ENTEREG- alvimopan capsule
Adolor Corporation
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ENTEREG safely and effectively. See full prescribing information for ENTEREG.
ENTEREG® (alvimopan) Capsules Initial U.S. Approval: 2008 WARNING: FOR SHORT-TERM HOSPITAL USE ONLYENTEREG is available only for short-term (15 doses) use in hospitalized patients. Only hospitals that have registered in and met all of the requirements for the ENTEREG Access Support and Education (E.A.S.E.™) program may use ENTEREG. INDICATIONS AND USAGEENTEREG is a peripherally acting µ-opioid receptor antagonist indicated to accelerate the time to upper and lower gastrointestinal recovery following partial large or small bowel resection surgery with primary anastomosis. (1) DOSAGE AND ADMINISTRATION12 mg administered 30 minutes to 5 hours prior to surgery followed by 12 mg twice daily for up to 7 days for a maximum of 15 doses. (2.1) DOSAGE FORMS AND STRENGTHSCapsules: 12 mg (3) CONTRAINDICATIONSTherapeutic doses of opioids for more than 7 consecutive days prior to ENTEREG (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (incidence ≥3% and ≥1% placebo) in patients undergoing bowel resection were anemia, dyspepsia, hypokalemia, back pain, and urinary retention. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Adolor Corporation at 1-866-4ADOLOR (1-866-423-6567) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION. Revised: 1/2012 |
FULL PRESCRIBING INFORMATION
WARNING: FOR SHORT-TERM HOSPITAL USE ONLY
ENTEREG is available only for short-term (15 doses) use in hospitalized patients. Only hospitals that have registered in and met all of the requirements for the ENTEREG Access Support and Education (E.A.S.E.) program may use ENTEREG. [see Warnings and Precautions (5.1 and 5.2)]
1 INDICATIONS AND USAGE
ENTEREG is indicated to accelerate the time to upper and lower gastrointestinal recovery following partial large or small bowel resection surgery with primary anastomosis.
2 DOSAGE AND ADMINISTRATION
2.1 Usual Dosage in Adults
For hospital use only. The recommended adult dosage of ENTEREG is 12 mg administered 30 minutes to 5 hours prior to surgery followed by 12 mg twice daily beginning the day after surgery for a maximum of 7 days or until discharge. Patients should not receive more than 15 doses of ENTEREG.
2.2 Special Populations
Geriatric Use: No dosage adjustment is necessary in elderly patients [see Use in Specific Populations (8.5)].
Hepatic Impairment: No dosage adjustment is necessary in patients with mild-to-moderate hepatic impairment (Child-Pugh Class A and B). ENTEREG is not recommended for use in patients with severe hepatic impairment (Child-Pugh Class C) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Renal Impairment: No dosage adjustment is necessary in patients with mild-to-severe renal impairment, but they should be monitored for adverse effects. ENTEREG is not recommended for use in patients with end-stage renal disease. [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
Race: No dosage adjustment is necessary in Black, Hispanic and Japanese patients, however, due to observed 2-fold greater ENTEREG plasma concentrations in healthy male Japanese subjects, Japanese patients should be monitored for possible adverse effects. [see Use in Specific Populations (8.8) and Clinical Pharmacology (12.3)]
3 DOSAGE FORMS AND STRENGTHS
12 mg blue, hard gelatin capsules with "ADL2698" printed on both the body and the cap of the capsule.
4 CONTRAINDICATIONS
ENTEREG is contraindicated in patients who have taken therapeutic doses of opioids for more than 7 consecutive days immediately prior to taking ENTEREG.
5 WARNINGS AND PRECAUTIONS
5.1 Myocardial Infarction in a 12 Month Study in Patients treated with Opioids for Chronic Pain
There were more reports of myocardial infarctions in patients treated with alvimopan 0.5 mg twice daily compared with placebo-treated patients in a 12-month study of patients treated with opioids for chronic pain. In this study, the majority of myocardial infarctions occurred between 1 and 4 months after initiation of treatment. This imbalance has not been observed in other studies of alvimopan, including studies in patients undergoing bowel resection surgery who received alvimopan 12 mg twice daily for up to 7 days. A causal relationship with alvimopan has not been established.
