BUTISOL SODIUM- butabarbital sodium tablet
Meda Pharmaceuticals
----------
BUTISOL SODIUM® CIII
(BUTABARBITAL SODIUM TABLETS, USP
AND BUTABARBITAL SODIUM ORAL SOLUTION, USP)
TABLETS & ORAL SOLUTION
IN-0110-13 REV. 5/2015
Description
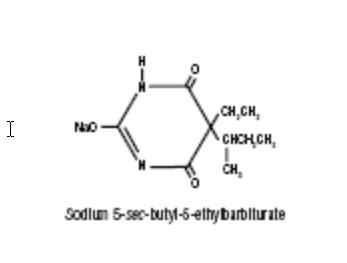
BUTISOL SODIUM® (butabarbital sodium tablets, USP and butabarbital sodium oral solution, USP) is a non-selective central nervous system depressant which is used as a sedative or hypnotic. It is available for oral administration as Tablets containing 30 mg or 50 mg butabarbital sodium; and as Oral Solution containing 30 mg/5 mL, with alcohol (by volume) 7%. Other ingredients in the Tablets are: calcium stearate, corn starch, dibasic calcium phosphate, FD&C Blue No. 1 (30 mg only), FD&C Yellow No. 5 (30 mg and 50 mg — see Precautions), FD&C Yellow No. 6 (50 mg only). Other ingredients in the Oral Solution are: D&C Green No. 5, edetate disodium, FD&C Yellow No. 5 (see Precautions), flavors (natural and artificial), propylene glycol, purified water, saccharin sodium, sodium benzoate. Butabarbital sodium occurs as a white, bitter powder which is freely soluble in water and alcohol, but practically insoluble in benzene and ether. The structural formula for butabarbital sodium is:
Clinical Pharmacology
BUTISOL SODIUM® (butabarbital sodium tablets, USP and butabarbital sodium oral solution, USP), like other barbiturates, is capable of producing all levels of CNS mood alteration from excitation to mild sedation, to hypnosis, and deep coma. Overdosage can produce death. Barbiturates depress the sensory cortex, decrease motor activity, alter cerebellar function, and produce drowsiness, sedation, and hypnosis.
Barbiturate-induced sleep differs from physiological sleep. Sleep laboratory studies have demonstrated that barbiturates reduce the amount of time spent in the rapid eye movement (REM) phase of sleep or dreaming stage. Also, Stages III and IV sleep are decreased. Following abrupt cessation of barbiturates used regularly, patients may experience markedly increased dreaming, nightmares, and/or insomnia. Therefore, withdrawal of a single therapeutic dose over 5 or 6 days has been recommended to lessen the REM rebound and disturbed sleep which contribute to drug withdrawal syndrome (for example, decrease the dose from 3 to 2 doses a day for 1 week).
In studies, secobarbital sodium and pentobarbital sodium have been found to lose most of their effectiveness for both inducing and maintaining sleep by the end of 2 weeks of continued drug administration even with the use of multiple doses. As with secobarbital sodium and pentobarbital sodium, other barbiturates might be expected to lose their effectiveness for inducing and maintaining sleep after about 2 weeks. The short-, intermediate-, and, to a lesser degree, long-acting barbiturates have been widely prescribed for treating insomnia. Although the clinical literature abounds with claims that the short-acting barbiturates are superior for producing sleep while the intermediate-acting compounds are more effective in maintaining sleep, controlled studies have failed to demonstrate these differential effects. Therefore, as sleep medications, the barbiturates are of limited value beyond short-term use.
Barbiturates are respiratory depressants. The degree of respiratory depression is dependent upon dose. With hypnotic doses, respiratory depression produced by barbiturates is similar to that which occurs during physiologic sleep with slight decrease in blood pressure and heart rate.
Barbiturates do not impair normal hepatic function, but have been shown to induce liver microsomal enzymes, thus increasing and/or altering the metabolism of barbiturates and other drugs (see Precautions-Drug interactions).
Pharmacokinetics: BUTISOL SODIUM® (butabarbital sodium tablets, USP and butabarbital sodium oral solution, USP) is the sodium salt of a weak acid. Barbiturates are weak acids that are absorbed and rapidly distributed to all tissues and fluids with high concentrations in the brain, liver, and kidneys. Barbiturates are bound to plasma and tissue proteins. The rate of absorption is increased if it is ingested as a dilute solution or taken on an empty stomach.
