Most adverse effects are mild, but 21% of patients discontinued therapy because of an adverse event, principally diarrhea, rash, nausea, vomiting, GI pain and neutropenia. Most adverse effects occur early in the course of treatment, but a new onset of adverse effects can occur after several months.
The incidence rates of adverse events listed in the following table were derived from multicenter, controlled clinical trials in stroke patients described above comparing TICLID, placebo and aspirin over study periods of up to 5.8 years. Adverse events considered by the investigator to be probably drug-related that occurred in at least 1% of patients treated with TICLID are shown in the following table:
| Event | TICLID (n = 2048) Incidence | Aspirin (n = 1527) Incidence | Placebo (n = 536) Incidence |
|---|---|---|---|
| Any Events | 60.0 (20.9) | 53.2 (14.5) | 34.3 (6.1) |
| Diarrhea | 12.5 (6.3) | 5.2 (1.8) | 4.5 (1.7) |
| Nausea | 7.0 (2.6) | 6.2 (1.9) | 1.7 (0.9) |
| Dyspepsia | 7.0 (1.1) | 9.0 (2.0) | 0.9 (0.2) |
| Rash | 5.1 (3.4) | 1.5 (0.8) | 0.6 (0.9) |
| GI Pain | 3.7 (1.9) | 5.6 (2.7) | 1.3 (0.4) |
| Neutropenia | 2.4 (1.3) | 0.8 (0.1) | 1.1 (0.4) |
| Purpura | 2.2 (0.2) | 1.6 (0.1) | 0.0 (0.0) |
| Vomiting | 1.9 (1.4) | 1.4 (0.9) | 0.9 (0.4) |
| Flatulence | 1.5 (0.1) | 1.4 (0.3) | 0.0 (0.0) |
| Pruritus | 1.3 (0.8) | 0.3 (0.1) | 0.0 (0.0) |
| Dizziness | 1.1 (0.4) | 0.5 (0.4) | 0.0 (0.0) |
| Anorexia | 1.0 (0.4) | 0.5 (0.3) | 0.0 (0.0) |
| Abnormal Liver Function Test | 1.0 (0.7) | 0.3 (0.3) | 0.0 (0.0) |
Incidence of discontinuation, regardless of relationship to therapy, is shown in parentheses.
Hematological:
Neutropenia/thrombocytopenia, TTP, aplastic anemia (see BOXED WARNING and WARNINGS), leukemia, agranulocytosis, eosinophilia, pancytopenia, thrombocytosis and bone-marrow depression have been reported.
Gastrointestinal:
TICLID therapy has been associated with a variety of gastrointestinal complaints including diarrhea and nausea. The majority of cases are mild, but about 13% of patients discontinued therapy because of these. They usually occur within 3 months of initiation of therapy and typically are resolved within 1 to 2 weeks without discontinuation of therapy. If the effect is severe or persistent, therapy should be discontinued. In some cases of severe or bloody diarrhea, colitis was later diagnosed.
Hemorrhagic:
TICLID has been associated with increased bleeding, spontaneous posttraumatic bleeding and perioperative bleeding including, but not limited to, gastrointestinal bleeding. It has also been associated with a number of bleeding complications such as ecchymosis, epistaxis, hematuria and conjunctival hemorrhage.
Intracerebral bleeding was rare in clinical trials in stroke patients with TICLID, with an incidence no greater than that seen with comparator agents (ticlopidine 0.5%, aspirin 0.6%, placebo 0.75%). It has also been reported postmarketing.
Rash:
Ticlopidine has been associated with a maculopapular or urticarial rash (often with pruritus). Rash usually occurs within 3 months of initiation of therapy with a mean onset time of 11 days. If drug is discontinued, recovery occurs within several days. Many rashes do not recur on drug rechallenge. There have been rare reports of severe rashes, including Stevens-Johnson syndrome, erythema multiforme and exfoliative dermatitis.
Less Frequent Adverse Reactions (Probably Related):
Clinical adverse experiences occurring in 0.5% to 1.0% of stroke patients in controlled trials include: Digestive System: GI fullness
Skin and Appendages: urticaria
Nervous System: headache
Body as a Whole: asthenia, pain
Hemostatic System: epistaxis
Special Senses: tinnitus
In addition, rarer, relatively serious and potentially fatal events associated with the use of TICLID have also been reported from postmarketing experience: Hemolytic anemia with reticulocytosis, immune thrombocytopenia, hepatitis, hepatocellular jaundice, cholestatic jaundice, hepatic necrosis, hepatic failure, peptic ulcer, renal failure, nephrotic syndrome, hyponatremia, vasculitis, sepsis, allergic reactions (including angioedema, allergic pneumonitis, and anaphylaxis), systemic lupus (positive ANA), peripheral neuropathy, serum sickness, arthropathy and myositis.
OVERDOSAGE:
One case of deliberate overdosage with TICLID has been reported by a foreign postmarketing surveillance program. A 38-year-old male took a single 6000-mg dose of TICLID (equivalent to 24 standard 250-mg tablets). The only abnormalities reported were increased bleeding time and increased SGPT. No special therapy was instituted and the patient recovered without sequelae.
Single oral doses of ticlopidine at 1600 mg/kg and 500 mg/kg were lethal to rats and mice, respectively. Symptoms of acute toxicity were GI hemorrhage, convulsions, hypothermia, dyspnea, loss of equilibrium and abnormal gait.
DOSAGE AND ADMINISTRATION:
HOW SUPPLIED:
TICLID is available in white, oval, film-coated 250-mg tablets, printed in blue with Ticlid on one side and 250 on the other. They are provided in unit of use bottles of 30 tablets (NDC 0004-0018-23) and 60 tablets (NDC 0004-0018-22) and 500 tablets (NDC 0004-0018-14).