5.2 Distribution Program for ENTEREG
ENTEREG is available only to hospitals that enroll in the E.A.S.E. program. To enroll in the E.A.S.E. program, the hospital must acknowledge that:
- -hospital staff who prescribe, dispense, or administer ENTEREG have been provided the educational materials on the need to limit use of ENTEREG to short-term, inpatient use;
- -patients will not receive more than 15 doses of alvimopan; and
- -ENTEREG will not be dispensed to patients after they have been discharged from the hospital.
Contact the E.A.S.E. program at 1-866-4ADOLOR (1-866-423-6567).
5.3 Opioid Tolerance and Gastrointestinal-Related Adverse Effects
Patients recently exposed to opioids are expected to be more sensitive to the effects of µ-opioid receptor antagonists, such as ENTEREG. Since ENTEREG acts peripherally, clinical signs and symptoms of increased sensitivity would likely be limited to the gastrointestinal tract (e.g., abdominal pain, nausea and vomiting, diarrhea). Patients receiving more than 3 doses of an opioid within the week prior to surgery were not studied in the postoperative ileus clinical trials; therefore, ENTEREG 12 mg capsules should be administered with caution to these patients.
5.4 Severe Hepatic Impairment
In patients with severe hepatic impairment, there is a potential for 10-fold higher plasma levels of drug [see Clinical Pharmacology (12.3)]. There are no studies of ENTEREG in patients with severe hepatic impairment undergoing bowel resection. Because of the limited data available, ENTEREG is not recommended for use in patients with severe hepatic impairment.
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice. The adverse event information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
The data described below reflect exposure to ENTEREG in 1,650 patients in 9 placebo-controlled studies worldwide. The population was 19 to 97 years old, 68% were female, and 83% were Caucasian; 61% were undergoing bowel resection surgery. The first dose of ENTEREG was administered 30 minutes to 5 hours before the scheduled start of surgery and then twice daily until hospital discharge (or for a maximum of 7 days of postoperative treatment).
Table 1 presents treatment-emergent adverse reactions reported in ≥3% patients treated with ENTEREG and for which the rate for ENTEREG was ≥1% than placebo. Treatment-emergent adverse reactions are those events occurring after the first dose of study medication treatment and within 7 days of the last dose of study medication or those events present at baseline that increased in severity after the start of study medication treatment.
| System Organ Class | Bowel Resection Patients | All Surgical Patients | |||
| Placebo (n = 986) % | ENTEREG (n = 999) % | Placebo (n = 1,365) % | ENTEREG (n = 1,650) % |
||
| Blood and lymphatic system disorders | |||||
| Anemia | 4.2 | 5.2 | 5.4 | 5.4 | |
| Gastrointestinal disorders | |||||
| Constipation | 3.9 | 4.0 | 7.6 | 9.7 | |
| Dyspepsia | 4.6 | 7.0 | 4.8 | 5.9 | |
| Flatulence | 4.5 | 3.1 | 7.7 | 8.7 | |
| Metabolism and nutrition disorders | |||||
| Hypokalemia | 8.5 | 9.5 | 7.5 | 6.9 | |
| Musculoskeletal and connective tissue disorders | |||||
| Back pain | 1.7 | 3.3 | 2.6 | 3.4 | |
| Renal and urinary disorders | |||||
| Urinary retention | 2.1 | 3.2 | 2.3 | 3.5 | |
7 DRUG INTERACTIONS
7.1 Potential for Drugs to Affect Alvimopan Pharmacokinetics
Based on in vitro data, alvimopan is not a substrate of CYP enzymes. Therefore, concomitant administration of ENTEREG with inducers or inhibitors of CYP enzymes is unlikely to alter the metabolism of alvimopan. No clinical studies have been performed to assess the effect of concomitant administration of inducers or inhibitors of cytochrome P450 enzymes on alvimopan pharmacokinetics.