Barbiturates are metabolized primarily by the hepatic microsomal enzyme system, and most metabolic products are excreted in the urine. The excretion of unchanged butabarbital in the urine is negligible. BUTISOL SODIUM® (butabarbital sodium tablets, USP and butabarbital sodium oral solution, USP) is classified as an intermediate-acting barbiturate. The average plasma half-life for butabarbital is 100 hours in the adult.
Although variable from patient to patient, butabarbital has an onset of action of about 3/4 to 1 hour, and a duration of action of about 6 to 8 hours.
Indications and Usage
BUTISOL SODIUM® (butabarbital sodium tablets, USP and butabarbital sodium oral solution, USP) is indicated for use as a sedative or hypnotic.
Since barbiturates appear to lose their effectiveness for sleep induction and sleep maintenance after 2 weeks, use of BUTISOL SODIUM® in treating insomnia should be limited to this time (see Clinical Pharmacology above).
Contraindications
Barbiturates are contraindicated in patients with known barbiturate sensitivity. Barbiturates are also contraindicated in patients with a history of manifest or latent porphyria.
Warnings
Because sleep disturbances may be the presenting manifestation of a physical and/or psychiatric disorder, symptomatic treatment of insomnia should be initiated only after a careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated.
Worsening of insomnia or the emergence of new thinking or behavior abnormalities may be the consequences of an unrecognized psychiatric or physical disorder. Such findings have emerged during the course of treatment with sedative-hypnotic drugs. Because some of the important adverse effects of sedative-hypnotics appear to be dose related (see PRECAUTIONS and DOSAGE AND ADMINISTRATION), it is important to use the smallest possible effective dose, especially in the elderly.
Complex behaviors such as “sleep driving” (i.e., driving while not fully awake after ingestion of a sedative-hypnotic, with amnesia for the event) have been reported. These events can occur in sedative-hypnotic-naive as well as in sedative-hypnotic-experienced persons. Although behaviors such as sleep-driving may occur with sedative-hypnotics alone at therapeutic doses, the use of alcohol and other CNS depressants with sedative-hypnotics appears to increase the risk of such behaviors, as does the use of sedative-hypnotics at doses exceeding the maximum recommended dose. Due to the risk to the patient and the community, discontinuation of sedative-hypnotics should be strongly considered for patients who report a “sleep driving” episode”.
Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a sedative-hypnotic. As with sleep-driving, patients usually do not remember these events.
Because BUTISOL SODIUM® (butabarbital sodium tablets, USP and butabarbital sodium oral solution, USP) can cause drowsiness and a decreased level of consciousness, patients, particularly the elderly, are at a higher risk of falls.
Severe anaphylactic and anaphylactoid reactions: Rare cases of angioedema involving the tongue, glottis or larynx have been reported in patients after taking the first or subsequent doses of sedative-hypnotics. Some patients have had additional symptoms such as dyspnea, throat closing, or nausea and vomiting that suggest anaphylaxis. Some patients have required medical therapy in the emergency department. If angioedema involves the tongue, glottis or larynx, airway obstruction may occur and be fatal. Patients who develop angioedema after treatment with sedative-hypnotics should not be rechallenged with the drug.
Habit forming: Barbiturates may be habit forming. Tolerance, psychological and physical dependence may occur with continued use (see Drug Abuse and Dependence below). Patients who have psychological dependence on barbiturates may increase the dosage or decrease the dosage interval without consulting a physician and may subsequently develop a physical dependence on barbiturates. To minimize the possibility of overdosage or the development of dependence, the prescribing and dispensing of sedative-hypnotic barbiturates should be limited to the amount required for the interval until the next appointment. Abrupt cessation after prolonged use in the dependent person may result in withdrawal symptoms, including delirium, convulsions, and possibly death. Barbiturates should be withdrawn gradually from any patient known to be taking excessive dosage over long periods of time. (See Drug Abuse and Dependence below.)
