FLUOROURACIL - fluorouracil injection, solution
Ingenus Pharmaceuticals, LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use FLUOROURACIL INJECTION safely and effectively. See full prescribing information for FLUOROURACIL INJECTION
FLUOROURACIL injection, for intravenous use Initial U.S. Approval:1962 RECENT MAJOR CHANGESDosage and Administration (2) 07/2016 INDICATIONS AND USAGEDOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONSNone (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSTo report SUSPECTED ADVERSE REACTIONS, contact Ingenus Pharmaceuticals, LLC. at 1-877-748-1970 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch (6) See 17 for PATIENT COUNSELING INFORMATION. Revised: 4/2019 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
Fluorouracil is a nucleoside metabolic inhibitor indicated for the treatment of patients with
2 DOSAGE AND ADMINISTRATION
2.1 General Dosage Information
Fluorouracil is recommended for administration either as an intravenous bolus or as an intravenous infusion. Do not inject the entire contents of the vial directly into patients. Individualize the dose and dosing schedule of fluorouracil based on tumor type, the specific regimen administered, disease state, response to treatment, and patient risk factors.
2.2 Recommended Dosage for Adenocarcinoma of the Colon and Rectum
- Double click here to open Word Application.
- Write unordered list in to word document.
- Close Word Application.
2.3 Recommended Dosage for Adenocarcinoma of the Breast
- The recommended dose of fluorouracil, administered as a component of a cyclophosphamide-based multidrug regimen, is 500 mg/m2or 600 mg/m2intravenously on Days 1 and 8 every 28 days for 6 cycles.
2.4 Recommended Dosage for Gastric Adenocarcinoma
- The recommended dose of fluorouracil, administered as a component of a platinum-containing multidrug chemotherapy regimen, is 200 mg/m2to 1000 mg/m2intravenously as a continuous infusion over 24 hours. The frequency of dosing in each cycle and the length of each cycle will depend on the dose of fluorouracil and the specific regimen administered.
2.5 Recommended Dosage for Pancreatic Adenocarcinoma
- The recommended dose of fluorouracil, administered as an infusional regimen in combination with leucovorin or as a component of a multidrug chemotherapy regimen that includes leucovorin, is 400 mg/m2intravenous bolus on Day 1, followed by 2400 mg/m2intravenously as a continuous infusion over 46 hours every two weeks.
2.6 Dose Modifications
Withhold fluorouracil for any of the following:
- Development of angina, myocardial infarction/ischemia, arrhythmia, or heart failure in patients with no history of coronary artery disease or myocardial dysfunction [see Warnings and Precautions (5.2)]
- Hyperammonemic encephalopathy [see Warnings and Precautions (5.3)]
- Acute cerebellar syndrome, confusion, disorientation, ataxia, or visual disturbances [see Warnings and Precautions (5.4)]
- Grade 3 or 4 diarrhea [see Warnings and Precautions (5.5)]
- Grade 2 or 3 palmar-plantar erythrodysesthesia (hand-foot syndrome) [see Warnings and Precautions (5.6)]
- Grade 3 or 4 mucositis [see Warnings and Precautions (5.8)]
- Grade 4 myelosuppression [see Warnings and Precautions (5.7)]
Upon resolution or improvement to Grade 1 diarrhea, mucositis, myelosuppression, or palmar-plantar erythrodysesthesia, resume fluorouracil administration at a reduced dose.
There is no recommended dose for resumption of fluorouracil administration following development of any of the following adverse reactions:
- Cardiac toxicity
- Hyperammonemic encephalopathy
- Acute cerebellar syndrome, confusion, disorientation, ataxia, or visual disturbances
2.7 Preparation for Administration
Fluorouracil is supplied in a single dose vial. The 10 mL and 20 mL vial are only intended for preparation under appropriate conditions for cytotoxic drugs [see References (15)]. Store vial at room temperature
Withdraw the calculated dose for an individual patient into a sterile syringe. Inspect the solution in syringe for particulate matter and discoloration prior to administration or further dilution. Discard syringe if the solution is discolored or contains particulate matter. Discard unused portion.