In vitro studies suggest that alvimopan and its 'metabolite' are substrates for p-glycoprotein. A population PK analysis did not reveal any evidence that alvimopan or 'metabolite' pharmacokinetics were influenced by concomitant medications that are mild-to-moderate p-glycoprotein inhibitors. No clinical studies of concomitant administration of alvimopan and strong inhibitors of p-glycoprotein (e.g., verapamil, cyclosporine, amiodarone, itraconazole, quinine, spirinolactone, quinidine, diltiazem, bepridil) have been conducted.
A population PK analysis suggests that the pharmacokinetics of alvimopan were not affected by concomitant administration of acid blockers or antibiotics. However, plasma concentrations of the 'metabolite' were lower in patients receiving acid blockers or preoperative oral antibiotics (49% and 81%, respectively). Because the 'metabolite' is not required for efficacy, no dosage adjustments are necessary in these patients.
7.2 Potential for Alvimopan to Affect the Pharmacokinetics of Other Drugs
Alvimopan and its 'metabolite' are not inhibitors of CYP 1A2, 2C9, 2C19, 3A4, 2D6, and 2E1 in vitro at concentrations far in excess of those observed clinically. Alvimopan and its 'metabolite' are not inducers of CYP 1A2, 2B6, 2C9, 2C19 and 3A4. In vitro studies also suggest that alvimopan and its 'metabolite' are not inhibitors of p-glycoprotein. These in vitro findings suggest that ENTEREG is unlikely to alter the pharmacokinetics of coadministered drugs through inhibition or induction of CYP enzymes or inhibition of p-glycoprotein.
Coadministration of alvimopan does not appear to alter the pharmacokinetics of morphine and its metabolite, morphine-6-glucuronide, to a clinically significant degree when morphine is administered intravenously. Dosage adjustment for intravenously administered morphine is not necessary when it is coadministered with alvimopan.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects: Pregnancy Category B
Reproduction studies have been performed in pregnant rats at about 68 to 136 times the recommended human oral dose based on the body surface area and intravenous doses of about 3.4 to 6.8 times the recommended human oral dose based on the body surface area and in pregnant rabbits at intravenous doses at about 5 to 10 times the recommended human oral dose based on the body surface area and have revealed no evidence of impaired fertility or harm to the fetus due to alvimopan. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
8.3 Nursing Mothers
Alvimopan and its 'metabolite' are detected in the milk of lactating rats. It is not known whether alvimopan is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when ENTEREG is administered to a nursing woman.
8.5 Geriatric Use
Of the total number of patients in 5 clinical efficacy studies treated with ENTEREG or placebo, 45% were 65 years of age and over, while 18% were 75 years of age and over. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. No dosage adjustment based on increased age is required [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Although there is a potential for higher plasma levels of drug in patients with mild-to-moderate hepatic impairment [see Clinical Pharmacology (12.3)], dosage adjustment in these patients is not required. Patients with mild-to-moderate hepatic impairment should be closely monitored for possible adverse effects (e.g., diarrhea, gastrointestinal pain, cramping) that could indicate high drug or 'metabolite' levels, and ENTEREG should be discontinued if adverse events occur. ENTEREG is not recommended for use in patients with severe hepatic impairment. [See Dosage and Administration (2.2), Warnings and Precautions (5.4), and Clinical Pharmacology (12.3)]
8.7 Renal Impairment
Alvimopan has not been studied in patients with end-stage renal disease and ENTEREG is not recommended for use in these patients. Patients with mild-to-severe renal impairment do not require dosage adjustment, but they should be monitored for adverse effects. [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. Patients with severe impairment should be closely monitored for possible adverse effects (e.g., diarrhea, gastrointestinal pain, cramping) that could indicate high drug or 'metabolite' levels, and ENTEREG should be discontinued if adverse events occur.