Store at 15° to 30°C (59° to 86°F).
IMPORTANT INFORMATION ABOUT TICLID (ticlopidine HCl)
TABLETS
The information in this leaflet is intended to help you use TICLID safely. Please read the leaflet carefully. Although it does not contain all the detailed medical information that is provided to your doctor, it provides facts about TICLID that are important for you to know. If you still have questions after reading this leaflet or if you have questions at any time during your treatment with TICLID, check with your doctor.
Why TICLID was Prescribed by Your Doctor:
Stroke Patients: TICLID is recommended to help reduce your risk of having a stroke, but only for patients who have had a stroke or early stroke warning symptoms while on aspirin, or for those who have these symptoms but are intolerant or allergic to aspirin.
Stent Patients: TICLID is recommended with aspirin for up to 30 days in patients who have had a stent implanted in their coronary arteries to reduce the risk of blood clots forming inside the stent.
Special Warning for Users of TICLID/Necessary Blood Tests: TICLID is not prescribed for those who can take aspirin to reduce the risk of stroke because TICLID can cause life-threatening blood problems. Getting your blood tests done and reporting symptoms to your doctor as soon as possible can avoid serious complications.
The white cells of the blood that fight infection may drop to dangerous levels (a condition called neutropenia). This occurs in about 2.4% (1 in 40) of people on ticlopidine. You should be on the lookout for signs of infection such as fever, chills or sore throat. If this problem is caught early, it can almost always be reversed, but if undetected it can be fatal.
Another problem that has occurred in some patients taking ticlopidine is a decrease in cells called platelets (a condition called thrombocytopenia). This may occur as part of a syndrome that includes injury to red blood cells, causing anemia, kidney abnormalities, neurologic changes and fever. This condition is called TTP and can be fatal.
Things you should watch for as possible early signs of TTP are yellow skin or eye color, pinpoint dots (rash) on the skin, pale color, fever, weakness on a side of the body, or dark urine. If any of these occur, contact your doctor immediately.
Both complications occur most frequently in the first 90 days after TICLID is started. To make sure you don't develop either of these problems, your doctor will arrange for you to have your blood tested before you start taking TICLID and then every 2 weeks for the first 3 months you are on TICLID. If detected, neutropenia and thrombocytopenia can almost always be reversed. It is essential that you keep your appointments for the blood tests and that you call your doctor immediately if you have any indication that you may have TTP or neutropenia. If you stop taking TICLID for any reason within the first 3 months, you will still need to have your blood tested for an additional 2 weeks after you have stopped taking TICLID.
Rarely, decreases in the white blood cells, red blood cells and platelets can occur together. This condition is called aplastic anemia and can be fatal.
Things you should watch for as possible early signs of aplastic anemia are feeling of excessive weakness and tiredness, paleness, bruising, and bleeding from areas such as your nose or gums. You may also develop signs of infection such as fever. If any of these occur, contact your doctor immediately.
Other Warnings and Precautions: A few people may develop jaundice while being treated with TICLID. The signs of jaundice are yellowing of the skin or the whites of the eyes or consistent darkening of the urine or lightening in the color of the stools. These symptoms should be reported to your physician promptly.
If any of the symptoms described above for neutropenia, TTP, aplastic anemia or jaundice occur, contact your doctor immediately.
TICLID should be used only as directed by your doctor. Do not give TICLID to anyone else. Keep TICLID out of reach of children!
Some people may have such side effects as diarrhea, skin rash, stomach or intestinal discomfort. If any of these problems are persistent, or if you are concerned about them, bring them to your doctor's attention.
It may take longer than usual to stop bleeding when taking TICLID. Tell your doctor if you have any more bleeding or bruising than usual, and, if you have emergency surgery, be sure to let your doctor or dentist know that you are taking TICLID. Also, tell your doctor well in advance of any planned surgery (including tooth extraction), because he or she may recommend that you stop taking TICLID temporarily.
How TICLID Works:
Stroke Patients: A stroke occurs when a clot (or thrombus) forms in a blood vessel in the brain or forms in another part of the body and breaks off, then travels to the brain (an embolus). In both cases the blood supply to part of the brain is blocked and that part of the brain is damaged. TICLID works by making the blood less likely to clot, although not so much less that it causes you to become likely to bleed, unless you have a bleeding disorder or some injury (such as a bleeding ulcer of the stomach or intestine) that is especially likely to bleed.
Stent Patients: A heart attack or angina (chest pain) can occur when fatty deposits block the arteries that carry oxygen and nutrient-rich blood to your heart. To decrease the chance of fatty deposits building up over time, your doctor may recommend the placement of a coronary stent. TICLID may be given to you with aspirin to make blood clots less likely to form inside the stent so that the artery remains open.
Who Should Not Take TICLID? Contact your doctor immediately and do not take TICLID if:
- you have an allergic reaction to TICLID
- you have a blood disorder or a serious bleeding problem, such as a bleeding stomach ulcer
- you have previously been told you had TTP or aplastic anemia
- you have severe liver disease or other liver problems
- you are pregnant or you are planning to become pregnant
- you are breast-feeding
Rx only
Distributed by:

27897570-0301
Copyright © 1999-2001 by Roche Laboratories Inc. All rights reserved.

| TICLID
ticlopidine hydrochloride tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| NDA | NDA019979 | 10/31/1991 | 07/18/2011 |
| Labeler - Genentech, Inc. (080129000) |
Revised: 08/2012 Genentech, Inc.