2.8 Administration
Do not administer in the same intravenous line concomitantly with other medicinal products.
For bolus administration, store undiluted fluorouracil in the syringe for up to 4 hours at room temperature (25°C). Administer fluorouracil as an intravenous bolus through an established intravenous line.
Store diluted solutions of fluorouracil for up to 4 hours at room temperature (25°C) prior to administration to the patient. For intravenous infusion regimens, administer through a central venous line using an infusion pump.
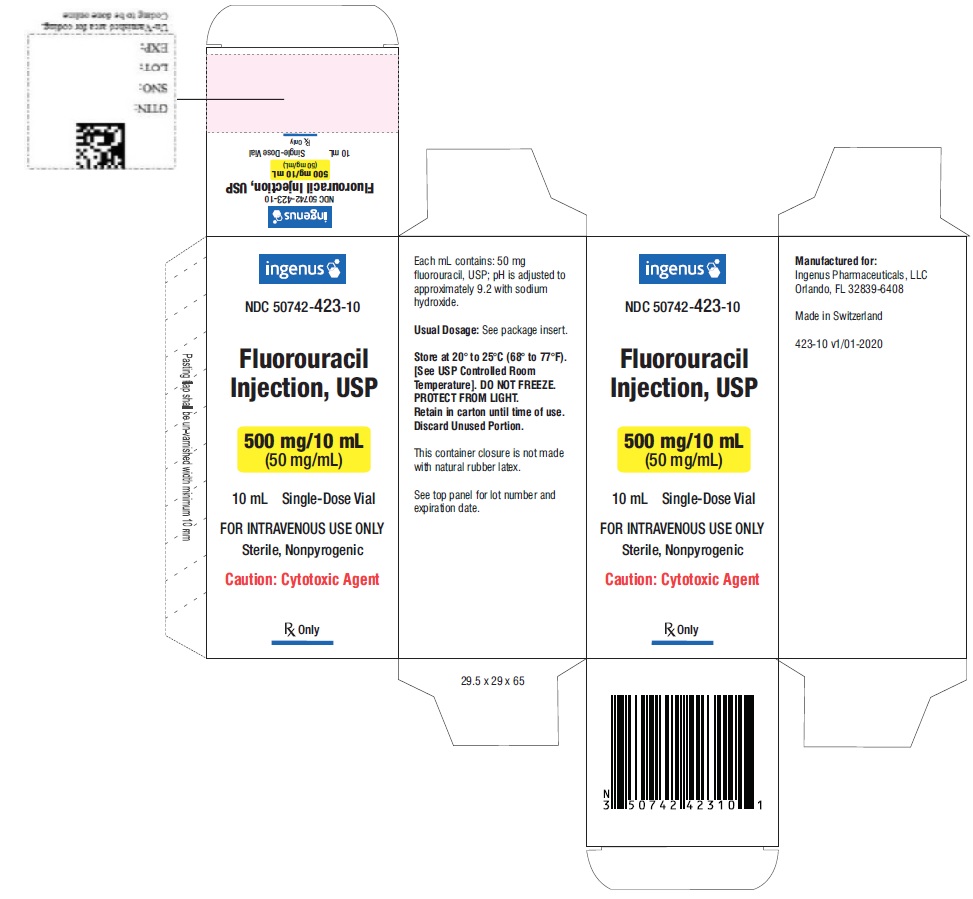
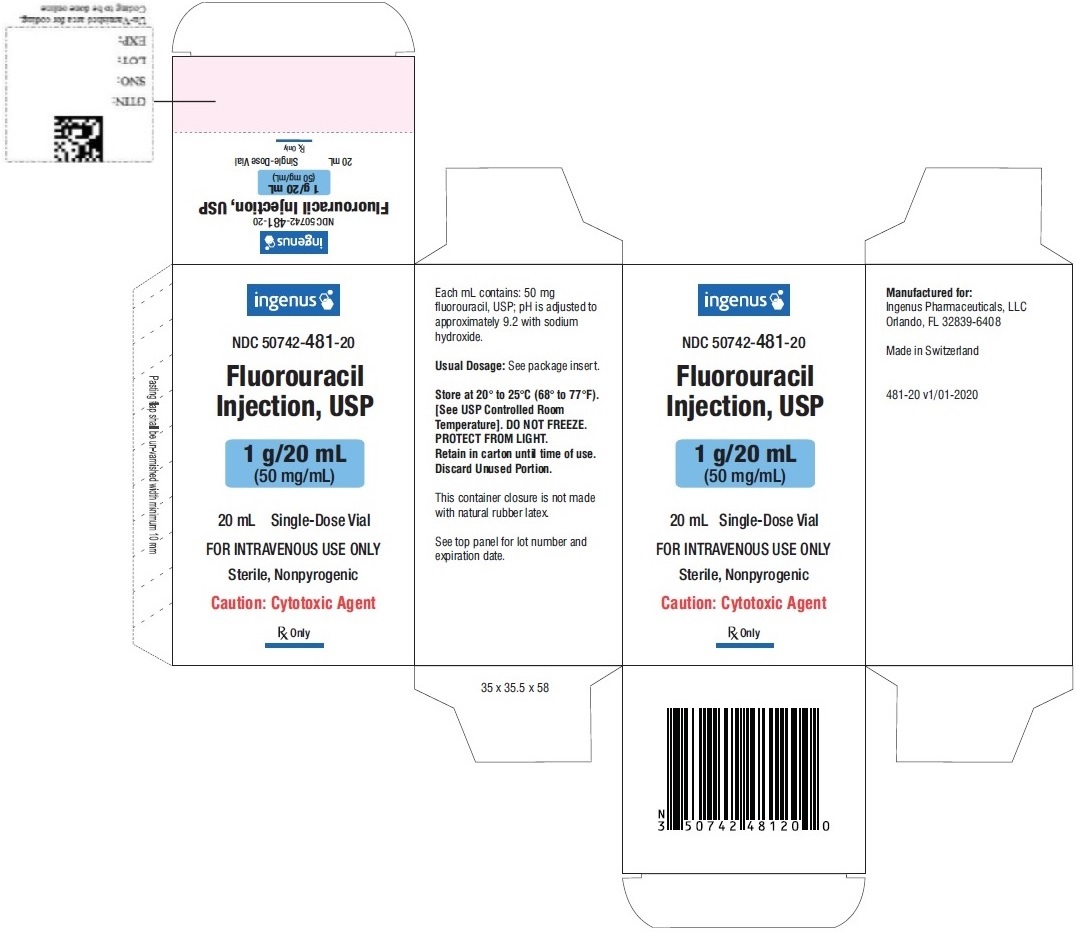
3 DOSAGE FORMS AND STRENGTHS
Fluorouracil injection is supplied as:
- Single dose vial containing 500 mg/10 mL (50 mg/mL) fluorouracil.
- Single dose vial containing 1 g/20 mL (50 mg/mL) fluorouracil.
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Serious or Fatal Adverse Reactions in Patients with Low or Absent Dipyrimidine Dehydrogenase (DPD) Activity
Based on postmarketing reports, patients with certain homozygous or certain compound heterozygous mutations in the DPD gene that result in complete or near complete absence of DPD activity are at increased risk for acute early-onset of toxicity and severe, life-threatening, or fatal adverse reactions caused by fluorouracil (e.g., mucositis, diarrhea, neutropenia, and neurotoxicity). Patients with partial DPD activity may also have increased risk of severe, life-threatening, or fatal adverse reactions caused by fluorouracil.