8.8 Race
No dosage adjustment is necessary in Black, Hispanic and Japanese patients. However, the exposure of ENTEREG in Japanese male healthy volunteers was approximately 2-fold greater than in Caucasian subjects. Japanese patients should be closely monitored for possible adverse effects (e.g., diarrhea, gastrointestinal pain, cramping) that could indicate high drug or 'metabolite' levels, and ENTEREG should be discontinued if adverse events occur. [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no specific antidote for overdosage with ENTEREG. Patients should be managed with appropriate supportive therapy. Single doses up to 120 mg and multiple doses up to 48 mg for 7 days have been administered to normal, healthy subjects in clinical studies. In these studies, alvimopan was well tolerated with no discontinuations due to adverse events and no reported serious adverse events or deaths.
11 DESCRIPTION
ENTEREG Capsules contain alvimopan, a peripherally-acting µ-opioid receptor (PAM-OR) antagonist. Chemically, alvimopan is the single stereoisomer [[2(S)-[[4(R)-(3-hydroxyphenyl)-3(R),4-dimethyl-1-piperidinyl]methyl]-1-oxo-3-phenylpropyl]amino]acetic acid dihydrate. It has the following structural formula:
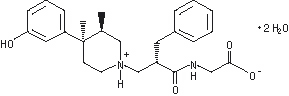
Alvimopan is a white to light beige powder with a molecular weight of 460.6, and the empirical formula is C25H32N2O4•2H2O. It has a solubility of <0.1 mg/mL in water or buffered solutions between pH 3.0 and 9.0, 1 to 5 mg/mL in buffered solutions at pH 1.2, and 10 to 25 mg/mL in aqueous 0.1 N sodium hydroxide. At physiological pH, alvimopan is zwitterionic, a property that contributes to its low solubility.
ENTEREG Capsules for oral administration contain 12 mg of alvimopan on an anhydrous basis suspended in the inactive ingredient polyethylene glycol.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Alvimopan is a selective antagonist of the cloned human µ-opioid receptor with a Ki of 0.4 nM (0.2 ng/mL) and no measurable opioid-agonist effects in standard pharmacologic assays. The dissociation of [3H]-alvimopan from the human µ-opioid receptor is slower than that of other opioid ligands, consistent with its higher affinity for the receptor. At concentrations of 1 to 10 µM, alvimopan demonstrated no activity at any of over 70 non-opioid receptors, enzymes, and ion channels.
Postoperative ileus is the impairment of gastrointestinal motility after intra-abdominal surgery or other non-abdominal surgeries. Postoperative ileus affects all segments of the gastrointestinal tract and may last from 5 to 6 days, or even longer. This may potentially delay gastrointestinal recovery and hospital discharge until its resolution. It is characterized by abdominal distention and bloating, nausea, vomiting, pain, accumulation of gas and fluids in the bowel, and delayed passage of flatus and defecation. Postoperative ileus is the result of a multifactorial process that includes inhibitory sympathetic input, release of hormones, neurotransmitters, and other mediators (e.g., endogenous opioids). A component of postoperative ileus also results from an inflammatory reaction and the effects of opioid analgesics. Morphine and other µ-opioid receptor agonists are universally used for the treatment of acute postsurgical pain; however, they are known to have an inhibitory effect on gastrointestinal motility and may prolong the duration of postoperative ileus.
Following oral administration, alvimopan antagonizes the peripheral effects of opioids on gastrointestinal motility and secretion by competitively binding to gastrointestinal tract µ-opioid receptors. The antagonism produced by alvimopan at opioid receptors is evident in isolated guinea pig ileum preparations where alvimopan competitively antagonizes the effects of morphine on contractility. Alvimopan achieves this selective gastrointestinal opioid antagonism without reversing the central analgesic effects of µ-opioid agonists.