Acute or chronic pain: Caution should be exercised when barbiturates are administered to patients with acute or chronic pain, because paradoxical excitement could be induced, or important symptoms could be masked. However, the use of barbiturates as sedatives in the postoperative surgical period, and as adjuncts to cancer chemotherapy, is well established.
Use in pregnancy: Barbiturates can cause fetal damage when administered to a pregnant woman. Retrospective, case-controlled studies have suggested a connection between the maternal consumption of barbiturates and a higher than expected incidence of fetal abnormalities. Following oral administration, barbiturates readily cross the placental barrier and are distributed throughout fetal tissues with highest concentrations found in the placenta, fetal liver, and brain.
Withdrawal symptoms occur in infants born to mothers who receive barbiturates throughout the last trimester of pregnancy (See Drug Abuse and Dependence). If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
Precautions
General: Barbiturates should be administered with caution, if at all, to patients who are mentally depressed, have suicidal tendencies, or a history of drug abuse.
Elderly or debilitated patients may react to barbiturates with marked excitement, depression, and confusion. In some persons, barbiturates repeatedly produce excitement rather than depression.
In patients with hepatic damage, barbiturates should be administered with caution and initially in reduced doses. Barbiturates should not be administered to patients showing the premonitory signs of hepatic coma.
BUTISOL SODIUM® (butabarbital sodium tablets, USP and butabarbital sodium oral solution, USP) Tablets and Oral Solution contain FD&C Yellow No. 5 (tartrazine) which may cause allergic-type reactions (including bronchial asthma) in certain susceptible individuals. Although the overall incidence of FD&C Yellow No. 5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin hypersensitivity.
Information for patients: Practitioners should give the following information and instructions to patients receiving barbiturates.
“Sleep Driving” and other complex behaviors: There have been reports of people getting out of bed after taking a sedative-hypnotic and driving their cars while not fully awake, often with no memory of the event. If a patient experiences such an episode, it should be reported to his or her doctor immediately, since “sleep driving” can be dangerous. This behavior is more likely to occur when sedative-hypnotics are taken with alcohol or other central nervous depressants (see WARNINGS). Other complex behaviors (e.g. preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a sedative-hypnotic. As with sleep-driving, patients usually do not remember these events.
The use of barbiturates carries with it an associated risk of psychological and/or physical dependence. The patient should be warned against increasing the dose of the drug without consulting a physician.
Barbiturates may impair mental and/or physical abilities required for the performance of potentially hazardous tasks, such as driving or operating machinery.
Advise patients that increased drowsiness and decreased consciousness may increase the risk of falls in some patients.
Alcohol should not be consumed while taking barbiturates. Concurrent use of the barbiturates with other CNS depressants, including other sedatives or hypnotics, alcohol, narcotics, tranquilizers, and antihistamines, may result in additional CNS depressant effects.
Laboratory tests: Prolonged therapy with barbiturates should be accompanied by periodic laboratory evaluation of organ systems, including hematopoietic, renal, and hepatic systems (See Precautions-General and Adverse Reactions).
Drug interactions: Most reports of clinically significant drug interactions occurring with the barbiturates have involved phenobarbital. However, the application of these data to other barbiturates appears valid and warrants serial blood level determinations of the relevant drugs when there are multiple therapies.
- 1.
- Anticoagulants. Phenobarbital lowers the plasma levels of dicumarol and causes a decrease in anticoagulant activity as measured by the prothrombin time. Barbiturates can induce hepatic microsomal enzymes resulting in increased metabolism and decreased anticoagulant response of oral anticoagulants (e.g., warfarin, acenocoumarol, dicumarol, and phenprocoumon). Patients stabilized on anticoagulant therapy may require dosage adjustments if barbiturates are added to or withdrawn from their dosage regimen.
- 2.
- Corticosteroids. Barbiturates appear to enhance the metabolism of exogenous corticosteroids probably through the induction of hepatic microsomal enzymes. Patients stabilized on corticosteroid therapy may require dosage adjustments if barbiturates are added to or withdrawn from their dosage regimen.
- 3.
- Griseofulvin. Phenobarbital appears to interfere with the absorption of orally administered griseofulvin, thus decreasing its blood level. The effect of the resultant decreased blood levels of griseofulvin on therapeutic response has not been established. However, it would be preferable to avoid concomitant administration of these drugs.