Withhold or permanently discontinue fluorouracil based on clinical assessment of the onset, duration and severity of the observed toxicities in patients with evidence of acute early-onset or unusually severe toxicity, which may indicate near complete or total absence of DPD activity. No fluorouracil dose has been proven safe for patients with complete absence of DPD activity. There is insufficient data to recommend a specific dose in patients with partial DPD activity as measured by any specific test.
5.2 Cardiotoxicity
Fluorouracil can cause cardiotoxicity, including angina, myocardial infarction/ischemia, arrhythmia, and heart failure, based on postmarketing reports. Reported risk factors for cardiotoxicity are administration by continuous infusion rather than intravenous bolus and presence of coronary artery disease. Withhold fluorouracil for cardiotoxicity. The risks of resumption of fluorouracil in patients with cardiotoxicity that has resolved have not been established.
5.3 Hyperammonemic Encephalopathy
Fluorouracil can cause hyperammonemic encephalopathy in the absence of liver disease or other identifiable cause, based on postmarketing reports. Signs or symptoms of hyperammonemic encephalopathy began within 72 hours after initiation of fluorouracil infusion; these included altered mental status, confusion, disorientation, coma, or ataxia, in the presence of concomitant elevated serum ammonia level. Withhold fluorouracil for hyperammonemic encephalopathy and initiate ammonia-lowering therapy. The risks of resumption of fluorouracil in patients with hyperammonemic encephalopathy that has resolved have not been established.
5.4 Neurologic Toxicity
Fluorouracil can cause neurologic toxicity, including acute cerebellar syndrome and other neurologic events, based on postmarketing reports. Neurologic symptoms included confusion, disorientation, ataxia, or visual disturbances. Withhold fluorouracil for neurologic toxicity. There are insufficient data on the risks of resumption of fluorouracil in patients with neurologic toxicity that has resolved.
5.5 Diarrhea
Fluorouracil can cause severe diarrhea. Withhold fluorouracil for Grade 3 or 4 diarrhea until resolved or decreased in intensity to Grade 1, then resume fluorouracil at a reduced dose. Administer fluids, electrolyte replacement, or antidiarrheal treatments as necessary.
5.6 Palmar-Plantar Erythrodysesthesia (Hand-Foot Syndrome)
Fluorouracil can cause palmar-plantar erythrodysesthesia, also known as hand-foot syndrome (HFS). Symptoms of HFS include a tingling sensation, pain, swelling, and erythema with tenderness, and desquamation. HFS occurs more commonly when fluorouracil is administered as a continuous infusion than when fluorouracil is administered as a bolus injection, and has been reported to occur more frequently in patients with previous exposure to chemotherapy. HFS is generally observed after 8-9 weeks of fluorouracil administration but may occur earlier. Institute supportive measures for symptomatic relief of HFS. Withhold fluorouracil administration for Grade 2 or 3 HFS; resume fluorouracil at a reduced dose when HFS is completely resolved or decreased in severity to Grade 1.
5.7 Myelosuppression
Fluorouracil can cause severe and fatal myelosuppression which may include neutropenia, thrombocytopenia, and anemia. The nadir in neutrophil counts commonly occurs between 9 and 14 days after fluorouracil administration. Obtain complete blood counts prior to each treatment cycle, weekly if administered on a weekly or similar schedule, and as needed. Withhold fluorouracil until Grade 4 myelosuppression resolves; resume fluorouracil at a reduced dose when myelosuppression has resolved or improved to Grade 1 in severity.
5.8 Mucositis
Mucositis, stomatitis or esophagopharyngitis, which may lead to mucosal sloughing or ulceration, can occur with fluorouracil. The incidence is reported to be higher with administration of fluorouracil by intravenous bolus compared with administration by continuous infusion. Withhold fluorouracil administration for Grade 3 or 4 mucositis; resume fluorouracil at a reduced dose once mucositis has resolved or decreased in severity to Grade 1.