12.2 Pharmacodynamics
In exploratory studies in healthy volunteers, alvimopan 3 mg three times daily appeared to reduce the delay in gastrointestinal transit produced by morphine 30 mg twice daily as measured by radio-opaque markers.
In a study designed to evaluate potential effects on cardiac conduction, alvimopan did not cause clinically significant QTc prolongation at doses up to 24 mg twice daily for 7 days. The potential for QTc effects at higher doses has not been studied.
12.3 Pharmacokinetics
Following oral administration of alvimopan, an amide hydrolysis compound is present in the systemic circulation, which is considered a product exclusively of intestinal flora metabolism. This compound is referred to as the 'metabolite'. It is also a µ-opioid receptor antagonist with a Ki of 0.8 nM (0.3 ng/mL).
Absorption: Following oral administration of ENTEREG Capsules in healthy volunteers, plasma alvimopan concentration peaked at approximately 2 hours postdose. No significant accumulation in alvimopan concentration was observed following twice daily (BID) dosing. The mean peak plasma concentration was 10.98 (±6.43) ng/mL and mean AUC0-12h was 40.2 (±22.5) ng•h/mL after dosing of alvimopan at 12 mg BID for 5 days. The absolute bioavailability was estimated to be 6% (range, 1% to 19%). Plasma concentrations of alvimopan increased approximately proportionally with increasing doses between 6 and 18 mg, but less than proportionally from 18 to 24 mg.
There was a delay in the appearance of the 'metabolite', which had a median Tmax of 36 hours following administration of a single dose of alvimopan. Concentrations of the 'metabolite' were highly variable between subjects and within a subject. The 'metabolite' accumulated after multiple doses of ENTEREG. The mean Cmax for the 'metabolite' after alvimopan 12 mg twice daily for 5 days was 35.73±35.29 ng/mL.
Concentrations of alvimopan and its metabolite are higher (~1.9-fold and ~1.4-fold, respectively) in POI patients than in healthy volunteers.
Food Effects: A high-fat meal decreased the extent and rate of alvimopan absorption. The Cmax and AUC were decreased by approximately 38% and 21%, respectively, and the Tmax was prolonged by approximately 1 hour. The clinical significance of this decreased bioavailability is unknown. In POI clinical trials, the preoperative dose of ENTEREG was administered in a fasting state. Subsequent doses were given without regard to meals.
Distribution: The steady state volume of distribution of alvimopan was estimated to be 30±10 L. Plasma protein binding of alvimopan and its 'metabolite' was independent of concentration over ranges observed clinically and averaged 80% and 94%, respectively. Both alvimopan and the 'metabolite' were bound to albumin and not to alpha-1 acid glycoprotein.
Metabolism and Elimination: The average plasma clearance for alvimopan was 402 (±89) mL/min. Renal excretion accounted for approximately 35% of total clearance. There was no evidence that hepatic metabolism was a significant route for alvimopan elimination. Biliary secretion was considered the primary pathway for alvimopan elimination. Unabsorbed drug and unchanged alvimopan resulting from biliary excretion were then hydrolyzed to its 'metabolite' by gut microflora. The 'metabolite' was eliminated in the feces and in the urine as unchanged 'metabolite', the glucuronide conjugate of the 'metabolite', and other minor metabolites. The mean terminal phase half-life of alvimopan after multiple oral doses of ENTEREG ranged from 10 to 17 hours. The terminal half-life of the 'metabolite' ranged 10 to 18 hours.
Special Populations:
Age: The pharmacokinetics of alvimopan, but not its 'metabolite', were related to age, but this effect was not clinically significant and does not warrant dosage adjustment based on increased age.