- 4.
- Doxycycline. Phenobarbital has been shown to shorten the half-life of doxycycline for as long as 2 weeks after barbiturate therapy is discontinued. This mechanism is probably through the induction of hepatic microsomal enzymes that metabolize the antibiotic. If phenobarbital and doxycycline are administered concurrently, the clinical response to doxycycline should be monitored closely.
- 5.
- Phenytoin, sodium valproate, valproic acid. The effect of barbiturates on the metabolism of phenytoin appears to be variable. Some investigators report an accelerating effect, while others report no effect. Because the effect of barbiturates on the metabolism of phenytoin is not predictable, phenytoin and barbiturate blood levels should be monitored more frequently if these drugs are given concurrently. Sodium valproate and valproic acid appear to decrease barbiturate metabolism; therefore, barbiturate blood levels should be monitored and appropriate dosage adjustments made as indicated.
- 6.
- Central nervous system. The concomitant use of other central nervous system depressants, including other sedatives or hypnotics, antihistamines, tranquilizers, or alcohol, may produce additive depressant effects.
- 7.
- Monoamine oxidase inhibitors (MAOI). MAOI prolong the effects of barbiturates probably because metabolism of the barbiturate is inhibited.
- 8.
- Estradiol, estrone, progesterone, and other steroid hormones. Pretreatment with or concurrent administration of phenobarbital may decrease the effect of estradiol by increasing its metabolism. There have been reports of patients treated with antiepileptic drugs (e.g., phenobarbital) who become pregnant while taking oral contraceptives. An alternate contraceptive method might be suggested to women taking phenobarbital.
Carcinogenesis, mutagenesis, impairment of fertility: No long-term studies in animals have been performed with butabarbital sodium to determine carcinogenic and mutagenic potential, or effects on fertility.
Nonteratogenic effects - Infants suffering from long-term barbiturate exposure in utero may have an acute withdrawal syndrome of seizures and hyperirritability from birth to a delayed onset of up to 14 days (see Drug Abuse and Dependence).
Labor and delivery: Hypnotic doses of barbiturates do not appear to significantly impair uterine activity during labor. Administration of sedative-hypnotic barbiturates to the mother during labor may result in respiratory depression in the newborn. Premature infants are particularly susceptible to the depressant effects of barbiturates. If barbiturates are used during labor and delivery, resuscitation equipment should be available.
Nursing mothers: Caution should be exercised when a barbiturate is administered to a nursing woman since small amounts of some barbiturates are excreted in the milk.
Geriatric use: Clinical studies of Butisol Sodium Tablets/Oral Solution did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Adverse Reactions
To report SUSPECTED ADVERSE REACTIONS, contact Meda Pharmaceuticals Inc. at 1-866-210-5952 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
The following adverse reactions have been observed with the use of barbiturates in hospitalized patients. Because such patients may be less aware of certain of the milder adverse effects of barbiturates, the incidence of these reactions may be somewhat higher in fully ambulatory patients.
More than 1 in 100 patients. The most common adverse reaction, somnolence, is estimated to occur at a rate of 1 to 3 patients per 100.
Less than 1 in 100 patients. The most common adverse reactions estimated to occur at a rate of less than 1 in 100 patients listed below, grouped by organ system, and by decreasing order of occurrence are:
Central nervous system/psychiatric: Agitation, confusion, hyperkinesia, ataxia, CNS depression, nightmares, nervousness, psychiatric disturbance, hallucinations, insomnia, anxiety, dizziness, thinking abnormality.
Respiratory: Hypoventilation, apnea.
Cardiovascular: Bradycardia, hypotension, syncope.
Gastrointestinal: Nausea, vomiting, constipation.
Other reported reactions: Headache, hypersensitivity (angioedema, skin rashes, exfoliative dermatitis), fever, liver damage.
Drug Abuse and Dependence
Abuse and dependence: Abuse and addiction are separate and distinct from physical dependence and tolerance. Abuse is characterized by misuse of the drug for non-medical purposes, often in combination with other psychoactive substances. Physical dependence is a state of adaptation that is manifested by a specific withdrawal syndrome that can be produced by abrupt cessation, rapid dose reduction, decreasing blood level of the drug and/or administration of an antagonist. Tolerance is a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug’s effects over time. Tolerance may occur to both the desired and undesired effects of drugs and may develop at different rates for different effects.