5.9 Increased Risk of Elevated International Normalized Ratio (INR) with Warfarin
Clinically significant elevations in coagulation parameters have been reported during concomitant use of warfarin and fluorouracil. Closely monitor patients receiving concomitant coumarin-derivative anticoagulants such as warfarin for INR or prothrombin time in order to adjust the anticoagulant dose accordingly [see Drug Interactions (7)].
5.10 Embryofetal Toxicity
Based on its mechanism of action, fluorouracil can cause fetal harm when administered to a pregnant woman. In animal studies, administration of fluorouracil at doses lower than a human dose of 12 mg/kg caused teratogenicity. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during and for 3 months following cessation of therapy with fluorouracil [see Use in Specific Populations (8.1, 8.6), Clinical Pharmacology (12.1), and Nonclinical Toxicology (13.1)]
6 ADVERSE REACTIONS
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Increased risk of serious or fatal adverse reactions in patients with low or absent dipyrimidine dehydrogenase activity [see Warnings and Precautions (5.1)]
- Cardiotoxicity [see Warnings and Precautions (5.2)]
- Hyperammonemic encephalopathy [see Warnings and Precautions (5.3)]
- Neurologic toxicity [see Warnings and Precautions (5.4)]
- Diarrhea [see Warnings and Precautions (5.5)]
- Palmar-plantar erythrodysesthesia (hand-foot syndrome) [see Warnings and Precautions (5.6)]
- Myelosuppression [see Warnings and Precautions (5.7)]
- Mucositis [see Warnings and Precautions (5.8)]
- Increased risk of elevated INR when administrated with warfarin [see Warnings and Precautions (5.9)]
6.1 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of fluorouracil. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hematologic: pancytopenia [see Warnings and Precautions (5.7)]
Gastrointestinal: gastrointestinal ulceration, nausea, vomiting
Allergic Reactions: anaphylaxis and generalized allergic reactions
Neurologic: nystagmus, headache
Dermatologic: dry skin; fissuring; photosensitivity, as manifested by erythema or increased pigmentation of the skin; vein pigmentation
Ophthalmic: lacrimal duct stenosis, visual changes, lacrimation, photophobia
Psychiatric: euphoria
Miscellaneous: thrombophlebitis, epistaxis, nail changes (including loss of nails)
7 DRUG INTERACTIONS
7.1 Anticoagulants and CYP 2C9 Substrates
Elevated coagulation times have been reported in patients taking fluorouracil concomitantly with warfarin. While pharmacokinetic data are not available to assess the effect of fluorouracil administration on warfarin pharmacokinetics, the elevation of coagulation times that occurs with the fluorouracil prodrug capecitabine is accompanied by an increase in warfarin concentrations. Thus, the interaction may be due to inhibition of cytochrome P450 2C9 by fluorouracil or its metabolites.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies with fluorouracil in pregnant women. Based on its mechanism of action, fluorouracil can cause fetal harm when administered to a pregnant woman. Administration of fluorouracil to rats and mice during selected periods of organogenesis, at doses lower than a human dose of 12 mg/kg, caused embryolethality and teratogenicity.
Malformations included cleft palate and skeletal defects. In monkeys, maternal doses of fluorouracil higher than an approximate human dose of 12 mg/kg resulted in abortion. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to a fetus [see Clinical Pharmacology (12.1)].
Animal Data
Malformations including cleft palate, skeletal defects and deformed appendages (paws and tails) were observed when fluorouracil was administered by intraperitoneal injection to mice at doses at or above 10 mg/kg (approximately 0.06 times a human dose of 12 mg/kg on a mg/m2 basis) for 4 days during the period of organogenesis. Similar results were observed in hamsters administered fluorouracil intramuscularly at doses lower than those administered in commonly used clinical treatment regimens. In rats, administration of fluorouracil by intraperitoneal injection at doses greater than 15 mg/kg (approximately 0.2 times a human dose of 12 mg/kg on a mg/m2 basis) for a single day during organogenesis resulted in delays in growth and malformations including micro anophthalmos. In monkeys, administration of fluorouracil during organogenesis at doses approximately equal to a human dose of 12 mg/kg on a mg/m2basis resulted in abortion; at a 50% lower dose, resorptions and decreased fetal body weights were reported.