Race: The pharmacokinetic characteristics of alvimopan were not affected by Hispanic or black race. Plasma 'metabolite' concentrations were lower in black and in Hispanic patients (by 43% and 82%, respectively) than in Caucasian patients following alvimopan administration. These changes are not considered to be clinically significant in surgical patients. Japanese male healthy volunteers had an approximately 2-fold increase in plasma alvimopan concentrations, but no change in metabolite pharmacokinetics. The pharmacokinetics of alvimopan have not been studied in subjects of other East Asian ancestry. Dosage adjustment in Japanese patients is not required [see Use in Specific Populations (8.8)].
Hepatic Impairment: Exposure to alvimopan following a single 12-mg dose tended to be higher (1.5 to 2 fold, on average) in patients with mild or moderate hepatic impairment (as defined by Child-Pugh Class A and B, n = 8 each) compared with healthy controls (n = 4). There were no consistent effects on the Cmax or half-life of alvimopan in patients with hepatic impairment. However, two of 16 patients with mild to moderate impairment had longer than expected half-lives of alvimopan indicating that some accumulation may occur upon multiple dosing. The Cmax of the 'metabolite' tended to be more variable in patients with mild or moderate hepatic impairment than in matched normal subjects. A study of 3 patients with severe hepatic impairment (Child-Pugh Class C), indicated similar alvimopan exposure in 2 patients and an approximately 10-fold increase in Cmax and exposure in 1 patient with severe hepatic impairment when compared with healthy control volunteers [see Warnings and Precautions (5.4) and Use in Specific Populations (8.6)].
Renal Impairment: There was no relationship between renal function (i.e., creatinine clearance [CrCl]) and plasma alvimopan pharmacokinetics (Cmax, AUC, or half-life) in patients with mild (CrCl 51-80 mL/min), moderate (CrCl 31-50 mL/min), or severe (CrCl <30 mL/min) renal impairment (n = 6 each). Renal clearance of alvimopan was related to renal function; however, because renal clearance was only a small fraction (35%) of the total clearance, renal impairment had a small effect on the apparent oral clearance of alvimopan. The half-lives of alvimopan were comparable in the mild, moderate and control renal impairment groups but longer in the severe renal impairment group. Exposure to the 'metabolite' tended to be 2- to 5-fold higher in patients with moderate or severe renal impairment compared to patients with mild renal impairment or control subjects. Thus, there may be accumulation of alvimopan and 'metabolite' in patients with severe renal impairment receiving multiple doses of ENTEREG. Patients with end-stage renal disease were not studied [see Warnings and Precautions (5.5) and Use in Specific Populations (8.7)].
Crohn's Disease: There was no relationship between disease activity in patients with Crohn's disease (measured as Crohn's Disease Activity Index or bowel movement frequency) and alvimopan pharmacokinetics (AUC or Cmax). Patients with active or quiescent Crohn's disease had increased variability in alvimopan pharmacokinetics and exposure tended to be 2-fold higher in patients with quiescent disease than in those with active disease or normal subjects. Concentrations of the 'metabolite' were lower in patients with Crohn's disease.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two year carcinogenicity studies have been conducted with alvimopan in CD-1 mice at oral doses up to 4000 mg/kg/day and in Sprague Dawley rats at oral doses up to 500 mg/kg/day. Oral administration of alvimopan for 104 weeks produced significant increases in the incidences of fibroma, fibrosarcoma and sarcoma in the skin/subcutis, and osteoma/osteosarcoma in bones of female mice at 4000 mg/kg/day (about 674 times the recommended human dose based on body surface area). In rats, oral administration of alvimopan for 104 weeks did not produce any tumor up to 500 mg/kg/day (about 166 times the recommended human dose based on body surface area).
Alvimopan was not genotoxic in the Ames test, the mouse lymphoma cell (L5178Y/TK+/-) forward mutation test, the Chinese Hamster Ovary (CHO) cell chromosome aberration test or the mouse micronucleus test. The pharmacologically active 'metabolite' ADL 08-0011 was negative in the Ames test, chromosome aberration test in CHO cells and mouse micronucleus test.