Addiction is a primary, chronic, neurobiological disease with genetic, psychosocial, and environmental factors influencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. Drug addiction is a treatable disease, utilizing a multidisciplinary approach, but relapse is common.
Barbiturates may be habit-forming. Tolerance, psychological dependence, and physical dependence may occur especially following prolonged use of high doses of barbiturates. Daily administration in excess of 400 milligrams (mg) of pentobarbital or secobarbital for approximately 90 days is likely to produce some degree of physical dependence. A dosage of from 600 to 800 mg taken for at least 35 days is sufficient to produce withdrawal seizures. The average daily dose for the barbiturate addict is usually about 1.5 grams. As tolerance to barbiturates developes, the amount needed to maintain the same level of intoxication increases; tolerance to a fatal dosage, however, does not increase more than two-fold. As this occurs, the margin between an intoxicating dosage and a fatal dosage becomes smaller.
Symptoms of acute intoxication with barbiturates include unsteady gait, slurred speech, and sustained nystagmus. Mental signs of chronic intoxication include confusion, poor judgment, irritability, insomnia, and somatic complaints. Symptoms of barbiturate dependence are similar to those of chronic alcoholism.
If an individual appears to be intoxicated with alcohol to a degree that is radically disproportionate to the amount of alcohol in his or her blood, the use of barbiturates should be suspected. The lethal dose of a barbiturate is far less if alcohol is also ingested.
The symptoms of barbiturate withdrawal can be severe and may cause death. Minor withdrawal symptoms may appear 8 to 12 hours after the last dose of a barbiturate. These symptoms usually appear in the following order: anxiety, muscle twitching, tremor of hands and fingers, progressive weakness, dizziness, distortion in visual perception, nausea, vomiting, insomnia, and orthostatic hypotension. Major withdrawal symptoms (convulsions and delirium) may occur within 16 hours and last up to 5 days after abrupt cessation of these drugs. Intensity of withdrawal symptoms gradually declines over a period of approximately 15 days.
Drug dependence to barbiturates arises from repeated administration of a barbiturate or agent with barbiturate-like effect on a continuous basis, generally in amounts exceeding therapeutic dose levels. The characteristics of drug dependence to barbiturates include: (a) a strong desire or need to continue taking the drug; (b) a tendency to increase the dose; (c) a psychic dependence on the effects of the drug related to subjective and individual appreciation for those effects; and (d) a physical dependence on the effects of the drug requiring its presence for maintenance of homeostasis and resulting in a definite, characteristic, and self-limited abstinence syndrome when the drug is withdrawn.
Treatment of barbiturate dependence consists of cautious and gradual withdrawal of the drug. Barbiturate-dependent patients can be withdrawn by using a number of different withdrawal regimens. In all cases, withdrawal takes an extended period of time. One method involves initiating treatment at the patient's regular dosage level, in 3 to 4 divided doses, and decreasing the daily dose by 10 percent if tolerated by the patient.
Infants physically dependent on barbiturates may be given phenobarbital 3 to 10 mg/kg/day. After withdrawal symptoms (hyperactivity, disturbed sleep, tremors, hyperreflexia) are relieved, the dosage of phenobarbital should be gradually decreased and completely withdrawn over a 2- week period.
Overdosage
Signs and symptoms: The toxic dose of barbiturates varies considerably. In general, an oral dose of 1 gram of most barbiturates produces serious poisoning in an adult. Death commonly occurs after 2 to 10 grams of ingested barbiturates. Symptoms of acute intoxication with barbiturates include unsteady gait, slurred speech, and sustained nystagmus. Mental signs of chronic intoxication include confusion, poor judgment, irritability, insomnia, and somatic complaints. Barbiturate intoxication may be confused with alcoholism, bromide intoxication, and with various neurological disorders.
Acute overdosage with barbiturates is manifested by CNS and respiratory depression which may progress to Cheyne-Stokes respiration, areflexia, constriction of the pupils to a slight degree (though in severe poisoning they may show paralytic dilation), oliguria, tachycardia, hypotension, lowered body temperature, and coma. Typical shock syndrome (apnea, circulatory collapse, respiratory arrest, and death) may occur.