8.3 Nursing Mothers
It is not known whether fluorouracil or its metabolites are present in human milk. Because many drugs are present in human milk and because of the potential for serious adverse reactions in nursing infants from fluorouracil, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.5 Geriatric Use
Reported clinical experience has not identified differences in safety or effectiveness between the elderly and younger patients.
8.6 Females and Males of Reproductive Potential
Females
Based on its mechanism of action, fluorouracil can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during treatment with fluorouracil and for up to 3 months following cessation of therapy [see Use in Specific Populations (8.1)].
Males
Fluorouracil may damage spermatozoa. Advise males with female partners of reproductive potential to use effective contraception during and for 3 months following cessation of therapy with fluorouracil [see Nonclinical Toxicology (13.1)].
Infertility
Females
Advise females of reproductive potential that, based on animal data, fertility may be impaired while receiving fluorouracil [see Nonclinical Toxicology (13.1)].
Males
Advise males of reproductive potential that, based on animal data, fertility may be impaired while receiving fluorouracil [see Nonclinical Toxicology (13.1)].
10 OVERDOSAGE
Administer uridine triacetate within 96 hours following the end of fluorouracil infusion for management of fluorouracil overdose.
11 DESCRIPTION
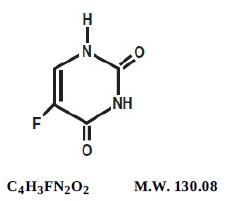
Fluorouracil injection, USP a nucleoside metabolic inhibitor, is a colorless to yellow, aqueous, sterile, nonpyrogenic injectable solution available in 10 mL and 20 mL, a sterile preparation that contains single dose for intravenous administration. Each mL contains 50 mg fluorouracil, USP in water for injection, USP. The pH is adjusted to approximately 9.2 with sodium hydroxide. Chemically, fluorouracil, a fluorinated pyrimidine, is 5-fluoro-2,4 (1H,3H)-pyrimidinedione. Its structural formula is:
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Fluorouracil is a nucleoside metabolic inhibitor that interferes with the synthesis of deoxyribonucleic acid (DNA) and to a lesser extent inhibits the formation of ribonucleic acid (RNA); these affect rapidly growing cells and may lead to cell death. Fluorouracil is converted to three main active metabolites: 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP), 5-fluorouridine-5′ triphosphate (FUTP) and 5-fluoro-2′-deoxyuridine-5′-triphosphate (FdUTP). These metabolites have several effects including the inhibition of thymidylate synthase by FdUMP, incorporation of FUTP into RNA and incorporation of FdUTP into DNA.
12.3 Pharmacokinetics
Following bolus intravenous injection, fluorouracil distributes throughout the body including the intestinal mucosa, bone marrow,liver, cerebrospinal fluid and brain tissue.
Elimination
Following bolus intravenous injection, 5 – 20 % of the parent drug is excreted unchanged in the urine in six hours. The remaining percentage of the administered dose is metabolized, primarily in the liver. The metabolites of fluorouracil (e.g., urea and α-fluoro-ß alanine) are excreted in the urine over 3 to 4 hours.
Following bolus intravenous injection of fluorouracil, as a single agent, the elimination half-life increased with dose from 8 to 20 minutes.
13 NONCLINICAL TOXICOLOGY
13.3 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with fluorouracil. Fluorouracil was mutagenic in vitro in the bacterial reverse mutation (Ames) assay and induced chromosomal aberrations in hamster fibroblasts in vitro and in mouse bone marrow in the in vivo mouse micronucleus assay.