Alvimopan at intravenous doses up to 10 mg/kg/day (about 3.4 to 6.8 times the recommended human oral dose based on the body surface area) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
13.2 Animal Toxicology and/or Pharmacology
A single oral dose of 500 mg/kg of alvimopan was not lethal to mice and rats.
Reproduction studies have been performed in pregnant rats at oral doses up to 200 mg/kg/day (about 68 to 136 times the recommended human oral dose based on the body surface area) and intravenous doses up to 10 mg/kg/day (about 3.4 to 6.8 times the recommended human oral dose based on the body surface area) and in pregnant rabbits at intravenous doses up to 15 mg/kg/day (about 5 to 10 times the recommended human oral dose based on the body surface area) and have revealed no evidence of impaired fertility or harm to the fetus due to alvimopan.
14 CLINICAL STUDIES
14.1 Postoperative Ileus
The efficacy of ENTEREG in the management of postoperative ileus was evaluated in 5 multicenter, randomized, double-blind, parallel-group, placebo-controlled studies: 4 US studies (Studies 1-4) and 1 non-US study (Study 5). Patients 18 years of age or older undergoing partial large or small bowel resection surgery with primary anastomosis or total abdominal hysterectomy under general anesthesia were randomly assigned to receive oral doses of ENTEREG 12 mg or matching placebo. The initial dose was administered at least 30 minutes and up to 5 hours prior to the scheduled start of surgery for most patients, and subsequent doses were administered twice daily beginning on the first postoperative day and continued until hospital discharge or a maximum of 7 days. There were no limitations on the type of general anesthesia used, but intrathecal or epidural opioids or anesthetics were prohibited.
All patients in the US studies were scheduled to receive intravenous patient-controlled opioid analgesia. In the non-US study, patients were scheduled to receive opioids either by intravenous patient-controlled opioid analgesia or bolus parenteral administration (intravenous or intramuscular). In all studies, there was no restriction on the type of opioid used or the duration of intravenous patient-controlled opioid analgesia. A standardized accelerated postoperative care pathway was implemented: early nasogastric tube removal (end of surgery); early ambulation (day following surgery); early diet advancement (liquids offered the day following surgery) and solids by the second day following surgery, as tolerated.
Patients who received more than 3 doses of an opioid (regardless of route) during the 7 days prior to surgery and patients with complete bowel obstruction or who were scheduled for a total colectomy, colostomy, or ileostomy were excluded.
The primary endpoint for all studies was time to achieve resolution of postoperative ileus, a clinically defined composite measure of both upper and lower gastrointestinal recovery. Although both 2-component (GI2: toleration of solid food and first bowel movement) and 3-component (GI3: toleration of solid food and either first flatus or bowel movement) endpoints were used in all studies, GI2 is presented as it represents the most objective and clinically relevant measure of treatment response in the bowel resection population. The time from the end of surgery to when the discharge order was written represented the length of hospital stay. In the 5 studies, 1,081 patients received placebo (157 for total abdominal hysterectomy) and 1,096 patients received ENTEREG (143 for total abdominal hysterectomy).
The efficacy of ENTEREG following total abdominal hysterectomy has not been established. Therefore, the following data are presented for the bowel resection population only.
Bowel Resection: A total of 1,877 patients underwent bowel resection. The average age was 61 years with equal proportions of males and females, and 88% were Caucasian. The most common indications for surgery were colon or rectal cancer and diverticular disease. In the non-US study (Study 5), average daily postoperative opioid consumption was approximately 50% lower and the use of non-opioid analgesics substantially higher, as compared with the US studies (Studies 1-4) for both treatment groups. During the first 48 hours postoperatively, the use of non-opioid analgesics was 69% compared with 4% for the non-US and US studies, respectively. In each of the 5 studies, ENTEREG accelerated the time to recovery of gastrointestinal function, as measured by the composite endpoint GI2, and time to discharge order written as compared with placebo. Hazard ratios greater than 1 indicate a higher probability of achieving the event during the study period with treatment with ENTEREG than with placebo. Table 2 provides the Hazard Ratios, Kaplan Meier means and the mean treatment differences (hours) in gastrointestinal recovery between ENTEREG and placebo.