In extreme overdose, all electrical activity in the brain may cease, in which case a “flat” EEG normally equated with clinical death cannot be accepted. This effect is fully reversible unless hypoxic damage occurs. Consideration should be given to the possibility of barbiturate intoxication even in situations that appear to involve trauma.
Complications: Pneumonia, pulmonary edema, cardiac arrhythmias, congestive heart failure, and renal failure may occur. Uremia may increase CNS sensitivity to barbiturates if renal function is impaired. Differential diagnosis should include hypoglycemia, head trauma, cerebrovascular accidents, convulsive states, and diabetic coma.
Treatment: Treatment of overdosage is mainly supportive and consists of the following:
- 1.
- Maintenance of an adequate airway, with assisted respiration and oxygen administration as necessary.
- 2.
- Monitoring of vital signs and fluid balance.
- 3.
- If the patient is conscious and has not lost the gag reflex, emesis may be induced with ipecac. Care should be taken to prevent pulmonary aspiration of vomitus. After completion of vomiting, 30 grams activated charcoal in a glass of water may be administered.
- 4.
- If emesis is contraindicated, gastric lavage may be performed with a cuffed endotracheal tube in place with the patient in the face down position. Activated charcoal may be left in the emptied stomach and a saline cathartic administered.
- 5.
- Fluid therapy and other standard treatment for shock, if needed.
- 6.
- If renal function is normal, forced diuresis may aid in the elimination of the barbiturate.
- 7.
- Although not recommended as a routine procedure, hemodialysis may be used in severe barbiturate intoxications or if the patient is anuric or in shock.
- 8.
- Appropriate nursing care, including rolling patients from side-to-side every 30 minutes, to prevent hypostatic pneumonia, decubiti, aspiration, and other complications of patients with altered states of consciousness.
- 9.
- Antibiotics should be given if pneumonia is suspected.
Dosage and Administration
Usual adult dosage:
Daytime sedative - 15 to 30 mg, 3 or 4 times daily.
Bedtime hypnotic - 50 to 100 mg.
Preoperative sedative - 50 to 100 mg, 60 to 90 minutes before surgery.
Usual pediatric dosage:
Preoperative sedative - 2 to 6 mg/kg maximum 100 mg.
Special patient population:
Dosage should be reduced in the elderly or debilitated because these patients may be more sensitive to barbiturates. Dosage should be reduced for patients with impaired renal function or hepatic disease (see Precautions).
How Supplied
BUTISOL SODIUM® (butabarbital sodium tablets, USP):
30 mg - colored green, scored, imprinted “BUTISOL SODIUM” and 37/113 in bottles of 100 (NDC 0037-0113-60).
Contains FD&C Yellow No. 5 (See Precautions).
Storage: Store at controlled room temperature 20°-25°C (68°-77°F).
Dispense in a tight container.
To report SUSPECTED ADVERSE REACTIONS, contact Meda Pharmaceuticals Inc. at 1-866-210-5952 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
BUTISOL SODIUM is a registered trademark of Meda Pharmaceuticals Inc., a Mylan company.
Distributed by:
Meda Pharmaceuticals®
Meda Pharmaceuticals Inc.
Somerset, NJ 08873-4120
Printed in U.S.A.
Rev. XX/2018
Package Label - Principal Display Panel - 100-count Bottle, 30 mg Tablets
NDC 0037-0113-60
100 Tablets
CIII
Butisol Sodium ®
(butabarbital sodium
tablets, USP)
30 mg
Rx Only
MEDA PHARMACEUTICALS
LB-011360-07 Rev. 8/07
Usual Dosage:Adults: One tablet
three or four times daily.
For complete information for use,
see accompanying product literature.
Contains FD&C Yellow No. 5
(tartrazine) as a color additive.
Store at controlled room temperature
20°-25°C (68°-77°F).
Dispense in a tight container.
MEDA PHARMACEUTICALS
Meda Pharmaceuticals Inc.
Somerset, New Jersey 08873-4120

| BUTISOL SODIUM
butabarbital sodium tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Meda Pharmaceuticals (051229602) |