Administration of fluorouracil intraperitoneally to male rats at dose levels equal to or greater than 1.7-fold the human dose of 12 mg/kg induced chromosomal aberrations in spermatogonia and inhibition of spermatogonia differentiation resulting in transient infertility. In female rats, intraperitoneal administration of fluorouracil during the pre-ovulatory phases of oogenesis at dose levels equal to or greater than 0.33 times a human dose of 12 mg/kg resulted in decreased incidence of fertile matings, increased pre- implantation loss, and fetotoxicity.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Supplied
Fluorouracil injection, USP is supplied in single dose vials, available in a box containing one vial, as listed below:
- NDC 50742-423-10: one box with one vial, containing 500 mg/10 mL (50 mg/mL) fluorouracil.
- NDC 50742-481-20: one box with one vial, containing 1 g/20 mL (50 mg/mL) fluorouracil.
16.2 Storage
Store at 20°C to 25°C (68°F to 77°F). [see USP Controlled Room Temperature]. DO NOT FREEZE. Protect from light. Retain in carton until time of use.
Fluorouracil is a cytotoxic drug. Follow applicable special handling and disposable procedures [see References (15)].
17 PATIENT COUNSELING INFORMATION
- Patients to notify their healthcare provider if they have a known DPD deficiency. Advise patients if they have complete or near complete absence of DPD activity, they are at an increased risk of severe and life-threatening mucositis, diarrhea, neutropenia and neurotoxicity [see Warnings and Precautions (5.1)] .
- Patients of the risk of cardiotoxicity. Advise patients to immediately contact their healthcare provider or to go to an emergency room for new onset of chest pain, shortness of breath, dizziness, or lightheadedness [see Warnings and Precautions (5.2)] .
- Patients to immediately contact their healthcare provider or go to an emergency room for new onset of confusion, disorientation, or otherwise altered mental status; difficulty with balance or coordination; or visual disturbances [see Warnings and Precautions (5.3, 5.4)] .
- Patients to contact their healthcare provider for severe diarrhea or for painful mouth sores with decreased oral intake of food or fluids [see Warnings and Precautions (5.5, 5.8)] .
- Patients to contact their healthcare provider for tingling or burning, redness, flaking, swelling, blisters, or sores on the palms of their hands or soles of their feet [see Warnings and Precautions (5.6)] .
- Patients of the importance of keeping appointments for blood tests. Instruct patients to monitor their temperature on a daily basis and to immediately contact their healthcare provider for fever or other signs of infection [see Warnings and Precautions (5.7)] .
- Patients to notify their healthcare provider of all drugs they are taking, including warfarin or other coumarin-derivative anticoagulants. Advise patients of the importance of keeping appointments for blood tests [see Warnings and Precautions (5.9)].
- Females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with fluorouracil and for up to 3 months after the last dose of fluorouracil. Instruct female patients to contact their healthcare provider if they become pregnant, if pregnancy occurs during fluorouracil treatment or during the 3 months following the last dose [see Warnings and Precautions (5.10), Use in Specific Populations (8.1 and 8.6), and Nonclinical Toxicology (13.1)] .
- Females and males of reproductive potential may have impaired fertility while receiving fluorouracil, based on animal data
- [see Use in Specific Populations (8.6) and Nonclinical Toxicology (13.1)] .
- Nursing mothers to discontinue nursing [see Use in Specific Populations (8.3)] .
Ingenus Pharmaceuticals, LLC
Orlando, FL 32839-6408
Made in Switzerland
Revised: 04/2019



| FLUOROURACIL
fluorouracil injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FLUOROURACIL
fluorouracil injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Ingenus Pharmaceuticals, LLC (833250017) |
| Registrant - Novast Laboratories , Ltd. (527695995) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ingenus Pharmaceuticals GmbH | 482730327 | ANALYSIS(50742-423, 50742-481) , LABEL(50742-423, 50742-481) , MANUFACTURE(50742-423, 50742-481) , PACK(50742-423, 50742-481) , STERILIZE(50742-423, 50742-481) | |