| Study No. | ENTEREG 12 mg Mean | Placebo Mean | Treatment Difference Mean | Hazard Ratio (95% CI) |
| 1 | 92.0 | 111.8 | 19.8 | 1.533 (1.293, 1.816) |
| 2 | 105.9 | 132.0 | 26.1 | 1.625 (1.256, 2.102) |
| 3 | 116.4 | 130.3 | 14.0 | 1.365 (1.057, 1.764) |
| 4 | 106.7 | 119.9 | 13.2 | 1.400 (1.035, 1.894) |
| 5 | 98.8 | 109.5 | 10.7 | 1.299 (1.070, 1.575) |
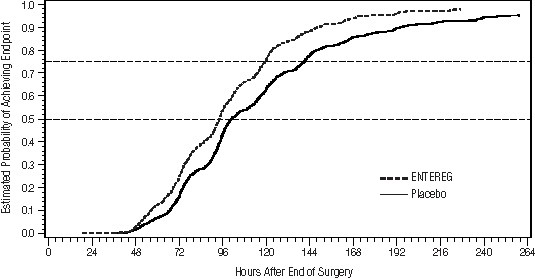
Gastrointestinal recovery began after approximately 48 hours post surgery. The proportion of patients receiving ENTEREG who achieved GI2 was higher at all times throughout the study observation period compared with those receiving placebo (Figure 1).
Figure 1. Time to GI2 Based on the Combined Data from Five Studies
Across studies 1-4, patients receiving ENTEREG had their discharge order written approximately 13 to 21 hours sooner compared to patients receiving placebo.
ENTEREG did not reverse opioid analgesia as measured by visual analog scale pain intensity scores and/or amount of postoperative opioids administered across all 5 studies.
There were no gender-, age-, or race-related differences in treatment effect.
The incidence of anastomotic leak was low and comparable in patients receiving either ENTEREG or placebo (0.8% and 1.1%, respectively).
16 HOW SUPPLIED/STORAGE AND HANDLING
ENTEREG Capsules, 12 mg, are blue, hard-gelatin capsules printed with "ADL2698" on both the body and the cap of the capsule. ENTEREG Capsules are available in unit-dose packs of 30 capsules (30 doses) (NDC 11227-010-30) for hospital use only.
17 PATIENT COUNSELING INFORMATION
17.1 Recent Use of Opioids
Patients should be informed that they must disclose long-term or intermittent opioid pain therapy, including any use of opioids in the week prior to receiving ENTEREG. They should understand that recent use of opioids may make them more susceptible to adverse reactions to ENTEREG, primarily those limited to the gastrointestinal tract (e.g., abdominal pain, nausea and vomiting, diarrhea).
17.2 Hospital Use Only
Patients should be informed that ENTEREG is for hospital use only for no more than 7 days after their bowel resection surgery.
17.3 Most Common Side Effects
Patients should be informed that the most common side effects with ENTEREG in patients undergoing bowel resection are constipation, dyspepsia, and flatulence.
Manufactured for Adolor Corporation
Exton, PA 19341-1127
US Patent Nos. 5,250,542; 5,434,171; 6,469,030
©2009, Adolor Corporation. All rights reserved.

| ENTEREG
alvimopan capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Adolor Corporation (884723966) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Piramal Healthcare (Canada) Ltd. | 244196473 | API MANUFACTURE, ANALYSIS | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharmaceutics International, Inc. | 878265586 | MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sharp Corporation | 002346625 | REPACK | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Central Glass Germany | 340011833 | API MANUFACTURE | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharmaceutical Manufacturing Research Services, Inc | 836225649 | MANUFACTURE | |